

Abstract
Covalent labeling with mass spectrometry is increasingly being used for the structural analysis of proteins. Diethylpyrocarbonate (DEPC) is a simple to use, commercially-available covalent labeling reagent that can readily react with a range of nucleophilic residues in proteins. We find that in intact proteins weakly nucleophilic side chains (Ser, Thr, and Tyr) can be modified by DEPC in addition to other residues such as His, Lys, and Cys, providing very good structural resolution. We hypothesize that the microenvironment around these side chains, as formed by a protein’s higher order structure, tunes their reactivity such that they can be labeled. To test this hypothesis, we compare DEPC labeling reactivity of Ser, Thr, and Tyr residues in intact proteins with peptide fragments from the same proteins. Results indicate that these residues almost never react with DEPC in free peptides, supporting the hypothesis that a protein’s local microenvironment tunes the reactivity of these residues. From a close examination of the structural features near the reactive residues, we find that nearby hydrophobic residues are essential, suggesting that the enhanced reactivity of certain Ser, Thr, and Tyr residues occurs due to higher local concentrations of DEPC.
Keywords: Covalent Labeling, Diethylpyrocarbonate, Mass Spectrometry, Liquid Chromatography, Protein Higher-Order Structure, Protein Microenvironment, Protein Structural Analysis
GRAPHICAL ABSTRACT
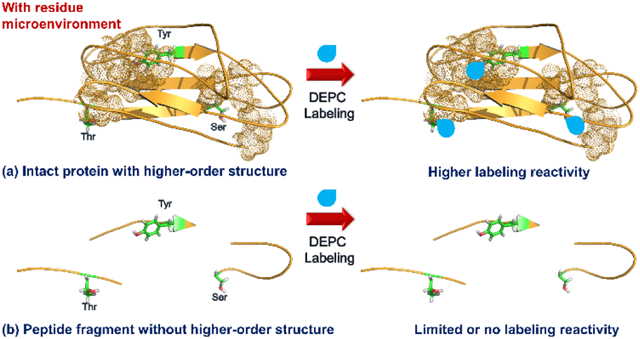
INTRODUCTION
Covalent labeling (CL) with mass spectrometry (MS) has been increasingly utilized for the higher order structural analysis of proteins and for studying protein-protein interactions. From reactions with labeling reagents, a protein’s structural properties or interactions can be encoded into the mass of protein, which can then be read-out in a straightforward site-specific manner via MS-based bottom-up analysis. The differential reactivity of proteins under different conditions can be used to distinguish and deduce protein conformations, topologies, and/or binding sites. Common applications of CL-MS techniques include protein folding/unfolding,1-4 amyloid-forming proteins,5-9 protein-ligand systems,10-13 and structural analysis of protein therapeutics,14-19 among others. Several review articles are dedicated to these relatively new MS-based structural techniques.20-26 The wider use of CL-MS techniques as an orthogonal method to probe structures of proteins results from their unique advantages over other analytical methods because of (a) a more rapid, sensitive, and sample-efficient analysis compared to NMR, X-ray crystallography, and cryo-EM, (b) the ability to obtain much higher structural resolution than optical spectroscopy, i.e. residue-level structural information can be obtained from CL-MS, and (c) the limited label loss and scrambling due to irreversible nature of labeling, compared to hydrogen-deuterium exchange (HDX) – MS. CL reagents modify solvent-exposed amino acid side chains either at specific residues (e.g. Lys labeling, Trp labeling, and carboxylic acid labeling) or a wide range of residues. In the latter case, non-specific reagents, e.g. hydroxyl radicals, carbenes, and diethylpyrocarbonate, can be used to gain greater structural resolution as they can probe a range of side chains simultaneously.
Diethylpyrocarbonate (DEPC) is a simple-to-use, commercially-available reagent that reacts with a range of nucleophilic residues and the N-terminus of proteins.20,21 Unlike radical and carbene reagents, DEPC can readily label proteins once added to solution and no specialized instrumentation is needed to generate a reactive species.20,21 CL-MS methods based on DEPC have been extensively developed by our group.27-30 We have shown its capability to obtain insight into protein-ligand and protein-protein interactions and conformational changes of therapeutic proteins with a structural resolution as low as 8 – 10 Å because up to 30% of surface exposed residues on proteins can be labeled.5-7, 12, 16 In addition to Cys, His and Lys which are good nucleophiles that readily react with DEPC and account for around 10% coverage of the average protein sequence,31 side chains of weakly nucleophilic residues, such as Ser, Thr, and Tyr, are also found to be modified by DEPC. We find that in many different proteins,16, 19, 27-29 these weak nucleophiles can be modified by DEPC to a significant extent in intact proteins. The reactivity of these residues is valuable because Ser, Thr, and Tyr are the third, seventh, and sixteenth most commonly-found amino acids in proteins, respectively, and the three of them account for about 17% of the average protein sequence.31 Indeed, the ability of DEPC to label Ser, Thr, and Tyr residues in proteins increases the resolution of structural analyses using DEPC-based CL-MS.
While Ser, Thr, and Tyr residues in proteins can be reactive with DEPC, we have found a relatively poor correlation between the solvent accessibility of these residues and the extent of their DEPC reactivity, which contrasts with the more reactive Lys and His residues. This contrast suggests that microenvironment might affect the reactivity of Ser, Thr, and Tyr residues.16, 19, 27, 29 Previous experimental and computational studies suggest that the reactivity of amino acid side chains in proteins can be tuned by the microenvironment in different ways. Microenvironment can influence the acid/base characteristics and the reactivity of side chains because of polar interactions (i.e. charge-charge, charge-dipole, or dipole-dipole)32, 33 or hydrophobic effects via Born or desolvation effects.32, 34-37 Steric hindrance as created by intra/intermolecular interactions of nearby side chains can limit reactivity.38 Also, certain microenvironments have been suggested to increase local concentrations of reactants and/or a reagent’s affinity for specific protein surface sites, thus enhancing the reactivity of some side chains.33, 39-41 In this work, we have investigated whether microenvironment effects explain the relatively poor correlation between DEPC reactivity and the solvent accessibility of Ser, Thr, and Tyr residues in proteins by comparing the DEPC reactivity of model peptides to proteins containing the same sequences but in a 3-dimensional context. We find that free peptides with Ser, Thr, and Tyr residues rarely react with DEPC under normal labeling conditions, whereas these residues in intact proteins can be very reactive. A careful study of the microenvironment around reactive Ser, Thr, and Tyr residues in proteins indicate that nearby hydrophobic patches increase the reactivity of these residues, presumably through an increased local concentration effect.
