

Abstract
We have cloned and characterized an intronic fragment of zebrafish lymphocyte cytosolic protein 1 (lcp1, also called L-plastin) that drives expression to the zebrafish enveloping layer (EVL). L-plastin is a calcium-dependent actin-bundling protein belonging to the plastin/fimbrin family of proteins, and is necessary for the proper migration and attachment of several adult cell types, including leukocytes and osteoclasts. However, in zebrafish lcp1 is abundantly expressed much earlier, during differentiation of the EVL. The cells of this epithelial layer migrate collectively, spreading vegetally over the yolk. L-plastin expression persists into the larval periderm, a transient epithelial tissue that forms the first larval skin. This finding establishes that L-plastin is activated in two different embryonic waves, with a distinct regulatory switch between the early EVL and the later leukocyte. To better study L-plastin expressing cells we attempted CRISPR/Cas9 homology-driven recombination (HDR) to insert a self-cleaving peptide (Cre-P2A-EGFP-CAAX) downstream of the native lcp1 promoter. This produced a stable zebrafish line expressing Cre recombinase in EVL nuclei and green fluorescence in EVL cell membranes. In vivo tracking of these labeled cells provided enhanced views of EVL migration behavior, membrane extensions, and mitotic events. Finally, we experimentally dissected key elements of the targeted lcp1 locus, discovering a ~300 bp intronic sequence sufficient to drive EVL expression. The lcp1: Cre-P2A-EGFP-CAAX zebrafish should be useful for studying enveloping layer specification, gastrulation movements and periderm development in this widely used vertebrate model. In addition, the conserved regulatory sequences we have isolated predict that L-plastin orthologs may have a similar early expression pattern in other vertebrate embryos.
Keywords: Actin, Cre recombinase, Cytoskeleton, Enhancer, Epiboly, Epidermis, Epithelium, Filopodia, Gastrulation, Lamellipodia, Microridges, Periderm, Promoter
1. Introduction
The major cytoskeletal protein of most cells is actin, which exists in both monomer and polymer form. Once polymerized, the resulting actin filaments are organized into parallel bundles and branching net-works by numerous accessory proteins. Most actin-organizing proteins are ubiquitous, appearing in all eukaryotic cells. However some are remarkably lineage-specific, which prompts the question of what unique functions these proteins provide and how their expression is controlled. In combination, both generalist and specialist actin organizers increase the capabilities of the cytoskeleton, expanding the cellular repertoire.
In this paper, we present our recent studies on a cell-specific actin-bundling protein called lymphocyte cytosolic protein 1 (lcp1). This is a highly-conserved, calcium-dependent phosphoprotein within the fim-brin family of proteins (Galkin et al., 2008; Zhang et al., 2016). Originally isolated from neoplastic human fibroblasts, it was then termed ‘p65’ for its molecular weight of 65kDa (Goldstein et al., 1985; Lin et al., 1988). Later, the same protein was identified as highly expressed in activated mouse macrophages, leading to the name of ‘leukocyte-specific plastin’ or ‘L-plastin’ (Shinomiya et al., 1994). Current research on L-plastin follows two well-traveled roads: its role in normal cells, and its role in tumor cells. In the normal context, L-plastin is characteristic of B-cells, T-cells, macrophages, monocytes, natural killer cells, and neutrophils (reviews by Delanote et al., 2005; Morley, 2012). In these cells, L-plastin is required for several critical functions including maturation, migration and immune synapse formation (Wang et al., 2010; Todd et al. 2011, 2016; Deady et al., 2014; Zhou et al., 2016). L-plastin is also expressed in osteoclasts, where it is necessary for cell-substrate contact and thus bone resorption (Ma et al., 2010; Chellaiah et al., 2018). Immune cells and osteoclasts are similar in that they are wandering cells that must make high-affinity contacts with specific extracellular targets. Thus, they likely require enhanced actin-bundling to stabilize cell membrane projections used in directed migration or substrate adhesion.
Enhanced migration is the hallmark of cancer, and most cancer deaths result from cellular spread from the primary tumor. L-plastin is thus additionally relevant due to its widespread expression in cancer cells of multiple origins. In animal models, overexpression of L-plastin contributes to apoptotic resistance, matrix invasion and metastatic spread (Janji et al., 2010), whereas siRNA suppression has the opposite effect (Chaijan et al., 2014; Inaguma et al., 2015). In humans, L-plastin has been suggested as diagnostic or prognostic marker for colon, kidney, nasopharyngeal, and oral cancers (Klemke et al., 2007; Samstag and Klemke, 2007; Li et al., 2010; Li and Zhao, 2011), and the significance of such expression is the subject of periodic reviews (Samstag and Klemke, 2007; Shinomiya, 2012). The question still unsolved from L-plastin’s discovery 30 years ago is how the same gene is regulated to be constitutive in selected lineages where it is useful, yet simultaneously repressed in all other cells. Therefore the regulatory control of L-plastin is also of interest, and will enhance the study of both normal and cancerous cells.
Here we show that L-plastin is not restricted to zebrafish leukocytes, but has an additional major expression domain during early embryonic development. Consistent with its reported mRNA expression, L-plastin protein is first detected at late blastula. Spatially, expression is restricted to the enveloping layer (EVL), an extraembryonic tissue with epithelial characteristics. Temporally, expression coincides with EVL differentiation and migration, when cells migrate rapidly over the yolk. Weak periderm expression persists to the tailbud stage, but quickly fades; by 24 h, expression is restricted to individual, wandering myeloid cells. These observations establish that zebrafish L-plastin is activated in two embryonic waves, with a distinct regulatory switch between the early, EVL and the later leukocyte.
To better study L-plastin expressing cells, we generated a novel stable transgenic zebrafish- Tg (lcp1:Cre-P2A-EGFP-CAAX)- containing a multifunction protein cassette downstream of a 990 bp lcp1 intronic fragment. This cassette drives nuclear-localized Cre recombinase and membrane-enriched EGFP in all L-plastin expressing cells. F2 embryos show Mendelian inheritance of the transgene, as well as triple expression of Cre, EGFP and L-plastin in the EVL. The fluorescent membrane label provides excellent detail of EVL migratory activity, membrane protrusions and mitotic events in vivo, as demonstrated by time-lapse confocal microscopy. To isolate the sequences that drive early embryonic expression in zebrafish, we dissected key elements of the targeted lcp1 locus and tested a series of promoter constructs in vivo. This yielded a ~300 bp intronic sequence sufficient to drive EVL and periderm expression, but unable to activate leukocyte expression. Our in silico analysis identifies this sequence as an alternative transcriptional start site (TSS) within the lcp1 first intron, which is active only during zebrafish gastrulation and segmentation (an 18-h interval). Overall, our novel, stable zebrafish line should be useful for studying enveloping layer specification, gastrulation movements and periderm development in this widely used vertebrate model. In addition, the regulatory sequences we have identified will enhance the study of how L-plastin expression is controlled in specific lineages such as skin, bone and the immune system.
2. Results
2.1. L-plastin protein is expressed in the enveloping layer and the embryonic periderm
When our study began, we knew that L-plastin mRNA was expressed in the EVL from late gastrulation to the early somite stage, and in wandering leukocytes at later stages (Thisse et al., 2001). To identify the earliest cells in which L-plastin protein is expressed, we performed whole-mount immunohistochemistry on wild type embryos. Our survey began at the 1–2 cell stage - a time at which L-plastin localization has not been well described - and ended at 2 dpf, when leukocyte expression is clearly established. All cleavage-stage embryos were negative (not shown), leading us to conclude that zebrafish L-plastin is not maternally deposited and that zygotic expression is weak or absent prior to mid-blastula.
As anticipated from previous reports of mRNA localization, anti-body signal first appeared at late blastula. At this stage, blastomeres contribute to three distinct populations: the enveloping layer (EVL), the deep cells (DC), and the nuclei of the yolk syncytial layer (YSL; Bruce, 2016). As epiboly proceeds, the EVL advances over the DC and encloses the yolk ball as a single layer of squamous epithelial cells forming the outer covering of the embryo. By 50% epiboly, L-plastin protein was detected exclusively in the EVL (Fig. 1A and B). This expression persisted through yolk plug closure and into somite-stage embryos (Fig. 1C and D). Interestingly, LCP1 expression within the EVL is not uniform, but appears variegated, such that cells are lighter or darker than their neighbors (Fig. 1E). Within each cell, fluorescent signals were distributed as dots or clumps throughout the cytoplasm (Fig. 1F). After the 15-somite stage, we saw immunoreactivity in broad areas of the zeb-rafish periderm, though at decreased intensity. By 1 dpf, periderm expression weakened and was replaced by intense, individual leukocyte staining (not shown).
Fig. 1.
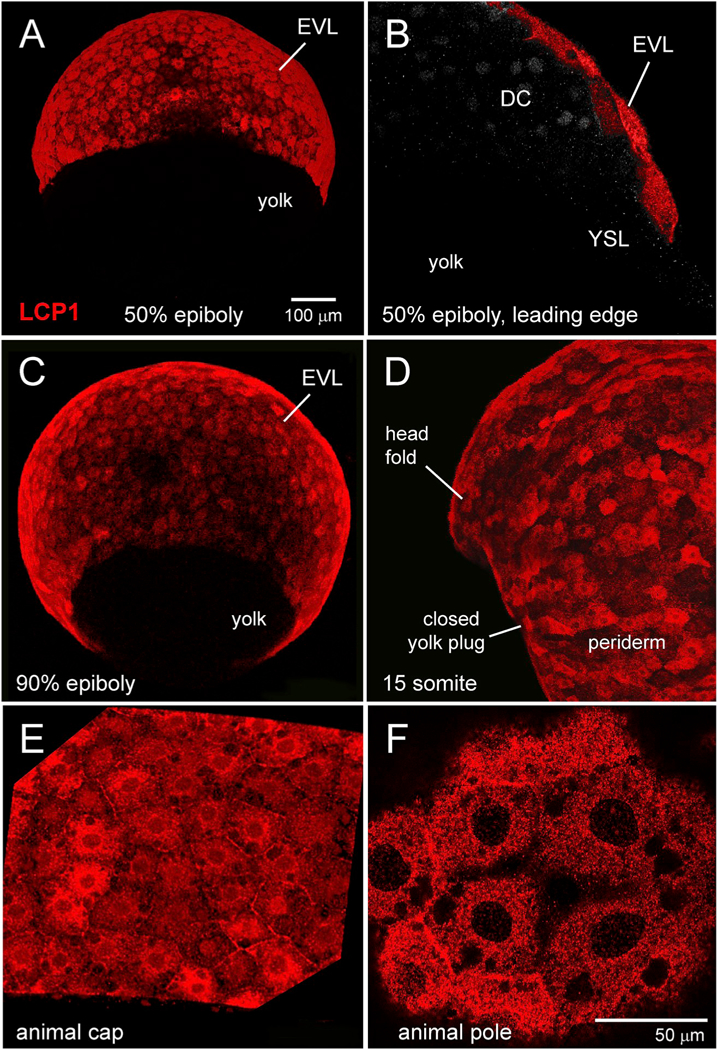
Immunohistochemical survey of L-plastin expression in early zebrafish development.
A. Enveloping layer (EVL) expression at 50% epiboly, seen in equatorial view.
B. EVL expression at 50% epiboly, seen in cutaway view of a single focal plane. The squamous EVL is strongly stained, in contrast to the deep cells (DC) and the yolk syncytial layer (YSL).
C. EVL expression at 90% epiboly, seen obliquely from the vegetal pole.
D. Periderm expression at the somite stage. Stain intensity is variegated, appearing brighter or darker in adjacent cells.
