

Abstract
Owing to the pervasiveness of hydroxyl groups in natural isolates, alcohol derivatives are alluring directing groups. Herein, an alcohol-derived sulfamate ester guides the light initiated xanthylation of primary, secondary, or tertiary centers. This process enables formal directed deuteration, azidation, thiolation, and vinylation reactions.
Graphical Abstract
INTRODUCTION
Alkyl C–H functionalization reactions1 enable pharmaceutical lead optimization.2 The utility of these processes is limited by insufficient control over which C–H bond in a molecule reacts, and over the types of functional groups that can be installed. To dictate the site of functionalization, we recently identified sulfamate esters3 as alcohol derivatives that can direct chlorination of an unactivated alkyl C(3)–H centers.4 To our knowledge, there are no general strategies to target these centers for functionalization by exogenous atom-transfer agents.5–7 To demonstrate the broad relevance of our approach,4 we envisioned the application of sulfamate esters to direct C–H xanthylation reactions.
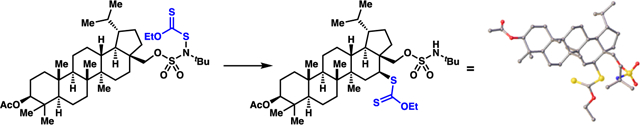
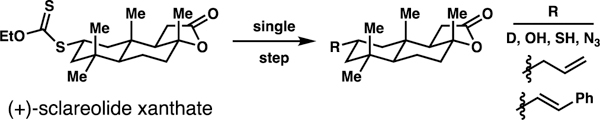
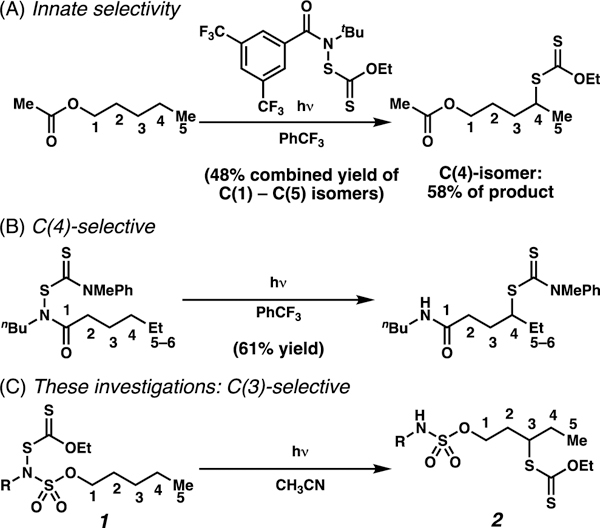
Alkyl xanthates (dithiocarbonates)8–9 are ideal precursors to diverse small molecule libraries because they are known platforms to affect formal deuteration,10 hydroxylation,11 thiolation,10 trifluoromethylthiolation,10 azidation,11 vinylation,12 or allylation13 reactions (Scheme 1). Motivated by these applications, processes have been developed to enable xanthylation of C(sp3)–H bonds that are etherial14 or unactivated,10a yet sterically accessible and electron-rich (Scheme 2A).1b, 1d, 15 Recently, Alexanian and co-workers have disclosed a directed C(4)–H dithiocarbamate-transfer reaction (Scheme 2B).16 Herein, we describe a reaction with complementary position-selectivity: a sulfamate ester-guided light-initiated reaction to xanthylate unactivated alkyl C(3)–H bonds (Scheme 2C). This approach enables generation of molecular diversity based on guided functionalization of a single C(sp3)–H center, with expected practical value to medicinal chemists.
Scheme 1.
Alkyl Xanthates are Precursors to Diverse Small Molecule Libraries
Scheme 2.
Prior Disclosures and These Investigations
The disclosed strategy for C(3)-xanthylation is informed by reinvigorated enthusiasm17–18 for the Hofmann-Löffler-Freytag (HLF) reaction.19–20 Conventional HLF processes rely on formation of an intermediate nitrogen-centered radical, which is poised to adopt a six-membered transition state to affect C–H abstraction, and thereby dictate the position selectivity of atom transfer.21–22 By contrast, we hypothesize that sulfamate esters are geometrically predisposed to adopt rare23–24 seven-membered transition states for C–H functionalization owing to elongated O–S and S–N bonds and compressed O–S–N bond angles.7a To date, there are few reports that engage seven-membered transition states for C–H abstraction with a removable linker,6b such as a sulfamate ester.25–26 In the process disclosed herein, sulfamate esters guide the generation of C(3)-centered radicals via 1,6-HAT and the resultant carbon-centered radicals are captured to furnish alkyl xanthate esters.
RESULTS and DISCUSSION
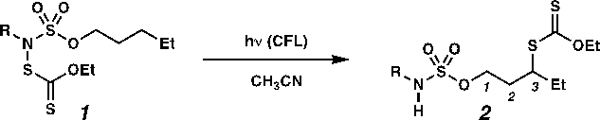
We chose to access the key nitrogen-centered radical intermediates by using light to homolyze the nitrogen–sulfur bonds of N-xanthylsulfamate esters 1. N-xanthylsulfamate esters 1 can be prepared by initial oxidation of sulfamate esters 3 with trichloroisocyanuric acid or tert-butyl hypochlorite. Subsequent treatment of N-chlorosulfamate esters with potassium O-ethyl xanthate,10a an inexpensive commercially available reagent, furnishes N-xanthylsulfamate esters 1 (see supporting information for details). As expected, photolysis of pentyl tert-butyl xanthylsulfamate ester 1a affects selective C(3)-xanthylation at a methylene center in 91% yield (Table 1, entry 1, C–H bond dissociation energy (BDE) ≈ 98 kcal mol–1).27 In the absence of light, xanthate transfer does not proceed, so light is required to initiate the transformation.
Table 1.
Xanthate-transfer Robust to N-Alkyl Variationsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | predicted pKab for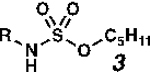
|
substrate | time (h) |
product | yield (%)c |
| 1 | tBu | 12.74 | 1a | 12 | 2a | 91 |
| 2 | Et | 12.76 | 1b | 24 | 2b | 91 |
| 3 | Me | 12.65 | 1c | 12 | 2c | 77 |
| 4 | CH2CF3 | 9.18 | 1d | 18 | 2d | 57 |
| 5 | 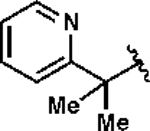 |
11.05 | 1e | 18 | 2e | 74 |
| Products derived from substrates that incorporate δ’-1,5-HAT sites | ||||||
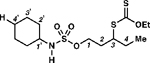 2f, 82% yieldd |
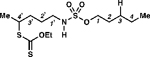 4, 85% yielde |
|||||
General reaction conditions: 1.0 equiv N-xanthylsulfamate ester 1, CH3CN (0.07 M), 22 °C, irradiated with compact fluorescent lights
(CFLs).
Aqueous ionization pKa values have been predicted using SPARC.23
Isolated yield.
12 h.
18 h.
Xanthate transfer proceeds upon photolysis of pentyl N-tert-butyl-, N-ethyl-, N-methyl-, N-trifluoroethyl-, N-2-(pyridine-2-yl)isopropyl, N-cyclohexyl-, and N-pentylxanthylsulfamate esters 1a–1g in acetonitrile at room temperature (Table 1). Resultant sulfamate esters 2a–2f and 4 differ in terms of steric encumbrance about the N–H bond and the predicted acidity.28 Unless otherwise noted, the mass balance for these reactions can be accounted for principally by recovered substrates 1, reduced sulfamate esters 3 and readily separable desired products 2.
This directed xanthate-transfer reaction overrides inductive deactivation of C–H bonds to oxidation, which is often used to induce site-selectivity in unguided transformations. Reactions of N-xanthylated 1a–f furnish xanthylated alkanes 2a–f with exquisite C(3)-selectivity (Table 1), even while functionalization of a more distal position, such as C(4), would be expected under conditions for the unguided xanthate-transfer process, which relies on inherent selectivity (c.f. Scheme 2).
We anticipated that the ability to vary N-alkyl substituent of the sulfamate ester would afford the opportunity to override this C(3)-selective functionalization process to engage a C(4’)-C–H center. Interestingly, pentyl N-cyclohexylxanthylsulfamate ester 1f engages in C(3)-xanthylation in lieu of remote transannular C–H functionalization at C(4’), which presumably would require a relatively high-energy boat transition state for C–H abstraction. By contrast N, O-dipentylxanthylsulfamate ester 1g undergoes selective xanthate transfer to furnish alkyl xanthate 4 to engage the C(4’)-C–H center, exclusive of the C(3)-C–H bond, presumably favoring a six- over a seven-membered transition state for C–H abstraction.
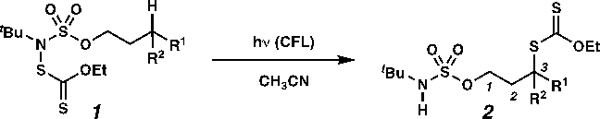
Xanthylation proceeds preferentially at methylene C(3)–H centers even when the C(3)–H bond is significantly stronger than other available C–H centers (Table 2). For example, C(3)-xanthate transfer proceeds without engaging the weak etherial C–H bond of silyl ether 1h (BDE ≈ 85 kcal mol–1, entry 1), a C–H bond that is weakened by an ester (i.e. 1i, BDE ≈ 96 kcal mol−1, entry 2), or the weak benzylic C–H bond in 1j (BDE ≈ 90 kcal mol−1, entry 3).27 Of course, the process remains effective even when the installed C–S bond is weakened by an adjacent aromatic ring (entry 4).