EXPERIMENTAL SECTION
Materials.
The proteins, peptides, and reagents used in this study are described in the Supporting Information.
Sample Preparation and DEPC Labeling Reactions.
β-2-microglobulin (β2m) and ubiquitin were prepared in 10 mM MOPS buffer (pH 7.4), while human growth hormone (hGH) was prepared in 10 mM phosphate buffer (pH 8.0). Stock solutions of DEPC at a concentration of 69 mM were freshly prepared in acetonitrile. DEPC labeling of β2m (50 μM) was initiated by adding DEPC in a molar excess of 4, and the solution was reacted for 1 min at 37 °C. DEPC labeling of ubiquitin (10 μM) was performed at 37 °C for 5 min at a DEPC to protein molar ratio of 4 to 1. DEPC labeling of hGH (30 μM) was performed at 5:1 (DEPC: protein) molar ratio for 1 min at 37 °C. In all cases, the reaction was quenched by the addition of imidazole at a 1:50 DEPC to imidazole molar ratio, and the final amount of acetonitrile present was 1%, which does not perturb the structure of protein during the labeling. Labeling experiments with these intact proteins were performed in triplicate or quadruplicate.
For the DEPC reactions of free peptides, peptides from the same proteins were first produced via proteolytic digestion (see Proteolytic Digestion). The N-termini of the peptides were acetylated before labeling them with DEPC. N-terminal blocking was achieved by reacting sulfo-NHS-acetate in a molar excess of 1,000 for 1 h at room temperature. The N-terminally blocked peptides were labeled and quenched under the same conditions as those used for the intact proteins. It was important to ensure that the digested peptide concentrations were as close as possible to the intact protein concentration so that the DEPC reactivity for the free peptide and intact protein could be appropriately compared. To ensure that the concentrations were as close as possible, the proteolytic digest was prepared using a stock solution with a higher protein concentration, so that upon the various dilutions steps associated with digestion and acetylation, the final concentration of digested peptides was identical to the intact protein concentration. Three or four replicate reactions and analyses were conducted on each protein digest.
Proteolytic Digestion.
After the DEPC reactions, labeled β2m and hGH samples were desalted and preconcentrated in 10 mM MOPS buffer (pH 7.4) and 10 mM phosphate buffer (pH 8.0), respectively, using a centrifugal filter with a 10 kDa molecular weight cutoff (MWCO). The resulting samples were incubated with 10% (v/v) acetonitrile at 50°C for 45 min to denature the intact protein. TCEP was added in a 40-fold molar excess to reduce the disulfide bonds, and iodoacetamide was simultaneously added in a 80-fold molar excess to alkylate the reduced Cys residues. The samples were kept in the dark at room temperature for 20 min. Subsequently, the proteolytic enzyme was added to the resulting samples at a 1:10 (w/w) enzyme to substrate, and the protein was digested at 37 °C. Digestion was performed with immobilized chymotrypsin and trypsin for β2m (3 h) and immobilized trypsin for hGH (5 h). For ubiquitin, the labeled samples were desalted and preconcentrated in 25 mM ammonium bicarbonate (pH 7.8) using a centrifugal filter with a 3 kDa MWCO. 80% (v/v) acetonitrile was added to the desalted samples to denature the protein for 45 min at 50 °C. Overnight digestion was then performed with immobilized trypsin at a 1:10 (w/w) enzyme to substrate. Following digestion, the immobilized enzyme was separated from the protein digest by centrifugation. The supernatant was collected for LC-MS/MS analysis.
To prepare peptides for DEPC labeling reactions, free peptides from the same proteins were produced via the same digestion conditions, except for ubiquitin preconcentration where 10 mM MOPS buffer (pH 7.4) was used instead of ammonium bicarbonate in order not to suppress the subsequent N-terminal blocking and DEPC labeling reactions.
Liquid Chromatography (LC).
For on-line LC-MS/MS analyses, a labeled sample containing approximately 0.5 μg ubiquitin peptides or 2 μg β2m or hGH peptides was loaded on a Thermo Scientific Easy-NanoLC 1000 system (Waltham, MA). Samples were loaded, trapped, and desalted on a Thermo Scientific Acclaim™ PepMap™ C18 trap column (2 cm × 75 μm, 3 μm particle size). Separation of peptides was performed using an Acclaim™ PepMap™ RSLC C18 nanocolumn (15 cm × 75 μm, 3 μm particle size; Thermo Scientific) with a flow rate of 300 nL/min. LC/MS-grade water (solvent A) and acetonitrile (solvent B), each containing 0.1% formic acid, were used as mobile phases. A linear gradient for separation of the peptides consisted of 0% B to 50% B over 60 min (β2m), 0% B to 50% B over 30 min (ubiquitin), and 0% B to 45% B over 35 min (hGH).
Mass Spectrometry (MS).
Mass spectra were acquired on a Thermo Scientific Orbitrap Fusion mass spectrometer (Waltham, MA). The nano-electrospray ionization (ESI) was operated using a positive mode at a needle voltage of 2100 V, and the ion transfer tube temperature was set to 325 °C. Mass spectra were acquired on an Orbitrap analyzer, with a resolution of 60,000. Tandem mass spectrometry (MS/MS) was conducted on a linear quadrupole ion trap using collision induced dissociation (CID) with a normalized collision energy of 35 %. Tandem mass spectra were collected for most intense peptides with ion abundances above 5000 from each mass spectral scan. Dynamic exclusion of 30 sec was activated after 3 spectra were acquired for any given precursor ion within 5 sec. Mass detection during MS and MS/MS was performed in centroid mode to simplify data analysis.
Chymotryptic digests obtained from the DEPC reactions on β2m intact proteins underwent LC-MS/MS analyses using a Thermo Scientific Dionex Ultimate 3000 HPLC (Waltham, MA) and a Bruker AmaZon quadrupole ion trap mass spectrometer (Billerica, MA). Details about these analyses can be found in the Supporting Information.
Peptide Identification and Peak Quantification.
