

Abstract
Despite significant progress in cancer research, current standard-of-care drugs fail to cure many types of cancers. Hence, there is an urgent need to identify better predictive biomarkers and treatment regimes. Conventionally, insights from hypothesis-driven studies are the primary force for cancer biology and therapeutic discoveries. Recently, the rapid growth of big data resources, catalyzed by breakthroughs in high-throughput technologies, has resulted in a paradigm shift in cancer therapeutic research. The combination of computational methods and genomics data has led to several successful clinical applications. In this review, we focus on recent advances in data-driven methods to model anticancer drug efficacy, and we present the challenges and opportunities for data science in cancer therapeutic research.
Keywords: big data, precision medicine, immunotherapy, drug resistance, response biomarker, combination therapy, toxicity
INTRODUCTION
Although oncologists started testing chemotherapy in patients during the 1940s, cancer remains one of the deadliest diseases in developed countries after eighty years. Research directed at understanding tumorigenesis and developing effective therapies has yielded significant successes. For example, the introduction of all-trans retinoic acid (ATRA) to treat acute promyelocytic leukemia driven by RARα translocation led to a cure in most patients suffering from this previously deadly disease (1). The use of imatinib in treating chronic myelogenous leukemia driven by the BCR-ABL fusion resulted in an ~80% decline in disease mortality (2). However, the success of ATRA, imatinib, and others including EGFR and ALKinhibitors (3, 4) in substantially improving long-term survival has been the exception rather than the norm. Currently, most tumors, once metastatic, remain incurable (5). Many targeted cancer drugs such as inhibitors of AKT and IGF1R, which show significant effects in preclinical models, fail to bring sufficient clinical benefits (6, 7). Many anticancer drugs also have debilitating side effects, and lowering the dose to control side effects can significantly limit therapy effectiveness (8, 9). Even with the significant progress of immunotherapies in recent years, only a minority of patients with specific cancer types benefit from the immune checkpoint blockade (ICB), and many patients relapse during ICB therapy (10). Therefore, there remains a significant unmet need for the scientific community to develop better anticancer treatments and predict patient responses.
Conventionally, hypothesis-driven studies are the driving force of cancer therapeutic discoveries. Recently, the rapid growth of big data resources has resulted in a paradigm shift (11). The application of omics technologies, from high-throughput sequencing to automated screening (Figure 1a), has generated large-scale data sets that capture different aspects of anticancer drug efficacy (Figure 1b). Computational methods are essential for the analysis of these big data resources (Figure 1c) to generate clinically useful results in predicting therapeutic response and side effects and in designing combination therapies (Figure 1d). Precision cancer medicine aims to understand the tumor microenvironment, host immunity, and the ecosystem at the molecular level to find treatments that best fit more patient subgroups. With fast-growing data and analytical resources, the scientific community is moving toward this goal of precision medicine. In this review, we focus on recent advances in data-driven approaches in modeling anticancer drug response and resistance. We also introduce readers to other reviews that cover the basic science and clinical aspects of anticancer drug response (5, 10, 12–17). The current review, as well as the literature cited within, provide an overview of the challenges and opportunities of data science in precision cancer medicine.
Figure 1.
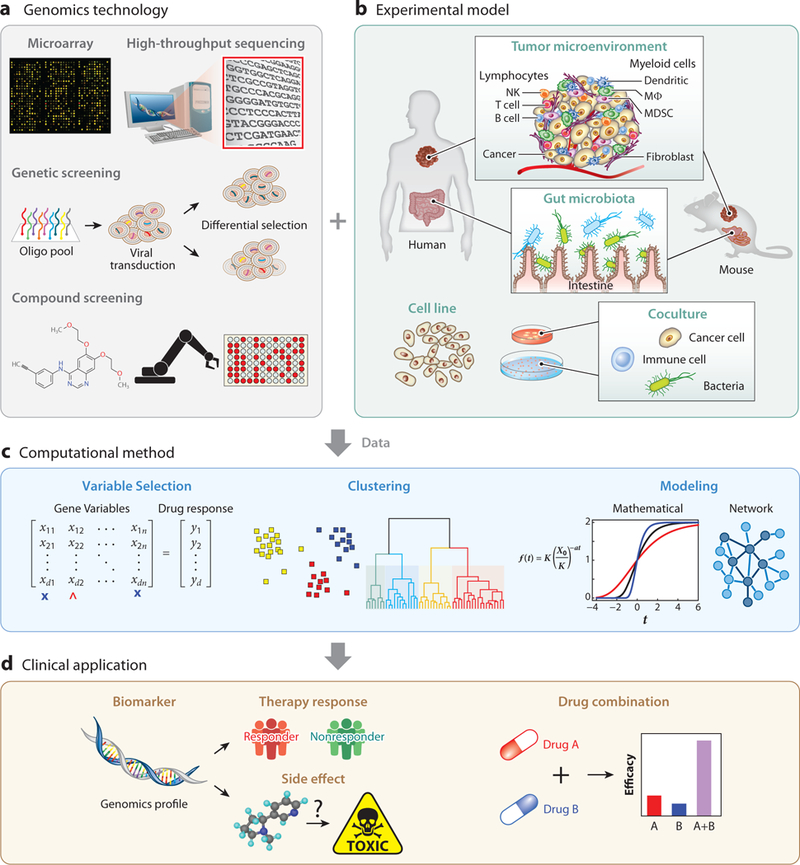
Data-driven approaches for modeling cancer therapy efficacy. Most data-driven studies of anticancer drug efficacy involve four components: genomics technology, experimental model, computational method, and clinical application. The use of genomics technology in experimental models generates data that can be analyzed by computational methods to generate results for clinical applications. (a) Microarray and high-throughput sequencing are widely used to study the DNA alterations and RNA transcriptomes in cancer samples. Genetics screens through RNAi or CRISPR technologies can study the effect of perturbing a gene in a cell line model (174). Compound screens based on automation frameworks can test the efficacy of many drugs on a cell line panel (29, 35, 36). (b) The most clinically relevant system is human, where both tumor microenvironment (10, 12) and gut microbiota (17) can determine anticancer drug efficacy. However, genetic experiments cannot be directly applied to humans, so mouse models are used as alternatives to study in vivo factors of drug response (43, 175, 176). Cancer cell lines are the most widely used research models. Cell lines can be cultured alone or cocultured either between cancer and immune cells (46–48) or between immune and bacteria cells (64, 69). (c) Most data analyses involve variable selection. Molecular alterations of genes across samples are input variables, and drug efficacy is the outcome (84). Variable selection methods can identify critical genes associated with anticancer drug efficacy. Clustering algorithms can be applied to identify patterns in a data set (115). Mathematical (97, 100) or network models (107) can be applied to explore the properties and mechanisms of a molecular circuit that mediate anticancer drug efficacy. (d) Many studies are designed to find biomarkers for therapy response prediction (177) or side effects (134–136) in clinical applications using the molecular profiles of patient samples. Data-driven models can also be applied to identify synergistic drug combinations to treat specific cancers (84). Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; NK, natural killer; MDSC, myeloid-derived suppressor cell; MΦ, macrophage; oligo, oligonucleotide; RNAi, RNA interference.
OMICS TECHNOLOGIES FOR MAKING DATA-DRIVEN DISCOVERIES
Omics technologies, especially fast and affordable next-generation sequencing, have resulted in a flood of big data in cancer research. The advances in genomics resources, catalyzed by the rapid technology development, have enabled discoveries on both actionable clinical solutions and therapy resistance mechanisms. In this section, we review several key genomics technologies and the data generated.
Genome-Scale Profiling of Clinical Samples
Several genomics approaches are enabling the molecular characterization of human cancer samples (Figure 1a). At the DNA level, whole-exome sequencing of over ~20,000 human genes can provide a systematic view of genetic alterations in protein-coding regions (18, 19). Alternatively, whole-genome sequencing can identify driver mutations in noncoding regions (e.g., noncoding RNAs, promoters, enhancers), and structural variations at high resolution (20, 21). In contrast, clinical assays usually target mutations on a panel of a few hundred genes recurrently mutated in cancer (22). At the RNA level, gene expression profiles can be measured with technologies such as microarrays, RNA sequencing (RNA-seq), or NanoString with varying degrees of gene focus. Together with methods to profile micro RNA expression, protein abundance, DNA methylation, and chromatin accessibility, these technologies have been applied to patient samples to generate large-scale cancer genomic and epigenomic data. For example, the Cancer Genome Atlas (TCGA) project generated 2.5 petabytes of genome-scale profiling data for cancer and matched normal tissues from more than 11,000 patients across 3 3 cancer types (23). Another example is the GENIE (genomics evidence neoplasia information exchange) project that released mutation profiles for more than 500 genes and a minimal set of clinical information for almost 30,000 cancer patients until the end of 2017 (24). Such data from many patients with diverse cancer types can inform the treatment decisions of other patients with similar mutations.
To model the mechanisms of intrinsic and acquired resistance to anticancer drugs (5), we need distinct experimental designs. For intrinsic resistance mechanisms, the gene expression or mutation profiles from pretreatment tumors of responders and nonresponders can be compared, while for acquired resistance mechanisms, recurrent genomic and transcriptomic alterations could be identified by comparing post- and pretreatment tumors. For example, a comprehensive study generated whole-exome sequences for 67 triplets of pretreatment tumors, post-treatment tumors, and normal tissues from melanoma patients treated with MAPK inhibitors (25). This study also generated the expression profiles of 48 pairs of pre- and post-treatment (drug-resistant) tumors. For a subset of these tumors, progression-free survival (PFS) data of the patients were available. The pretreatment tumor profiles and PFS information could be used to implicate molecular alterations associated with intrinsic resistance to MAPK inhibitors, while post-treatment profiles could reveal somatic mutation and gene expression drivers of acquired resistance to MAPK inhibitors. This study identified several transcriptomic alterations, such as MET, YAP1, and LEF1 dysregulation, as indicators of acquired resistance to MAPK inhibitors. Meanwhile, the drug-resistant tumors recurrently lose CD8 T cell numbers and cancer cell antigen presentation (25).