E. 70% epiboly, intermediate magnification of animal cap EVL.
F. 70% epiboly, high magnification of animal cap EVL. In this single focal plane, L-plastin expression appears as tightly packed cytoplasmic dots or clumps.
DC = deep cells; EVL = enveloping layer; YSL = yolk syncytial layer.
These observations support a model in which L-plastin protein is not maternally deposited and expression is activated at late blastula stage. After differentiation of the blastomere lineages into EVL, DC and YSL, L-plastin is a highly specific marker for the EVL and its immediate derivative, the embryonic periderm.
2.2. Construction and validation of a stable transgenic L-plastin reporter line
Based on our previous work, we knew that fish carrying one or two L-plastin null alleles (lcp1+/− and lcp1−/−) were viable and fertile (Kell et al., 2018). This allowed us to plan a homology-driven recombination (HDR) experiment in which one copy of L-plastin would be replaced with an engineered construct downstream of the native promoter.
We first designed a CRISPR guide RNA to target the first coding exon (Fig. 2A; Table 1). This reagent was highly efficient, producing indels in > 95% of injected embryos (Fig. 2B and C). Next, we prepared a transgenic cassette for insertion. Key features of this cassette were 1) an optimized Kozak sequence surrounding the translational start site, 2) a single open reading frame containing both a Cre recombinase and a membrane-targeted green fluorescent protein (EGFP-CAAX), and 3) two ~1 kB homology arms flanking the target exon (Fig. 2D). For a detailed explanation of cassette features and assembly procedures, see Methods. Finally, cassette DNA was combined with the validated guide RNA and purified Cas9 protein to induce HDR (Fig. 2E).
Fig. 2.
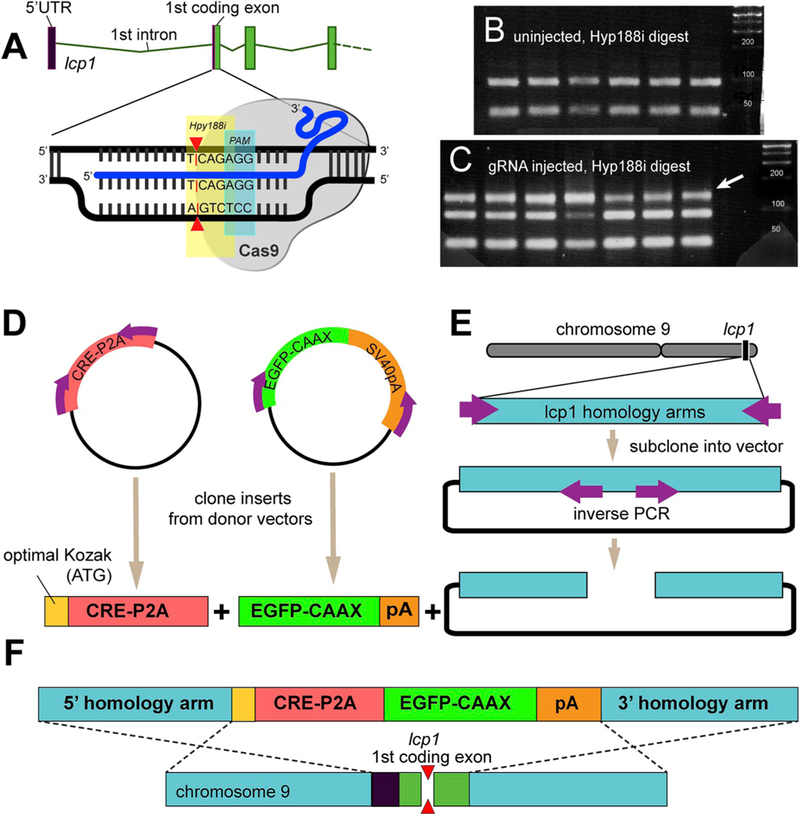
Targeted modification of zebrafish L-plastin.
A. Plan for a guide RNA (blue) targeting the first coding exon of lcp1. The predicted CRISPR/Cas9 double strand break (red triangle) interrupts a Hpy188I restriction site (yellow). PAM = protospacer adjacent motif. For guide RNA and genotyping primer sequences, see Table 1.
B. Wild type Hpy188I restriction pattern. The unmodified amplicon appears as a doublet.
C. High-efficiency indel activity at the gRNA target site. Compared to uninjected controls, 100% of injected embryos show an upper, undigested band (white arrow) indicating loss of the targeted Hpy188I restriction site.
D. Construction of a transgenic cassette targeting the first coding exon of lcp1. The cassette features an optimized Kozak sequence (yellow) followed by a Cre recombinase/P2A self-cleaving peptide (red), a membrane-targeted EGFP-CAAX (green) and a SV40 polyA (orange).
E. The cassette open reading frame is flanked by ~1kB 5’ and 3’ homology arms (blue) comprising chromosomal sequences isogenous to the injected fish population. For assembly details, see Methods 4.3.
F. Planned homology-driven recombination (HDR) in which the transgenic cassette displaces the first coding exon of lcp1. Following recombination, modified cells will have one allele encoding full-length L-plastin, and one allele encoding the inserted transgene.
Table 1.
Key oligos and primers for the knock-in construct.
| guide RNA targeting zebrafish lcp1 exonl | |
| LPLASTIN CRISPR E1 T7 | 5’-TAA TAC GAC TCA CTA TAG GAG ATG GAG GAG CTC AGA GGT TTT AGA GCT AGA AAT AGC- 3’ |
| lcp1 exonl genotyping primer, ~195 bp fragment | |
| L-plastin CRISPR_E1_T7 geno FWD | 5’-TCA ACG TGC TCT GAC TCT GG-3’ |
| L-plastin CRISPR_E1_T7 geno REV | 5’-TGC GCA CAT GTC TCT GTA CT-3’ |
| Gibson assembly primers for lcp1 (Cre-P2A-EGFP-CAAX-SV40polyA) | |
| LCP1_CRE_FWD | 5’-CAG GTG ATC ATC ATA GCG GGC AAA CAT GGC GAA TTT ACT GAC GGT AC-3’ |
| CRE_P2A_REV | 5’-CAG CTT GCT TCA GTA GGC TGA AGT TAG TAG CTC CGC TTC CAT CGC CAT CTT CCA GCA GGC-3’ |
| P2A_EGFP_FWD | 5’-CAG CCT ACT GAA GCA AGC TGG AGA CGT GGA GGA GAA CCC TGG ACC TCT GGT GAG CAA GGG CGA GGA G-3’ |
| EGFP_LPC1_REV | 5’-CTG ACC GAC TTT AGT GAA GGC GAG GAT CAT AAT CAG CCA TAC CA-3’ |
| lcp1 genomic region, ~2.8 kB on chromosome 9 | |
| LCP1 LARGE F1 | 5’-AGC TTT CAG ATG ATG CAG ATA TCT CGT G-3’ |
| LCP1 LARGE R1 | 5’-GAG TAT GAG TGC TGT GTT CCC GAC TG-3’ |
Three months after injecting this mixture into several hundred wild type embryos, we outcrossed surviving F0 fish to screen for germline transmission. Using epifluorescence microscopy at 70% epiboly, we confirmed intense EGFP+ EVL cells in the progeny of a single female. Approximately 20 of these transgenic embryos were then grown to adulthood, producing F1s. Although all F1 individuals showed the expected 50% transmission, two founder males provided the greatest signal intensity. These males were mated weekly with wild type females and we used their F2 progeny to analyze transgenic signals in vivo.
2.3. Tg(lcp1:Cre-P2A-EGFP-CAAX) embryos recapitulate early L-plastin expression
Consistent with the protein expression of our target gene (Fig. 1), transgenic embryos first showed weak fluorescent EVL cells at 30% epiboly (Fig. 3A). Over the next 4–6 h this signal intensified, high-lighting the vegetal migration of the EVL over the yolk (Fig. 3B and Movie S1). Full-thickness confocal stacks confirmed that EGFP + cells were limited to a single layer (Fig. 3C), whereas the deep cells, YSL and yolk mass were negative for transgene activation.
Fig. 3.
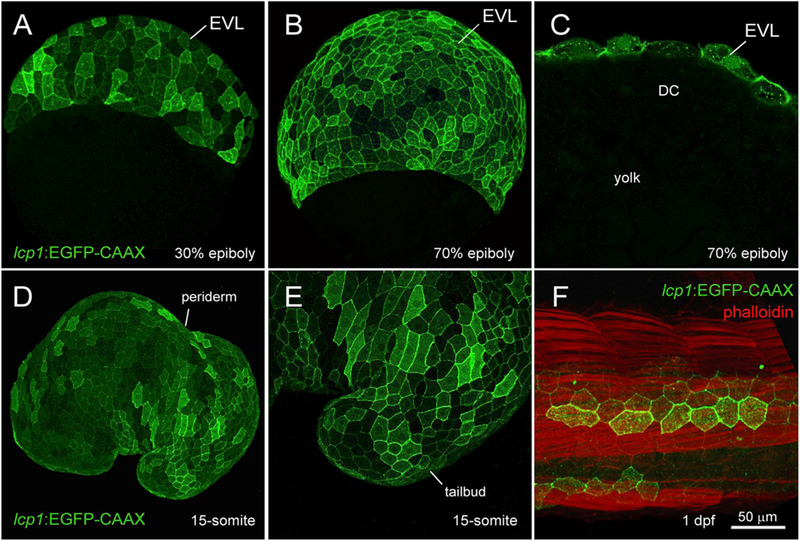
Transgenic expression in lcp1:EGFP-CAAX embryos.
A. 30% epiboly, transgenic embryo in equatorial view. The EVL shows weak, variegated EGFP expression.
B. 70% epiboly, transgenic embryo in equatorial view. Signal intensifies as the EVL spreads over the yolk.
C. 70% epiboly, cutaway view of a single focal plane. Only EVL cells are EGFP +, in contrast to the deep cells (DC) and yolk syncytial layer (YSL).
D. Live embryo at the 15-somite stage. lcp1:EGFP-CAAX labels the periderm, the outermost epithelial layer.
E. Magnification of the tailbud in D, showing variegated EGFP in adjacent peridermal cells.
F. Fixed embryo at 1 dpf, with phal-loidin-568 counterstain. A patch of label-retaining peridermal cells (green) lies over the developing trunk musculature (red).
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gep.2019.03.001.
After yolk plug closure, the EVL ceases migration and differentiates in place, forming the embryonic periderm (Fig. 3D). Although every peridermal cell was still fluorescent to some degree, the intensity was variegated from cell to cell (Fig. 3E). By somite stage (~24 hpf) widespread peridermal label had decreased significantly. In its place we observed regional labeling of smaller cells groups, often in clusters or rows. Although this signal was variable from embryo to embryo, by repeated sampling we were able to document transgenic epithelial cells covering almost every body region, including the cornea, pericardium, tailbud, and trunk (Fig. 3F). These label-retaining periderm cells, or LRPCs, provided crisp confocal images with well-defined membrane features, including EGFP accumulations in the labyrinthine apical mi-croridges typical of zebrafish epidermis (Bereiter-Hahn et al., 1979; Uehara et al., 1991; Pinto et al., 2018). Scattered LRPCs remained visible, though rarer, up to the mid-pharyngula stage (~30 hpf). By 2 dpf, EGFP signals were undetectable (not shown).
To verify that the transgenic EGFP + cells in these embryos also expressed endogenous L-plastin, we immunostained several hundred F2 gastrulae with the previously-used primary antibody (anti-zebrafish LCP1). This procedure yielded equivalent sets of EGFP+ and LCP1 + cells (Fig. 4). As expected, the transgenic signal was brightest in the EVL plasma membrane, consistent with the CAAX-labeled fluorophore, while the antibody signal was brightest in the EVL cytoplasm, consistent with L-plastin’s cytoskeletal role.