Thermo Scientific Xcalibur™ software was used to visualize total ion chromatograms (TICs) from raw mass spectral data files obtained from the LC-MS/MS analyses using Thermo Scientific Orbitrap Fusion. Extracted ion chromatograms (XICs) of specific masses of unmodified and modified peptides were generated. Peptide sequencing and labeling site identification were achieved using CID tandem mass spectra. Assignments of b and y ions were performed with a mass tolerance of 0.5 Da. A custom software pipeline described previously16,19 and specifically designed for protein CL-MS studies with DEPC was used to identify labeling sites and quantify peak areas. The search parameters were set as follows: a precursor mass tolerance of 10 ppm, carbamidomethylation of Cys as a variable modification, DEPC modification of His, Lys, Ser, Thr, Tyr, and N-terminus as a user variable modification. DEPC modification results in mass addition of 72.0211 Da (Scheme S1). For N-terminally blocked peptides, acetylation of N-termini was set as a fixed modification, while its side reaction at Lys was set as a variable modification. Acetylation of N-terminus or Lys leads to mass addition of 42.0106 Da (Scheme S2). Details about how the extents of DEPC modification were determined are described in the Supporting Information.
RESULTS AND DISCUSSION
DEPC Labeling Reactivity of Weakly Nucleophilic Residues
Solvent accessible nucleophilic side chains (Cys, His, Lys, Thr, Tyr, Ser) and N-termini of proteins can be modified by DEPC via a nucleophilic substitution at a carbonyl group (Scheme S1) to yield a mass addition of +72.021 Da. The specific modification sites in proteins can be identified and semi-quantified using MS-based bottom-up sequence analysis (Figure S1). As indicated in the introduction, Ser, Thr, and Tyr residues in proteins are readily modified by DEPC. Indeed, from four proteins studied previously, including equine myoglobin,29 human β2m,16, 29 hGH,16 murine IgG1,16 and rituximab,19 which have a total of 1,770 amino acids, 154 out of 198 Ser residues, 100 out of 136 Thr residues, and 48 out of 71 Tyr residues were labeled in intact proteins at relative levels ranging from 0.01 to 90%. In stark contrast, these residues in small peptides are relatively unreactive after 5 min with DEPC at a DEPC:peptide molar ratio of 5:1, which is the commonly used time and labeling ratio for intact proteins. As an example, Ser and Thr residues in the model peptide sequence of Fmoc-DGXGG-amide, where X = Ser or Thr, are completely unreactive (Figure 1a and b), whereas the same peptide with Tyr (i.e. Fmoc-DGYGG-amide) produces a relative modification extent of 5% (Figure 1c). By comparison, the same peptide sequence with His (Fmoc-DGHGG-amide) and Lys (Fmoc-DGKGG-amide) are much more reactive, having modification percentages of 88% and 43%, respectively, under the same labeling conditions (Figure 1d and e). Modification of Ser and Thr residues in this peptide is still negligible (0% and 0.3%, respectively) even with a 50-fold molar excess of DEPC (Figure S3). The same lack of reactivity at Ser, Thr, and Tyr residues is observed in other peptides too, including bradykinin and preproenkephalin, even when the N-terminus is blocked to reduce competitive reaction with DEPC (Figure S4).
Figure 1.
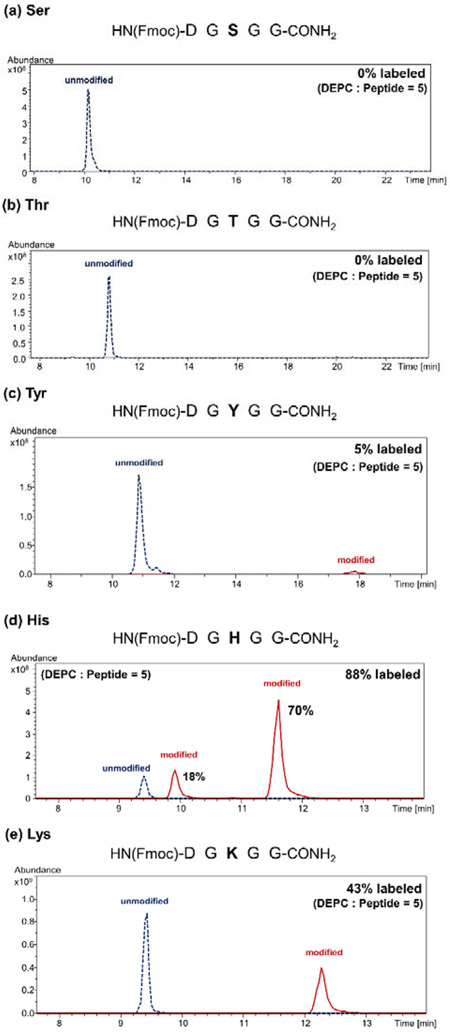
DEPC labeling of model peptides. Extracted ion chromatograms (XICs, left) of the +1 ions of the unmodified and DEPC-modified peptides Fmoc-DGXGG-amide, where X is a DEPC modifiable residue (a) Ser, (b) Thr, (c) Tyr, (d) His, or (e) Lys, after allowing the peptide to react with DEPC at a DEPC:peptide molar ratio of 5:1. Modification percentages are calculated from peak areas in XICs. Tandem mass spectra acquired after CID of the labeled precursor ions are used to confirm the site of modification. MS/MS assignments of the modified peptides are shown in Figure S2. Note that two chromatographic peaks for the labeled peptide in (d) are observed because the His side chain has two nitrogens that are separately labeled to produce isomers that can be separated by LC.
Influence of Higher-Order Structure on the Covalent Labeling Based Structural Analysis of Proteins
One possible reason for the significant reactivity of Ser, Thr, and Tyr residues in proteins but not in free peptides could be due to local sequence effects that are not fully accounted for in the model peptides. To test this possibility, we compared the labeling reactivity of intact proteins with their peptide fragments as produced by proteolysis (Figure 2). In the first scenario (Figure 2a), three proteins, β2m, ubiquitin, and hGH, were reacted with DEPC under native conditions (37 °C, pH ~ 7.5) using a DEPC molar excess of 4 or 5, which has previously been found to prevent labeling-induced structural perturbations to proteins during the labeling reaction.12, 16, 27 In the second scenario (Figure 2b), peptide fragments from the same three proteins were first produced via the same digestion conditions and then reacted with DEPC under the same labeling conditions. In this second set of experiments, the N-termini of the peptide fragments were blocked via acetylation with sulfo-NHS-acetate (Scheme S2) before labeling them with DEPC because many new N-termini are produced upon protein digestion and these newly-formed N-termini could potentially outcompete Ser, Thr, and Tyr residues.
Figure 2.