Table 2.
Xanthate-Transfer Proceeds with High Position Selectivity at Tertiary, Benzylic or Secondary Centersa
 | ||||
|---|---|---|---|---|
| entry | product | time (h) | yield (%)b |
|
| 1 | 2h | 18 | 74 | |
| 2 | 2i | 18 | 83 | |
| 3 | 2j | 18 | 80 | |
| 4 | 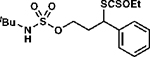 |
2k | 26 | 76 |
| 5 | 2l | 12 | 61 | |
| 6 | 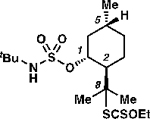 |
2m | 16 | 64 |
| 7 | 2n | 15 | 48c | |
| 8 | 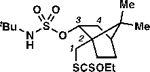 |
2o | 19 | 46d |
| 9 | 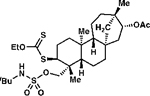 |
2p | 18 | 76e |
General reaction conditions: 1.0 equiv N-xanthylulfamate ester 1, CH3CN (0.07 M), 35—38 °C, irradiated with compact fluorescent lights (CFLs).
Isolated yield. c C(2)-alkyl xanthate (not depicted) isolated in 18% yield.
C(4)-alkyl xanthate (not depicted) isolated in 29% yield.
Isolated yield of major diastereomer.
Xanthylation proceeds preferentially at methylene C(3)–H centers even when the C(3)–H bond is significantly stronger than other available C–H centers (Table 2). For example, C(3)-xanthate transfer proceeds without engaging the weak etherial C–H bond of silyl ether 1h (BDE ≈ 85 kcal mol–1, entry 1), a C–H bond that is weakened by an ester (i.e. 1i, BDE ≈ 96 kcal mol−1, entry 2), or the weak benzylic C–H bond in 1j (BDE ≈ 90 kcal mol−1, entry 3).27 Of course, the process remains effective even when the installed C–S bond is weakened by an adjacent aromatic ring (entry 4).
Surprisingly, this protocol can transform C(3)–H tertiary centers (BDE ≈ 96 kcal mol–1)27 to provide tertiary alkyl xanthates (Table 2, entries 5–6), which can be difficult to prepare through other methods as they are prone to elimination.29–31 Moreover, the reaction remains guided even in the presence of alternative tertiary C–H bonds that are innately more reactive. For example, the C(3)–H bond of 3,7-dimethyloctanol-derived 1l is processed preferentially, even though this bond is inductively deactivated to oxidation relative to the more distal C(7)–H tertiary center32 (entry 5). Further, a sulfamate ester directs xanthylation to the C(8)–H bond of menthol-derived 1m, though the electronically similar C(5)–H bond is often oxidized preferentially in processes that rely on innate selectivity (entry 6).33 Analogous position selectivity has been observed in intramolecular amination reactions, and in sulfamate ester-guided halogen-transfer processes by White,7g Che,7d Du Bois,34 Burns and Zare,25b and by our laboratory.4
A slight erosion of selectivity occurs when the only available C(3)–H bonds are stronger primary centers (BDE ≈ 101 kcal mol−1).27 Xanthyl transfer to propyl tert-butylsulfamate ester 1n and (–)-borneol derivative 1o occurs principally through guided functionalization (entries 7–8). This recapitulates the selectivity induced in White’s manganese-catalyzed intramolecular amination with sulfamate esters7g and our sulfamate ester-guided chlorine-transfer reaction.4 Nevertheless, with each of these substrates, a single isomeric product can be detected, evidencing a decrease in selectivity when the reaction is guided to a primary center.
To our delight, the reactivity and site-selectivity observed with relatively simple substrates is retained in more elaborate small molecules, which proceed with substrate-induced diastereoselectivity (Table 2, entry 9). Oxidation of isosteviol derivative 1p generates secondary xanthate ester 2p in 76% isolated yield of major diastereomer (entry 9). Moreover, an even more challenging test of this method is betulinic acid derived 1q, which incorporates C(3)–H methylene centers on both the five- and six-membered rings (Scheme 3). Upon exposure, C–H xanthylation proceeds to furnish 68% isolated yield of major diastereomer of functionalized 2q. The relative stereochemistry of this isolable major diastereomer has been confirmed single crystal x-ray diffraction.
Scheme 3.
Xanthate Transfer to Betulinic Acid Derivative
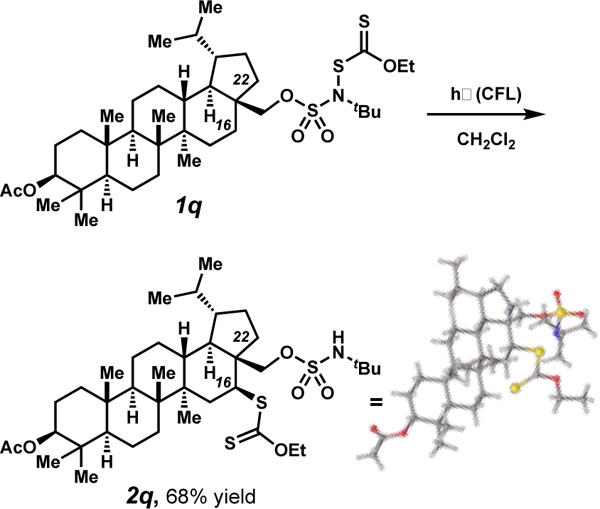
Theoretically, this xanthate-transfer process could occur through a closed-cycle and/or a radical chain propagation mechanism (Scheme 4). In these processes, light initiates N–S bond homolysis9b to convert N-xanthate 1a into a xanthate radical and sulfamyl radical 5. This sulfamyl radical is poised for an intramolecular C–H abstraction through a seven-membered transition state to produce carbon-centered radical 6, imparting position-selectivity to this transformation. At this point, the proposed reaction pathways diverge. In a closed process, alkyl radical 6 recombines with the xanthate radical to terminate the reaction. By contrast, in a chain propagation mechanism, alkyl radical 6 reacts with another substrate molecule to furnish alkyl xanthate 2a and another equivalent of sulfamyl radical 5, which would then propagate this chain reaction.
Scheme 4.
Xanthate transfer proceeds via light-initiated radical chain propagation
These mechanistic hypotheses differ in terms of quantum yield (Φ), which is defined by quantity of product formed per photon of light absorbed by the substrate. In the closed-cycle, absorption of a single photon of light can produce a maximum of a single equivalent of product (Φ ≤ 1). By contrast, in the radical chain propagation mechanism, when the substrate absorbs a single photon of light, multiple equivalents of alkyl xanthate can form (Φ > 1).
Quantum yield measurements demonstrate that the reaction proceeds through a light-initiated chain propagation mechanism.35 In a common process, chemical actinometry using potassium ferrioxalate can be used to determine the photon flux of a fluorimeter at 313 nm.35–36 After 4 hours of irradiation of N-xanthylated 1a in acetonitrile at 313 nm, alkylxanthate 2a can be isolated in 50–52% yield. This yield corresponds to 3 equivalents of product formed per absorbed photon (Φ = 3) and confirms that this reaction proceeds through a chain propagation mechanism.
Produced alkyl xanthates can be employed in procedures for C(3)–thiolation, azidation, deuteration, trifluoromethylthiolation, vinylation, or allylation. To highlight these opportunities, we have converted alkyl xanthate 2a to thiol 7,10 azide 8,11 trifluoromethylthiol 9,10 deuterio D-3a,10 allyl 10,13 and vinyl 1112 (Table 3).
Table 3.
Alkyl Xanthates are Precursors to Diverse Small Molecule Librariesa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | yield (%)a | entry | R | yield (%)a | ||
| 1 | SH | 7 | 81b | 4 | D | d-3a | 81e |
| 2 | N2 | 8 | 79c | 5 |  |
10 | 62f |
| 3 | SCF3 | 9 | 76d | 6 | 11 | 73g | |
Isolated yield.
4-Methylpiperidine, EtOH, 21 °C.
Ethyl sulfonyl azide, dilauroyl peroxide, PhCl, 100 °C.
((2-Phenylpropan-2-yl)oxy)(trifluoromethyl)sulfane, dilauroyl peroxide, PhCl, 100 °C.
BEt3 (1.0 M in THF), dodecanethiol, d4-DCE:D2O (2:1), O2, 21 °C.
Allyl ethyl sulfone, dilauroyl peroxide, PhCl, 100 °C.
Styryl ethyl sulfone, tBuOOtBu, PhCl, 130 °C.
In combination with known strategies to displace sulfamate esters with an iodide,25b azide,25b or acetate nucleophile (Table 4), xanthate ester diversification provides a powerful strategy to access a broad variety of 1,3-difunctionalized small molecules.
Table 4.
Alkyl xanthates are precursors to diverse small molecule librariesa
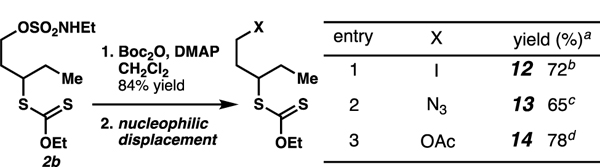 |
Isolated yield.
3.3 equiv NaI, acetone, 35 °C, 6 h.
3.3 equiv NaN3, DMSO, 35 °C, 6 h.
3.3 equiv LiOAc, DMSO, 40 °C, 18 h.