Fig. 4.
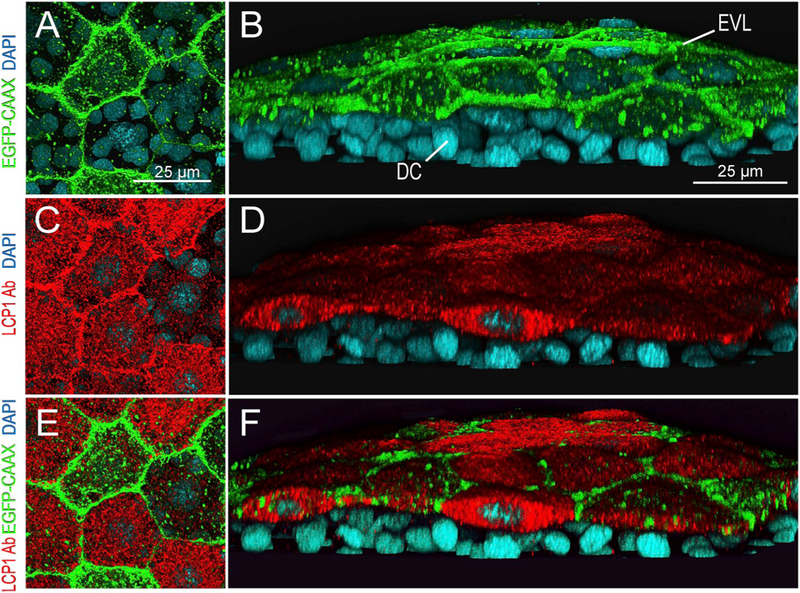
lcp1:EGFP-CAAX and endogenous LCP1 protein co-label the EVL in transgenic embryos.
A. Z-projection showing EGFP signals in EVL cells of a lcp1 :EGFP-CAAX gastrulating embryo. Signals are strongest in the membrane, with scattered foci in cytoplasmic vesicles and perinuclear Golgi.
B. Tilted stack of the same volume as (A), showing EGFP in the EVL but not in the underlying deep cells (DC).
C,D.Red channel views of the same volume, showing whole-mount immunostaining with anti-zebrafish L-plastin. Stain intensity is variegated, but is limited to the EVL.
E. Merged image of A and C.
F. Merged image of B and D. As expected, the two labels are in different compartments: transgenic EGFP-CAAX highlights the EVL membrane, while endogenous L-plastin (an actin-bundling protein) fills the EVL cytoplasm. Neither signal appears in the deep cells.
Overall, these observations show that our novel transgenic line recapitulates the expected early pattern of L-plastin protein expression in the transparent zebrafish embryo. The fluorescent label provides effective live imaging of EVL and periderm cells up to the mid-pharyngula stage.
2.4. Time-lapse observation of membrane-labeled EVL documents baso-lateral fluttering, radial protrusive activity and heritable levels of EGFP intensity
By designing our construct with a CAAX-tagged EGFP we wished to draw attention to the migratory and protrusive activities of L-plastin expressing cells. To document this activity in the EVL during epiboly, we collected time lapse images of transgenic embryos for up to 18 h in vivo. In the following description we summarize our observations of lateral protrusive activity, radial protrusive activity, and mitotic events in EVL cells.
2.4.1. Lateral and radial protrusive activity
The EVL has a distinct anatomical position, cellular morphology and cytoskeletal specializations (Ho, 1992; Bruce, 2016). Phenotypically, it resembles a simple squamous epithelium. However, a basal lamina is absent, meaning that EVL cells crawl directly on the underlying deep cells. Adjacent EVL cells are connected laterally by apical tight junctions (Kiener et al., 2008; Fukazawa et al., 2010) and their peripheral cytoplasm is enriched with filamentous actin (Zalik et al., 1999; Cheng et al., 2004). In contrast, the underlying deep cells are morphologically rounded, loosely connected, and lack a dense F-actin perimeter. In combination, these features allow one to assign blastomeres to either the EVL or DC rather unambiguously.
Without an attached lamina, the basal membranes of the EVL can be rather free-moving. Protrusions that extend underneath adjacent EVL cells have been documented in fixed embryos using labeled phalloidin (Zalik et al., 1999) and transmission electron microscopy (Slanchev et al., 2009). However, these structures have not, to our knowledge, been examined in live embryos. We therefore took advantage of our membrane-enhanced transgenics to record EVL protrusive behavior at both the animal cap and equatorial margin of gastrulating embryos between 50% and 70% epiboly. Time lapse sequences (30–40 min) showed that local membrane extensions were extremely dynamic, both in lateral extent and elongation velocity (Fig. 5A & Movie S2). Individual membrane shapes varied greatly, flowing between lamellipodial and filopodial forms. In some cases we saw distal membranes break off from the cell proper, forming one or several short-lived vesicles. In addition to apical Z-projections showing the lateral extent of EVL membranes, we used 4D rendering to view our time series data from the basal, rather than the apical surface. By coloring these images according to depth, we identified numerous radially-directed protrusions repeatedly probing the underlying deep cells (Fig. 5B & Movie S3).
Fig. 5.
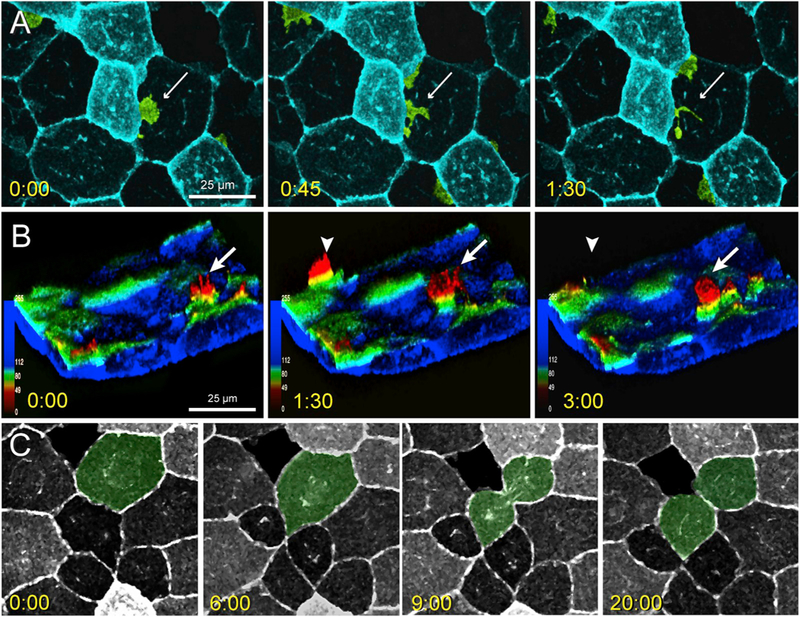
Lateral protrusive activity, radial protrusive activity, and mitotic events in lcp1:EGFP-CAAX EVL cells.
A. Time-lapse imaging of EVL lateral protrusive activity (Movie S2). The view is of the animal cap, and several membrane projections are false-colored green. A bright EVL cell extends a la-mellipodial projection (white arrow) under the nucleus of an adjacent dark cell. The projection then breaks up into smaller filopods and vesicles.
B. Time-lapse imaging of EVL radial protrusive activity (Movie S3). The confocal volume has been segmented to remove the deep cell layer, inverted to show EVL membranes extending upwards, and recolored using an automated depth-coding scheme. Blue voxels = apical extreme; red voxels = basal extreme. Radial protrusions appear as yellow-to-red spikes (white arrows) that extend and retreat every few minutes.
C. Time-lapse imaging of EVL mitosis. The dividing cell is false-colored green. EGFP expression levels vary from cell to cell but are stable throughout the cell cycle. When mitoses occur, daughters inherit the parental signal. Timestamp for all panels = minutes:seconds.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gep.2019.03.001.
2.4.2. EGFP intensity is persistent through the cell cycle
An unusual feature of L-plastin mRNA, protein and transgene expression is the spatially-variegated intensity in adjacent EVL cells. Although we thought initially that these intensity variations might be short-term and transient, time lapse imaging showed that single-cell fluorescence intensity was extremely durable, persisting up to 8 h in vivo. Dividing EVL cells, although rare, were seen easily due to a marked concentration of EGFP in the perinuclear Golgi, and the rearrangement of this organelle during different phases of the cell cycle (Fig. 5C). Interestingly, these mitotic events always produced daughters with identical EGFP levels, similar to the parent cell.
2.5. Transgenic embryos express active Cre in EVL cells, allowing lineage tracing of isogenic groups
After the experiments above, we were confident that the engineered lcp1 :EGFP-CAAX was an effective visual marker for L-plastin-expressing cells. To confirm that the lcp1:Cre recombinase was also present, we performed whole-mount immunohistochemistry with an anti-Cre anti-body. By outcrossing F1s to wild types, we generated 50:50 wildtype: transgenic clutches and fixed these at several stages up to 1 dpf. Beginning at 30% epiboly we detected weak nuclear Cre in 50% of the offspring, consistent with Mendelian inheritance of the transgene. By 70% epiboly this signal brightened, clearly marking transgenic versus wild type siblings (Fig. 6A and B). As expected, both EGFP and Cre signals co-localized in the EVL and the intensities were correlated positively (Fig. 6C). High magnification Z-stacks confirmed strong anti-Cre signals in the large, flattened nuclei of EVL cells, and not in the deep cells, which have small, rounded nuclei (Fig. 6D).
Fig. 6.
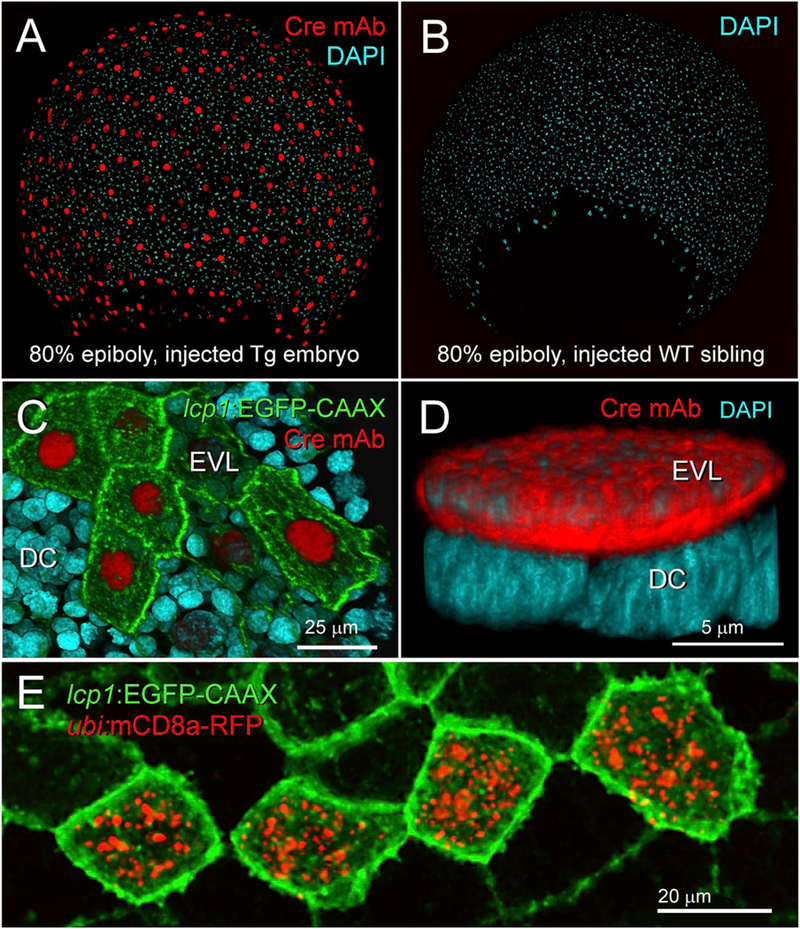
Expression and activity of Cre recombinase in lcp1(Cre-P2A-EGFP-CAAX) transgenic embryos.