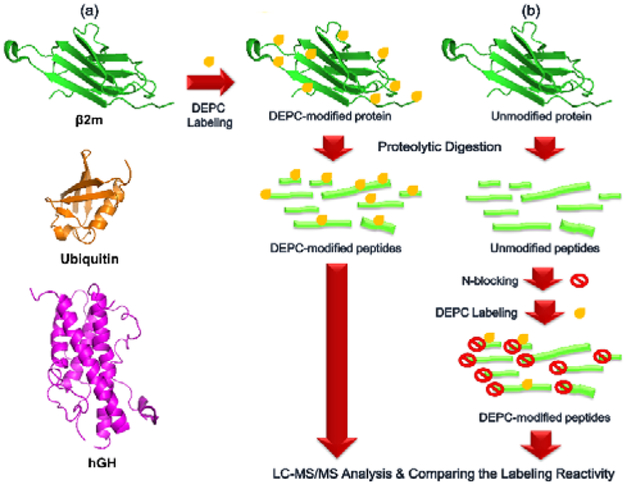
Experimental workflow to evaluate the influence of higher-order structure (i.e. microenvironment effects) on covalent labeling by comparing the DEPC labeling reactivity of (a) intact proteins with that of (b) their N-terminally acetylated peptide fragments.
Upon comparing the modification extents of Ser, Thr, and Tyr residues under the two DEPC reaction scenarios shown in Figure 2, we find that these residues are rarely modified in free peptides but are modified in the intact proteins (Table 1). When intact proteins are reacted with DEPC, 18 out of 67 Ser, Thr, and Tyr residues (9 in β2m, 2 in ubiquitin, and 7 in hGH) are labeled at levels ranging from 0.02% to 64%. When the same proteins are first digested into peptide fragments, N-terminally blocked, and then reacted with DEPC under the same labeling conditions, only two residues in the peptide fragments are found to be modified, and these are modified at very low levels (< 3%) (see MS/MS data in Figure S5). Moreover, only one of these residues (Tyr136 in hGH) is modified in the intact protein. All modifiable Ser, Thr, and Tyr residues along with all the modified His and Lys sites (His, Lys, Ser, Thr, Tyr) and their labeling levels in β2m, ubiquitin, and hGH are listed in Tables S1, S2, and S3, respectively. Together, the results indicate that the weak nucleophilic side chains, without the influence of protein higher-order structure, are poorly reactive with DEPC. These observations imply that the microenvironment around specific Ser, Thr, and Tyr residues in proteins tune their reactivity such that they can only be labeled in the 3D context of the protein.
Table 1.
DEPC modification percentages of Ser, Thr, and Tyr residues in intact proteins and peptide fragments for β2m, ubiquitin, and hGH. Each experiment was performed in triplicate or quadruplicate (n = 3 or 4). Error bars shown in a table are standard deviations. Listed are side chain solvent accessible surface areas (SASA) and brief details of the microenvironment around each residue. The presence (✔) or absence (✖) of nearby charged polar contact(s) and hydrophobic residue(s) within 4 Å and 6 Å, respectively, are presented in a table. (+) and (−) represent positively charged and negatively charged polar contacts, respectively.
| Residues | DEPC modification (%) | SASA (%)* | Microenvironment | ||
|---|---|---|---|---|---|
| Intact protein | Peptide fragments | Charged polar contact(s) [within 4 Å] |
Hydrophobic residue(s) [within 6 Å] |
||
| β-2-microglobulin (β2m) | |||||
| T4 | 59 ± 5 (N-term & T4) |
N.D.ǂ | 81.5 | ✖ | ✔ |
| Y10 | 15 ± 2 | N.D. | 41.6 | ✖ | ✔ |
| S11 | 3.0 ±0.5 | N.D. | 10.3 | ✖ | ✔ |
| S20 | N.D. | 2.6 ± 0.6 | 61.6 | H-bonding with Glu (−) | ✖ |
| S33 | 1.6 ± 0.4 | N.D. | 70.1 | ✖ | ✔ |
| S55 | 0.6 ± 0.3 | N.D. | 55.3 | ✖ | ✔ |
| S57 | 1.8 ± 0.9 | N.D. | 34.9 | H-bonding with Asp (−) | ✔ |
| S61 | 10 ± 1 | N.D. | 28.1 | H-bonding with Arg (+) | ✔ |
| Y63 | 0.3 ± 0.2 | N.D. | 27.7 | ✖ | ✔ |
| Y66 | 0.3 ± 0.2 | N.D. | 7.5 | ✖ | ✔ |
| Ubiquitin | |||||
| T7 | 13 ± 2 | N.D. | 14.2 | ✖ | ✔ |
| S65 | 2.4 ± 0.2 | N.D. | 9.2 | ✖ | ✔ |
| Human growth hormone (hGH) | |||||
| T28 | 1.3 ± 0.6 | N.D. | 10.7 | H-bonding with Asp (−) | ✔ |
| Y29 | 0.2 ± 0.1 | N.D. | 4.1 | H-bonding with Lys (+) | ✔ |
| Y36 | 0.4 ± 0.3 | N.D. | 37.2 | ✖ | ✔ |
| S56 | 0.02 ± 0.01 | N.D. | 20.8 | ✖ | ✔ |
| S58 | 64 ± 4 | N.D. | 3.3 | ✖ | ✔ |
| T136 | 0.15 ± 0.09 | 0.1 ± 0.1 | 35.9 | H-bonding with Arg (+) | ✔ |
| Y144 | 0.66 ± 0.05 | N.D. | 31.1 | ✖ | ✔ |
SASA calculation is explained in detail in the Supplemental Experimental Section.
N.D. = not detected (less than DEPC labeling threshold of 0.01%19)
Identifying Structural Features that Tune the Reactivity of Weak Nucleophiles
To identify the protein structural factors that affect the reactivity of Ser, Thr, and Tyr residues, we considered SASA and microenvironment effects such as polar and hydrophobic interactions. SASAs of the side chains were calculated using GETAREA,42 and insight into the local microenvironment around each side chain was obtained from the protein’s 3D structure using the molecular visualization program PyMOL.43 SASA is typically considered an important criteria for residue reactivity in CL-MS, but we find a relatively poor correlation between SASA and Ser, Thr, and Tyr reactivity. Typically, side chains with %SASA below 20% are considered buried;42, 44 however, we do not see a significant difference in the number of Ser, Thr, and Tyr residues having %SASA values above 20% between the sets of labeled and unlabeled residues in the three proteins. Indeed, of the 18 Ser, Thr, and Tyr residues labeled in the proteins, only 11 (or 61%) have SASA percentages above 20%, whereas in the unlabeled Ser, Thr, and Tyr residues, 34 of the 44 (or 77%) have SASA percentages above 20% (Figure S6). Hence, solvent accessibility does not significantly influence the reactivity of these weakly nucleophilic residues, which is consistent with the previously observed poor correlation between SASA and reactivity of these residues.16,19,27,29 We also considered if protein secondary structure affects the reactivity of these side chains, but we do not find a preference for α-helix, β-strand, or random coil structure among the residues that are found to be labeled (see Tables S1 to S3).