High-Throughput Screening on Preclinical Models
The study of anticancer therapy efficacy would ideally include both tumor molecular profiles and drug response information across a large cohort of patients. However, the expense and effort of collecting such data have limited the number of examples where this has been done. A research alternative to clinical profiling is to use preclinical models, such as immortalized cell lines and mouse models (Figure 1b). For example, drug-resistant cell lines derived from long-term treatment of drug-sensitive parental cell lines are frequently used to study drug resistance mechanisms (Supplemental Figure 1). Additionally, automation can enable the conduct of compound Supplemental Material screens across many cancer cell lines to discover new anticancer drugs and resistance mechanisms (Figure 2). An early example of the compound screen was developed in the late 1980s for the National Cancer Institute 60 human cancer cell lines project (NCI60) as an in vitro alternative to the use of animal tumors (26). The NCI60 screen supported the development of several anticancer drugs, such as paclitaxel and bortezomib, which were approved by the US Food and Drug Administration (FDA) for cancer treatment (26, 27). Data mining on the NCI60 screen result also led to many findings. For example, association analysis between gene mutation status and drug efficacy on the NCI60 panel discovered that the BRAF mutation is a predictor of MEK inhibitor sensitivity (28). Since then, cell line screening has rapidly become a popular platform for cancer research.
Figure 2.
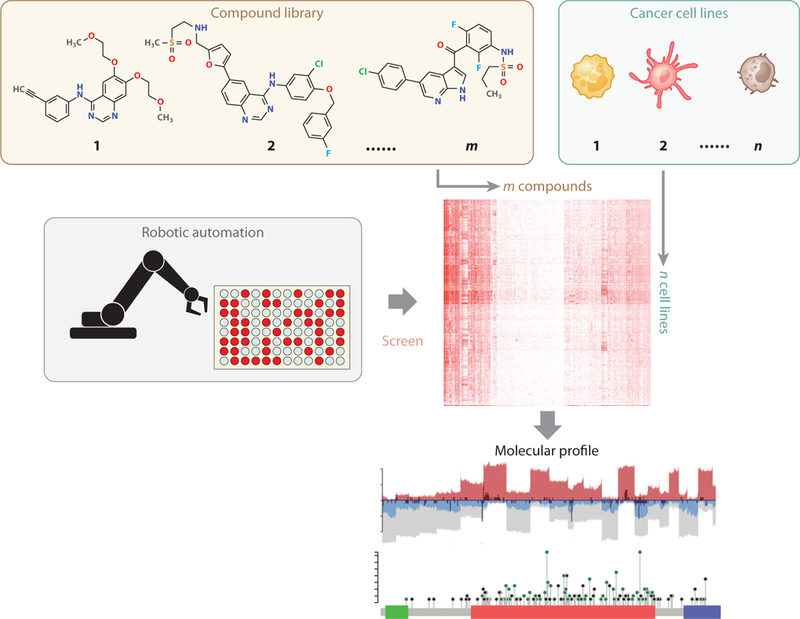
Compound screening in cancer cell lines. Automation frameworks can be utilized to test the growth inhibition effects of a library of compounds across many cancer cell lines with diverse genetic backgrounds. Most compound screen projects also profiled the molecular features (e.g., gene expression, copy number, mutation status) of cell lines. The final data output is the growth inhibition effects of compounds on cell lines, together with cell line molecular profiles.
Recently, data on three large-scale compound-screen projects became publicly available. The Cancer Cell Line Encyclopedia (CCLE) (29) project is a collaboration between the Broad Institute and Novartis Institutes for Biomedical Research (NIBR) (30). The investigators collected ~1,000 human cancer cell lines and completed the acquisition of comprehensive molecular profiling data, including gene expression, copy number alteration, and somatic mutation. The CCLE released the growth inhibition profiles of 24 anticancer drugs across 504 cell lines (29). The Broad and NIBR also independently released gene essentiality measurements, defined as the impact of gene loss on cell growth, on CCLE cell lines through CRISPR and short hairpin RNA (shRNA) screens (31–34). The CCLE screens reported that AHR expression determines MEK inhibitor efficacy in NRAS-mutant lines, and SLFN11 expression predicts sensitivity to topoisomerase inhibitors. Similar to the CCLE project, the Cancer Therapeutics Response Portal (CTRP) screened more than 500 compounds and their combinations on the CCLE cell lines (35). The Genomics of Drug Sensitivity in Cancer (GDSC) project profiled the sensitivity of about 1,000 COSMIC cell lines to 250 compounds and identified many genetic alterations associated with drug efficacy (36).
Even with automation, large-scale cellular compound screens are labor intensive and expensive. To overcome this challenge, researchers developed a technique called PRISM to perform pooled screens on barcoded cell line mixtures (37). The Luminex microspheres could detect the different growth rates of cell lines under the treatment of either a test compound or a DMSO control by quantifying their barcode fractions in pools (38–40). The difference in the barcode fraction between the treatment and control conditions reflects the inhibition effect of a compound on a given cell line. PRISM was used to screen a large set of compounds in ~100 cancer cell lines and was extended to screening erlotinib sensitivity in 23 lung cancer cell lines in mouse xenografts (37). A potential limitation is that the interactions among different cell lines in a pool may confound the drug sensitivity measurements, so the utility of PRISM awaits further evaluation.
A limitation of compound screens is that cultured cell line models cannot reflect the tumor microenvironment (41). Many anticancer drugs not only exert cytotoxic effects but also induce immunological responses (12). Moreover, the effects of antibody drugs, such as trastuzumab, depend on the antibody-dependent cell-mediated cytotoxicity effects from natural killer cells (42). Therefore, numerous murine models were developed to better approximate human tumors. One model is the xenotransplantation of human tumors (xenografts) in immunocompromised mice, in which human tumor cells are transplanted either ectopically under the skin or orthotopically into the organs where the tumor originated. One study established ~1,000 patient-derived tumor xenograft (PDX) models with a diverse set of driver mutations (43). With these PDX models, the authors performed in vivo screens to assess the responses to 62 compounds with about 2,000 drug response measurements. Many conclusions from this study regarding the factors influencing targeted therapy efficacy are highly consistent with the results from human clinical trials. This PDX screen also provided further validation for both gene mutation and expression biomarkers generated from cell line studies. Moreover, the PDX platform demonstrated the ability to evaluate the clinical efficacy of combination therapies, which may not be faithfully reflected in in vitro assays.
A limitation of the PDX model is that immunocompromised mice cannot simulate the immune response, a critical factor of anticancer drug response (12, 44). Also, there are numerous instances where murine and human ligands and receptors do not cross-signal (45). Genetically engineered and syngeneic recipient mice could help in both of these areas. They would preserve a competent immune system, although compound screens on murine models cannot scale up due to the lack of automation frameworks. Also, it is not yet clear how to create murine models in which the complexity of the genetics of human cancers is modeled more robustly.
A further assessment of immune-active agents can be conducted using the cytotoxic T cell killing assay that could both simulate the effect of immune systems and scale up with robotics. In this assay, cancer cells with a specific antigen are cocultured with the cytotoxic T cells with the corresponding T cell receptor (46–48). Automated image analysis measures cancer cell death in the presence of both lymphocytes and a candidate compound. A recent study screened 850 compounds for synergistic drugs of T cell-mediated killing using two patient-derived melanoma cell lines and their autologous T cells (47). In principle, the automation frameworks for the cellular compound screens (e.g., CCLE, CTRP, GDSC) could adapt to the format of coculture assays to study the effects of compounds on cancer cells in the presence of a simplified immune system.
Moving from the Bulk Tumor to Single Cells
Most previous studies profiled the cancer genome from bulk tumor tissues, which will give the mixture profiles of cancer, stromal, and immune cells in the tumors. However, the acquisition of drug resistance during the treatment may depend on variations in rare populations (49). Moreover, cell composition, location, and interactions within each tumor play critical roles in determining therapy response (50, 51). For example, patient survival in colorectal cancer depends on the location and density of T cells in the cores and margins of tumors (52). In murine models of breast cancer, the relative spatial distribution in the tumors of macrophage and cancer cells of different phenotypes can explain the tumor immune evasion and immunotherapy resistance (53). Therefore, the conventional technologies of bulk tumor profiling may not be adequate to resolve the heterogeneity and complexity of cancer therapy response.
The past fewyears have seen the rapid development of single-cell technologies for investigating the cellular heterogeneity in DNA (54), RNA (55), proteins (56), and metabolites (57). For example, a recent study generated the single-cell gene expression profiles in temporal specimens of ovarian cancer patients with acquired platinum resistance (58). This study observed an accumulation of genetically identical cells with distinct transcriptome states, indicating epigenetic mechanisms of treatment resistance. In another study with almost 5,000 single-cell RNA-seq profiles from 19 melanoma patients, investigators found that all tumors harbored cancer cells in a drug-resistant state, indicating the eventual tumor progression to resistance during treatment (59). Recently, a large-scale study profiled the transcriptomes of ~6,000 single cells from 18 head and neck cancer patients and identified a subset of cancer cells enriched with a partial signature of epithelial-to-mesenchymal transition (p-EMT) (60). These cells localized to the leading edge of tumors, where the interactions between cancer-associated fibroblast and malignant cells may promote the p-EMT program and induce tumor invasion. All the studies mentioned above demonstrated the new insights that single-cell technology can bring compared to the conventional bulk tumor profiling.
Both the number and quality of single-cell data sets have significantly increased recently. However, the gene dropout rate, the heterogeneity of populations, and the lack of spatial-temporal context present significant challenges to single-cell genomics (61). Integrative analysis of single-cell and bulk tumor data and cell signatures from previous studies may ameliorate some limitations of single-cell data (59, 62). Meanwhile, single-cell imaging technologies may provide spatial information for different cells and genes in a tumor (63). We foresee that the development of single-cell technologies will rapidly generate rich data sets for understanding the complexity and heterogeneity of cancer drug response and will provide many new opportunities for computational method development.