A. Fixed transgenic embryo at 80% epiboly. Immunohistochemical labeling shows robust nuclear signal in all EVL cells. red = anti-Cre monoclonal anti-body, blue = DAPI nuclear stain.
B. Fixed wild type sibling embryo co-stained with the specimen in (A). Although exposed to the same primary and secondary antibody solutions, no nuclei stain red.
C. Fixed transgenic embryo, animal cap at 80% epiboly. EGFP and Cre signals are positively correlated. Cells with bright membranes have bright nuclei; cells with dim membranes have dim nuclei.
D. High magnification view of a single, Cre-positive EVL nucleus (red) overlying multiple Cre-negative deep cell nuclei (blue).
E. In vivo confirmation of Cre activity. After transient transgenesis with a Cre reporter plasmid, a live cluster of transgenically labeled periderm cells (green) expresses recombinant RFP-tagged mouse CD8a (red). For details, see Methods.
After these observations in fixed tissues, we confirmed Cre activity in vivo by injecting a customized expression construct containing a floxed stop cassette upstream of a red-tagged transmembrane protein (mouse CD8a-TagRFP-T). TagRFP-T is a slow-folding monomeric red fluorophore with excellent brightness and stability (Shaner et al., 2008). To improve signal further, we postponed tissue collection until segmentation stage, allowing new RFP molecules to accumulate. Using this assay, we observed bright red puncta in clusters or rows of EGFP + peridermal cells, consistent with clonal expansion from a recombined parent cell (Fig. 6E). In contrast, RFP was absent from both uninjected transgenics and injected wild-type controls (not shown).
Summarizing our observations, we conclude that the lcp1 :Cre-P2A-EGFP-CAAX transgenic line faithfully recapitulates L-plastin protein expression within the first 24 h of zebrafish development. Using a single open reading frame containing the P2A self-cleaving peptide, we were able to express two separate proteins-membrane EFP and nuclear Cre-in all EVL cells. In future studies, this combination should be useful to visualize, trace and genetically modify the EVL and its lineage derivative, the embryonic periderm.
2.6. In silico identification of an evolutionarily-conserved lcp1 intronic element with stage-specific transcriptional activation in zebrafish
Our experiments thus far had confirmed stable Mendelian inheritance and strong expression of the transgenic construct in the EVL and periderm. However the lack of fluorescent label in leukocytes plus the inability to detect targeted insertions in chromosome 9 by PCR raised the possibility that cassette integration had been not sequence-specific, but random. We reasoned that a random insertion could only recapitulate EVL expression in one of two states. One state would be if integration had occurred, just by chance, near another EVL-active locus. The other state would be if the construct itself contained both a core promoter and an EVL-specific enhancer and could act as a ‘mini-gene’. To test the mini-gene hypothesis, we injected several clutches of wild type embryos with plasmid DNA only, omitting the guide RNA and Cas9 protein (see 4.3.4). Within 6 h, we observed that approximately 50% of the injected embryos had fluorescent EVL cells (not shown). Having definitive evidence for endogenous promoter activity, we next analyzed the cassette’s homology arms for candidate regulatory regions.
There are no previous studies on the zebrafish L-plastin promoter, though several groups have examined the human and mouse sequences (Lin et al. 1993, 1997; Peng et al., 2001). Despite high amino acid conservation among these species’ proteins (~82%), comparing their intron-exon structures is more challenging. The zebrafish first intron is large (> 13 kB) and contains over 67% repeating elements, predominantly DNA transposons. The human and mouse first introns are even larger (> 20 kB) and also highly repetitive. Predictably, these introns do not align well, confounding the search for shared functional regions.
Considering that other fish genomes might provide a better structural match for the zebrafish sequence under study, we examined the L-plastin locus in several other teleosts. Three species in particular- the stickleback, platyfish, and amazon molly-shared a first intron of only 4kB (Fig. 7A). We reasoned that these ‘minimal introns’ might show key areas of regulatory conservation. Using these 4 kB sequences in a multiple alignment (Table 2), we identified four peaks of > 50% sequence similarity (Fig. 7B). These were termed evolutionarily conserved regions, or ECRs. Notably, the fourth ECR was located at the extreme 3’ end of the intron. By searching the zebrafish genome with this short consensus ECR, we found that the corresponding region fell within our previously-constructed ~1 kB 5’ homology arm.
Fig. 7.
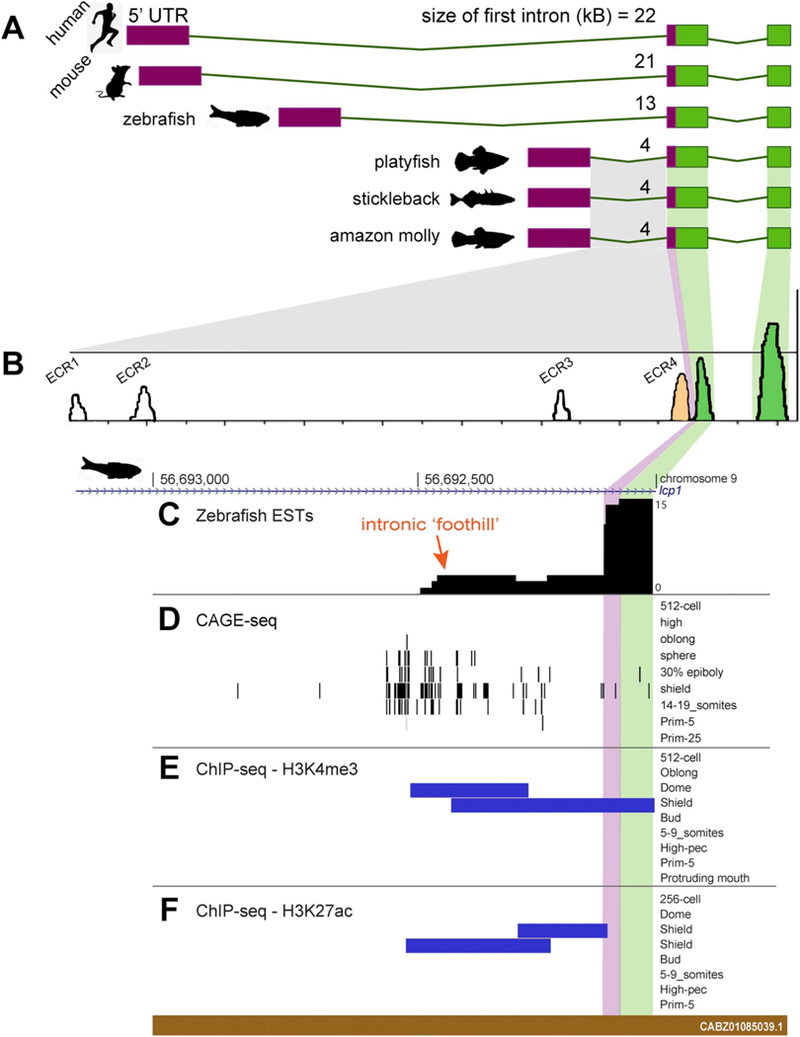
Phylogenetic comparison of lcp1 first exon size and discovery of several evolutionarily-conserved regions (ECRs).
A. Comparative size of the lcp1 first intron in selected vertebrate genomes. 5’UT1 = purple; protein-coding regions = green.
B. mVISTA alignment of the lcp1 first intron in the amazon molly, stickleback, and pla-tyfish reveals 4 evolutionarily conserved regions (> 50% similarity), labeled ECR1 through ECR4.
C. Spliced ESTs show an intronic ‘foothill’ (orange arrow) adjacent to the canonical exon boundary, indicating transcriptional activity. These foothill ESTs are stage-specific to shield or segmentation stage embryos (see Table 3).
D. A CAGE-seq cluster active between sphere and somite stages suggests a devel-opmentally-regulated alternative transcriptional start site (TSS) approximately 500 bp upstream of the canonical exon boundary.
E&F. Activating histone modifications (H3K4me3 and H3K27ac) overlap the ESTs and the CAGE-seq cluster. These ontogenetic marks appear in only a narrow stage-specific window, from dome through shield. 5’ UTR = 5’ untranslated region; ECR = evolutionarily conserved region; lcp1 = L-plastin.
Table 2.
Teleost genomic regions.
Realizing that this ECR was the likely source of EVL-specific promoter activity, we sought additional information at both genetic and epigenetic levels. Because more than a year had passed since the design of our original construct, we were able to exploit new public resources in this attempt. Specifically, we used the DANIO-CODE track hub (Tan et al., 2016), which annotates the GRCz10 zebrafish assembly with both predicted and experimental gene regulatory information (July 2018, danio-code.zfin.org). The following sections describe four specific annotations-ESTs, CAGE-seq, histone modifications and RNA-seq- that were most informative in our target region.
2.6.1. Expressed sequence tags (ESTs)
ESTs are short sequence of cDNA that have been mapped to the genome (Boguski et al., 1993). As such, they record transcriptional activity in the tissue and stage examined. ESTs can be classified as spliced or unspliced, and those that are spliced should match only exons. Accumulations of spliced ESTs thus document alternative exons used within a locus. Within the zebrafish first intron, we found a minor peak of spliced ESTs (N = 4; Fig. 7C). Although this peak was inconspicuous relative to the EST coverage in the adjacent exon, the expectation of observing four hits at random in a < 100 bp window is extremely small- less than one in 1012 (van Arensbergen et al., 2017). Of the four sequences represented, three were from a shield-stage library and one was from a segmentation-stage library (Table 3). The narrow ontogenetic range of these ESTs, plus the experimentally-confirmed promoter activity of the lcp1 5’ homology arm, led us to consider that this sequence contained an alternative transcriptional start site (TSS) active only in the early embryo.
Table 3.
Spliced ESTs in the 5’ homology arm.
| EST | Tissue Source | Library | Clone | Genomic Position |
| BI892175 | shield stage, 6 h post-fertilization | LIBEST_009773 | MPMGp637_20L5 | chr9:56669317–56692132 |
| BI889201 | shield stage, 6 h post-fertilization | LIBEST_009773 | MPMGp637_22C18 | chr9:56689976–56692323 |
| BI889183 | shield stage, 6 h post-fertilization | LIBEST_009773 | MPMGp637_22J17 | chr9:56689766–56692352 |
| BQ169336 | segmentation stage embryo | LIBEST_009662 | CB245 | chr9:56689765–56692332 |
2.6.2. CAGE-seq peaks and activating histone modifications
Cap analysis of gene expression (CAGE) allows the sensitive identification of transcriptional start sites (Fort and Fish, 2017). Active TSS contain CAGE-seq clusters, and we detected just such a cluster within the lcp1 first intron (Fig. 7D). In chromosomal coordinates, the maximum CAGE signal was located at the 5’ end of the EST ‘foothill’. On-togenetically, peaks became detectable at 30% epiboly and persisted through segmentation (14–19 somite stage). A similar pattern was observed for two types of activating histones-H3K4me and H3K27ac - that were present from dome to shield stage, but absent at prior and later stages (Fig. 7E and F). These histone marks are associated with RNA polymerase II occupancy and generally predict transcriptionally active or ‘poised’ sequences (Aday et al., 2011).
2.6.3. Stage-specific RNA-seq reads
Finally, by reviewing RNA-seq reads that map to the lcp1 first intron and first coding exon, we detected an ontogenetically bivalent transcriptional landscape with distinct differences between early and later stages (Fig. 8). Beginning at dome stage, embryos activate the intronic TSS, generating RNA-seq reads that cross the intron-exon boundary. However by late segmentation (prim-5 and later) reads map only to the canonical first coding exon. This exon boundary is maintained at later larval stages and in many adult tissues.