CONCLUSION
In summary, we have developed a process to enable sulfamate esters as alcohol derivatives to guide xanthate-transfer and furnish C(3)-functionalized products. The research confirms the recently disclosed basic scientific insight that N-functionalized sulfamate esters can guide position-selective C(sp3)–H functionalization, thereby suggesting that this may prove to be a broadly applicable strategy to alter the site-selectivity of known HLF processes. This guided reaction can be combined with known technologies to affect formal C(sp3)–H thiolation, azidation, allylation, vinylation, trifluoromethylthiolation, or deuteration over multiple steps. This approach enables the generation of molecular diversity based on guided functionalization of a single C(sp3)–H center, with expected practical value to lead optimization.
EXPERIMENTAL SECTION
General Information.
Moisture-sensitive reactions were performed using flame-dried glassware under an atmosphere of dry nitrogen (N2). Air- and water-sensitive reactions, where noted, were performed in an MBraun MB200 glove box held under an atmosphere of nitrogen gas (working pressure 2–6 mbar). Flame-dried equipment was stored in a 130 °C oven before use and either allowed to cool in a cabinet desiccator or assembled hot and allowed to cool under an inert atmosphere. Air- and moisture-sensitive liquids and solutions were transferred via plastic or glass syringe or by stainless steel cannula. Chromatographic purification of products was accomplished using Teledyne Isco CombiFlashRf system employing 12–220 g Redi-Sep Rf normal phase silica columns (particle size 40–63 μm, 230–400 mesh) or manual flash chromatography using Alfa Aesar Florisil (100–200 mesh). Thin layer chromatography was performed on EMD Millipore silica gel 60 F254 glass-backed plates (layer thickness 250 μm, particle size 10–12 μm, impregnated with a fluorescent indicator). Visualization of the developed chromatogram was accomplished by fluorescence quenching under shortwave UV light and/or by staining with p-anisaldehyde, or KMnO4 stains. Room temperature is 22 °C. NMR spectra were obtained at 20–23 °C on Varian iNOVA spectrometers operating at 400, 500 or 700 MHz for 1H NMR, 101, 126, 176 MHz for 13C NMR, and 376 MHZ for 19F NMR, and are reported as chemical shifts (δ) in parts per million (ppm). Spectra were referenced internally according to residual solvent signals (1H: CDCl3, 7.26 ppm; CD3CN, 1.94 ppm; 13C: CDCl3, 77.0 ppm, CD3CN: 118.3 ppm). Data for NMR spectra use the following abbreviations to describe multiplicity: s, singlet; br s, broad singlet; d, doublet; t, triplet; dd, doublet of doublets; td, triplet of doublets; tt, triplet of triplets; ddd, doublet of doublet of doublets; dddd, doublet of doublet of doublet of doublets; ddt, doublet of doublet of triplets; app dd, apparent doublet of doublets; m, multiplet. Coupling constants (J) are reported in units of Hertz (Hz). IR spectra were obtained on a Nicolet 6700 FT-IR system. Peaks are reported in cm–1 with indicated relative intensities: s (strong, 0–33% T); m (medium, 33–67% T); w (weak, 67–95% T); and br (broad). High resolution mass spectra (HRMS, m/z) were recorded on an Agilent LCMS-TOF-DART spectrometer using electrospray ionization (ESI, Duke University Department of Chemistry Instrumentation Center). UV-Vis spectra were obtained on an Agilent Cary 5000 UV-Vis-NIR Spectrophotometer. GE helical 26W 1600 lumen compact fluorescent light bulbs (CFL) of broad visible light spectrum were employed without filters. The microwave vials and disposable crimp caps containing Teflon-lined septa were obtained from ChemGlass (Item numbers CG-4920–01 and CG-4920–10, respectively). http://chemglass.com/microwave-reaction-vials-complete-packages?sku=CG-4920
Preparation of Reagents.
(–)-16-acetoyxy-18-hydroxy-13-methyl-17-norkaurane (S2).
A flame-dried flask with stirbar was charged with stevia extract (2.9 g, 3.6 mmol, 1 equiv) and deionized water (115 mL). Upon dissolution of stevia extracts, concentrated HCl (2.5 mL) was carefully added and the flask was fitted with a reflux condenser. The resulting clear solution was refluxed for 2 hours upoon which the solution turned into an off-white suspension. The suspension was extracted with EtOAc (4 × 50 mL). The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude isosteviol, a brown solid, was used in the next step without further purification. The crude isosteviol, dissolved in dry THF (60 mL), was added dropwise to a suspension of lithium aluminum hydride (1.36 g, 35.8 mmol, 10 equiv) in THF (60 mL) that had been cooled at 0 °C in an ice water bath. The reaction was left to stir at 0 °C for 15 minutes and then refluxed overnight. The resulting grey suspension was carefully quenched with water until no effervescence was observed. The off-white suspension was filtered through a pad of celite and the aqueous layer was extracted with ether (3 × 50 mL). The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure to afford crude diol as yellow solid. The crude diol was used in the next step without further purification.The crude diol was dissolved in anhydrous CH2Cl2 (25 mL). To the clear yellow solution, acetic anhydride (2.84 g, 27.8 mmol, 7.8 equiv), DMAP (43.5 mg, 0.36 mmol, 0.1 equiv), and triethylamine (5.3 mL, 37.7 mmol, 10.6 equiv) were added in that order. The yellow solution was the stirred at room temperature for 12 hours. The reaction was quenched with deionized water (100 mL) and the resulting biphasic mixture was cooled to 0 °C using ice/water bath. The solution was then acidified using 6.2 N H2SO4 (18.6 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with sat. NaHCO3 until pH 7 was reached. Then, the organic layer was dried with MgSO4, filtered, and concentrated under reduced pressure to afford crude bis-acetate as yellow oil. The crude oil was used in the next step without further purification.The yellow oil was taken in MeOH (3 mL)/THF (12 mL) and cooled to 0 °C in an ice/water bath. To the chilled solution, KOH (202 mg, 3.6 mmol, 1.0 equiv) was added and stirred for 30 min. The clear solution was then concentrated under reduced pressure and diluted with deionized water (50 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL) and the combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The product was obtained as a white foam (530 mg, 46% yield, over four steps) following silica gel column chromatography with hexanes:EtOAc (98:2). 1H NMR (700 MHz, CDCl3) δ 4.72 (dd, J = 9.1, 6.1 Hz, 1H), 3.75 (d, J = 10.9 Hz, 1H), 3.39 (d, J = 10.8 Hz, 1H), 2.06 (s, 3H), 1.80–1.76 (m, 4H), 1.68–1.63 (m, 1H), 1.59–1.49 (m, 7H), 1.40–1.32 (m, 4H), 1.24–1.16 (m, 3H), 1.06–1.03 (m, 2H), 0.95 (s, 3H), 0.90 (s, 3H), 0.85 (s, 3H); 13C{1H} NMR (176 MHz, CD3CN) δ 171.4, 81.7, 65.5, 56.9, 56.5, 55.0, 42.3, 41.7, 41.5, 40.8, 39.6, 38.5, 37.6, 35.4, 34.5, 27.1, 24.9, 21.2, 20.2, 20.0, 18.0, 15.5; IR (neat) ν 3525 (br), 2924 (m), 2845 (w), 1737 (w), 1712 (m), 1451 (w), 1382 (w), 1369 (w), 1253 (m), 1206 (w), 1152 (w), 1056 (w), 1031 (m), 973 (w), 850 (w), 739 (w), 629 (w), 607 (w) cm–1; TLC Rf = 0.22 (hexanes/EtOAc, 8:2), HRMS (ESI-TOF) m/z: [M + K]+ Calcd for C22H36O3•K 387.2296; Found 387.2298.
(E)-(2-(ethylsulfonyl)vinyl)benzene (S3).
The compound was prepared according to a modified literature procedure.37 A flame-dried flask with stir bar, fitted with a reflux condenser, was charged with sodium ethyl sulfinate salt (2.32 g, 20 mmol, 3.0 equiv) and sodium acetate (2.46 g, 30 mmol, 1.5 equiv) and the flask was evacuated and backfilled with N2. Acetonitrile (68 mL) was added via syringe followed by styrene (0.76 mL, 6.7 mmol, 1.0 equiv). Iodine (7.61 g, 30 mmol, 1.5 equiv) was then added by briefly removing the reflux condenser and adding the solid in a single portion. The reaction was then heated to reflux for 1 h. After 1 h, the reaction was removed from heat and allowed to cool to room temperature and quenched with 10% aqueous sodium thiosulfate. The reaction was then diluted with saturated aqueous sodium bicarbonate solution (50 mL) and transferred to a separatory funnel rinsing the flask with EtOAc (ca. 50 mL). An additional 50 mL EtOAc was added and the organic phase was removed. The aqueous was extracted twice more with EtOAc (3 × 50 mL). The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The product was obtained as a white solid (706 mg, 54% yield) following silica gel column chromatography with hexanes:EtOAc (98:2). The spectroscopic data was in accordance with that previously published in the literature.38 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 15.5 Hz, 1H), 7.53 (d, J = 5.5 Hz, 2H), 7.46–7.43 (m, 3H), 6.81 (d, J = 15.5 Hz, 1H), 3.10 (q, J = 7.4 Hz, 2H), 1.39 (t, J = 7.4 Hz, 3H).
Preparation and Characterization of Triethylammonium Sulfamate Salts:
General Procedure A.