Extending from Tumor Microenvironment to Host Microbiota
Recently, it has been reported that human microbiota, especially the gut microbiota, modulates the response and side effects from chemotherapies to immunotherapies (17). The human microbiota is the ensemble of bacteria and other microorganisms that inhabit the epithelial barrier surfaces. Several studies in mice demonstrated that the murine gut microbiota regulates the response to different chemotherapies (64–66). For example, the efficacy of cyclophosphamide (CTX) relies on intestinal bacteria (64, 65). CTX treatment can damage the gut mucus layer, allowing bacteria to penetrate the lamina barrier and translocate to secondary lymphoid organs. Translocated bacteria, such as Enterococcus hirae or Barnesiella intestinihominis, may activate the innate and adaptive immune cells and initiate antitumor immunity. Recently, several studies have shown that the gut microbiome may significantly influence response to ICB targeting the CTLA4 and PD1/PDL1 proteins (67–71). Moreover, favorable gut microbiota from ICB-responding patients can enhance the antitumor immunity when transplanted into the gut of mice, highlighting the potential value of fecal transplantation (69, 70).
Gut microbiota is a key modulator not only of therapy response but also of drug toxicity. For example, one of the side effects of irinotecan is the intestinal toxicity (severe diarrhea, weight loss, and anorexia) resulting from gut microbiota metabolism. Irinotecan is transformed into its active form, SN-38, in the liver and small intestine and then detoxified in the liver into inactive SN-38-G before being secreted into the gut. Gut bacterial |3-glucuronidases can reconvert SN-38-G into active SN-38, which induces significant intestinal toxicity and diarrhea (72).
Two standard approaches for microbiome profiling include high-throughput sequencing of either the whole-metagenome or the genomic DNA sequences of 16S ribosomal RNA genes in the microbe population (73). Whole-metagenome shotgun sequencing provides species-level resolution of bacteria, and with adequate sequencing depth, can quantify the near-complete genomic content of the collection of microbes in a sample. 16S ribosomal RNA sequencing is a cheaper alternative for studying phylogeny and taxonomy of microbes in a sample. In addition to direct microbiome sequencing, computational methods can also infer microbiome compositions by identifying nonhuman nucleic acids from sequencing data of human samples (74–76).
Characterizing specific bacteria strains and understanding their functions might offer insights leading to the discovery of novel therapeutics. For example, metagenomic profiling of bacteria strains in the fecal samples of patients with favorable therapy outcomes identified bacteria strains that enhanced the efficacy of ICB in mice (69, 70). Another example is that identification of microbial sequences in colon cancer RNA-seq data found Fusobacterium to be associated with distant metastases of tumors (77). Moreover, antibiotic treatment in PDX models can decrease both Fusobacterium load and cancer cell proliferation. Therefore, profiling and targeting the microbiota in response to cancer therapies may become one of the next frontiers of cancer precision medicine.
COMPUTATIONAL METHODS FOR MODELING DRUG EFFICACY
The selection of computational methods is a critical step in transforming genomic data into biological insight and clinical application (Figure 1c). The choice of data analysis methods depends on many factors, such as the quality, complexity, and sample size of a data set. Most studies of anticancer drug efficacy involve the variable selection among many gene expression or mutation features, with drug response as the outcome on a limited number of samples. Also, mathematical and network models can help understand the quantitative properties and potential biological mechanisms of drug response and resistance. In this section, we introduce some commonly used computational methods and models for cancer therapeutic discoveries.
Modeling Associations in Cancer Genomics Data
In many studies, a common practice is to primarily model the linear relationships between gene features and drug efficacy outcomes (Figure 3a, left panel). For example, most cancer biomarker studies explored whether the somatic mutation status or expression level of a gene set can predict therapy outcomes (78). Such problems can be solved with variable selection methods under a unified framework of linear models (Figure 3b). If drug response is a vector of continuous values across samples, least squares regression can be applied to identify gene mutation or expression features associated with drug efficacy (79). If drug response is a vector of binary responder status, logistic regression can be applied (79). If drug response is a vector of patient survival with censorship to remove patients after a follow-up time, Cox proportional hazard (Cox-PH) regression can be applied to identify essential features (80). Each regression method has assumptions and requirements on input data; therefore, data preprocessing and a sanity check of results are necessary to ensure analysis reliability (81).
Figure 3.
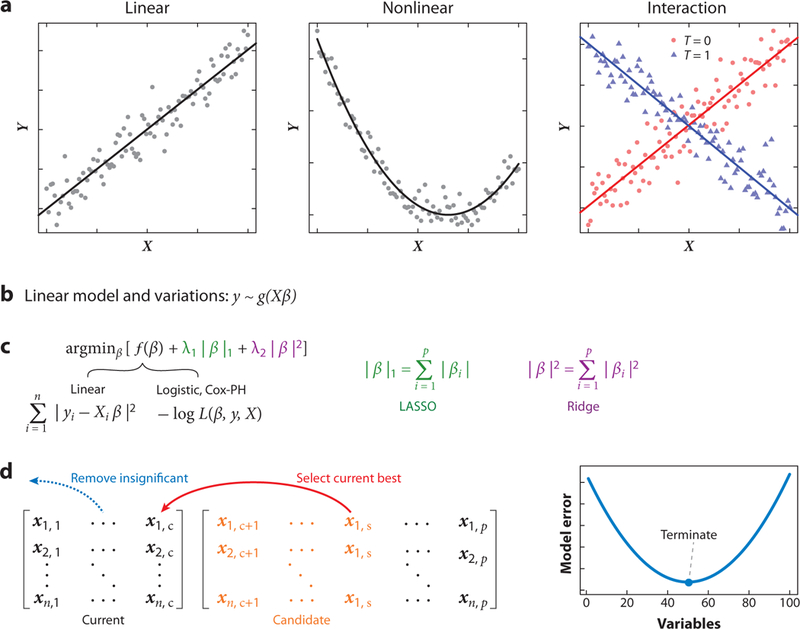
Variable selection in high-dimensional data. (a) Three common relationships between variable matrices (X) and outcomes (Y). (b) The unified framework of linear models y ~ g(Xβ) for n samples and p variables (for p > n), variable matrix X = n × p, and coefficient vector β = p. The number of samples n may range from 10 to 1,000 in most studies, representing the number of profiled patients. The number of variables p is about 20,000 in most studies, representing the number of human genes. (c) High-dimensional regression through regularization. The coefficients of most high-dimensional regressions can be solved under a unified framework of minimizing the objective function f (β) together with a combination of L1 (LASSO) and L2 (ridge) penalties (where λ1, λ2 ≥ 0). The objective function of linear regression is the sum of least squares across all samples. The objective functions of logistic and Cox-PH regressions are the negative log of the likelihood function L(β, y, X). (d) High-dimensional regression through stepwise forward selection. At each step, the best variable is selected from a candidate pool to minimize the model error, such as cross-validation error. The procedure will terminate if any further variable selection increases the model error. Some previously selected variables may become insignificant during the stepwise process and get removed from the model. Abbreviations: Cox-PH, Cox proportional hazard; LASSO, least absolute shrinkage and selection operator.
In some cases, the modeling of nonlinear relationships might be critical to inferring gene function in cancer (Figure 3a, middle panel). For example, tumors with high levels of PDL1, a T cell exhaustion driver, resist killing by cytotoxic T cells (82). However, the level of PDL1 expression in melanoma tumors is associated with improved survival, which contradicts the protumor role of PDL1 (Supplemental Table 1, top rows). This counterintuitive association arises from the positive correlation between the level of PDL1 and lymphocyte infiltration in melanoma tumors, and patients with higher lymphocyte infiltration in tumors have longer survival than those with lower infiltration (83). In a Cox-PH regression that models nonlinear relationships, the quadratic term of the PDL1 (variable X) is associated with higher death risk Y (Supplemental Table 1, bottom rows). A significant quadratic term represents a U-shape correlation between a variable X and outcome Y (Figure 3a, middle panel) (81), which associates a higher PDL1 level with worse patient survival among lymphocyte-high tumors with high PDL1 levels.
Another critical variable relationship is the interaction. The concept of statistical interaction between variables is different from physical or genetic interactions between proteins or genes. In statistics, interaction occurs when the association between a variable X and outcome Y depends on the status of another variable T (79). In a hypothetical example (Figure 3a, right panel), variable X could have either a positive or negative correlation with Y when the variable T is 1 or 0, respectively. The variable interaction could be tested by a multiplication term in a multivariate regression (79). In some cases, the interaction between variables, rather than the individual variables, might be predictive of anticancer drug efficacy. For example, we developed a method named CARE (computational analysis of resistance) to model how the drug-targeted gene interacts with other genes to affect drug efficacy in cellular compound screens (84). When evaluated using clinical data of targeted therapies, the CARE signatures of gene variable interactions can predict patient outcomes better than signatures of individual gene effects (84).
Selecting Variables in High-Dimensional Data
Many clinical data sets in cancer research have small sample sizes (e.g., fewer than 100 patients) but a vast number of features (e.g., the expression or mutation status of 20,000 human genes). This type of data sets is termed high-dimensional. In these settings, classical regression methods will fail, including least squares, logistic, and Cox-PH regressions. Each classical regression method computes an optimal coefficient vector to minimize an objective function that measures the coherence between the model and the training data. However, a unique optimal coefficient vector does not exist in a high-dimensional setting because many sets of coefficients could make the model perfectly fit the training data, even when the variables are completely unrelated to the response (79). Moreover, the fitted models may not have reliable prediction performance on an independent test data set, a problem known as overfitting. Nonetheless, the classical methods can be modified with several techniques to perform variable selection in high-dimensional data (Figure 3c,d).