The microenvironment of a side chain can influence the acid/base characteristics of side chains because of polar interactions (charge-charge interactions, charge-dipole interactions, dipole-dipole interactions)32, 33 or hydrophobic effects (i.e. Born or desolvation effect).32, 34-37 In chemical labeling experiments conducted with monofunctional NHS esters, O-acylation of Ser, Thr, and Tyr was suggested to be dependent on protein/peptide conformation and intramolecular interactions.33, 45-47 Hydrogen bonding and other non-covalent interactions can cause changes in a side chain’s protonation state and thus affect its nucleophilicity.33, 48, 49 To explore this possible effect, we examined adjacent charged residues (Arg, Lys, Asp, Glu) and hydrogen bonding interactions between these residues and Ser, Thr, and Tyr. Nearby positively-charged residues (Arg, Lys) can presumably stabilize deprotonated forms of these weak nucleophiles, increasing their nucleophilicity, and negatively-charged residues (Asp, Glu) that form an ionic hydrogen bond with the hydroxyl group of Ser, Thr, and Tyr side chains might also increase their nucleophilicity (Scheme S3). Only interactions between these acidic and basic groups and Ser, Thr, or Tyr residues within 4 Å were considered as this is a common distance over which such interactions typically have an effect.50-53 From our results, though, there seems to be very little correlation between nearby charged residues and Ser, Thr, and Tyr labeling in proteins. Only 28% of the labeled Ser, Thr, and Tyr residues have nearby charged polar contacts, which is very similar to the 20% of the unlabeled residues that have nearby charged residues (Figure S6). Perhaps, the charged polar contacts cannot sufficiently tune the nucleophilicity of Ser, Thr, and Tyr residues to a degree that it can enhance reactivity of these side chains.
The presence of nearby hydrophobic residues was also considered as such groups might increase the local concentration of DEPC, thereby enhancing the reactivity of Ser, Thr, and Tyr residues towards chemical labeling.33, 39 Even though DEPC is readily soluble in aqueous solutions at moderate concentrations (up to 40 mM) which is useful for labeling proteins,54 this molecule is quite hydrophobic and has limited solubility in water (~ 0.1%). We hypothesized that perhaps the hydrophobicity of a protein’s local structure may enhance Ser, Thr, and Tyr reactivity by increasing DEPC’s local concentration. To study the potential influence of a hydrophobic microenvironment, we searched for the presence of hydrophobic side chains (A1a, Ile, Leu, Met, Pro, Val) and aromatic sidechains (Phe, Trp, Tyr) within 6 Å. A distance of 6 Å was chosen because of the molecular dimensions of DEPC, which is a symmetrical molecule having two electrophilic carbonyls, one of which could be positioned close enough to the hydroxyl group of Ser, Thr, or Tyr if it interacted with a hydrophobic group 6 Å away (Scheme 1). Interestingly, each of the Ser, Thr, and Tyr residues that are found to be labeled in these model proteins sits close to at least one hydrophobic side chain (Figures 3 and S6). In contrast, only half of the unlabeled Ser, Thr, and Tyr residues have nearby hydrophobic residues (Figure S6). These results strongly suggest that hydrophobicity is an important factor that enhances the labeling reactivity of Ser, Thr, and Tyr residues in intact proteins. We propose that elevated local DEPC concentrations near these hydrophobic groups explain this enhanced reactivity. An analogous idea was suggested by Zenobi et al. to explain the enhanced cross-linking reactivity of Ser, Thr, and Tyr with bifunctional NHS esters to nearby Lys or His residues.33 They argued that the high local concentration of the NHS cross-linker after formation of an N-acylated intermediate at His or Lys would allow the O-acylation of weak nucleophiles to occur at higher reaction yield. Similarly, a recent study performed by Tetin and coworkers suggested that a fluorescent label AlexaFluor488 was predominantly conjugated to Lys residues located in the proximity of Trp and Tyr residues.39
Scheme 1.
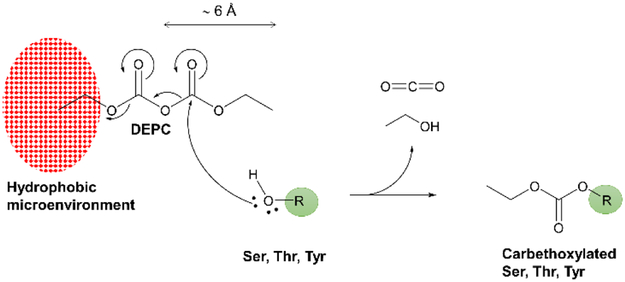
Mechanism of the nucleophilic substitution at the carbonyl group for DEPC covalent labeling. Our study indicates that hydrophobicity contributes to the higher labeling reactivity of certain Ser, Thr, and Tyr residues, due to the increase in local concentration of DEPC near these residues.
Figure 3.
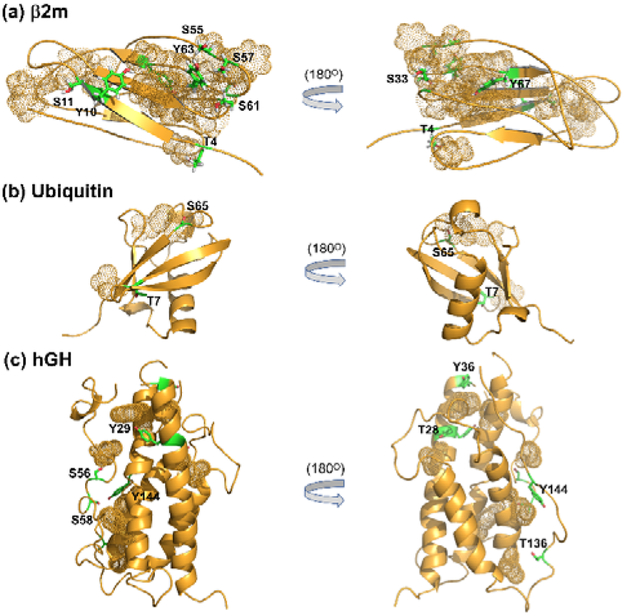
Ser, Thr, and Tyr residues that are found to be labeled by DEPC in the proteins (a) β2m, (b) ubiquitin, and (c) hGH. Side chains of the labeled residues are shown in green. Dotted spheres represent the nearby hydrophobic side chains that contribute to the higher DEPC labeling reactivity of these residues.