Sulfamic acid salts were prepared according to a modified literature procedure.3 A round bottom flask equipped with magnetic stir bar was charged with sulfur trioxide pyridine complex (SO3•pyr, 1.0 equiv). Acetonitrile (0.33 M) was then added in a single portion without taking any precautions to exclude air or moisture. The suspension was stirred at 22 °C until all of the SO3•pyr had dissolved. Upon complete dissolution, the reaction flask was cooled at 0 °C in an ice water bath and capped with a rubber septum fitted with a nitrogen inlet. Amine (R1–NH2, 1.0 equiv) was then added dropwise via syringe. Following complete addition of amine, Et3N (1.1 equiv) was added dropwise. The reaction was removed from the ice bath and stirred for 0.5 h. Upon completion, the solvent was removed under reduced pressure to give a triethylammonium sulfamate salt, which was used without further purification.
Triethylammonium cyclohexylsulfamate (S3f).
Prepared from sulfur trioxide pyridine complex (3.18 g, 20 mmol) and cyclohexylamine (1.98 g, 20 mmol) following general procedure A. The product was obtained as a yellow solid (5.16 g, 92% yield).1H NMR (400 MHz, CDCl3) δ 10.17 (br s, 1H), 3.73 (br s, 1H), 3.20 (tt, J = 10.5, 4.0 Hz, 1H), 3.18–3.11 (m, 6H), 2.08 (dd, J = 12.1, 4.2 Hz, 2H), 1.67 (dt, J = 13.1, 4.0 Hz, 2H), 1.57–1.51 (m, 1H), 1.35 (t, J = 7.3 Hz, 9H), 1.31–1.02 (m, 5H); 13C{1H} NMR (126 MHz, CD3CN) δ 53.9, 47.1, 34.6, 26.5, 25.8, 9.0; IR (neat) ν 3261 (br), 2986 (w), 2929 (w), 2850 (w), 2692 (w), 1546 (w), 1478 (w), 1449 (w), 1393 (w), 1243 (w), 1219 (m), 1142 (m), 1095 (m), 890 (w), 865 (w), 799 (w), 609 (m), 590 (m), 553 (m) cm–1; HRMS (ESI-TOF) m/z: [M – HNEt3]– Calcd for C6H12NO3S– 178.0543; Found 178.0544.
Triethylammonium pentylsulfamate (S3g).
Prepared from sulfur trioxide pyridine complex (3.18 g, 20 mmol) and pentylamine (1.74 g, 20 mmol) following general procedure A. The product was obtained as a viscous yellow oil (5.15 g, 96% yield). 1H NMR (400 MHz, CDCl3) δ 10.14 (br s, 1H), 4.37 (br s, 1H), 3.19–3.12 (m, 6H), 3.05 (t, J = 7.4 Hz, 2H), 1.59–1.52 (m, 1H), 1.36 (t, J = 7.3 Hz, 9H), 1.32–1.28 (m, 4H), 0.89–0.85 (m, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 77.1, 46.3, 44.3, 29.3, 29.2, 22.4, 14.1, 8.7; IR (neat) ν 3421 (br), 3265 (br), 2955 (w), 2931 (w), 2860 (w), 2708 (w), 1641 (w), 1467 (w), 1400 (w), 1219 (m), 1161 (s), 1061 (m), 1030 (s), 616 (m), 548 (m) cm–1; HRMS (ESI-TOF) m/z: [M – HNEt3]– Calcd for C5H12NO3S– 166.0543; Found 166.0540.
Preparation and Characterization of Sulfamate Esters:
General Procedure B.
Novel sulfamate esters were prepared according to a modified literature procedure.3 A flame-dried round bottom flask equipped with magnetic stir bar was charged with triphenylphosphine oxide (1.65 equiv) and fitted with a rubber septum. The flask was evacuated and backfilled with N2. Anhydrous CH2Cl2 (3.0 mL per mmol of triphenylphosphine oxide) was added and the flask was cooled at 0 °C in an ice water bath. Trifluoromethanesulfonic anhydride (1.5 equiv) that had been freshly removed from the glovebox was then added to the cooled solution dropwise via syringe. The reaction was allowed to stir at 0 °C in an ice water bath for 15 minutes. A solution of sulfamate salt (1.5 equiv) in CH2Cl2 (1.0 mL per mmol of sulfamic acid salt) was added to the solution of triphenylphosphine ditriflate via cannula transfer. The flask was rinsed with an additional CH2Cl2 (ca. 0.5 mL per mmol of sulfamic acid salt) to achieve quantitative transfer. The resulting colorless to pale yellow solution was stirred for 15 minutes at 0 °C. During this time, a clean, flame-dried round bottom flask equipped with stir bar was fitted with a rubber septum and subsequently evacuated and backfilled with N2. This process was repeated two more times. The flask was then charged with Et3N (3.0 equiv) and CH2Cl2 (1.33 mL per mmol of Et3N) and the mixture was cooled at –78 °C in an iPrOH/dry ice bath. The sulfamate salt solution was transferred dropwise to the Et3N solution via cannula (during which time a yellow to intense red color often developed), rinsing the flask with an additional CH2Cl2 (ca. 0.5 mL per mmol of alcohol) to achieve quantitative transfer. The resultant solution was stirred at –78 °C for 15 minutes. The alcohol (R2–OH, 1.0 equiv) was then added as a solution in CH2Cl2 (1 mL per mmol of alcohol) to the Et3N solution via canula. The alcohol-containing flask was rinsed with an additional CH2Cl2 (ca. 0.5 mL per mmol of alcohol) to achieve quantitative transfer. Without removing the cooling bath, the reaction was then stirred for 18 h, during which time no additional dry ice was added, and the mixture warmed to room temperature. After 18 h, the reaction was diluted with 1 M HCl (0.1 M) and H2O (0.2 M). The biphasic mixture was transferred to a separatory funnel, rinsing the flask with CH2Cl2 to achieve quantitative transfer. The organic phase was separated and the aqueous was extracted with CH2Cl2 (2 X 25 mL). The combined organic phases were dried with MgSO4, filtered, and concentrated. The resulting crude material was then purified by silica gel flash chromatography by dry loading the samples and eluting with hexanes:EtOAc as noted below. Deviations from the reported procedure are specified below. The preparation and characterization of sulfamate esters 3a–e, 3h, 3k–l, and 3n–o have been published previously.3
Pentyl cyclohexylsulfamate ester (3f).
Prepared from salt (S3f) (0.84 g, 3.0 mmol) and n-amyl alcohol (0.22 mL, 2.0 mmol) following general procedure B. The product was obtained as a colorless oil (428 mg, 86% yield) after silica gel column chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 4.30–4.26 (m, 1H), 4.11 (t, J = 6.6 Hz, 2H), 3.35–3.26 (m, 1H), 2.04–1.98 (m, 2H), 1.75–1.69 (m, 4H), 1.64–1.55 (m, 1H), 1.40–1.32 (m, 6H), 1.29–1.14 (m, 4H), 0.91 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 70.5, 53.4, 33.5, 28.5, 27.6, 25.1, 24.6, 22.1, 13.8; IR (neat) ν 3294 (br), 2930 (m), 2856 (w), 1451 (m), 1341 (w), 1301 (w), 1170 (S), 1080 (m), 965 (s), 912 (s), 887 (S), 842 (m), 808 (m), 758 (m), 724 (s), 579 (s) cm–1; TLC Rf = 0.20 (hexanes:/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C11H23NO3S•NH4 267.1737; Found 267.1742.
Pentyl pentylsulfamate ester (3g).
Prepared from salt (S3g) (0.81 g, 3.0 mmol) and n-amyl alcohol (0.22 mL, 2.0 mmol) following general procedure B. The product was obtained as a colorless oil (399 mg, 84% yield) after silica gel column chromatography using hexanes:EtOAc (9:1).1H NMR (400 MHz, CDCl3) δ 4.29 (br s, 1H), 4.12 (t, J = 6.6 Hz, 2H), 3.12 (q, J = 7.4 Hz, 2H), 1.76–1.69 (m, 2H), 1.61–1.54 (m, 2H), 1.40–1.30 (m, 8H), 0.91 (td, J = 7.1, 2.1 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 70.8, 43.9, 29.3, 28.7, 28.6, 27.7, 22.3, 22.2, 14.0, 13.9; IR (neat) ν 3303 (br), 2957 (w), 2931 (w), 2861 (w), 1428 (w), 1342 (m), 1169 (s), 1083 (m), 964 (m), 909 (m), 832 (m), 758 (w), 725 (m), 579 (m), 538 (m) cm–1; TLC Rf = 0.43 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C10H23NO3S•NH4 255.1737; Found 255.1737.
5-((tert-butyldimethylsilyl)oxy)pentyl tert-butylsulfamate ester (3h).
Prepared from triethylammonium tert-butylsulfamate salt (0.76 g, 3.0 mmol) and 5-((tert-butyldimethylsilyl)oxy)pentan-1-ol39 (0.44 g, 2.0 mmol) following general procedure B. The product was obtained as a colorless oil (550 mg, 78% yield) after silica gel column chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 4.30 (s, 1H), 4.11 (t, J = 6.6 Hz, 2H), 3.61 (t, J = 6.2 Hz, 2H), 1.78–1.71 (m, 2H), 1.58–1.49 (m, 2H), 1.49–1.42 (m, 2H), 1.35 (s, 9H), 0.89 (s, 9H), 0.04 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 70.3, 62.7, 54.5, 32.2, 29.6, 28.6, 25.9, 22.1, 18.3, –5.3; IR (neat) ν 3295 (br), 2954 (m), 2972 (m), 2855 (m), 1496 (w), 1472 (w), 1394 (w), 1349 (m), 1255 (w), 1162 (s), 1096 (m), 1005 (w), 974 (w), 835 (m), 775 (m), 617 (w) cm–1; TLC Rf = 0.25 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H36NO4SSi 354.2129; Found 354.2136.