A popular technique is regularized regression, which optimizes a linear combination of the objective function and convex penalty terms on coefficients (Figure 3c) (85). These penalties help find coefficients of the optimal solution in high-dimensional settings while preventing the regression procedure from overfitting the training data (86). One common penalty, named L1 or LASSO (least absolute shrinkage and selection operator) shrinkage, controls the sum of absolute values of all coefficients (Figure 3c). LASSO regression achieves variable selection by setting most coefficients to zero and leaving the coefficients of essential variables as the only nonzero coefficients (87). Another common penalty, named L2 or ridge shrinkage, controls the sum of squares of all coefficients (Figure 3c). Although ridge regression assigns nonzero coefficients to most variables and thus does not select variables, it can achieve better prediction performance than LASSO when the variables are highly colinear (85).
Elastic net regression combines the advantage of LASSO and ridge regressions by optimizing the linear combination of the objective function and the two penalties (85). The penalty weights (λ1 and λ2 in Figure 3c) are those that give the lowest cross-validation error. This regularization method could be applied to least squares, logistic, and Cox-PH regressions. Elastic net is very popular in cancer genomics data analysis. For example, elastic net of least squares regression is used in compound screen projects to select gene features (e.g., mutation, copy number, expression) associated with drug efficacy (29, 88).
Another popular approach in high-dimensional variable selection is the stepwise forward regression (8i). This method utilizes a greedy approach to select the current best variable from the candidate pool to minimize the model error in a stepwise manner (Figure 3d). The model error can be computed through cross-validation or statistical metrics such as the Bayesian information criterion (81). At each forward step, some previously selected variables may become insignificant, and a backward removal step may eliminate these variables from the model. Compared to elastic net, forward selection in least squares regression is more computationally efficient through a highly optimized implementation (89). Forward selection and its variations are widely used. For example, the elucidation of the EndoPredict® biomarker, a predictor of disease risk in breast cancer, involved the forward-backward selection to identify gene expression features of disease recurrence (90).
Besides elastic net and forward selection, many other approaches, such as linear support vector machine (91) and random forest (81), are also applicable to high-dimensional data. The consortium of Dialogue on Reverse Engineering Assessment and Methods (DREAM) hosted a challenge that evaluated 44 algorithms on their performance of predicting drug sensitivities in compound cell line screens (92). This DREAM challenge reported several interesting observations. First, all top solutions modeled nonlinear relationships. Second, predictive power benefited from prior knowledge of biological pathways. Third, gene expression data provided the highest predictive power among all data types, and performance could be further improved by including other data types. Fourth, integrating predictions from independent methods produced the most robust results because different methods had complementary advantages in examining different aspects of the data.
What makes high-dimensional variable selection possible is the assumption that most regression coefficients are zero, where nonzero values indicate the essential variables (93). However, the colinearity among variables in biological data often fails to meet these criteria (94). Especially when the number of variables is much higher than the sample size, any variable can be well approximated by a couple of spurious variables due to chance correlation (81, 93). In such cases, we may choose the wrong variables and draw false conclusions. A critical procedure to overcome the colinearity issue is to train the model parameters through cross-validation and to evaluate the quality of fitted models on independent test data. Meanwhile, some prior knowledge may inform the grouping of correlated variables into one. For example, the gene expression values of many immune cell markers are highly correlated in bulk tumor profiles. They could be bundled as one feature (95). Notably, inferring immune infiltration in tumors should be taken with extra caution because gene signatures of different cell types might be highly correlated (96). Other complementary resources may also help us to significantly reduce the data dimensionality by limiting the variable selection on smaller gene subsets. For example, when searching for regulators of drug efficacy in a clinical data set, we could focus on the top hits in genetic screens, where the gene knockdown effects on drug sensitivity are evaluated at genome scale in cancer models.
Applying Systems Biology Models
In addition to variable selection, mathematical models are useful approaches in cancer research because of their ability to explore the quantitative properties of drug response (97). For example, previous work by Norton & Simon that modeled tumor growth patterns found that cancer cell growth may follow an S-shaped curve, where the growth rate is lowest for both small and large tumors but highest at an intermediate tumor size (98). Since some chemotherapies may preferentially kill proliferating cells, a dose capable of depleting a tumor of intermediate size may not be sufficient to cure a small or large tumor due to the growth rate difference. Therefore, chemotherapy might have reduced efficacy if an insufficient dose is administered at a time when the tumor is kinetically less sensitive to treatment. This kinetic resistance is different from the acquired and intrinsic resistance caused by molecular mechanisms. Later, a clinical trial validated the results from Norton & Simon’s mathematical model, finding that intense and prolonged doses are necessary for the clinical efficacy of chemotherapies (99). This example highlighted the utility of mathematical models in guiding therapy delivery schedules (100–102).
Besides mathematical models, biological network models are another class of promising approaches, especially for finding regulators. Often, gene features identified by variable selection methods (discussed above) may be associated only with drug efficacy, but not the regulators. Even though genetics screens, such as CRISPR and shRNA screens, can systematically identify the regulators of drug efficacy in cell line models, these technologies cannot be easily applied to patient tumors. Network models have the advantage of integrating the genomic profiles of patient tumors and inferring the potential regulators and pathways (103–105). A previous study demonstrated that, when integrated with gene expression or histone marks, biological networks are predictive of the regulator genes of cancer cell vulnerability (106). Hypothetically, similar network methods could also be applied to identify regulators of anticancer drug response and resistance. Furthermore, network models can be combined with mathematical models to study the quantitative properties of drug combinations (107) and design synergistic drug combinations (108). Therefore, we foresee system biology models playing a more significant role in finding effective cancer therapies in the future.
TRANSLATION: FROM DATA ANALYTICS TO CLINICAL APPLICATIONS
The development of high-throughput technologies is accelerating the translation of basic cancer research discoveries into clinical practice (Figure 1d). The previous decade has witnessed the translation of several research results from genomics data to FDA-approved or -marketed biomarker tests in the clinic. Meanwhile, many recently developed data-driven approaches have also shown promising potential for clinical application.
Identifying Prognostic Biomarkers
Drug response biomarkers are of critical clinical value because patients who do not benefit from a therapy not only waste time and money but also may suffer severe side effects. Early discoveries of cancer biomarkers mainly depended on biological understanding and empirical observations. With the rapid development of genomics resources, data-driven approaches can be used to identify reliable biomarkers. The classic examples are genomics tests for predicting recurrence risk in early-stage estrogen receptor (ER)/progesterone receptor (PR)-positive, HER2-negative breast cancer patients. Since such patients enjoy good clinical benefit from adjuvant endocrine therapy alone (109), it would be ideal for low-risk patients to avoid the unnecessary side effects of additional chemotherapy. The earliest genomic biomarker of disease recurrence in breast cancer, the Oncotype DX® assay, was developed by combining prior knowledge and heuristic gene selection (110–112). The development of later biomarkers, such as MammaPrint®, EndoPredict, and Prosigna®, all utilized variable selection methods on clinical data cohorts (Figure 4a).
Figure 4.
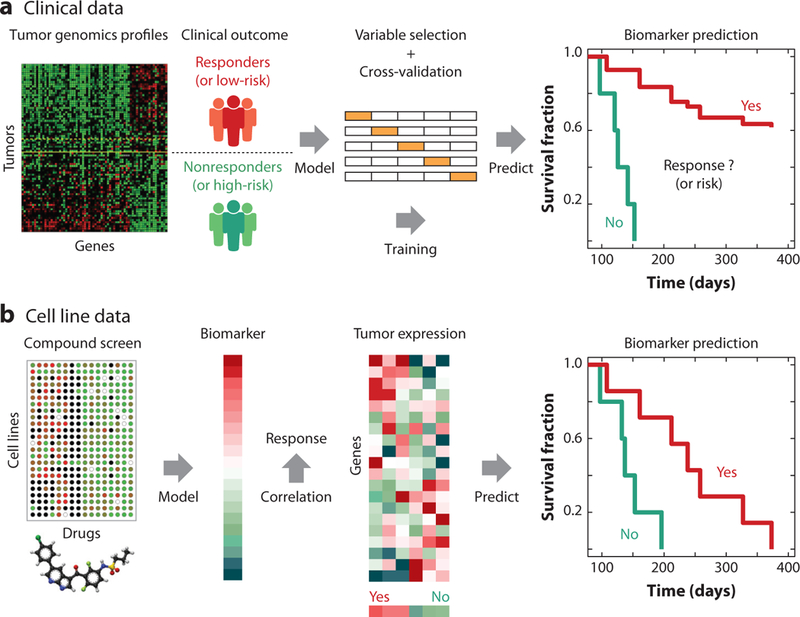
Biomarker training using clinical and cell line data. (a) The training of a multigene biomarker to guide treatment decisions starts from a collection of tumor genomics profiles paired with the patients’ clinical outcomes. The association between gene profiles and patients’ clinical outcomes is tested by statistical models, and a subset of genes are selected through a cross-validation procedure to optimize prediction accuracy. The accuracy of the gene biomarker will be evaluated in clinical trials for Food and Drug Administration approval or commercialization. (b) Computational methods can identify response biomarkers from compound screen data. Statistical methods can identify genes whose molecular status is significantly associated with drug efficacy across screened cell lines. The identified biomarker could be a subset of genes or a genome-wide vector of scores with one value per gene. In the latter case, the therapy response of each patient could be predicted by correlating between tumor gene expression values and biomarker scores.
The authors of MammaPrint conducted a microarray transcriptome profiling of 78 tumors and found that the expression levels of 231 genes were correlated with recurrence risk (113). They finalized a 70-gene biomarker set by sequentially selecting genes from the list ordered by the magnitude of correlation and evaluating the classification accuracy using leave-one-out cross-validation. For each tumor, MammaPrint computes the Pearson correlation between the tumor expression profiles and the average profile from the good prognosis groups to predict recurrence risk using a threshold determined from the training data. The MINDACT trial, which investigated the utility of biomarkers in predicting chemotherapy benefits, confirmed the accuracy of MammaPrint (114).