Fig. 8.
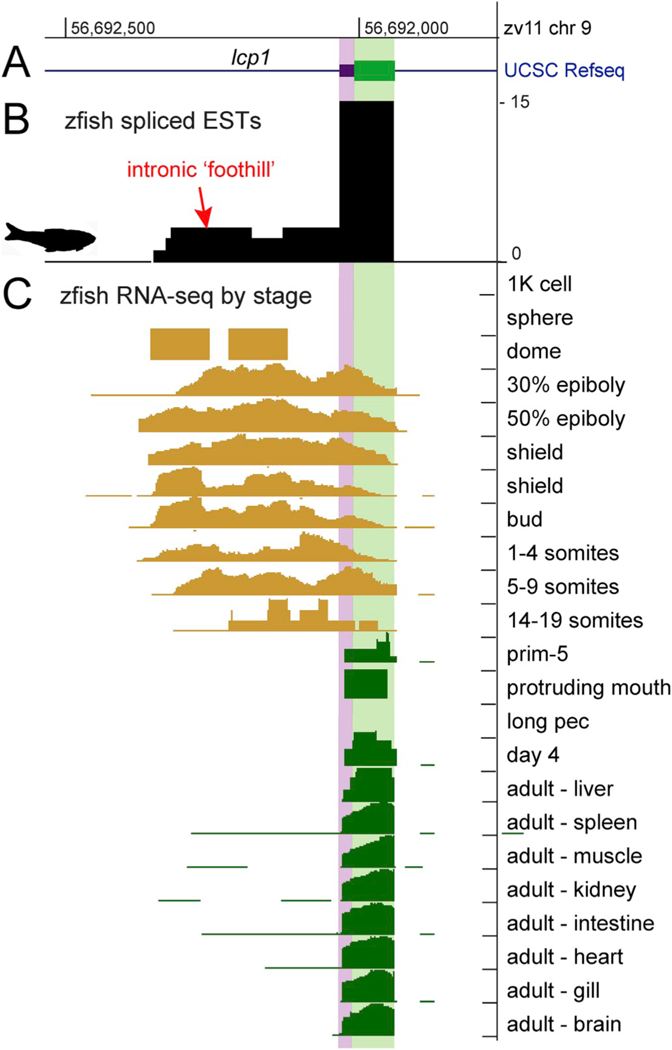
Stage-specific transcriptional activity of zebrafish lcpl ECR4.
A. Schematic of the first coding exon of lcp1. 5’ UTR = purple; protein-coding region = green.
B. Spliced ESTs form an inconspicuous ‘foothill’ upstream of the first coding exon.
C. RNA-seq reads by stage. Intronic expression (orange) is absent prior to dome stage but becomes strong at 30% epiboly and persists until segmentation (14–19 somites). Intronic read activity is absent at later stages (dark green) which show only canonical expression of the first coding exon. Source: UCSC Genome Browser, DANIO-CODE track hub
In summary, alignment of several fish genomes reveals multiple evolutionarily conserved elements (ECRs) within the first intron of lcpl. Regulatory annotations of the zebrafish genome reveal that, in this species, the intron’s 3’ end contains a stage-specific TSS that is maximally active from epiboly through segmentation. At later stages, its transcriptional activity is shut down. This bivalent chromatin state is consistent with the switch in L-plastin expression from early EVL cells to later leukocytes, and reveals a possible intronic mechanism for this gene’s two-stage regulation.
2.7. In vivo assessment of lcp1 promoter activity
From our original HDR construct containing both 5’ and 3’ homology arms we produced five modified fragments for promoter analysis in vivo. The 3’ homology arm was removed completely, as few regulatory motifs were found there. Within the 5’ homology arm we designed a six-construct deletion series (Fig. 9A). Because the start of transcription was not yet defined, each construct was named according to the number of bases 5’ of the translational start codon (ATG) within our engineered Kozak sequence. After subcloning, sequencing and purification, each construct was injected separately into zebrafish zygotes, and healthy embryos were screened at 50% epiboly for EGFP expression in EVL cells.
Fig. 9.
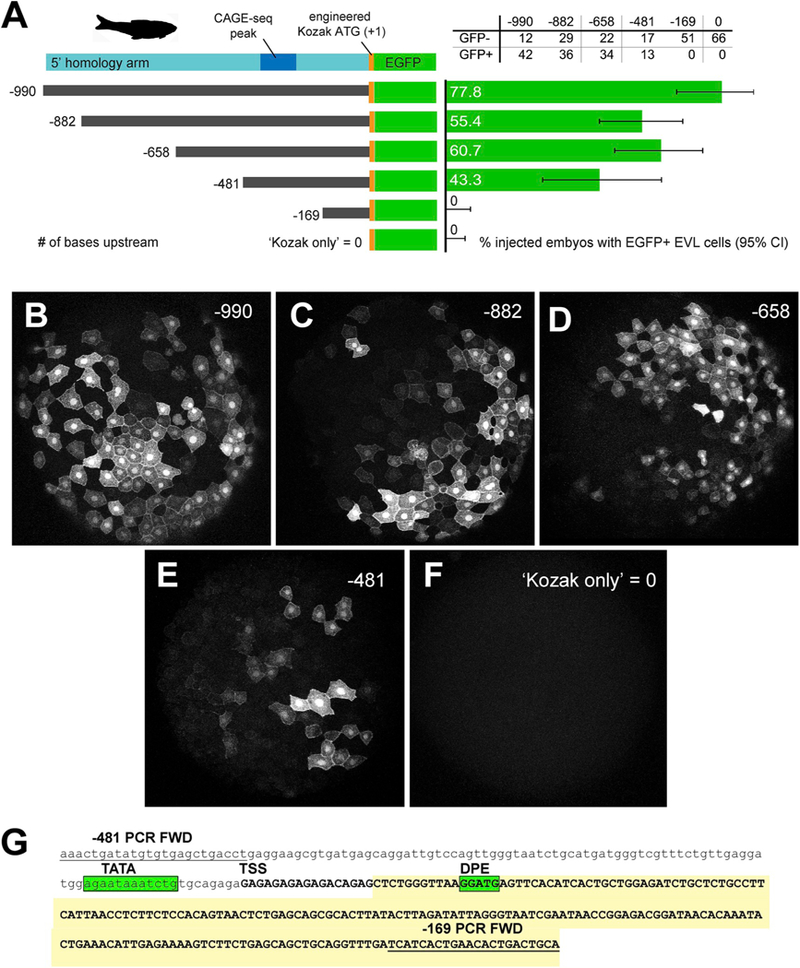
In vivo validation of zebrafish first intron promoter and enhancer activity.
A. LEFT: 5’ deletion series of the zebrafish 5’ homology arm upstream of the transgenic cassette (lcp1:Cre-P2A-EGFP-CAAX, abbreviated ‘EGFP’). Constructs are numbered according to the number of bases upstream of the engineered translation start site (ATG, where A = +1.) A. RIGHT: Results of deletion-series injections. Green bars = proportion of injected embryos with EGFP + EVL cells. Gray lines = 95% confidence interval on the proportion.
B-F. Animal cap views of injected, EGFP + embryos collected at 50% epiboly.
G. Annotation of the minimal intronic region (−481 to −169) sufficient for EVL activity. The deletion-construct forward PCR primers are underlined. The predicted transcript is in UPPER CASE. TATA = TATA box motif; TSS = transcription start site; DPE = downstream promoter element. The EST ‘foothill’ (See Figs. 7 and 8) is shaded yellow.
Of the six constructs tested, the four longest fragments (−990, −882, −658 and −481) produced similar distributions of brightly-labeled EVL cells (Fig. 9B-E). Within each treatment, the proportion of transformed embryos was highly variable (43–77%), as was the number of labeled cells per embryo. The two shortest fragments (−169 and 0) produced no signals (Fig. 9F). We conclude that an approximately ~300 bp region, between −481 and −169, is sufficient to drive EVL expression. In silico analysis of this region using a eukaryotic promoter predictor (YAPP,www.bioinformatics.org/yap; Gershenzon and Ioshikhes, 2004; Jin et al., 2006) confirmed a synergistic pair of core elements (TATA/DPE) in this interval (Fig. 9G). The predicted transcript, which begins between these two elements, matches both the EST ‘foothill’ and CAGE-seq cluster in the same region. The discovery of these elements within the 5’ homology arm explains in part how the lcpl:Cre-P2A-EGFP-CAAX cassette can function as a mini-gene, driving EVL expression in zebrafish embryos.
3. Discussion
3.1. Characterization of L-plastin expression in early zebrafish development
L-plastin’s best known role is as a leukocyte marker (Morley, 2012). Less well understood is its expression in other contexts. Here, we extend previous work on this gene’s mRNA localization and show that L-plastin is a highly specific marker for the EVL and its derivative, the embryonic periderm. Using a zebrafish-specific L-plastin antibody, we find that protein expression in these cells is punctate, with clusters or dots spread throughout the cytoplasm. This spatial pattern is consistent with that reported for human cells, including T-cells in vitro (Freeley et al., 2012). Similar patterns have been observed in C. albicans yeast buds (Zhang et al., 2016) and C. elegans zygotes (Ding et al., 2017), indicating broad conservation of plastin/fimbrin protein localization in diverse eukaryotes. Within zebrafish, L-plastin’s early restriction to the EVL resembles that of other EVL-localized genes including the epithelial cell adhesion molecule EpCAM (Slanchev et al., 2009) and various cyto-keratins (Imboden et al., 1997; Gong et al., 2002; Sagerstrom et al., 2005; Chen et al., 2011; Krens et al., 2011), extending the list of genetic markers for this embryonic cell population.
The initial goal of our study was to produce a L-plastin reporter expressing membrane-enhanced GFP in all leukocytes, allowing detailed observation of their protrusive and phagocytic activities. Unexpectedly, we produced a L-plastin reporter that drives GFP to the EVL. This novel, stable line provides an excellent opportunity to visualize EVL migration, intercalation, and differentiation in vivo. During zebrafish development, the EVL is the first tissue to achieve a distinctive morphology. Its flattened cells form a protective barrier that is essential for regulating osmotic pressure and surface tension. Yet this covering is not stationary, but migrating. Successful epiboly depends on EVL cells crawling from the animal pole over the yolk with traction forces provided by a leading-edge actomyosin ring (Ho, 1992; Solnica-Krezel et al., 1996; Yin et al., 2008). While maintaining lateral connectivity, EVL cells also mutually intercalate to reduce the number of leading-edge cells. By labeling the EVL with membrane-localized EGFP, the lcp1 :Cre-P2A-EGFP-CAAX transgenic line provides a convenient optical platform for observing these characteristic structures and movements. At low magnification, we could image live embryos up to 18 h without any loss of signal. At high magnification, durations of 30–40 min still provided good contrast; beyond this, degradation of signal could be corrected by mild image processing.
An unexpected observation in our line is that lcp1 :EGFP-CAAX expression is heterogeneous, appearing brighter or darker in adjacent EVL cells. This heterogeneity occurs at three levels— after in situ localization of mRNA, after immunostaining of protein, and after genomic integration of our lcp1 promoter construct. What causes this effect? Initially, we considered calcium waves. During gastrulation, calcium waves occur randomly but repeatedly in EVL cell clusters (Slusarski et al., 1997; Webb and Miller, 2003). In support of this concept, a recently-published transgenic reporter highlights patches of calcium-rich EVL cells similar in size and shape to what we observe (Chen et al., 2017). Other factors that could influence expression of our construct include multiple integration sites and variable copy numbers within the two F1 males. However, as we have observed Mendelian inheritance of this EVL variegation in three generations of their descendants, it is unlikely that these factors apply. Our working hypothesis is that individual EVL cells differentially express the transgene based on preexisting internal regulatory states that affect the inherited promoter. These states, as yet unknown, may lie beneath the observation that many EVL-specific genes are expressed in a mosaic or ‘salt-and-pepper’ pattern (Fukazawa et al., 2010; Kotkamp et al., 2014; Liu et al., 2015).