To further investigate the idea that the increased local DEPC concentration can drive the reactivity of Ser, Thr, and Tyr residues, we labeled β2m, ubiquitin, and hGH peptides at 10-fold higher DEPC concentrations (i.e. 50:1 DEPC:peptide). Whereas only two Ser, Thr, or Tyr residues out of 67 are labeled at low DEPC concentration (i.e. 4:1 or 5:1 DEPC:peptide), we find seven residues are modified at the higher DEPC concentration (Table 2). Obviously, this number of labeled Ser, Thr, and Tyr residues is still lower than in intact proteins, suggesting that the environment of the folded protein is still probably important for forming the necessary hydrophobic microenvironment to increase the DEPC local concentration sufficiently.
Table 2.
DEPC modification percentages of weakly nucleophilic residues in intact proteins and peptide fragments of β2m, ubiquitin, and hGH. DEPC to protein/peptide molar ratios used in the study are specified in the column headers. Each experiment was performed in triplicate or quadruplicate (n = 3 or 4). Error bars shown in a table are standard deviations.
| Residues | DEPC modification (%) | ||
|---|---|---|---|
| Intact protein (CL at 4X/5X molar excess) |
Peptide fragments (CL at 4X or 5X molar excess) |
Peptide fragments (CL at 50X molar excess) |
|
| β-2-microglobulin (β2m) | |||
| T4 | 59 ± 5 (N-term & T4) |
N.D.ǂ. | 0.02 ± 0.01 |
| Y10 | 15 ± 2 | N.D. | 3.9 ± 0.8 |
| S11 | 3.0 ± 0.5 | N.D. | 0.02 ± 0.01 |
| S20 | N.D. | 2.6 ± 0.6 | N.D. |
| S33 | 1.6 ± 0.4 | N.D. | N.D. |
| S55 | 0.6 ± 0.3 | N.D. | N.D. |
| S57 | 1.8 ± 0.9 | N.D. | N.D. |
| S61 | 10 ± 1 | N.D. | N.D. |
| Y63 | 0.3 ± 0.2 | N.D. | N.D. |
| Y66 | 0.3 ± 0.2 | N.D. | N.D. |
| Y67 | N.D. | N.D. | 1.3 ± 0.6 |
| Ubiquitin | |||
| T7 | 13 ± 2 | N.D. | N.D. |
| S65 | 2.4 ± 0.2 | N.D. | N.D. |
| Human growth hormone (hGH) | |||
| T28 | 1.3 ± 0.6 | N.D. | N.D. |
| Y29 | 0.2 ± 0.1 | N.D. | N.D. |
| Y36 | 0.4 ± 0.3 | N.D. | N.D. |
| S56 | 0.02 ± 0.01 | N.D. | N.D. |
| S58 | 64 ± 4 | N.D. | N.D. |
| S107 | N.D. | N.D. | 0.2 ± 0.1 |
| T136 | 0.15 ± 0.09 | 0.1 ± 0.1 | 0.08 ± 0.02 |
| Y144 | 0.66 ± 0.05 | N.D. | 0.02 ± 0.03 |
N.D. = not detected (less than DEPC labeling threshold of 0.01%19)
The importance of nearby hydrophobic groups for enhancing Ser, Thr, or Tyr reactivity may explain why the SASA of side chains does not correlate well with extent of reactivity for these residues. This fact can be illustrated by considering the distribution of %SASA values for both labeled and unlabeled Ser, Thr, and Tyr residues in the three proteins (Figure 4). Most of the labeled residues have SASA percentages between 5% and 45%, and only 17% of the residues have SASA values above 45%. In contrast, almost half of the unlabeled residues have SASA above 45%. As expected, we see a decrease in the average number of nearby hydrophobic sites when SASA increases (red bars in Figure 4). Between SASA values of 25% and 45%, there is a ‘sweet spot’ where many Ser, Thr, and Tyr residues are found to be labeled. In this range, the Ser, Thr, and Tyr residues have enough nearby hydrophobic sites, yet are still solvent accessible enough for DEPC to react with them. The unlabeled side chains are too exposed to the aqueous environment, and lack nearby hydrophobic groups to enhance their reactivity. Together, our findings indicate that a hydrophobic microenvironment influences Ser, Thr, and Tyr reactivity towards DEPC labeling in intact proteins more than SASA. Additional studies are needed to further verify this effect, but the reactivity of specific Ser, Thr, and Tyr residues could be used to provide insight into the local microenvironment around these residues and could be useful in CL-MS based protein structural prediction. For example, the reactivity of specific Ser, Thr, and Tyr residues could indicate the presence of nearby hydrophobic residues, and this information could be used to improve confidence in certain structural models.
Figure 4.
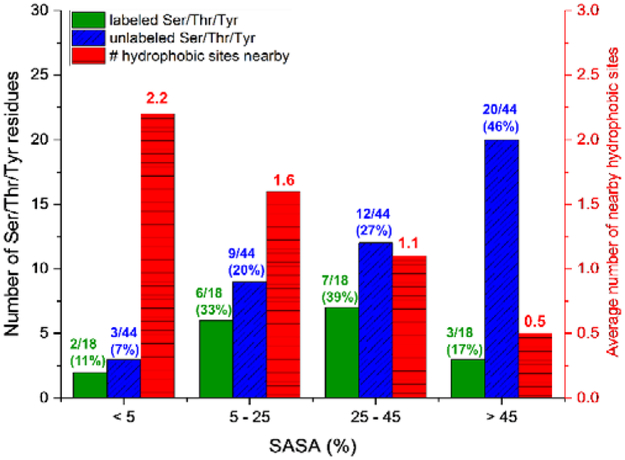
Histograms illustrating the distribution of %SASA values for both labeled (green) and unlabeled (blue) Ser, Thr, and Tyr residues in the proteins β2m, ubiquitin, and hGH. (See left axis.) Also included is the distribution of the average number of nearby hydrophobic sites (red) per Ser/Thr/Tyr residue in each bin of SASA values. (See right axis.)