Methyl 6-hydroxyhexanoate tert-butylsulfamate ester (3i).
Prepared from triethylammonium tert-butylsulfamate salt (1.91 g, 7.5 mmol) and methyl 6-hydroxyhexanoate40 (0.73 g, 5.0 mmol) following general procedure B. The product was obtained as a colorless oil (1.29 g, 92% yield) after silica gel column chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 4.33 (s, 1H), 4.11 (t, J = 6.5 Hz, 2H), 3.67 (s, 3H), 2.33 (t, J = 7.3 Hz, 2H), 1.78–1.70 (m, 2H), 1.69–1.63 (m, 2H), 1.49–1.40 (m, 2H), 1.35 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3) δ 173.7, 69.6, 54.2, 51.3, 33.5, 29.3, 28.2, 24.9, 24.1; IR (neat) ν 3294 (br), 2952 (w), 2871 (w), 1734 (m), 1435 (w),1394 (m), 1344 (m), 1229(m), 1156 (s), 1103 (w), 999 (m), 953 (m), 873 (m), 811 (m), 725 (m), 614 (m), 584 (m) cm–1; TLC Rf = 0.24 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C11H23NO5S•NH4 299.1635; Found 299.1627.
5-phenylpentyl tert-butylsulfamate ester (3j).
Prepared from triethylammonium tert-butylsulfamate salt (1.9 g, 7.5 mmol) and 5-phenylpentanol41 (0.82 g, 5.0 mmol) following general procedure B. The product was obtained as a colorless oil (1.20 g, 78% yield) after silica gel column chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 7.28–7.24 (m, 2H), 7.18–7.15 (m, 3H), 4.33 (s, 1H), 4.08 (t, J = 6.9 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 1.77–1.70 (m, 2H), 1.68–1.61 (m, 2H), 1.47–1.39 (m, 2H), 1.33 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 142.2, 128.3, 128.3, 125.7, 70.2, 54.5, 35.7, 30.9, 29.6, 28.7, 25.2; IR (neat) ν 3296 (br), 2972 (w), 2934 (w), 2858 (w), 1495 (w), 1452 (w), 1429 (w), 1393 (m), 1368 (w), 1341 (m), 1230 (w), 1157 (s), 952 (m), 875 (m), 800 (m), 746 (m), 698 (m), 614 (m), 583 (m) cm–1; TLC Rf = 0.54 (hexanes/EtOAc, 7:3); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C15H24NO3S 298.1482; Found 298.1484.
(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl tert-butylsulfamate (3m).
Prepared from triethylammonium tert-butylsulfamate salt (0.76 g, 3.0 mmol) and (–)-menthol (2.0 mmol, 312.5 mg) following general procedure B. The product was obtained as a colorless oil (443 mg, 76% yield) after silica gel chromatography using hexanes:EtOAc (9:1).1H NMR (400 MHz, CDCl3) δ 4.48 (br d, 1H), 4.37 (td, J = 10.9, 4.5 Hz, 1H), 2.42–2.37 (m, 1H), 2.15 (qd, J = 6.9, 2.4 Hz, 1H), 1.72–1.63 (m, 2H), 1.35 (s, 9H), 1.18 (q, J = 12 Hz, 2H), 1.09–0.98 (m, 2H), 0.91 (t, J = 7.0 Hz, 6H), 0.87–0.83 (m, 1H), 0.81 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 83.2, 54.8, 47.7, 41.7, 33.9, 31.6, 29.8, 25.4, 23.0, 21.9, 20.9, 15.6; IR (neat) ν 3288 (br), 2955 (w), 2870 (w), 1456 (w), 1433 (w), 1393 (w), 1368 (m), 1330 (m), 1233 (w), 1159 (s), 1003 (w), 942 (m), 907 (s), 873 (s), 799 (m), 639 (m), 620 (m), 594 (m), 575 (m) cm–1; TLC Rf = 0.43 (hexane/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M – H]− Calcd for C14H28NO3S 290.1795; Found 290.1799.
(–)-16-acetoxy-13-methyl-17-norkaurane tert-butylsulfamate (3p).
Prepared from triethylammonium tert-butylsulfamate salt (303 mg, 0.89 mmol) and (–)-16-acetoyxy-18-hydroxy-13-methyl-17-norkaurane (S2, 206 mg, 0.59 mmol) following general procedure B. The product was obtained as a white foamy solid (234 mg, 82% yield) after silica gel chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 4.73 (t, J = 7.6 Hz, 1H), 4.27 (d, J = 9.3 Hz, 1H), 4.22 (s, 1H), 3.81 (d, J = 9.3 Hz, 1H), 2.06 (s, 3H), 1.82–1.76 (m, 3H), 1.70–1.62 (m, 3H), 1.59–1.48 (m, 5H), 1.41–1.37 (m, 3H), 1.35 (s, 9H), 1.26–1.17 (m, 3H), 1.07–1.01 (m, 3H), 1.00 (s, 3H), 0.90 (s, 3H), 0.87 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 171.2, 81.4, 72.9, 56.7, 56.3, 54.9, 54.4, 42.1, 41.4, 40.6, 39.1, 37.4, 37.0, 35.5, 34.3, 29.8, 29.6, 27.4, 24.8, 21.1, 20.0, 19.9, 17.7, 15.4; IR (neat) ν 3250 (br), 2929 (w), 2846 (w), 1716 (m), 1453 (w), 1438 (w), 1349 (m), 1285 (w), 1259 (m), 1157 (m), 1051 (w), 1035 (w), 1008 (w), 983 (m), 966 (m), 935 (m), 875 (m), 852 (w), 825 (m), 713 (w), 671 (w), 598 (m), 547 (w), 531 (w) cm–1; TLC Rf = 0.42 (hexanes:/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + Cl]– Calcd for C26H45NO5S•Cl 518.2713; Found 518.2718.
3β-acetoxydihydrobetulinic tert-butylsulfamate (3q).
Prepared from triethylammonium tert-butylsulfamate salt (0.50 g, 2.0 mmol) and (–)-3β,28-diacetoxydihydrobetulin42 (0.63 g, 1.3 mmol) following general procedure B. The product was obtained as a white foamy solid (502 mg, 62% yield) after silica gel chromatography using hexanes:EtOAc (9:1). 1H NMR (400 MHz, CDCl3) δ 4.47 (dd, J = 10.4, 5.8 Hz, 1H), 4.28 (d, J = 9.2 Hz, 1H), 4.20 (br s, 1H), 3.81 (d, J = 9.2 Hz, 1H), 2.04 (s, 3H), 1.92–1.83 (m, 3H), 1.74–1.45 (m, 12H), 1.42–1.19 (m, 11H), 1.36 (s, 9H), 1.05 (s, 3H), 0.95 (s, 3H), 0.87–0.82 (m, 12H), 0.77 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 171.0, 80.9, 69.0, 55.3, 54.6, 49.9, 48.1, 46.7, 44.5, 42.8, 40.9, 38.3, 37.8, 37.3, 37.0, 34.3, 34.1, 30.3, 29.7, 29.4, 27.9, 26.7, 26.7 23.6, 22.9, 21.5, 21.3, 20.7, 18.1, 16.5, 16.0, 16.0, 14.8, 14.6; IR (neat) ν 3313 (br), 2949 (w), 2873 (w), 1743 (w), 1455 (w), 1427 (w), 1392 (w), 1368 (w), 1337 (w), 1235 (w), 1163 (w), 1082 (w), 1045 (w), 1012 (w), 977 (w), 958 (w), 934 (w), 876 (w), 821 (w), 613 (w), 604 (w), 595 (w), 558 (w) cm–1; TLC Rf = 0.55 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M – H]– C36H62NO5S Calcd for 620.4354; Found 620.4366.
Preparation and Characterization of N-Chlorosulfamate Esters:
General Procedure C.
A flame-dried round bottom flask equipped with magnetic stir bar was charged with sulfamate ester (1.0 equiv), and the flask was evacuated and backfilled with N2. Anhydrous CH2Cl2 (0.2 M) was then added followed by dropwise addition of tert-butyl hypochlorite43 (tBuOCl, 2.0 equiv) or by single addition of trichloroisocyanuric acid (TCICA, 1.2 equiv). The solution was stirred at 22 °C and monitored by TLC until complete consumption of starting material was observed, (generally 1–2 hours, see below for specific details). Upon complete consumption of starting material, all volatiles were removed under reduced pressure. The resulting crude material was purified by silica gel column chromatography, eluting with a hexanes:EtOAc solvent system as noted below. The N-chlorosulfamate esters were stored in a freezer at –20 °C in the dark. Deviations from the reported procedure are specified below. The preparation and characterization of N-chlorosulfamate esters S1a, S1c–d, S1k–l, and S1n have been published previously.4
Pentyl ethylchlorosulfamate ester (S1b).