L-plastin is significant because it is one of the most abundantly-expressed proteins in normal leukocytes, but is misexpressed in many non-hematopoietic cancer cells. What features would unite the EVL, leukocytes, and cancer cells? Although leukocytes and cancer cells have some obvious phenotypic overlap, the shared features of leukocytes and the EVL are less clear. The former are individual, amoeboid mesodermal cells, whereas the latter is a squamous epithelial sheet. Despite these differences, the fact that both express high levels of the same cytoplasmic actin-bundler may point to a shared functional requirement. Because actin bundling by L-plastin is thought to stabilize membrane projections in moving or probing cells, we hypothesize that the EVL uses L-plastin to stabilize its baso-lateral and radially-directed filopods during epiboly. Experiments to test this hypothesis are now under way, crossing the lcp1 :Cre-EGFP transgenic with our recently-developed lcp1 knockout lines (Kell et al., 2018).
By choosing a large (~1 kB) homology arm for our planned HDR construct, we inadvertently dissected out a novel intronic regulatory sequence. We find that a ~300 bp fragment of the zebrafish lcp1 first intron is sufficient to recapitulate endogenous L-plastin expression in the gastrula and pharyngula. Although promoter elements are classically thought to cluster at the 5’ ends of loci, genome-wide studies of transcription, translation and enhancer activity show that regulation is more diffuse than formerly appreciated. Most genes have multiple TSS - four, on average - that are regulated in complex ways (Carninci et al., 2006; Nepal et al., 2013; review by Bagchi and Iyer, 2016). First introns are also known to have strong regulatory roles. In particular, the 5’ and 3’ ends of such introns tend to be protected from interspersed repeats and other mutations, reflecting selection for key motifs adjacent to splice sites (Majewski and Ott, 2002). This model applies well to the lcp1 sequence isolated here, in which an alternative TSS lies immediately adjacent to the first coding exon. Therefore introns should be examined for rare but possibly significant elements of regulatory control.
3.2. Concluding remarks
Although early work on L-plastin identified this gene in myeloid lineages and cancer cells, we have verified that it is also expressed during zebrafish gastrulation. By inserting a multifunction transgenic cassette downstream of a lcp1 intronic sequence, we were able to express both a site-specific recombinase and a fluorescent tracer in the EVL and periderm, providing a novel tool for the study of these cell populations. Given that the zebrafish embryo is an outstanding optical platform for assessing cell behavior in vivo, it is likely that this will become a completely new context for studying L-plastin protein function. The ease of chemical and genetic modification of the embryo, plus the availability of numerous transgenic and mutant lines will also accelerate research in this area.
4. Materials and methods
4.1. Fish strains and breeding
The DePaul University Institutional Animal Care and Use Committee (IACUC) approved all animal protocols. Adult fish were kept in a heated, recirculating rack system at 28 °C on a 14:10 light-dark cycle with a maximum stocking density of 1 fish/200 mL. The adult diet was a 50:50 mix of finely ground flake food (TetraMin) and decapsulated brine shrimp eggs (American Brine Shrimp) given once or twice a day.
Fertilized eggs were obtained by placing adult pairs in a 500 mL crossing tank filled with system water. Clutches were collected the following morning and unfertilized embryos removed. After 5–6 days of development, small fry were transferred to a stand-alone rotifer coculture system (Best et al., 2010) for up to one week before transitioning to standard housing. Larvae were fed live rotifers and small amounts of purchased powdered diet (Golden Pearl™ 50–100 and 100–300 μm food).
4.2. . Whole-mount immunohistochemistry
The procedures used in this study followed our previous published reports (Figueroa et al., 2015; Kell et al., 2018). Rabbit anti-zebrafish L-plastin, the kind gift of Dr. Michael Redd, was used at 1:20,000–1:50,000 (Redd et al., 2006). Mouse anti-Cre recombinase (MAB3120, Sigma-Aldrich) was used at 1:2000–1:4000. Fluorescent secondaries (goat anti-rabbit Cy3 and/or goat anti-mouse Cy3; Jackson ImmunoResearch) were applied at 1:200, either with or without DAPI (1 mg/mL) to stain nuclei.
4.3. Construction of the lcp1:Cre-P2A-EGFP-CAAX transgenic line
4.3.1. Genomic DNA isolation
Fish were anesthetized using 0.015% μg/mL buffered Tricaine (Sigma-Aldrich: E102521) in system water, and then the distal half of the caudal fin was removed using a razor blade. Fish were allowed to recover in system water plus methylene blue overnight, before being returned to the recirculating system. Individual caudal fin clips were placed in PCR tubes containing 50 μL of freshly-made lysis buffer (10 mM Tris-HCl, 50 mM KCl, 0.3% Tween-20 and 1% Triton-X 100) and incubated at 98 °C for 10 min. After addition of Proteinase K (10 U), lysis proceeded at 55 °C for 2–3 h or overnight. After heat-inactivation at 98 °C for 10 min, the unpurified lysates were vortexed, spun down and stored at −20 °C. For PCR, we used 2–4 μL of lysate supernatant in a typical reaction volume of 20–50 μL.
4.3.2. Guide RNA design and validation
Within lcp1 exon1, we identified a convenient protospacer adjacent motif (PAM) within a Hpy188I restriction site. Using this sequence, we designed a guide RNA against the bases immediately upstream (Fig. 2A). gRNA synthesis and purification followed methods in our previous report (Kell et al., 2018). Table 1 lists the primers used.
We verified efficient Cas9 cleavage at the target site by injecting into 1–2 cell embryos a mixture of 50 ng/μL guide RNA, 150 ng/μL Cas9 mRNA, 300 mM KCl and 0.5% phenol red. Twenty-four hours after injection, individual embryos were genotyped with a diagnostic PCR primer pair flanking the Hpy188I target site (Table 1). These primers make an amplicon of 193 bp which, when cut with Hpy188I, yields four fragments per wild type allele: 93 bp, (2 × 44) bp and 12 bp. The 12 bp band is difficult to see, so in practice the wild type restriction pattern appears as a doublet (Fig. 2B). Indel-modified embryos are likely to have one cut site abolished, producing a unique fragment of (93 + 44) = 137 bp. Thus, the mosaic restriction pattern appears as a triplet with a diagnostic larger band (Fig. 2C).
4.3.3. Donor vector construction
The (lcp1:Cre-P2A-EGFP-CAAX) donor vector was planned as a Gibson assembly of three PCR products: a Cre-P2A fragment, an EGFP-CAAX-SV40-polyA fragment, and a vector backbone (pGEM-T Easy) containing two ~1 kB homology arms flanking lcp1 exon 1 (Fig. 2D). All fragments were amplified using high-fidelity polymerase (Q5 Hi-Fi 2× Master Mix, New England Biolabs), confirmed by gel electrophoresis, and purified using standard columns (Zymoclean™ Gel DNA Recovery Kit, Zymo Research). For primer combinations, see Table 1.
We subcloned the Cre-P2A and EGFP-SV40-polyA fragments by single-step subcloning from existing source plasmids (Fig. 2D). Using the primers shown, the Cre fragment was 1100 bp long and the EGFP-CAAX fragment was 1392 bp long. We produced the lcp1 homology arms by two-step subcloning (Fig. 2E). First, we designed a primer pair to amplify a 2804 bp region of zebrafish genomic DNA centered on the lcp1 target exon (zv11, chr9: 56,257,654–56,260,457). We then T/A cloned this product into pGEM-T Easy forming a 6 kB circle. After sequence confirmation, we designed a second primer pair for inverse PCR. Using the primers shown, this produced a ~6 kB linear molecule. Finally, we assembled all three fragments for 60 min at 50 °C using at 1:2:2 M ratio of backbone:insert:insert (NEBuilder kit, New England Biolabs).
We added 2 μL of this assembly reaction to chemically competent E. coli cells (NEB-5α, New England BioLabs) followed by heat shock and plating on selective media. After colony PCR, we expanded several clones and had them sequenced by an outside vendor (Genewiz, South Plainfield, NJ). Using DNADynamo alignment software (v1.484) we trimmed and aligned the supplied trace files to find clones with a correct open reading frame. We grew a single clone (‘1.1.18’) to midiprep volume (Zyppy™ Plasmid Midiprep Kit, Zymo Research), then stored the purified plasmid in single-use aliquots at −20 °C.
4.3.4. Embryo transgenesis and progeny screening
Egg collection and mounting followed standard methods (Yuan and Sun, 2009). We mixed circular donor vector (15–60 ng/μL), lcp1 exon 1 guide RNA (100–150 ng/μL), and recombinant nuclear-localized Cas9 protein (600 ng/μL) in a solution of 0.5% phenol red and 300 μM KCl. After mixing, this solution was kept at room temperature throughout the experiment. We then performed yolk injections at the one-cell stage using a positive-displacement microinjector (Nanoject III, Drummond Scientific), adding approximately 2 nL per embryo.
We raised surviving mosaic founders (F0) to maturity, then out-crossed these to wild types. Using epifluorescence to screen clutches at 70% epiboly, we manually selected the brightest green progeny, then grew these to produce F1s. When outcrossed to wild types, all F1s produced the expected 50:50 ratio of transgenic:wild type progeny. These clutches were again screened and selected, establishing F2s. When outcrossed to wild types, all F2s produced 50% bright, transgenic progeny, confirming stable transmission over four generations.
4.4. Confocal microscopy and image processing
All images were obtained with a Leica SP5 laser scanning confocal microscope. Fixed embryos were mounted in glass-bottom Petri dishes (MatTek P35G-0–20-C) in Tris-buffered 50% glycerol (pH 7.2–7.6). Live embryos were transferred to large glass Petri dishes containing 1× E3 medium, then dechorionated manually. The live imaging chamber was a glass-bottom Petri dish to which was added a thin (1–2 mm) layer of high-melt agarose in 1× PBS. After this layer had set, we used small-gauge blunt dispensing needles to punch out vertical cylinders of agar over the cover slip, forming a ‘golf course’ of 10–12 holes. Using a fire-polished glass pipette, we loaded 2 or 3 dechorionated embryos into each hole such that the embryos were lightly touching each other and the surrounding agar. We then flooded the plate with additional E3. In this preparation, the submerged embryos were stabilized, but could still complete morphogenetic movements. Exposure to molten or viscous mounting media was avoided, so as not to damage the EVL nor restrict diffusion.
For time lapse imaging, the prepared agar plates were covered and placed on a humidified microscope stage at 28 °C. To document epiboly, we used a 20× air lens to capture images every 1–2 min for up to 8 h. To document EVL protrusive activity, we used 40× or 60× oil-immersion lenses to capture images every 30–45 s for 40–60 min. To minimize bleaching, all scans were at a medium resolution (512 × 512) using the minimum laser power necessary. 3D and 4D datasets were exported as.lif files and processed in FIJI and FluoRender (Schindelin et al., 2012; Wan et al., 2012) using a defined workflow of built-in commands (despeckle, Z-projection, brightness/contrast adjustment, background subtraction, and drift correction.) Processed images were exported as high-resolution TIFF files for figure preparation (Adobe Creative Suite 6), or as AVI files for video editing (Movavi Video Suite).