The training of MammaPrint only used data from one expression cohort. However, the development of most genomics biomarkers usually integrates data from several independent cohorts with hundreds of patients profiled. For example, the training data of the EndoPredict assay comprised both newly collected and published microarray cohorts, which included 964 tumors from patients treated with adjuvant tamoxifen (90). The authors searched for gene probes from microarray platforms with sufficient expression dynamics and selected 104 candidate genes through Cox-PH regression with recurrence as the outcome. Then, a forward-backward feature selection procedure (81) identified genes whose level could significantly predict recurrence risk (Figure 3d). The authors selected the parameters in their model through cross-validation, and the final predictor comprises eleven genes, including eight cancer risk genes and three normalization controls. For each tumor, the EndoPredict assay assigns a risk score using a linear model that combined the expression level of eleven predictor genes and several clinical parameters. Thresholds for the risk score are determined using the training data to discriminate patients into low- and high-risk groups.
The training methods of both MammaPrint and EndoPredict were supervised procedures with disease recurrence status as the outcome. Predictive biomarkers can also come from unsupervised procedures on data sets without clinical outcome data. For example, Prosigna (previously known as PAM50) is a widely used genomic test used to classify breast tumor subtypes (115). The authors collected microarray cohorts from both public domain and in-house collections that included 189 breast tumors and 29 normal samples. Hierarchical clustering of the expression profiles identified clusters representing the intrinsic subtypes (e.g., luminal A, luminal B, HER2, basal, normal). The gene expression profile of a patient’s tumor was compared with each of the pretrained signatures to determine the subtype. In the MA.12 study, the PAM50 classification was superior to immunohistochemistry assay in predicting both overall survival and tamoxifen benefit (116).
There are many other similar biomarkers for predicting disease recurrence risk and therapeutic benefits in ER/PR-positive breast cancer, such as the breast cancer index (117, 118) and Mammostrat® (119). An analysis comparing several expression biomarkers for breast cancer found that despite little gene overlap, the different biomarkers showed significant prediction agreement (120). Similar biomarkers also exist for other cancer types, such as ColoPrint® (121), Oncotype Dx for colon cancer (122), Decipher® (123), Oncotype Dx for prostate cancer (124), and Pervenio™ for early-stage lung cancer (125).
Despite the rapid advance of predictive biomarkers driven by genomics data, there are still significant challenges. Most current commercial biomarker efforts have focused on diseases with a favorable clinical outcome. For example, among ER/PR-positive breast cancer patients tested by MammaPrint and Oncotype Dx, the five-year disease-free rate without chemotherapy is higher than 90% in the low-risk group as determined by conventional clinical measures (114). Most newly diagnosed prostate cancer cases tested by Oncotype Dx represent low-risk disease, with less than 3% of men dying from prostate cancer. The most critical metric for biomarkers is the negative predictive value (NPV), which is the probability that patients with negative results truly will not benefit from the therapy (126). False negative predictions will prevent the patient from benefitting from treatment. In the cancer types with a very favorable outcome, it is much easier for biomarkers to achieve very high NPV using conventional clinical measures. However, compared to prostate cancer and luminal breast cancer patients, patients with other cancers such as glioblastoma and liver cancer have much worse clinical outcomes. In these cancer types, either there is no effective therapy available, making the biomarker of less value, or genomics biomarkers are not accurate enough for therapies with moderate efficacy. With the rapid development of potent anticancer agents and increasing amounts of clinical genomics data, we foresee that more and better drug response biomarkers in most cancer types will become available for patients and doctors over time.
Recently, an enormous amount of effort has been focused on the development of response biomarkers for ICB (127). While ICB may lead to remarkable clinical responses, for most cancer types, the majority of patients do not respond (10). Multiple factors have been associated with ICB effectiveness, including the degree of cytotoxic T cell infiltration, mutation or neo-antigen load, checkpoint molecule expression, antigen presentation defects, interferon signaling, tumor aneuploidy, some oncogenic signatures, and intestinal microbiota (10, 67, 128–132). However, none of these factors is sufficiently robust to achieve accurate outcome prediction (133). We foresee that computational methods have the potential to identify robust response biomarkers by integrating ICB clinical data with other complementary immuno-oncology data.
Predicting Therapy Toxicity
Toxicity is a primary concern for many anticancer drugs. The therapeutic window of a drug is the range of dosages that can treat disease effectively without having intolerable toxicity. Many anticancer drugs have a narrow therapeutic window, with a small difference between the doses for antitumor effects and significant toxicity. However, for certain drugs, the therapeutic window may be very different depending on the patient’s genetic background. For example, 6-mercaptopurine (6-MP) is a drug that treats acute lymphocytic leukemia and chronic myeloid leukemia. The side effects of 6-MP depend on genetic polymorphisms of TPMT, NUDT15, and ITPA (134–136). In this instance, before treatment, genetic tests are necessary to screen patients with specific allele variants, especially the homozygous variants.
Hypothetically, with sufficient training data, genomic biomarkers could be developed to predict the toxicity of a drug in each patient (Figure 1d). A DREAM challenge demonstrated that computational methods could predict cytotoxicity phenotype based on the genetic profiles of lymphoblastoid cell lines (137). Although this study was on cell line models and environmental chemicals (138), it provided a proof-of-concept example that the genotype data together with compound structural attributes might predict individualized toxicity. Currently, there are still no successful data-driven toxicity models that are clinically deployed to predict personalized side effects. With growing data and better computational methods, such models may become feasible in the future.
In addition to predicting personalized drug toxicity, computational models are essential tools in the early stage of drug discovery to screen low-toxicity compounds. With specific toxicity endpoints (e.g., median lethal dose values, tissue-specific toxicity events), quantitative structure-activity relationship (QSAR) models are useful for toxicity prediction through regressions (139, 140). For each chemical, the predictor variables of regression comprise chemical and molecular properties; the response variable could be a toxicity endpoint. Through regression, the QSAR model fits a relationship between chemical structures and toxicity that can predict the activities of new chemicals. A recent study developed a data integration framework named PrOCTOR to predict drug toxicity through the integration of data from drug target expression in tissues, gene network connectivity, chemical structures, and toxicity annotations from clinical trials (141). Intriguingly, PrOCTOR predicted that many FDA-approved anticancer drugs are unpromising for clinical development due to their cytotoxicity. Therefore, cancer-specific models with distinct schemes from general toxicity prediction might be necessary to predict cancer drug toxicity. Furthermore, such models should consider the more rapid recovery of normal tissue versus tumor tissue after treatment and the ability to mitigate drug toxicity by differences in dose schedule.
It is worth noting that drug toxicity studies are often limited by a lack of sufficient training data. However, there is a wealth of data buried in the archives of the pharmaceutical industry in formats that are difficult to harmonize and analyze. The eTOX project involved collaborations among thirteen pharmaceutical companies, eleven academic institutions, and six small- and medium-sized enterprises (142). The goal is to build a comprehensive toxicity database and to enable reliable modeling of drug safety endpoints through data sharing. We foresee that the eTOX project will significantly facilitate the development of computational toxicology methods.
Designing Combination Therapies
The emergence of therapeutic resistance together with the frequent incomplete response to primary therapy underscores the importance of effective drug combinations. Currently, most clinically approved combinations, such as dual BRAF and MEK inhibition in BRAF-mutant tumors, are developed from observations in drug-resistant samples or empirical evaluation of drug combinations (143, 144). Alternatively, combinatorial drug screens may identify effective combinations (145). However, the current screening platforms still cannot test all pairwise drug combinations across a broad panel of tumor models to investigate the vast space of potential drug combinations. Thus, data-driven approaches are essential to complement the current experimental methods.
Many data-driven approaches to identify resistance regulators and design combination therapies depend on compound screening data. For example, the molecular characterization of ATP-binding cassette (ABC) transporters across the NCI60 cell line panel identified the transporters that are essential for in vitro drug resistance to certain agents (146). A later analysis of the NCI60 data revealed that the cell-killing effects of thiosemicarbazone significantly correlate with the ABC transporter expression levels (146). This result implicated thiosemicarbazone as a lead compound for targeting multiple chemotherapy resistance (147). Recently, we developed a statistical framework, named CARE, to determine potential regulators of targeted therapy resistance (84). CARE analyzes how drug target genes interact with other genes to affect the drug efficacy in screened cell lines through multivariate regressions. When finding genes regulating lapatinib resistance from both compound screens and clinical data, CARE identified PRKD3 as the top candidate. Later experiments validated that PRKD3 inhibition, through either small interfering RNA or compounds, significantly sensitized HER2 inhibition by lapatinib in HER2-positive breast cancer cells.
The examples above focused on finding synergistic drugs that can overcome the resistance to a primary drug. Many studies also aim to discover cotargeting strategies against targets without known inhibitors. For example, a large body of work is identifying drug combinations to mimic RAS (e.g., NRAS, KRAS) inhibition, since direct pharmacological inhibition of RAS has been unfeasible. MEK is the key downstream component of RAS signaling; however, single-agent MEK inhibition has been ineffective against tumor cells with activating RAS mutations (148). To identify the difference between targeting MEK and RAS, one study investigated genes whose expression was differentially regulated by eliminating NRAS but showed either no change or change in the opposite direction by MEK inhibition (149). This study collected gene expression data based on an inducible NRAS Q61K-driven mouse model of melanoma, as well as public data sets measuring the transcriptome response of human melanoma cells under various treatments. A statistical model was developed to test the difference of transcriptomic effects between NRAS and MEK inhibition. The authors further applied a network modeling approach, named TRAP, to identify the key transcriptional regulators and found CDK4 as a synergistic target with MEK inhibition (149). Combined treatment of MEK and CDK4 inhibitors in mouse models showed significant synergy, which was consistent with earlier studies in cell lines (150).
THE CHALLENGES OF BIG DATA RESEARCH IN CANCER
Given the recent advances in data-driven discoveries catalyzed by the genomics revolution, we may anticipate a significant burst in research productivity. However, big data can also bring significant challenges instead of breakthroughs. The current data resources in cancer research are far from adequate to answer many important questions about drug response and resistance. Our future efforts should focus on resolving the big data challenges to achieve impactful discoveries.