Prepared from pentyl ethylsulfamate (293 mg, 1.5 mmol) and tert-butyl hypochlorite (326 mg, 3.0 mmol) following general procedure C with a 2 h reaction time. The product was obtained as a coloroless oil (225 mg, 74% yield) after silica gel flash column chromatography using hexanes:EtOAc (20:1). 1H NMR (400 MHz, CDCl3) δ 4.37 (t, J = 6.5 Hz, 2H), 3.58 (q, J = 7.0 Hz, 2H), 1.82–1.75 (m, 2H), 1.43–1.39 (m, 4H), 1.35 (t, J = 7.0 Hz, 3H), 0.92 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 75.1, 52.6, 28.5, 27.3, 22.0, 13.7, 12.3; IR (neat) ν 2959 (w), 2935 (w), 2873 (w), 1461 (w), 1373 (m), 1184 (s), 1150 (w), 1040 (m), 951 (s), 915 (s), 856 (m), 808 (m), 781 (m), 724 (w), 618 (s), 587 (m), 555 (m) cm–1; TLC Rf = 0.42 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C7H16ClNO3S•NH4 247.0878; Found 247.0876.
Methyl 6-hydrohexanoate tertbutylchlorosulfamate ester (S1i).
Prepared from methyl 6-hydroxyhexanoate tert-butylsulfamate ester (422 mg, 1.5 mmol) and trichloroisocyanuric acid (418 mg, 1.8 mmol) following general procedure C with a 1.5 h reaction time. The product was obtained as a colorless oil (473 mg, ≥ 98% yield) after silica gel flash column chromatography using hexanes:EtOAc (5:1). 1H NMR (400 MHz, CDCl3) δ 4.30 (t, J = 6.2 Hz, 2H), 3.67 (s, 3H), 2.33 (t, J = 7.3 Hz, 2H), 1.82–1.75 (m, 2H), 1.71–1.64 (m, 2H), 1.50 (s, 9H), 1.50–1.43 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 173.8, 73.9, 67.5, 51.5, 33.7, 28.5, 28.5, 25.0, 24.3; IR (neat) ν 2947 (w), 1734 (m), 1462 (w), 1436 (w), 1365 (s), 1235 (m), 1189 (s), 1164 (s), 1102 (w), 1060 (w), 1009 (w), 946 (m), 885 (m), 786 (w), 729 (w), 614 (s), 589 (m) cm–1; TLC Rf = 0.37 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C11H22ClNO5S•NH4 333.1246; Found 333.1247.
Preparation and Characterization of N-Xanthylsulfamate Esters.
General Procedure D.
Adapted from a procedure reported by Alexanian and coworkers.10a In a flame-dried round bottom flask, potassium ethyl xanthate (1 equiv) was dissolved in anhydrous MeCN (42 mL per 1 mmol of potassium ethyl xanthate) and the flask was wrapped with aluminium foil. To this yellow solution, a solution of N-chlorosulfamate ester (1.0 equiv), in anhydrous MeCN (9 mL per 1 mmol of N-chlorosulfamate ester), was added dropwise via cannula over 10 minutes. At this point, the solution turned to a cloudy white suspension which was stirred at 22 °C and monitored by TLC until complete consumption of starting material was observed (generally within 30 minutes, see below for specific details). Upon complete consumption of starting material, the suspension was concentrated under reduced pressure and then diluted with H2O (ca. 50 mL). The crude material was extracted with dichloromethane (3 X 25 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting crude material was purified by silica gel column chromatography, eluting with a hexanes:EtOAc solvent system as noted below. The N-xanthylsulfamate esters were stored in a freezer at –20 °C in the dark. Deviations from the reported procedure are specified below.
General Procedure E.
A flame-dried round bottom flask equipped with a magnetic stir bar was charged with sulfamate ester (1.0 equiv) and fitted with a rubber septum. The flask was evacuated and backfilled with N2. To the flask were added sequentially anhydrous CH2Cl2 (10 mL per mmol of sulfamate ester), and tert-butyl hypochlorite (tBuOCl, 1.2 equiv, dropwise addition). The solution was stirred at 22 °C and monitored by TLC until complete consumption of starting material was observed (generally 1–6 hours, see below for specific details). Upon complete consumption of starting material, to the reaction flask was added a suspension of potassium ethyl xanthate (1.5 equiv) in anhydrous CH2Cl2 (50 mL per 1 mmol of potassium ethyl xanthate) dropwise via cannula over 10 minutes. A white precipitate was observed at the outset of this transfer. The cloudy white suspension was stirred at 22 °C. Upon complete consumption of starting material as judged by TLC (generally within 30 minutes, see below for specific details), the suspension was concentrated under reduced pressure and then diluted with H2O (ca. 50 mL). The crude was extracted with dichloromethane (3 X 25 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting crude material was purified by flash column chromatography on silica gel column, eluting with a hexanes:EtOAc solvent system as noted below. The N-xanthylsulfamate esters were stored in a freezer at –20 °C in the dark. Deviations from the reported procedure are specified below.
Pentyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1a).
Prepared either according to general procedure D or general procedure E. The compound was obtained as a colorless oil after silica gel column chromatography using hexanes:EtOAc (99:1).
The compound was prepared from pentyl tert-butyl(chloro)sulfamate ester (0.89 g, 3.48 mmol) following general procedure D in 83% yield (0.99 g).
The compound was prepared from pentyl tert-butylsulfamate ester (112 mg, 0.50 mmol) following general procedure E in 59% yield (102 mg).
The compound was prepared from pentyl tert-butylsulfamate ester (893 mg, 4.0 mmol) following general procedure E in 65% yield (891 mg). 1H NMR (400 MHz, CDCl3) δ 4.77–4.71 (m, 2H), 4.24–4.19 (m, 1H), 4.12–4.07 (m, 1H), 1.74–1.69 (m, 2H), 1.52 (s, 9H), 1.49 (t, J = 7.2 Hz, 3H), 1.38–1.35 (m, 4H), 0.91 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.1, 71.7, 70.4, 66.3, 29.7, 28.4, 27.5, 22.0, 13.8, 13.5; IR (neat) ν 2958 (w), 2871 (w), 1465 (w), 1363 (m), 1245 (m), 1185 (m), 1162 (m), 1112 (w), 1032 (s), 997 (w), 962 (m), 942 (m), 918 (m), 876 (s), 822 (m), 761 (m), 724 (w), 660 (w), 628 (m), 580 (w) cm–1; TLC Rf = 0.49 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C12H25NO4S3•NH4 361.1284; Found 361.1286.
Pentyl ethyl((ethoxycarbonothioyl)thio)sulfamate (1b).
Prepared either according to general procedure D or general procedure E. The compound was obtained as a colorless oil after silica gel column chromatography using hexanes:EtOAc (99:1)
The compound was prepared from pentyl ethylchlorosulfamate ester (0.41 g, 1.75 mmol) following general procedure D in 75% yield (0.41 g).
The compound was prepared from pentyl ethylsulfamate ester (100 mg, 0.51 mmol) following general procedure E in 69% yield (112 mg).
The compound was prepared from pentyl ethylsulfamate ester (976 mg, 5.0 mmol) following general procedure E in 71% yield (1.12 g).
1H NMR (400 MHz, CDCl3) δ 4.74 (q, J = 7.1 Hz, 2H), 4.20–4.12 (m, 2H), 3.80 (q, J = 7.1 Hz), 1H), 3.77–3.60 (m, 1H), 1.76–1.69 (m, 2H), 1.48 (t, J = 7.1 Hz, 3H), 1.42–1.32 (m, 4H), 1.30 (t, J = 7.2 Hz, 3H), 0.91 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 211.9, 72.1, 70.6, 50.5, 28.4, 27.5, 22.0, 13.9, 13.8, 13.6; IR (neat) ν 2957 (w), 2933 (w), 2871 (w), 1462 (w), 1368 (m), 1246 (m), 1179 (s), 1111 (m), 1093 (m), 1033 (s), 997 (m), 959 (m), 917 (m), 878 (s), 822 (m), 767 (s), 725 (m), 675 (w), 647 (m), 579 (m), 530 (m) cm–1; TLC Rf = 0.44 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H22NO4S3 316.0706; Found 316.0706.
Pentyl methyl((ethoxycarbonothioyl)thio)sulfamate (1c).
Prepared from pentyl methylchlorosulfamate ester (3c, 0.52 g, 2.4 mmol) following general procedure D. The compound was obtained as a colorless oil (412 mg, 57% yield) after silica gel column chromatography using hexanes:EtOAc (99:1). 1H NMR (400 MHz, CDCl3) δ 4.75 (q, J = 7.1 Hz, 2H), 4.16 (t, J = 6.6 Hz, 2H), 3.38 (s, 3H), 1.77–1.70 (m, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.42–1.33 (m, 4H), 0.92 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 211.8, 72.4, 70.5, 42.9, 28.4, 27.5, 22.1, 13.8, 13.7; IR (neat) ν 2957 (w), 2933 (w), 2871 (w), 1464 (w), 1368 (m), 1246 (s), 1171 (s), 1112 (m), 1089 (m), 1034 (s), 997 (m), 958 (s), 917 (m), 840 (s), 763 (m), 726 (m), 685 (w), 656 (s), 579 (m) cm−1; TLC Rf = 0.34 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H20NO4S3 302.0549; Found 302.0554.
Pentyl (2,2,2-trifluoroethyl)((ethoxycarbonothioyl)thio)sulfamate (1d).