4.5. Cre recombinase reporter assay
Our reporter plasmid for Cre recombinase was assembled as a ubiquitin-driven open reading frame (ubtloxp-STOP-loxp-mCD8a-TagRFP-T) within a standard Tol2 plasmid vector (pDEST-Tol2-p2A2). A floxed stop cassette was placed between the ubi promoter and a coding region comprising a mouse T-cell surface glycoprotein (mCD8a, from Addgene clone #17745) and a monomeric red fluorescent protein (TagRFP-T, see Shaner et al., 2008). Recombination removes the upstream stop cassette, allowing synthesis of the fusion protein. For extended examples of similar ubiquitin-driven constructs in the Tol2 vector system, see (Mosimann et al., 2011).
After standard DNA purification, circular plasmid was stored in single-use aliquots at −20 °C. Injection into one-cell embryos followed the general procedures in 4.3.4. For transient transgenesis, the reaction mixture contained 75–100 ng/μL plasmid DNA.
4.6. Analysis of evolutionarily conserved elements
To detect interspecific evolutionarily-conserved regions (ECRs) we used the mVISTA suite of programs (genome.lbl.gov/vista/). This suite aligns multiple input sequences, then extracts regions that rise above a defined similarity threshold (Mayor et al., 2000; Frazer et al., 2004). Input sequences were obtained from the reference genomes in Ensembl. Alignments were produced without repeat-masking using the LAGAN alignment option (Brudno et al., 2003). The default p-value threshold was 0.5.
4.7. Promoter deletion series and expression screening
To assess the regulatory activity of the lcp1 5’ homology arm we made a series of deletion constructs (Fig. 9). We designed five different forward primers against a common reverse primer (Table 4), amplified the fragments with a proofreading polymerase mixture (Q5 Hi-Fi, New England Biolabs) and purified the fragments from agarose gels. DNA recovery followed a modified “freeze and squeeze” method (Thuring et al., 1975). Briefly, we collected each slice of 1% agarose in a 1.5 mL plastic microfuge tube, crushed it with a micropestle and froze it at −80 °C for 1–2 h. After thawing, we crushed again and spun at 20,000 g for 15 min to recover the cleared supernatant. After purification by sodium acetate/ethanol precipitation, the product was modified for T/A cloning using Bullseye™ Taq (MidSci Scientific). A-tailed fragments were then ligated into the pGEM-T Easy cloning vector, the same backbone as the source sequence.
Table 4.
Primers and PCR conditions for promoter subcloning.
| Construct Prefix (_lcpl :Cre-P2A-EGFP-CAAX) | Forward Primers | Primer Pair Annealing Temp (Ta)* |
|---|---|---|
| −822 | 5’ TGC ATT ACA TAC AGG AAC AGC GG 3’ | 68 °C |
| −658 | 5’ AAA CTG CAT GCT GTG ATT ACT TC 3’ | 64 °C |
| −481 | 5’ AAA CTG ATA TGT GTG AGC TGA CCT 3’ | 67 °C |
| −169 | 5’ TCA TCA CTG AAC ACT GAC TGC A 3’ | 67 °C |
| 0, ‘Kozak only’ | 5’ AAA CAT GGC GAA TTT ACT GAC GG 3’ | 68 °C |
| Reverse Primer | ||
| All constructs | 5’ GTC TCT GTA CTG ACC GAC TTT AGT GA 3’ | |
We calculated annealing temperatures for these reactions using the manufacturer’s on-line calculator (tmcalculator.neb.com) which is recommended for the specific
polymerase used (Q5 High-Fidelity 2× Master Mix, New England Biolabs).
After bacterial transformation, clonal expansion, DNA isolation and sequence confirmation, each construct was injected into 1-cell embryos (see 4.3.4). The injection solution contained 75 ng/μL circular plasmid DNA, 300 mM KCl, and 0.5% phenol red in nuclease-free water. Control embryos were injected with a ‘Kozak only’ construct that contained the complete open reading frame (Cre-P2A-EGFP-CAAX) but no 5’ flanking sequence. Injected embryos were returned to egg water at 28.5 °C and fixed at 50% epiboly. Fixed embryos were mounted in 1% high-melt agar in egg water, such that the animal cap was facing the cover slip. Confocal images were collected as 75-μm stacks starting from the animal pole inwards. Image acquisition and processing were as described in 4.4.
4.8. Data graphing and null hypothesis statistical testing
We graphed the deletion-series results using Prism software and examined within- and among-treatment variation using 95% confidence intervals on the proportion (Newcombe, 1998).
4.9. Key Resources
Additional data regarding experimental reagents, resources and suppliers are provided in the Key Resources document.
Supplementary Material
KEY RESOURCES TABLE.
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti zebrafish L-plastin | Michael Redd (Redd et al., 2006) | N/A |
| Mouse monoclonal anti-Cre recombinase (clone 2D8) | Millipore | Cat#MAB3120; RRID: AB_2085748 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: Tg(lcp1:CRE-P2A-EGFP-CAAX) | This paper | ZFIN: ZDB-TGCONSTRCT-180718-1 |
| Oligonucleotides | ||
| CRISPR gRNA targeting zebrafish lcp1 first coding exon, see Table 1 | This paper | N/A |
| Genotyping primers for zebrafish lcp1 first coding exon, see Table 1 | This paper | N/A |
| Gibson assembly primers for making the CRE-P2A-EGFP-CAAX cassette, see Table 1 | This paper | N/A |
| Primers for amplifying lcp1 genomic region for 5′ and 3′ homology arms, see Table 1 | This paper | N/A |
| Primers for constructing lcp1 5′ deletion series, see Table 4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: HDR donor vector cassette (CRE-P2A-EGFP-CAAX) with 5′ and 3′ homology arms in pGEM-T Easy backbone | This paper | N/A |
| Plasmid: CRE recombinase reporter (ubi-loxp-STOP-loxp-Cd8a-TagRFP-T) in pDestTol2pA2 backbone | This paper | N/A |
| Software and Algorithms | ||
| mVISTA | European Molecular Biology Laboratory | genome.lbl.gov/vista/mvista/ |
| Other | ||
Acknowledgements
Financial support was provided by grants from the National Institute of General Medical Sciences (R15-GM120664, to E. E. LeClair) and the DePaul University Research Council (URC). Confocal analysis was made possible in part by FluoRender software funded by the National Institutes of Health (NIH R01-GM098151 and P41-GM103545). We thank Janine Kirin, J.D. Davis, Jesse Hacker and Jordan Johnson for their contributions to fish care, and Mason Posner (Ashland University, OH) for valuable pre-submission review.
Footnotes
Declarations of interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gep.2019.03.001.
References
- Aday AW, Zhu LJ, Lakshmanan A, Wang J, Lawson ND, 2011. Identification of cis regulatory features in the embryonic zebrafish genome through large-scale profiling of H3K4me1 and H3K4me3 binding sites. Dev. Biol. 357 (2), 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi DN, Iyer VR, 2016. The determinants of directionality in transcriptional initiation. Trends Genet. 32 (6), 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Osborn M, Weber K, Vöth M, 1979. Filament organization and formation of microridges at the surface of fish epidermis. J. Ultrastruct. Res. 69 (3), 316–330. [DOI] [PubMed] [Google Scholar]
- Best J, Adatto I, Cockington J, James A, Lawrence C, 2010. A novel method for rearing first-feeding larval zebrafish: polyculture with Type L saltwater rotifers (Brachionus plicatilis). Zebrafish 7 (3), 289–295. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Lowe TM, Tolstoshev CM, 1993. dbEST-database for “expressed sequence tags”. Nat. Genet. 4 (4), 332–333. [DOI] [PubMed] [Google Scholar]
- Bruce AE, 2016. Zebrafish epiboly: spreading thin over the yolk. Dev. Dynam. 245 (3), 244–258. [DOI] [PubMed] [Google Scholar]
- Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S, Program NCS, 2003. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13 (4), 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engström PG, Frith MC, 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38 (6), 626. [DOI] [PubMed] [Google Scholar]
- Chaijan S, Roytrakul S, Mutirangura A, Leelawat K, 2014. Matrigel induces L-plastin expression and promotes L-plastin-dependent invasion in human cholangiocarcinoma cells. Oncol Lett 8 (3), 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellaiah MA, Majumdar S, Aljohani H, 2018. Peptidomimetic inhibitors of L-plastin reduce the resorptive activity of osteoclast but not the bone forming activity of osteoblasts in vitro. PLoS One 13 (9), e0204209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CF, Chu CY, Chen TH, Lee SJ, Shen CN, Hsiao CD, 2011. Establishment of a transgenic zebrafish line for superficial skin ablation and functional validation of apoptosis modulators in vivo. PLoS One 6 (5), e20654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xia L, Bruchas MR, Solnica-Krezel L, 2017. Imaging early embryonic calcium activity with GCaMP6s transgenic zebrafish. Dev. Biol. 430 (2), 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JC, Miller AL, Webb SE, 2004. Organization and function of microfilaments during late epiboly in zebrafish embryos. Dev. Dynam. 231 (2), 313–323. [DOI] [PubMed] [Google Scholar]
- Deady LE, Todd EM, Davis CG, Zhou JY, Topcagic N, Edelson BT, Ferkol TW, Cooper MA, Muenzer JT, Morley SC, 2014. L-plastin is essential for alveolar macrophage production and control of pulmonary pneumococcal infection. Infect. Immun. 82 (5), 1982–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delanote V, Vandekerckhove J, Gettemans J, 2005. Plastins: versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 26 (7), 769–779. [DOI] [PubMed] [Google Scholar]
- Ding WY, Ong HT, Hara Y, Wongsantichon J, Toyama Y, Robinson RC, Nedelec F, Zaidel-Bar R, 2017. Plastin increases cortical connectivity to facilitate robust polarization and timely cytokinesis. J. Cell Biol. 216 (5), 1371–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa F, Singer SS, LeClair EE, 2015. Making maxillary barbels with a proximaldistal gradient of Wnt signals in matrix-bound mesenchymal cells. Evol. Dev. 17 (6), 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort A, Fish RJ, 2017. Deep cap analysis of gene expression (CAGE): genome-wide identification of promoters, quantification of their activity, and transcriptional net-work inference. Methods Mol. Biol. 1543, 111–126. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I, 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 (Web Server issue), W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeley M, O’Dowd F, Paul T, Kashanin D, Davies A, Kelleher D, Long A, 2012. L-plastin regulates polarization and migration in chemokine-stimulated human T lymphocytes. J. Immunol. 188 (12), 6357–6370. [DOI] [PubMed] [Google Scholar]
- Fukazawa C, Santiago C, Park KM, Deery WJ, de la Torre Canny SG, Holterhoff CK, Wagner DS, 2010. poky/chuk/ikkl is required for differentiation of the zebrafish embryonic epidermis. Dev. Biol. 346 (2), 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Cherepanova O, Lebart MC, Egelman EH, 2008. High-resolution cryo-EM structure of the F-actin-fimbrin/plastin ABD2 complex. P Natl Acad Sci USA 105 (5), 1494–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenzon NI, Ioshikhes IP, 2004. Synergy of human Pol II core promoter elements revealed by statistical sequence analysis. Bioinformatics 21 (8), 1295–1300. [DOI] [PubMed] [Google Scholar]
- Goldstein D, Djeu J, Latter G, Burbeck S, Leavitt J, 1985. Abundant synthesis of the transformation-induced protein of neoplastic human fibroblasts, plastin, in normal lymphocytes. Cancer Res. 45 (11 Pt 2), 5643–5647. [PubMed] [Google Scholar]