Inconsistencies Between Data Sets
A common challenge to interpreting the data from clinical studies is that independent cohorts aiming to answer the same question may reach different conclusions. For example, the gene signature of anti-PDl therapy response identified in one study (151) was not predictive in another study (152). Similarly, many anti-BRAF resistance drivers identified in the literature were not reproducibly found in independent clinical studies (25). From the Gene Expression Omnibus database from the National Center for Biotechnology Information, we collected pairs of human melanoma expression profiles between post-treatment tumors that are resistant to BRAF inhibitors and pretreatment-sensitive tumors. A hierarchical clustering of differential expression profiles between drug-resistant and parental tumors identified 16 distinct clusters with negative correlations between each of the two groups (Supplemental Figure 2). Expression profiles even from the same study appeared in several anticorrelated clusters. One possible interpretation of the inconsistency is that there might be many drug resistance mechanisms, as reflected by the many clusters. Another possibility is that the expression data may reflect passenger alterations instead of drivers. Therefore, it may be premature to draw conclusions from an analysis of a single data cohort without corroborating results from other cohorts, experimental validation, and mechanistic insights.
Another cause of inconsistency arises when data sets from two different technologies measuring the same biological signal lead to different results. For example, the winners from a DREAM challenge in predicting essential genes from shRNA screens failed to predict the top genes from CRISPR screens (106). Further complicating the issue, in high-throughput studies, genomic measurements might correlate with batch effects such as processing platform or date instead of clinical features (153). Therefore, to ensure reliable discoveries, researchers must conduct analyses under robust standards, such as consistent control samples, batch effect removal, and systematic evaluation of independent computational methods, parameters, and cohorts.
Incomplete Clinical Information
In many cancer genomic resources, the lack of treatment information is a particular limitation to data utility. For example, most patients profiled in the TCGA project do not have treatment information. For some cancer types, we may assume that most patients received the standard-of-care therapy. For example, luminal breast cancer patients should get hormone therapies, while HER2 + patients should get trastuzumab treatments. On the other hand, many might have been treated with surgical resection only. However, such treatment information is not explicitly available in TCGA to enable modeling of therapy response and resistance.
Many ongoing efforts are trying to overcome the limitation of available clinical information. For example, an industry collaboration collected about 20,000 patients, for whom both Flatiron electronic health records (EHRs) and Foundation Medicine mutational profiles from next-generation sequencing (NGS) are available (154). The EHR-to-NGS integration linked the longitudinal clinical information with the genomic data and recapitulated findings regarding prognostic biomarkers and therapeutic implications. Another example is the 100,000 Genomes Project that aims to sequence 100,000 whole exomes of diseased and healthy cells from cancer patients and rare disease patients documented in the United Kingdom’s National Health Service (NHS) system. The NHS system provides detailed medical records and health data of all patients for further analysis. Currently, these genomics resources with detailed clinical information are either proprietary (e.g., EHR-NGS integration; see Reference 154) or in progress (e.g., 100,000 Genomes Project) and thus unavailable for public analysis. However, we foresee that the availability of clinical information should improve in future data cohorts.
The Bottleneck of Data Dimensionality
Recent years have seen many successful examples of big data analytical systems with enormous financial and social impacts, such as the consumer recommendation systems of Amazon, Netflix, and Facebook. However, data science in studying cancer drug effectiveness has only shown limited clinical success. Most clinical studies of anticancer drugs contained profiles of only a small number of patients. For example, several recent studies of ICB released the gene expression profiles of about 30 patients (151, 152, 155, 156), which are not sufficient for selecting response features among all human genes and pathways. In contrast, ImageNet, a data set widely used in computer vision research, contains about 15 million images with detailed hierarchical annotations across 20,000 semantic terms (157). Such a large, well-annotated cohort provided a solid platform to develop deep learning models for image classification, localization, and detection (158). Therefore, there is a significant gap of data dimensionality between cancer biology and other data science fields.
The bottleneck of data dimensionality in cancer research lies in the unique difficulties in sample collection and annotation. ImageNet was able to collect pictures from several internet engines and conduct semantic annotation with the crowdsourcing platform Amazon Mechanical Turk (157). Since image understanding is a natural ability of most people, this crowdsourcing strategy can leverage human power around the world. In contrast, for most cancer types, biopsies through surgical removal of tumors may not happen after metastasis. Even when noninvasive biopsy options are available, genome-scale profiling ofcancer samples still incurs a high cost not often reimbursed by medical insurance. Therefore, most data sets of anticancer drug response have small sample sizes (e.g., fewer than 100 patients) compared to the variable dimensionality (e.g., about 20,000 human genes). In the section titled Selecting Variables in High-Dimensional Data, we discussed several algorithmic solutions in analyzing high-dimensional data. However, many limitations in high-dimensional data, such as variable colinearity, may prevent any computational methods from giving robust results. Therefore, other nonalgorithmic solutions are necessary to overcome the bottleneck of data dimensionality in cancer research.
A strategy to overcome the data dimensionality limitation might lie in the data integration. Even though each study may not provide enough information, analyses integrating all studies together can increase the confidence in the results. For example, the cBioPortal platform has integrated 168 cancer genomics data sets with the molecular profiles of 47,135 samples across over 20 cancer types (159). For each gene of interest, this data integration effort enables interactive exploration of molecular alteration patterns and clinical relevance across thousands of samples and neighbor genes in various types of biological networks (160). Similarly, Oncomine integrates 715 cancer genomics data sets across 86,733 samples to enable interactive exploration and analysis of gene functions in cancer (161). Such data integration efforts represent a cost-effective approach to increase sample size through efficient reuse of published resources.
Another strategy to resolve the limitation of clinical data dimensionality is to utilize the large-scale data sets from preclinical models. Especially, the data sets on cancer cell lines can be generated across a much larger number of samples than patient clinical data. Despite some studies questioning whether cell line data could capture clinical relevance (162, 163), several studies demonstrated that the data from compound screens could derive reliable biomarkers to predict clinical response to therapies (84, 164, 165). Moreover, through downsampling analysis, one study demonstrated that the prediction reliability of biomarkers from preclinical models really benefited from the large sample number (~ 1,000 cell lines) of compound screen data (84).
A third solution for breaking the data bottlenecks is through collaborations between industry and academia. There are many large-scale data sets generated in the industry. Even though these resources are primarily proprietary, some companies release their data for scientific research. For example, Novartis released many data sets of pharmacological and genetic screens, such as the CCLE (29), DRIVE (deep RNA interference interrogation of viability effects in cancer) (33), and PDX Encyclopedia (43) cohorts. Recently, the IBM Watson and Broad Institute launched a five-year, $50 million initiative to collect genomics data from about 10,000 drug-resistant samples. Similarly, collaborations among multiple research institutions can also provide large-scale clinical genomics data sets. For example, the GENIE project is an international data sharing initiative among eight institutions that released mutation profiles for more than 500 genes and a minimal set of clinical information for almost 30,000 cancer patients until the end of 2017 (24).
Lastly, there are also several efforts in creating new resources from published data sets (166–168). Even though they may not focus on anticancer drug efficacy, some of them provide good examples of public data reuse to answer specific questions. For example, the CREEDS (crowd-extracted expression of differential signatures) project collected thousands of drug and gene perturbation signatures using the crowdsourcing approach through an online Coursera course with about 70 participants across 25 countries (168). Such crowdsourcing strategies devised by experts may enable efficient reuse of public data to create larger data sets for cancer research.
CONCLUSION
In this review, we summarized the literature on high-throughput technologies and data-driven approaches that model the efficacy of anticancer drugs. Despite the abundant literature and a few successful clinical applications, there are still many unsolved problems and new challenges. Our review primarily focused on small molecule or antibody drugs. However, there are many other types of anticancer therapies, such as radiotherapy, cell therapy (169, 170), personalized vaccines (171, 172), nanoparticles (173), and fecal transplantation (17, 69, 70). Genomic profiling efforts for these conventional and emerging treatment modalities may bring new challenges and opportunities to data science.
The success of precision cancer medicine hinges on using data science to better characterize the interactions between the tumor microenvironment, host immunity, and the ecosystem. Meanwhile, the translation from analytic results to prognosis and treatment regimens in the clinics requires the collaboration of the whole scientific community, including data scientists, molecular biologists, and clinical oncologists. With the increasing availability of big data resources and computational methods, we envision that big data approaches will significantly contribute to the future development of precision cancer medicine.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant U24 CA224316, Department of Defense grant PC140817P1, and the Breast Cancer Research Foundation (to X.S.L.). The authors thank colleagues, including Wenbin Li, Cliff Meyer, Deng Pan, Jun Liu, Ethan Cerami, Jon Aster, Myles Brown, and Kai Wucherpfennig for their insightful discussions. The authors also thank Qiu Wu, Sailing Shi, Tong Han, Ying Li, Ziyi Li, and Taiwen Li for reading the relevant literature.
Footnotes
DISCLOSURE STATEMENT
W.R.S. is a former employee and shareholder of Novartis Pharmaceuticals and a patent holder of EGFR mutation testing. X.S.L. is a co-founder and shareholder of GV20 Oncotherapy.