Prepared from pentyl (2,2,2-trifluorethyl) chlorosulfamate ester (3d, 1.10 g, 3.88 mmol) following general procedure D. The compound was obtained as a colorless oil (312 mg, 22% yield) after silica gel column chromatography using hexanes:EtOAc (99:1). This N-xanthate ester was used immediately, as it was found to degrade even when stored at –20 °C in the dark. 1H NMR (400 MHz, CD3CN) δ 4.76 (q, J = 7.1 Hz, 2H), 4.36 (br s, 2H), 4.25 (t, J = 6.5 Hz, 2H), 1.74–1.70 (m, 2H), 1.45 (t, J = 7.1 Hz, 3H), 1.38–1.32 (m, 4H), 0.91 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CD3CN) δ 211.5, 124.9 (q, J = 278.8 Hz), 75.1, 72.7, 55.7 (q, J = 34.6 Hz), 29.1, 28.1, 22.8, 14.2, 13.9; 19F NMR (376 MHz, CD3CN) δ – 71.89 (t, J = 8.7 Hz); IR (neat) ν 2960 (w), 2934 (w), 2873 (w), 1466 (w), 1371 (m), 1311 (m), 1268 (s), 1155 (s), 1112 (m), 1034 (s), 952 (s), 921 (m), 809 (s), 764 (m), 727 (m), 710 (m), 651 (m), 572 (s) cm–1; TLC Rf = 0.61 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H19F3NO4S3 370.0423; Found 370.0423.
Pentyl 2-(pyridine-2-yl)propan-2-yl) ((ethoxycarbonothioyl)thio)sulfamate (1e).
Prepared from pentyl 2-(pyridine-2-yl)propan-2-yl) sulfamate ester (3e, 430 mg, 1.5 mmol) following general procedure E. The compound was obtained as a yellow solid (421 mg, 69% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 8.55 (dd, J = 4.9, 1.5 Hz, 1H), 7.67 (app td, J = 7.9, 1.5 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.18 (dd, J = 7.9, 4.9 Hz, 1H), 4.76 (q, J = 7.0 Hz, 2H), 4.3 (dt, J = 9.4, 6.6 Hz, 1H), 4.19 (dt, J = 9.4, 6.7 Hz, 1H), 1.96 (s, 3H), 1.82 (s, 3H), 1.73–1.65 (m, 2H), 1.54 (t, J = 7.1 Hz, 3H), 1.36–1.32 (m, 4H), 0.91 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.2, 163.3, 148.5, 136.4, 122.2, 119.8, 72.3, 71.1, 70.4, 28.4, 28.0, 27.4, 22.0, 13.8, 13.6; IR (neat) ν 2960 (w), 2925 (w), 2857 (w), 1588 (w), 1456 (w), 1434 (w), 1394 (w), 1364 (m), 1292 (m), 1276 (w), 1245 (m), 1176 (m), 1152 (m), 1122 (m), 1092 (w), 1035 (m), 958 (m), 942 (m), 918 (m), 878 (m), 820 (m), 784 (m), 764 (m), 745 (m), 728 (m), 657 (m), 622 (w), 585 (m), 567 (m) cm–1; TLC Rf = 0.43 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H27N2O4S3 407.1128; Found 407.1122.
Pentyl cyclohexyl((ethoxycarbonothioyl)thio)sulfamate (1f).
Prepared from pentyl cyclohexyl sulfamate ester (3f, 250 mg, 1.0 mmol) following general procedure E. The compound was obtained as a colorless oil (237 mg, 64% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.73 (q, J = 7.2 Hz, 2H), 4.21 (dt, J = 9.1, 6.6 Hz, 1H), 4.08 (dt, J = 9.6, 6.5 Hz, 1H), 3.92–3.86 (m, 1H), 2.01 (d, J = 11.3 Hz, 1H), 1.83–1.61 (m, 6H), 1.64–1.52 (m, 2H), 1.47 (t, J = 7.1 Hz, 3H), 1.40–1.31 (m, 6H), 1.09 (dd, J = 13.0, 3.7 Hz, 1H), 0.91 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 212.7, 72.0, 70.5, 62.8, 31.7, 31.0, 28.5, 27.5, 25.6, 25.4, 24.8, 22.1, 13.8, 13.6; IR (neat) ν 2930 (m), 2856 (w), 1452 (w), 1369 (m), 1268 (m), 1247 (m), 1177 (s), 1155 (m), 1112 (m), 1031 (s), 990 (m), 962 (m), 914 (s), 868 (s), 845 (m), 820 (m), 761 (m), 728 (m), 705 (w), 625 (m), 593 (s) cm–1; TLC Rf = 0.59 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C14H28NO4S3 370.1175; Found 370.1178.
Pentyl pentyl((ethoxycarbonothioyl)thio)sulfamate (1g).
Prepared from pentyl pentyl sulfamate ester (3g, 356 mg, 1.5 mmol) following general procedure E. The compound was obtained as a colorless oil (327 mg, 61% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.74 (q, J = 7.1 Hz, 2H), 4.15 (br s, 2H), 3.58 (s, 2H), 1.76–1.66 (m, 4H), 1.48 (t, J = 7.1 Hz, 3H), 1.41–1.26 (m, 8H), 0.91 (td, J = 7.1, 2.9 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 211.9, 72.1, 70.4, 55.3, 28.4, 28.3, 28.1, 27.4, 22.1, 22.0, 13.8, 13.7, 13.6; IR (neat) ν 2956 (w), 2931 (w), 2871 (w), 1465 (w), 1369 (m), 1246 (m), 1177 (s), 1113 (m), 1078 (w), 1034 (s), 997 (m), 959 (m), 917 (m), 883 (s), 821 (S), 762 (m), 726 (m), 677 (w), 646 (m), 563 (m) cm–1; TLC Rf = 0.43 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H28NO4S3 358.1175; Found 358.1174.
5-((tert-butyldimethylsilyl)oxy)pentyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1h).
Prepared from 5-((tert-butyldimethylsilyl)oxy)pentyl tert-butyl sulfamate ester (3h, 178 mg, 0.5 mmol) following general procedure E. The compound was obtained as a colorless oil (142 mg, 60% yield) after silica gel column chromatography using hexanes:EtOAc (99:1). 1H NMR (400 MHz, CDCl3) δ 4.78–4.70 (m, 2H), 4.22 (dt, J = 9.5, 6.6 Hz, 1H), 4.10 (dt, J = 9.5, 6.5 Hz, 1H), 3.61 (t, J = 6.2 Hz, 2H), 1.78–1.71 (m, 2H), 1.58–1.41 (m, 4H), 1.51 (s, 9H), 1.49 (t, J = 7.1 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.2, 71.7, 70.4, 66.4, 62.7, 32.1, 29.8, 28.7, 25.9, 22.0, 18.3, 13.6, 5.3; IR (neat) ν 2928 (w), 2855 (w), 1471 (w), 1364 (m), 1247 (w), 1186 (m), 1163 (m), 1093 (m), 1034 (s), 1003 (m), 939 (m), 880 (m), 831 (s), 773 (m), 733 (m), 660 (m), 629 (m), 581 (w) cm–1; TLC Rf = 0.31 (hexanes/EtOAc, 95:5); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H39NO5S3Si•Na 496.1652; Found 496.1655.
Methyl 6-hydroxyhexanoate tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1i).
Prepared from methyl 6-hydrohexanoate tertbutylchlorosulfamate ester (3i, 226 mg, 0.72 mmol) following general procedure D. The compound was obtained as a colorless oil (210 mg, 73% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.79–4.67 (m, 2H), 4.20 (dt, J = 9.6, 6.6 Hz, 1H), 4.09 (dt, J = 9.6, 6.5 Hz, 1H), 2.32 (s, 3H), 2.32 (t, J = 7.4 Hz, 2H), 1.77–1.71 (m, 2H), 1.70–1.62 (m, 2H), 1.50 (s, 9H), 1.47 (t, J = 7.2 Hz, 3H), 1.44–1.38 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.0, 173.6, 71.3, 70.4, 66.4, 51.4, 33.6, 29.7, 28.5, 25.0, 24.2, 13.5; IR (neat) ν 2980 (w), 2948 (w), 1734 (m), 1462 (w), 1436 (w), 1363 (s), 1268 (m), 1246 (m), 1185 (m), 1160 (s), 1111 (m), 1031 (s), 997 (m), 941 (m), 876 (s), 824 (m), 730 (m), 660 (m), 628 (s), 581 (m) cm–1; TLC Rf = 0.31 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C14H27NO6S3•NH4 419.1339; Found 419.1329.
5-phenylpentyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1j).
Prepared from 5-phenylpentyl tert-butyl sulfamate ester (3j, 300 mg, 1.0 mmol) following general procedure E. The compound was obtained as a colorless oil (255 mg, 61% yield) after silica gel column chromatography using hexanes:EtOAc (98:2). 1H NMR (400 MHz, CDCl3) δ 7.28–7.24 (m, 2H), 7.18–7.14 (m, 3H), 4.76–4.67 (m, 2H), 4.19 (dt, J = 9.6, 6.6 Hz, 1H), 4.08 (dt, J = 9.7, 6.6 Hz, 1H), 3.61 (t, J = 7.7 Hz, 2H), 1.77–1.70 (m, 3H), 1.68–1.59 (m, 3H), 1.49 (s, 9H), 1.46 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.1, 142.0, 128.3, 128.3, 125.7, 71.5, 70.4, 66.3, 35.6, 30.8, 29.7, 28.7, 25.1, 13.6; IR (neat) ν 2979 (w), 2934 (w), 2857 (w), 1495 (W), 1453 (w), 1364 (m), 1267 (m), 1246 (m), 1185 (m), 1161 (s), 1112 (m), 1031 (s), 997 (m), 941 (m), 877 (s), 822 (m), 747 (m), 698 (m), 660 (m), 628 (s), 581 (m) cm–1; TLC Rf = 0.68 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H30NO4S3 420.1332; Found 420.1331.