CONCLUSIONS
DEPC is capable of modifying weakly nucleophilic residues such as Ser, Thr, and Tyr in intact proteins, allowing for higher structural resolution. The same reagent, however, is essentially unreactive with Ser, Thr, and Tyr residues in small peptides, suggesting that the 3D context of the protein’s structure is able to tune the reactivity of these residues with DEPC. By comparing the DEPC reactivity of three intact proteins and their proteolytic fragments, we are able to identify the chemical environments that most influence the reactivity of these side chains in intact proteins. Interestingly, we find evidence that the presence of nearby hydrophobic residues increases the DEPC reactivity of Ser, Thr, and Tyr residues in intact proteins. This finding suggests that the reactivity of certain Ser, Thr, and Tyr residues could be used to indicate the presence of nearby hydrophobic groups and thus could be used as constraints in protein structure prediction.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH) under Grant R01 GM075092. The authors acknowledge Prof. Stephen J. Eyles and Dr. Adam Graichen for their help with the Thermo Scientific Orbitrap Fusion mass spectrometer. The data described herein were acquired on an Orbitrap Fusion mass spectrometer funded by National Institutes of Health grant S10OD010645. Eric M. Graban and Dr. John E. Hale (QuarryBio Inc.) are gratefully thanked for a permission to use their custom software pipeline for data analysis. P.L. also acknowledges his doctoral fellowship from the Faculty of Pharmaceutical Sciences, Chulalongkorn University (Bangkok, Thailand).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Materials; experimental procedures for DEPC labeling of model peptides; additional methods for LC-MS/MS analyses of protein digests; determination of modification percentages; SASA calculations; supplemental schemes and figures for the results and discussion; tables summarizing DEPC modification percentages and structural features for nucleophilic residues in the proteins β2m, ubiquitin, and hGH.
The authors declare no competing financial interest.
REFERENCES
- 1.Stocks BB; Konermann L, Structural Characterization of Short-Lived Protein Unfolding Intermediates by Laser-Induced Oxidative Labeling and Mass Spectrometry. Anal. Chem 2009, 81 (1), 20–27. [DOI] [PubMed] [Google Scholar]
- 2.Vahidi S; Stocks BB; Liaghati-Mobarhan Y; Konermann L, Submillisecond Protein Folding Events Monitored by Rapid Mixing and Mass Spectrometry-Based Oxidative Labeling. Anal. Chem 2013, 85 (18), 8618–8625. [DOI] [PubMed] [Google Scholar]
- 3.Chen J; Rempel DL; Gross ML, Temperature Jump and Fast Photochemical Oxidation Probe Submillisecond Protein Folding. J. Am. Chem. Soc 2010, 132 (44), 15502–15504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J; Rempel DL; Gau BC; Gross ML, Fast Photochemical Oxidation of Proteins and Mass Spectrometry Follow Submillisecond Protein Folding at the Amino-Acid Level. J. Am. Chem. Soc 2012, 134 (45), 18724–18731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srikanth R; Mendoza VL; Bridgewater JD; Zhang G; Vachet RW, Copper Binding to β-2-Microglobulin and Its Pre-Amyloid Oligomers. Biochemistry 2009, 48 (41), 9871–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendoza VL; Antwi K; Barón-Rodríguez MA; Blanco C; Vachet RW, Structure of the Preamyloid Dimer of β-2-Microglobulin from Covalent Labeling and Mass Spectrometry. Biochemistry 2010, 49 (7), 1522–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendoza VL; Barón-Rodríguez MA; Blanco C; Vachet RW, Structural Insights into the Pre-Amyloid Tetramer of β-2-Microglobulin from Covalent Labeling and Mass Spectrometry. Biochemistry 2011, 50 (31), 6711–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinger AL; Kiselar J; Ilchenko S; Komatsu H; Chance MR; Axelsen PH, A Synchrotron-Based Hydroxyl Radical Footprinting Analysis of Amyloid Fibrils and Prefibrillar Intermediates with Residue-Specific Resolution. Biochemistry 2014, 53 (49), 7724–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li KS; Rempel DL; Gross ML, Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J. Am. Chem. Soc 2016, 138 (37), 12090–12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H; Gau BC; Jones LM; Vidavsky I; Gross ML, Fast Photochemical Oxidation of Proteins for Comparing Structures of Protein–Ligand Complexes: The Calmodulin-Peptide Model System. Anal. Chem 2011, S3 (1), 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manzi L; Barrow AS; Scott D; Layfield R; Wright TG; Moses JE; Oldham NJ, Carbene footprinting accurately maps binding sites in protein-ligand and protein-protein interactions. Nat. Commun 2016, 7, 13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu T; Marcinko TM; Kiefer PA; Vachet RW, Using Covalent Labeling and Mass Spectrometry To Study Protein Binding Sites of Amyloid Inhibiting Molecules. Anal. Chem 2017, 89 (21), 11583–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo C; Cheng M; Gross ML, Protein-Metal-Ion Interactions Studied by Mass Spectrometry-Based Footprinting with Isotope-Encoded Benzhydrazide. Anal. Chem 2019, 91 (2), 1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watson C; Sharp JS, Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. AAPS J. 2012, 14 (2), 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deperalta G; Alvarez M; Bechtel C; Dong K; McDonald R; Ling V, Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs 2013, 5 (1), 86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borotto NB; Zhou Y; Hollingsworth SR; Hale JE; Graban EM; Vaughan RC; Vachet RW, Investigating Therapeutic Protein Structure with Diethylpyrocarbonate Labeling and Mass Spectrometry. Anal. Chem 2015, 87 (20), 10627–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y; Wecksler AT; Molina P; Deperalta G; Gross ML, Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (5), 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan LY; Salas-Solano O; Valliere-Douglass JF, Localized conformational interrogation of antibody and antibody-drug conjugates by site-specific carboxyl group footprinting. mAbs 2017, 9 (2), 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limpikirati P; Hale JE; Hazelbaker M; Huang Y; Jia Z; Yazdani M; Graban EM; Vaughan RC; Vachet RW, Covalent labeling and mass spectrometry reveal subtle higher order structural changes for antibody therapeutics. mAbs 2019, 11 (3), 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Limpikirati P; Liu T; Vachet RW, Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 2018, 144, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendoza VL; Vachet RW, Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev 2008, 28 (5), 785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu G; Chance MR, Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev 2007, 107 (8), 3514–3543. [DOI] [PubMed] [Google Scholar]