LITERATURE CITED
- 1.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, et al. 1988. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72:567–72 [PubMed] [Google Scholar]
- 2.Deininger M, Buchdunger E, Druker BJ. 2005. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105:2640–53 [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, et al. 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–500 [DOI] [PubMed] [Google Scholar]
- 4.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, et al. 2014. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. New Engl. J. Med. 371:2167–77 [DOI] [PubMed] [Google Scholar]
- 5.Holohan C, Van Schaeybroeck S, Longley DB,Johnston PG. 2013. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13:714–26 [DOI] [PubMed] [Google Scholar]
- 6.Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, et al. 2016. Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (review). Int. J. Oncol. 48:869–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, et al. 2015. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 16:426–35 [DOI] [PubMed] [Google Scholar]
- 8.Widakowich C, de Castro G Jr., de Azambuja E, Dinh P, Awada A 2007. Review: side effects of approved molecular targeted therapies in solid cancers. Oncologist 12:1443–55 [DOI] [PubMed] [Google Scholar]
- 9.June CH, Warshauer JT, Bluestone JA. 2017. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 23:540–47 [DOI] [PubMed] [Google Scholar]
- 10.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168:707–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang P, Liu XS. 2015. Big data mining yields novel insights on cancer. Nat. Genet. 47:103–4 [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. 2015. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28:690–714 [DOI] [PubMed] [Google Scholar]
- 13.LopezJS Banerji U. 2017. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 14:57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahoney KM, Rennert PD, Freeman GJ. 2015. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14:561–84 [DOI] [PubMed] [Google Scholar]
- 15.Sheng Z, Sun Y, Yin Z, Tang K, Cao Z. 2017. Advances in computational approaches in identifying synergistic drug combinations. Briefings Bioinform. 2017:bbx047 [DOI] [PubMed] [Google Scholar]
- 16.Hu X, Zhang Z. 2016. Understanding the genetic mechanisms of cancer drug resistance using genomic approaches. Trends Genet. 32:127–37 [DOI] [PubMed] [Google Scholar]
- 17.Roy S, Trinchieri G. 2017. Microbiota: a key orchestrator of cancer therapy. Nat. Rev. Cancer 17:271–85 [DOI] [PubMed] [Google Scholar]
- 18.Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, et al. 2014. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 20:682–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, et al. 2017. Integrative clinical genomics of metastatic cancer. Nature 548:297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, et al. 2015. Whole-genome characterization of chemoresistant ovarian cancer. Nature 521:489–94 [DOI] [PubMed] [Google Scholar]
- 21.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zehir A, Benayed R, Shah RH, Syed A, Middha S, et al. 2017. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23:703–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Res. Netw., Weinstein JN, Collisson EA, Mills GB, Shaw KR, et al. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45:1113–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.AACR Proj. GENIE Consort. 2017. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7:818–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hugo W, Shi H, Sun L, Piva M, Song C, et al. 2015. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162:1271–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoemaker RH. 2006. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6:813–23 [DOI] [PubMed] [Google Scholar]
- 27.Cragg GM. 1998. Paclitaxel (Taxol®): a success story with valuable lessons for natural product drug discovery and development. Med. Res. Rev. 18:315–31 [DOI] [PubMed] [Google Scholar]
- 28.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, et al. 2006. BRAF mutation predicts sensitivity to MEK inhibition. Nature 439:358–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483:603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, et al. 2006. An efficient rapid system for profiling the cellular activities of molecular libraries. PNAS 103:3153–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, et al. 2016. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 6:914–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, et al. 2017. Defining a cancer dependency map. Cell 170:564–76.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald ER III, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, et al. 2017. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170:577–92.e10 [DOI] [PubMed] [Google Scholar]
- 34.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, et al. 2017. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49:1779–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, et al. 2015. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5:1210–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, et al. 2016Alandscape of pharmacogenomic interactions in cancer. Cell 166:740–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, et al. 2016. High-throughput identification of genotype-specific cancervulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol 34:419–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J. 2006Amethodforhigh-throughput gene expression signature analysis. Genome Biol. 7:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, et al. 2009. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 27:77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, et al. 2011. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 7:787–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sellers WR. 2011. A blueprint for advancing genetics-based cancer therapy. Cell 147:26–31 [DOI] [PubMed] [Google Scholar]
- 42.Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, et al. 2006. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 94:259–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, et al. 2015. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 21:1318–25 [DOI] [PubMed] [Google Scholar]
- 44.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. 2008. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 8:59–73 [DOI] [PubMed] [Google Scholar]
- 45.Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, et al. 2006. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 281:13964–71 [DOI] [PubMed] [Google Scholar]
- 46.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, et al. 2017. Identification of essential genes for cancer immunotherapy. Nature 548:537–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mbofung RM, McKenzie JA, Malu S, Zhang M, Peng W, et al. 2017. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 8:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay R, et al. 2018. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359:770–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, et al. 2017. Rare cell variability and druginduced reprogramming as a mode of cancer drug resistance. Nature 546:431–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meacham CE, Morrison SJ. 2013. Tumour heterogeneity and cancer cell plasticity. Nature 501:328–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee AJ, Swanton C. 2012. Tumour heterogeneity and drug resistance: personalising cancer medicine through functional genomics. Biochem. Pharmacol. 83:1013–20 [DOI] [PubMed] [Google Scholar]
- 52.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, et al. 2006. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–64 [DOI] [PubMed] [Google Scholar]
- 53.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, et al. 2017. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 77:3982–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gawad C, Koh W, Quake SR. 2016. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17:175–88 [DOI] [PubMed] [Google Scholar]
- 55.Wu AR, Wang J, Streets AM, Huang Y. 2017. Single-cell transcriptional analysis. Annu. Rev. Anal. Chem. 10:439–62 [DOI] [PubMed] [Google Scholar]
- 56.Spitzer MH, Nolan GP. 2016. Mass cytometry: single cells, many features. Cell 165:780–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zenobi R 2013. Single-cell metabolomics: analytical and biological perspectives. Science 342:1243259 [DOI] [PubMed] [Google Scholar]
- 58.Izar B, Tirosh I, Stover E, Rotem A, Shah P, et al. 2017. Dissecting treatment resistance in patients with ovarian cancer and PDX-models using single-cell RNA-sequencing Proc. Am. Assoc. Cancer Res., Washington, DC, 1–5 April Philadelphia: Am. Assoc. Cancer Res. [Google Scholar]
- 59.Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, et al. 2016. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352:189–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, et al. 2017. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171:1611–24.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuan GC, Cai L, Elowitz M, Enver T, Fan G, et al. 2017. Challenges and emerging directions in single-cell analysis. Genome Biol. 18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, et al. 2017. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169:1342–56.e16 [DOI] [PubMed] [Google Scholar]
- 63.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, et al. 2014. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20:436–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, et al. 2016. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45:931–43 [DOI] [PubMed] [Google Scholar]
- 65.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, et al. 2013. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342:971–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, et al. 2013. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342:967–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, et al. 2015. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350:1084–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, et al. 2015. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350:1079–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, et al. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97 [DOI] [PubMed] [Google Scholar]
- 70.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, et al. 2018. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, et al. 2018. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 359:104–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stringer AM, Gibson RJ, Logan RM, Bowen JM, Yeoh AS, Keefe DM. 2008. Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol. Ther. 7:1919–25 [DOI] [PubMed] [Google Scholar]
- 73.Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, et al. 2015. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 13:360–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, et al. 2011. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 29:393–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, et al. 2012. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22:292–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, et al. 2012. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, et al. 2017. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358:1443–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mabert K, Cojoc M, Peitzsch C, Kurth I, Souchelnytskyi S, Dubrovska A. 2014. Cancer biomarker discovery: current status and future perspectives. Int. J. Radiat. Biol. 90:659–77 [DOI] [PubMed] [Google Scholar]
- 79.Freedman D 2009. Statistical Models: Theory and Practice. Cambridge, UK: Cambridge Univ. Press [Google Scholar]
- 80.Kleinbaum DG. 1998. Survival analysis, a self-learning text. Biometr. J. 40:107–8 [Google Scholar]
- 81.James G, Witten D, Hastie T, Tibshirani R.2013. An Introduction to Statistical Learning: With Applications in R. New York: Springer-Verlag [Google Scholar]
- 82.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. 2008. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26:677–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cancer Genome Atlas Netw. 2015. Genomic classification of cutaneous melanoma. Cell 161:1681–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang P, Lee W, Li X, Johnson C, Liu JS, et al. 2018. Genome-scale signatures of gene interaction from compound screens predict clinical efficacy of targeted cancer therapies. Cell Syst. 6:343–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zou H, Hastie T. 2005. Regularization and variable selection via the elastic net. F. R. Stat. Soc. B 67:301–20 [Google Scholar]
- 86.Hastie T, Tibshirani R, Friedman JH. 2009. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. New York: Springer-Verlag [Google Scholar]
- 87.Tibshirani R 1996. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. B 58:267–88 [Google Scholar]
- 88.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, et al. 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483:570–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang P, Freedman ML, Liu JS, Liu XS. 2015. Inference of transcriptional regulation in cancers. PNAS 112:7731–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Filipits M, Rudas M, Jakesz R, Dubsky P, Fitzal F, et al. 2011. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin. Cancer Res. 17:6012–20 [DOI] [PubMed] [Google Scholar]
- 91.Smola AJ, Scholkopf B. 2004. A tutorial on support vector regression. Stat. Comput. 14:199–222 [Google Scholar]
- 92.Costello JC, Heiser LM, Georgii E, Gonen M, Menden MP, et al. 2014. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 32:1202–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fan J, Lv J 2010. A selective overview of variable selection in high dimensional feature space. Stat. Sin. 20:101–48 [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao P, Yu B. 2006. On model selection consistency of Lasso. J. Mach. Learn. Res. 7:2541–63 [Google Scholar]