- Gong Z, Ju B, Wang X, He J, Wan H, Sudha PM, Yan T, 2002. Green fluorescent protein expression in germ-line transmitted transgenic zebrafish under a stratified epithelial promoter from keratin8. Dev. Dynam. 223 (2), 204–215. [DOI] [PubMed] [Google Scholar]
- Ho RK, 1992. Cell Movements and Cell Fate during Zebrafish Gastrulation. Development, pp. 65–73. [PubMed] [Google Scholar]
- Imboden M, Goblet C, Korn H, Vriz S, 1997. Cytokeratin 8 is a suitable epidermal marker during zebrafish development. C R Acad Sci III 320 (9), 689–700. [DOI] [PubMed] [Google Scholar]
- Inaguma S, Riku M, Ito H, Tsunoda T, Ikeda H, Kasai K, 2015. GLI1 orchestrates CXCR4/CXCR7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 6 (32), 33648–33657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janji B, Vallar L, Al Tanoury Z, Bernardin F, Vetter G, Schaffner-Reckinger E, Berchem G, Friederich E, Chouaib S, 2010. The actin filament cross-linker L-plastin confers resistance to TNF-alpha in MCF-7 breast cancer cells in a phosphorylation-dependent manner. J. Cell Mol. Med. 14 (6), 1264–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin VX, Singer GA, Agosto-Pérez FJ, Liyanarachchi S, Davuluri RV, 2006Genome-wide analysis of core promoter elements from conserved human and mouse orthologous pairs. BMC Bioinf. 7 (1), 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kell MJ, Riccio RE, Baumgartner EA, Compton ZJ, Pecorin PJ, Mitchell TA, Topczewski J, LeClair EE, 2018. Targeted deletion of the zebrafish actin-bundling protein L-plastin (lcp1). PLoS One 13 (1), e0190353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiener TK, Selptsova-Friedrich I, Hunziker W, 2008. Tjp3/zo-3 is critical for epidermal barrier function in zebrafish embryos. Dev. Biol. 316 (1), 36–49. [DOI] [PubMed] [Google Scholar]
- Klemke M, Rafael MT, Wabnitz GH, Weschenfelder T, Konstandin MH, Garbi N, Autschbach F, Hartschuh W, Samstag Y, 2007. Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. Int. J. Cancer 120 (12), 2590–2599. [DOI] [PubMed] [Google Scholar]
- Kotkamp K, Mössner R, Allen A, Onichtchouk D, Driever W, 2014. A Pou5f1/Oct4 dependent Klf2a, Klf2b, and Klf17 regulatory sub-network contributes to EVL and ectoderm development during zebrafish embryogenesis. Dev. Biol. 385 (2), 433–447. [DOI] [PubMed] [Google Scholar]
- Krens SF, Möllmert S, Heisenberg CP, 2011. Enveloping cell-layer differentiation at the surface of zebrafish germ-layer tissue explants. Proc. Natl. Acad. Sci. U. S. A. 108 (3) E9–10;author reply E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao R, 2011. Expression and clinical significance of L-plastin in colorectal carcinoma. J. Gastrointest. Surg. 15 (11), 1982–1988. [DOI] [PubMed] [Google Scholar]
- Li MX, Xiao ZQ, Chen YH, Peng F, Li C, Zhang PF, Li MY, Li F, Duan CJ, Li DJ, Yao HX, Chen ZC, 2010. Proteomic analysis of the stroma-related proteins in nasopharyngeal carcinoma and normal nasopharyngeal epithelial tissues. Med. Oncol. 27 (1), 134–144. [DOI] [PubMed] [Google Scholar]
- Lin CS, Aebersold RH, Kent SB, Varma M, Leavitt J, 1988. Molecular cloning and characterization of plastin, a human leukocyte protein expressed in transformed human fibroblasts. Mol. Cell Biol. 8 (11), 4659–4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Chang CH, Huynh T, 1997. The murine L-plastin gene promoter: identifycation and comparison with the human L-plastin gene promoter. DNA Cell Biol. 16 (1), 9–16. [DOI] [PubMed] [Google Scholar]
- Lin CS, Chen ZP, Park T, Ghosh K, Leavitt J, 1993. Characterization of the human L-plastin gene promoter in normal and neoplastic cells. J. Biol. Chem. 268 (4), 2793–2801. [PubMed] [Google Scholar]
- Liu H, Leslie EJ, Jia Z, Smith T, Eshete M, Butali A, Dunnwald M, Murray J, Cornell RA, 2015. Irf6 directly regulates Klf17 in zebrafish periderm and Klf4 in murine oral epithelium, and dominant-negative KLF4 variants are present in patients with cleft lip and palate. Hum. Mol. Genet. 25 (4), 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Sadashivaiah K, Madayiputhiya N, Chellaiah MA, 2010. Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. J. Biol. Chem. 285 (39), 29911–29924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Ott J, 2002. Distribution and characterization of regulatory elements in the human genome. Genome Res. 12 (12), 1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I, 2000. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16 (11), 1046–1047. [DOI] [PubMed] [Google Scholar]
- Morley S, 2012. The actin-bundling protein L-plastin: a critical regulator of immune cell function. Int J Cell Biol 2012 935173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI, 2011. Ubiquitous transgene expression and Cre-based recombination driven by the ubi-quitin promoter in zebrafish. Development 138 (1), 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal C, Hadzhiev Y, Previti C, Haberle V, Li N, Takahashi H, Suzuki AMM, Sheng Y, Abdelhamid RF, Anand S, 2013. Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Res. 23 (11), 1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcombe RG, 1998. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat. Med. 17 (8), 873–890. [DOI] [PubMed] [Google Scholar]
- Peng XY, Won JH, Rutherford T, Fujii T, Zelterman D, Pizzorno G, Sapi E, Leavitt J, Kacinski B, Crystal R, Schwartz P, Deisseroth A, 2001. The use of the L-plastin promoter for adenoviral-mediated, tumor-specific gene expression in ovarian and bladder cancer cell lines. Cancer Res. 61 (11), 4405–4413. [PubMed] [Google Scholar]
- Pinto CS, Khandekar A, Rajasekaran B, Kiesel P, Pigino G, Sonawane M, 2018. Microridges are apical projections formed of branched F-actin networks that organize the glycan layer. bioRxiv 442871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redd M, Kelly G, Dunn G, Way M, Martin P, 2006. Imaging macrophage chemotaxis in vivo: studies of microtubule function in zebrafish wound inflammation. Cell Motil. Cytoskelet. 63 (7), 415–422. [DOI] [PubMed] [Google Scholar]
- Sagerström CG, Gammill LS, Veale R, Sive H, 2005. Specification of the enveloping layer and lack of autoneuralization in zebrafish embryonic explants. Dev. Dynam. 232 (1), 85–97. [DOI] [PubMed] [Google Scholar]
- Samstag Y, Klemke M, 2007. Ectopic expression of L-plastin in human tumor cells: diagnostic and therapeutic implications. Adv. Enzym. Regul. 47, 118–126. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T,Preibisch S, Rueden C, Saalfeld S, Schmid B, 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9 (7), 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY, 2008. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5 (6), 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya H, 2012. Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int J Cell Biol 2012 213492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya H, Hirata H, Saito S, Yagisawa H, Nakano M, 1994. Identification of the 65-kDa phosphoprotein in murine macrophages as a novel protein: homology with human L-plastin. Biochem Bioph Res Co 202 (3), 1631–1638. [DOI] [PubMed] [Google Scholar]
- Slanchev K, Carney TJ, Stemmler MP, Koschorz B, Amsterdam A, Schwarz H, Hammerschmidt M, 2009. The epithelial cell adhesion molecule EpCAM is required for epithelial morphogenesis and integrity during zebrafish epiboly and skin development. PLoS Genet. 5 (7), e1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarski DC, Yang-Snyder J, Busa WB, Moon RT, 1997. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev. Biol. 182 (1), 114–120. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, Stemple DL, Mountcastle-Shah E, Rangini Z, Neuhauss SC, Malicki J, Schier AF, Stainier DY, Zwartkruis F, Abdelilah S, Driever W, 1996. Mutations affecting cell fates and cellular rearrangements during gastrulation in zebrafish. Development 123, 67–80. [DOI] [PubMed] [Google Scholar]
- Tan H, Onichtchouk D, Winata C, 2016. DANIO-CODE: toward an encyclopedia of DNA elements in zebrafish. Zebrafish 13 (1), 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Pflumio S, Fürthauer M, Loppin B, Heyer V, Degrave A, Woehl R, Lux A, Steffan T, Charbonnier X, Thisse C, 2001. Expression of the zebrafish genome during embryogenesis (NIH R01 RR15402). ZFIN direct data submission. http://zfin.org. [Google Scholar]
- Thuring R, Sanders J, Borst P, 1975. A freeze-squeeze method for recovering long DNA from agarose gels. Anal. Biochem. 66 (1), 213–220. [DOI] [PubMed] [Google Scholar]
- Todd EM, Deady LE, Morley SC, 2011. The actin-bundling protein L-plastin is essential for marginal zone B cell development. J. Immunol. 187 (6), 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EM, Zhou JY, Szasz TP, Deady LE, D’Angelo JA, Cheung MD, Kim AH, Morley SC, 2016. Alveolar macrophage development in mice requires L-plastin for cellular localization and retention within alveoli. Blood 128 (24), 2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara K, Miyoshi M, Miyoshi S, 1991. Cytoskeleton in microridges of the oral mucosal epithelium in the carp, Cyprinus carpio. Anat. Rec. 230 (2), 164–168. [DOI] [PubMed] [Google Scholar]
- van Arensbergen J, FitzPatrick VD, de Haas M, Pagie L, Sluimer J, Bussemaker HJ, van Steensel B, 2017. Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol. 35 (2), 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Otsuna H, Chien CB, Hansen C, 2012. FluoRender: an application of 2D image space methods for 3D and 4D confocal microscopy data visualization in neurobiology research. IEEE Pac Vis Symp 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Morley SC, Donermeyer D, Peng I, Lee WP, Devoss J, Danilenko DM, Lin Z, Zhang J, Zhou J, Allen PM, Brown EJ, 2010. Actin-bundling protein L-plastin regulates T cell activation. J. Immunol. 185 (12), 7487–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb SE, Miller AL, 2003. Imaging intercellular calcium waves during late epiboly in intact zebrafish embryos. Zygote 11 (2), 175–182. [DOI] [PubMed] [Google Scholar]
- Yin C, Kiskowski M, Pouille PA, Farge E, Solnica-Krezel L, 2008. Cooperation of polarized cell intercalations drives convergence and extension of presomitic mesoderm during zebrafish gastrulation. J. Cell Biol. 180 (1), 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Sun Z, 2009. Microinjection of mRNA and morpholino antisense oligonucleotides in zebrafish embryos. JoVE 27 10.3791/1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalik SE, Lewandowski E, Kam Z, Geiger B, 1999. Cell adhesion and the actin cytoskeleton of the enveloping layer in the zebrafish embryo during epiboly. Biochem. Cell Biol. 77 (6), 527–542. [PubMed] [Google Scholar]
- Zhang B, Yu Q, Wang Y, Xiao C, Li J, Huo D, Zhang D, Jia C, Li M, 2016a. The Candida albicans fimbrin Sac6 regulates oxidative stress response (OSR) and morphogenesis at the transcriptional level. Biochim. Biophys. Acta 1863 (9), 2255–2266. [DOI] [PubMed] [Google Scholar]
- Zhang R, Chang M, Zhang M, Wu Y, Qu X, Huang S, 2016b. The structurally plastic CH2 domain is linked to distinct functions of fimbrins/plastins. J. Biol. Chem. 291 (34), 17881–17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JY, Szasz TP, Stewart-Hutchinson PJ, Sivapalan J, Todd EM, Deady LE, Cooper JA, Onken MD, Morley SC, 2016. L-Plastin promotes podosome long-evity and supports macrophage motility. Mol. Immunol. 78, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.