- 23.Wang L; Chance MR, Structural Mass Spectrometry of Proteins Using Hydroxyl Radical Based Protein Footprinting. Anal. Chem 2011, 83 (19), 7234–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiselar J; Chance MR, High-Resolution Hydroxyl Radical Protein Footprinting: Biophysics Tool for Drug Discovery. Annu. Rev. Biophys 2018, 47 (1), 315–333. [DOI] [PubMed] [Google Scholar]
- 25.Li KS; Shi L; Gross ML, Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Acc. Chem. Res 2018, 51 (3), 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B; Cheng M; Rempel D; Gross ML, Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods 2018, 144, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendoza VL; Vachet RW, Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem 2008, 80 (8), 2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y; Vachet RW, Diethylpyrocarbonate Labeling for the Structural Analysis of Proteins: Label Scrambling in Solution and How to Avoid It. J. Am. Soc. Mass Spectrom 2012, 23 (5), 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y; Vachet RW, Increased Protein Structural Resolution from Diethylpyrocarbonate-based Covalent Labeling and Mass Spectrometric Detection. J. Am. Soc. Mass Spectrom 2012, 23 (4), 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borotto NB; Degraan-Weber N; Zhou Y; Vachet RW, Label Scrambling During CID of Covalently Labeled Peptide Ions. J. Am. Soc. Mass Spectrom 2014, 25 (10), 1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trinquier G; Sanejouand YH, Which effective property of amino acids is best preserved by the genetic code? Protein Eng., Des. Sel 1998, 11 (3), 153–169. [DOI] [PubMed] [Google Scholar]
- 32.Harris TK; Turner GJ, Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 2002, 53 (2), 85–98. [DOI] [PubMed] [Google Scholar]
- 33.Mädler S; Bich C; Touboul D; Zenobi R, Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J. Mass Spectrom 2009, 44 (5), 694–706. [DOI] [PubMed] [Google Scholar]
- 34.Mehler EL; Fuxreiter M; Simon I; Garcia-Moreno E,B, The role of hydrophobic microenvironments in modulating pKa shifts in proteins. Proteins: Struct., Funct., Bioinf 2002, 48 (2), 283–292. [DOI] [PubMed] [Google Scholar]
- 35.Isom DG; Castañeda CA; Cannon BR; García-Moreno E,B, Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. 2011, 108 (13), 5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peck MT; Ortega G; De Luca-Johnson JN; Schlessman JL; Robinson AC; García-Moreno E,B, Local Backbone Flexibility as a Determinant of the Apparent pKa Values of Buried Ionizable Groups in Proteins. Biochemistry 2017, 56 (40), 5338–5346. [DOI] [PubMed] [Google Scholar]
- 37.Robinson AC; Majumdar A; Schlessman JL; García-Moreno E,B, Charges in Hydrophobic Environments: A Strategy for Identifying Alternative States in Proteins. Biochemistry 2017, 56 (1), 212–218. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y; Wu Y; Yao M; Liu Z; Chen J; Chen J; Tian L; Han G; Shen J-R; Wang F, Probing the Lysine Proximal Microenvironments within Membrane Protein Complexes by Active Dimethyl Labeling and Mass Spectrometry. Anal. Chem 2016, 88 (24), 12060–12065. [DOI] [PubMed] [Google Scholar]
- 39.Ruan Q; Zhao C; Ramsay CS; Tetin SY, Characterization of Fluorescently Labeled Protein with Electrospray Ionization-MS and Fluorescence Spectroscopy: How Random is Random Labeling? Anal. Chem 2018, 90 (16), 9695–9699. [DOI] [PubMed] [Google Scholar]
- 40.Jumper CC; Schriemer DC, Mass Spectrometry of Laser-Initiated Carbene Reactions for Protein Topographic Analysis. Inal. Chem 2011, 83 (8), 2913–2920. [DOI] [PubMed] [Google Scholar]
- 41.Jumper CC; Bomgarden R; Rogers J; Etienne C; Schriemer DC, High-Resolution Mapping of Carbene-Based Protein Footprints. Anal. Chem 2012, 84 (10), 4411–4418. [DOI] [PubMed] [Google Scholar]
- 42.Fraczkiewicz R; Braun W, Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem 1998, 19 (3), 319–333. [Google Scholar]
- 43.Schrodinger LLC The PyMOL Molecular Graphics System, Version 1.8, 2015.
- 44.Negi S; Zhu H; Fraczkiewicz R; Braun W Solvent Accessible Surface Areas, Atomic Solvation Energies, and Their Gradients for Macromolecules. 2015. April 17 [accessed 2019 Feb 9]. http://curie.utmb.edu/area_man.html.
- 45.Miller BT; Collins TJ; Nagle GT; Kurosky A, The occurrence of O-acylation during biotinylation of gonadotropin-releasing hormone and analogs. Evidence for a reactive serine. J. Biol. Chem 1992, 267 (8), 5060–9. [PubMed] [Google Scholar]
- 46.Miller BT; Kurosky A, Elevated Intrinsic Reactivity of Seryl Hydroxyl Groups within the Linear Peptide Triads His-Xaa-Ser or Ser-Xaa-His. Biochem. Biophys. Res. Commun 1993, 196 (1), 461–467. [DOI] [PubMed] [Google Scholar]
- 47.Miller BT; Collins TJ; Rogers ME; Kurosky A, Peptide Biotinylation with Amine-Reactive Esters: Differential Side Chain Reactivity. Peptides 1997, 18 (10), 1585–1595. [DOI] [PubMed] [Google Scholar]
- 48.Guo X; Bandyopadhyay P; Schilling B; Young MM; Fujii N; Aynechi T; Guy RK; Kuntz ID; Gibson BW, Partial Acetylation of Lysine Residues Improves Intraprotein Cross-Linking. Anal. Chem 2008, 80 (4), 951–960. [DOI] [PubMed] [Google Scholar]
- 49.Thurlkill RL; Grimsley GR; Scholtz JM; Pace CN, pK values of the ionizable groups of proteins. Protein Sci. 2006, 15 (5), 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z; Browne SJ; Vachet RW, Exploring Salt Bridge Structures of Gas-Phase Protein Ions using Multiple Stages of Electron Transfer and Collision Induced Dissociation. J. Am. Soc. Mass Spectrom 2014, 25 (4), 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petsko GA; Ringe D, Protein Structure and Function. New Science Press Ltd: London, England, United Kingdom, 2004; pp 10–11. [Google Scholar]
- 52.Whitford D, Proteins: Structure and Function. John Wiley & Sons Ltd: West Sussex, England, United Kingdom, 2005; pp 53–58. [Google Scholar]
- 53.Finkelstein AV; Ptitsyn OB, Protein Physics: A Course of Lectures. Academic Press: London, England, United Kingdom, 2002; pp 33–42. [Google Scholar]
- 54.Miles EW, Modification of histidyl residues in proteins by diethylpyrocarbonate In Methods in Enzymology, Academic Press: Cambridge, Massachusetts, United States, 1977; Vol. 47, pp 431–442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.