- 95.Siemers NO, Holloway JL, Chang H, Chasalow SD, Ross-MacDonald PB, et al. 2017. Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLOS ONE 12:e0179726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li B, Liu JS, Liu XS. 2017. Revisit linear regression-based deconvolution methods for tumor gene expression data. Genome Biol. 18:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Altrock PM, Liu LL, Michor F. 2015. The mathematics of cancer: integrating quantitative models. Nat. Rev. Cancer 15:730–45 [DOI] [PubMed] [Google Scholar]
- 98.Norton L, Simon R. 1977. Tumor size, sensitivity to therapy, and design of treatment schedules. Cancer Treat. Rep. 61:1307–17 [PubMed] [Google Scholar]
- 99.Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, et al. 2003. Randomized trial of dosedense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. F. Clin. Oncol. 21:1431–39 [DOI] [PubMed] [Google Scholar]
- 100.Michor F, Beal K. 2015. Improving cancer treatment via mathematical modeling: surmounting the challenges is worth the effort. Cell 163:1059–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, et al. 2011. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 3:90ra59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leder K, Pitter K, LaPlant Q, Hambardzumyan D, Ross BD, et al. 2014. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell 156:603–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, et al. 2014. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 159:402–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. 2007. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hofree M, Shen JP, Carter H, Gross A, Ideker T. 2013. Network-based stratification of tumor mutations. Nat. Methods 10:1108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang P, Wang H, Li W, Zang C, Li B, et al. 2015. Network analysis of gene essentiality in functional genomics experiments. Genome Biol. 16:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nelander S, Wang W, Nilsson B, She QB, Pratilas C, et al. 2008. Models from experiments: combinatorial drug perturbations of cancer cells. Mol. Syst. Biol. 4:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Korkut A, Wang W, Demir E, Aksoy BA, Jing X, et al. 2015. Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. eLife 4:e04640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Early Breast Cancer Trialists’ Collab. Group. 2005. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365:1687–717 [DOI] [PubMed] [Google Scholar]
- 110.Paik S, Shak S, Tang G, Kim C, Baker J, et al. 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New Engl. J. Med. 351:2817–26 [DOI] [PubMed] [Google Scholar]
- 111.Paik S, Tang G, Shak S, Kim C, Baker J, et al. 2006. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 24:3726–34 [DOI] [PubMed] [Google Scholar]
- 112.Albain KS, Barlow WE, Shak S, Hortobagyi GN, Livingston RB, et al. 2010. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 11:55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530–36 [DOI] [PubMed] [Google Scholar]
- 114.Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, et al. 2016. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. New Engl.F. Med. 375:717–29 [DOI] [PubMed] [Google Scholar]
- 115.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, et al. 2009. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27:1160–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chia SK, Bramwell VH, Tu D, Shepherd LE, Jiang S, et al. 2012. A 50-gene intrinsic subtype classifier for prognosis and prediction of benefit from adjuvant tamoxifen. Clin. Cancer Res. 18:4465–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sgroi DC, Carney E, Zarrella E, Steffel L, Binns SN, et al. 2013. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J. Natl. CancerInst. 105:1036–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sanft T, Aktas B, Schroeder B, Bossuyt V, DiGiovanna M, et al. 2015. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res. Treat. 154:533–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bartlett JM, Bloom KJ, Piper T, Lawton TJ, van de Velde CJ, et al. 2012. Mammostrat as an immuno-histochemical multigene assay for prediction of early relapse risk in the tamoxifen versus exemestane adjuvant multicenter trial pathology study. J. Clin. Oncol. 30:4477–84 [DOI] [PubMed] [Google Scholar]
- 120.Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, et al. 2006. Concordance among gene-expression-based predictors for breast cancer. New Engl. J. Med. 355:560–69 [DOI] [PubMed] [Google Scholar]
- 121.Salazar R, Roepman P, Capella G, Moreno V, Simon I, et al. 2011. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 29:17–24 [DOI] [PubMed] [Google Scholar]
- 122.Yamanaka T, Oki E, Yamazaki K, Yamaguchi K, Muro K, et al. 2016. 12-Gene recurrence score assay stratifies the recurrence risk in stage II/III colon cancer with surgery alone: the SUNRISE study. J. Clin. Oncol. 34:2906–13 [DOI] [PubMed] [Google Scholar]
- 123.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, et al. 2013. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLOS ONE 8:e66855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Knezevic D, Goddard AD, Natraj N, Cherbavaz DB, Clark-Langone KM, et al. 2013Analytical validaion of the oncotype DX prostate cancer assay—a clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genom. 14:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kratz JR, He J, VanDen Eeden SK, Zhu ZH, Gao W, et al. 2012A practical molecular assay to predict survival in resected non-squamous, non-small-cell lung cancer: development and international validation studies. Lancet 379:823–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simon R 2015. Sensitivity, specificity, PPV, and NPV for predictive biomarkers. J. Natl. Cancer Inst. 107:djv153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sharma P, Allison JP. 2015. Immune checkpoint targeting in cancertherapy: toward combination strategies with curative potential. Cell 161:205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Masucci GV, Cesano A, Hawtin R, Janetzki S, Zhang J, et al. 2016. Validation of biomarkers to predict response to immunotherapy in cancer: volume I—pre-analytical and analytical validation. J. Immunother. Cancer 4:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Davoli T, Uno H, Wooten EC, Elledge SJ.2017. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355:eaaf8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cogdill AP, Andrews MC, Wargo JA. 2017. Hallmarks of response to immune checkpoint blockade. Br. J. Cancer 117:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. 2014. Genetic basis for clinical response to CTLA-4 blockade in melanoma. New Engl. J. Med. 371:2189–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, et al. 2015. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl. J. Med. 372:2509–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nishino M, Ramaiya NH, Hatabu H, Hodi FS. 2017. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 14:655–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang JJ, Landier W, Yang W, Liu C, Hageman L, et al. 2015. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol. 33:1235–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dean L 2012. Mercaptopurine therapy and TPMT genotype In Medical Genetics Summaries, ed. Pratt V, McLeod H, Dean L, Malheiro A, Rubinstein W. Bethesda, MD: Natl. Cent. Biotechnol. Inform; https://www.ncbi.nlm.nih.gov/books/NBK100660/ [Google Scholar]
- 136.Adam de Beaumais T, Fakhoury M, Medard Y, Azougagh S, Zhang D, et al. 2011. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br. J. Clin. Pharmacol. 71:575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eduati F, Mangravite LM, Wang T, Tang H, Bare JC, et al. 2015. Prediction of human population responses to toxic compounds by a collaborative competition. Nat. Biotechnol. 33:933–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Abdo N, Xia M, Brown CC, Kosyk O, Huang R, et al. 2015. Population-based in vitro hazard and concentration-response assessment of chemicals: the 1000 genomes high-throughput screening study. Environ. Health Perspect. 123:458–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Patlewicz G, Fitzpatrick JM. 2016. Current and future perspectives on the development, evaluation, and application of in silico approaches for predicting toxicity. Chem. Res. Toxicol. 29:438–51 [DOI] [PubMed] [Google Scholar]
- 140.Raies AB, Bajic VB. 2016. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 6:147–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gayvert KM, Madhukar NS, Elemento O. 2016. A data-driven approach to predicting successes and failures of clinical trials. Cell Chem. Biol. 23:1294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sanz F, Pognan F, Steger-Hartmann T, Diaz C, eTox, et al. 2017. Legacy data sharing to improve drug safety assessment: the eTOX project. Nat. Rev. Drug Discov. 16:811–12 [DOI] [PubMed] [Google Scholar]
- 143.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, et al. 2009. MEK1 mutations confer resistance to MEK and B-RAF inhibition. PNAS 106:20411–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, et al. 2010. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 102:1724–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Held MA, Langdon CG, Platt JT, Graham-Steed T, Liu Z, et al. 2013. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 3:52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, et al. 2004. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 6:129–37 [DOI] [PubMed] [Google Scholar]
- 147.Ludwig JA, Szakacs G, Martin SE, Chu BF, Cardarelli C, et al. 2006. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 66:4808–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, et al. 2012. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 18:555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, et al. 2012. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 18:1503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li J, Xu M, Yang Z, Li A, Dong J. 2010. Simultaneous inhibition of MEK and CDK4 leads to potent apoptosis in human melanoma cells. Cancer Investig. 28:350–56 [DOI] [PubMed] [Google Scholar]
- 151.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, et al. 2016. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, et al. 2017. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171:934–49.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, et al. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11:733–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Singal G, Miller PG, Agarwala V, He J, Gossai A, et al. 2017. Development and validation of a real-world clinico-genomic database. Am. Soc. Clin. Oncol. 35:2514 [Google Scholar]
- 155.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, et al. 2015. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207–11. Erratum. 2016. Science 352:aaf8264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, et al. 2017. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multi-omic analysis. PLOSMed. 14:e1002309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Deng J, Dong W, Socher R, Li L-J, Li K, Fei-Fei L. 2009. Imagenet: a large-scale hierarchical image database Proc. Comput. Vis. Pattern Recognit., Miami, Fla., 20–25 June, pp. 248–55 New York: IEEE [Google Scholar]
- 158.Krizhevsky A, Sutskever I, Hinton GE. 2012. Imagenet classification with deep convolutional neural networks Proc. Int. Conf. Neurallnf. Process. Syst., Lake Tahoe, Nev., 3–6 December, ed. Pereira F, Burgess CJC, Bottou L, Weinberger KQ, pp. 1097–105. Red Hook, NY: Curran Assoc. [Google Scholar]
- 159.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, et al. 2011. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 39:D685–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, et al. 2004. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, et al. 2011. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. PNAS 108:18708–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gillet JP, Varma S, Gottesman MM. 2013. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 105:452–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, et al. 2013. Modeling precision treatment of breast cancer. Genome Biol. 14:R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Geeleher P, Cox NJ, Huang RS. 2014. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 15:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, et al. 2015. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21:938–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Feng C, Araki M, Kunimoto R, Tamon A, Makiguchi H, et al. 2009. GEM-TREND: a web tool for gene expression data mining toward relevant network discovery. BMC Genom. 10:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang Z, Monteiro CD, Jagodnik KM, Fernandez NF, Gundersen GW, et al. 2016. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 7:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rosenberg SA, Restifo NP. 2015. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lim WA, June CH. 2017. The principles of engineering immune cells to treat cancer. Cell 168:724–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, et al. 2017. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547:217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, et al. 2017. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547:222–26 [DOI] [PubMed] [Google Scholar]
- 173.Davis ME, Chen ZG, Shin DM. 2008. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug Discov. 7:771–82 [DOI] [PubMed] [Google Scholar]
- 174.Shalem O, Sanjana NE, Zhang F. 2015. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16:299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, et al. 2017. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547:413–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M,et al. 2016A biobank of breast cancer explants with preserved intra tumor heterogeneity to screen anticancer compounds. Cell 167:260–74.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Trifiletti DM, Sturz VN, Showalter TN, Lobo JM. 2017Towards decision-making using individualized risk estimates for personalized medicine: a systematic review of genomic classifiers of solid tumors. PLOS ONE 12:e0176388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.