(3-phenyl)propyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1k).
Prepared from (3-phenyl)propyl tert-butylchlorosulfamate ester (3k, 311 mg, 1.02 mmol) following general procedure D. The compound was obtained as a colorless oil (295 mg, 74% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 7.34–7.28 (m, 2H), 7.23–7.18 (m, 3H), 4.77–4.68 (m, 2H), 4.23 (dt, J = 9.7, 6.3 Hz, 1H), 4.12 (dt, J = 10.0, 6.4 Hz, 1H), 2.74 (t, J = 7.6 Hz, 2H), 2.09–2.00 (m, 2H), 1.51 (s, 9H), 1.47 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.1, 140.3, 128.5, 128.4, 126.2, 70.7, 70.5, 66.5, 31.6, 30.5, 29.8, 13.6; IR (neat) ν 3020 (w), 2992 (w), 2974 (w), 2940 (w), 2870 (w), 1495 (w), 1474 (w), 1453 (W), 1388 (w), 1359 (s), 1263 (s), 1184 (w), 1159 (s), 1074 (m), 1049 (m), 1027 (s), 997 (m), 963 (s), 936 (s), 916 (s), 872 (s), 844 (s), 812 (m), 802 (m), 752 (s), 700 (s), 628 (s), 594 (m), 574 (s), 554 (m) cm–1; TLC Rf = 0.53 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H25NO4S3•Na 414.0838; Found 414.0834.
3,7-dimethyloctyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1l).
Prepared from 3,7-dimethyloctyl tert-butylchlorosulfamate ester (3l, 180 mg, 0.55 mmol) following general procedure D. The compound was obtained as a colorless oil (162 mg, 71% yield) after silica gel column chromatography using hexanes:EtOAc (99:1). The 1H NMR and 13C NMR data listed for this compound were obtained at 80 °C. At 25 °C, the 1H NMR and 13C NMR spectra indicated the compound exists as a mixture of rotamers. 1H NMR (500 MHz, CD3CN, 80 °C) δ 4.74–4.83 (m, 2H), 4.32–4.20 (m, 2H), 1.81–1.72 (m, 1H), 1.62–1.56 (m, 2H), 1.54 (s, 9H), 1.47 (t, J = 7.1 Hz, 3H), 1.38–1.30 (m, 4H), 1.23–1.18 (m, 3H), 0.95 (d, J = 6.5 Hz, 3H), 0.91 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (126 MHz, CD3CN, 80 °C) δ 215.5, 72.2, 72.1, 67.9, 40.3, 38.0, 37.0, 30.7, 30.5, 29.0, 25.6, 23.3, 23.2, 20.1, 14.3.
1H NMR (500 MHz, CD3CN, 25 °C) δ 4.81–4.68 (m, 2H), 4.28–4.15 (m, 2H), 1.76–1.72 (m, 1H), 1.60–1.51 (m, 2H), 1.50 (s, 9H), 1.35–1.29 (m, 4H), 1.181.13 (m, 3H), 0.91 (dd, J = 6.5 Hz, 2.8 Hz, 3H), 0.88–0.86 (m, 6H); 13C{1H} NMR (126 MHz, CD3CN, 25 °C) δ 214.7, 71.8, 71.4, 71.3, 67.2, 39.7, 37.4, 37.4, 36.2, 36.2, 30.0, 30.0, 29.8, 28.5, 25.1, 25.1, 22.9, 22.8, 19.6, 19.5, 13.8.
IR (neat) ν 2954 (w), 2926 (w), 2868 (w), 1463 (w), 1364 (m), 1267 (m), 1245 (m), 1186 (m), 1163 (m), 1112 (w), 1033 (s), 998 (w), 942 (m), 876 (s), 810 (m), 760 (m), 661 (w), 629 (m), 588 (w) cm–1; TLC Rf = 0.55 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C17H35NO4S3•NH4 431.2067; Found 431.2070.
(1R, 2S, 5R)-2-isopropyl-5-methylcyclohexyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1m).
Prepared from (1R, 2S, 5R)-2-isopropyl-5-methylcyclohexyl tert-butyl sulfamate ester (3m, 437 mg, 1.5 mmol) following general procedure E. The compound was obtained as a colorless oil (284 mg, 46% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). By comparison to 1l, 13C NMR spectral analysis suggests the compound exists as a mixture of rotamers at 25 °C. Unlike 1l, rotameric peaks did not coalesce when variable temperature NMR experiments were performed. The NMR data listed below were obtained at 25 °C and are presumed to characterize a mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 4.79–4.68 (m, 2H), 4.51 (m, 1H), 2.34 (app dd, J = 38.8, 12.0 Hz, 1H), 2.18–2.09 (m, 1H), 1.74–1.65 (m, 2H), 1.53 (s, 9H), 1.49 (t, J = 7.3 Hz, 3H), 1.39–1.33 (m, 1H), 1.24–1.03 (m, 4H), 0.95–0.89 (m, 6H), 0.83 (app. ddd, J = 9.5, 6.9, 1.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.6, 85.3, 85.2, 70.4, 70.3, 66.7, 66.6, 47.9, 47.8, 41.8, 41.4, 33.8, 31.6, 30.1, 25.6, 25.5, 23.0, 22.9, 22.0, 21.9, 20.9, 20.9, 15.8, 15.5, 13.6, 13.6; IR (neat) ν 2955 (w), 2869 (w), 1455 (w), 1364 (m), 1266 (m), 1245 (s), 1186 (m), 1162 (m), 1112 (m), 1034 (s), 999 (m), 939 (m), 916 (m), 858 (s), 818 (m), 798 (m), 773 (m), 734 (w), 659 (m), 634 (m), 594 (w), 574 (m) cm–1; TLC Rf = 0.25 (hexanes/EtOAc, 95:5); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H34NO4S3 412.1645; Found 412.1646.
Propyl tert-butyl((ethoxycarbonothioyl)thio)sulfamate (1n).
Prepared from propyl tert-butylchloro sulfamate ester (3n, 750 mg, 3.26 mmol) following general procedure D. The compound was obtained as a colorless oil (744 mg, 72% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.78–4.70 (m, 2H), 4.19 (dt, J = 9.5, 6.6 Hz, 1H), 4.07 (dt, J = 9.5, 6.5 Hz, 1H), 1.76 (qdd, J = 9.5, 6.6, 7.2 Hz, 2H), 1.52 (s, 9H), 1.49 (t, J = 7.2 H, 3H), 1.00 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.0, 73.0, 70.3, 66.1, 29.5, 22.1, 13.4, 10.0; IR (neat) ν 2976 (w), 2938 (w), 2880 (w), 1463 (w), 1363 (s), 1266 (m), 1245 (s), 1185 (m), 1162 (s), 1112 (m), 1031(s), 874 (m), 941 (s), 874 (s), 839 (s), 741 (m), 659 (m), 627 (s), 578 (m) cm–1; TLC Rf = 0.43 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C10H21NO4S3•NH4 333.0971; Found 333.0972.
(1R, 2S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl tert-butyl((ethoxycarbonothioyl)thio) sulfamate (1o).
Prepared from (1R, 2S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl tert-butyl sulfamate ester (3o, 434 mg, 1.5 mmol) following general procedure E. The compound was obtained as a colorless oil (209 mg, 34% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). By comparison to 1l, the 13C NMR spectral analysis suggests the compound exists as a mixture of rotamers at 25 °C. Unlike 1l, rotameric peaks did not coalesce when variable temperature NMR experiments were performed. The NMR data listed below were obtained at 25 °C and are presumed to characterize a mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 4.75 (m, 3H), 2.39–2.28 (m, 1H), 1.88–1.81 (m, 1H), 1.77–1.70 (m, 2H), 1.54 (s, 9H), 1.49 (t, J = 7.1 Hz, 3H), 1.43–1.36 (m, 2H), 1.33–1.29 (m, 1H), 0.93 (d, J = 4.9 Hz, 3H), 0.88 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.5, 89.7, 89.3, 70.4, 66.7, 66.5, 49.7, 49.7, 47.7, 47.6, 44.7, 44.6, 36.2, 35.6, 30.1, 30.0, 27.93, 27.9, 26.7, 26.6, 19.7, 18.8, 13.6, 13.4, 13.1; IR (neat) ν 2956 (w), 2875 (w), 1453 (w), 1365 (m), 1326 (w), 1266 (w), 1246 (m), 1187 (m), 1158 (m), 1112 (m), 1034 (s), 1003 (m), 985 (m), 941 (m), 917 (m), 862 (s), 836 (m), 807 (m), 780 (w), 736 (s), 661 (m), 632 (m), 593 (w), 565 (m) cm–1; TLC Rf = 0.43 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C17H31NO4S3•NH4 427.1754; Found 427.1750.
(–)-16-acetoyxy-13-methyl-17-norkaurane tert-butyl((ethoxycarbonthioyl)thio) sulfamate (1p).
Prepared from 3p (265 mg, 0.55 mmol) following general procedure E. The compound was obtained as a white foamy solid (100 mg, 30% yield) after silica gel column chromatography using hexanes:EtOAc (98:2). The NMR data listed below were obtained at 25 °C and are presumed to characterize a mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 4.72–4.62 (m, 3H), 4.32 (d, J = 9.2 Hz, 0.5H), 4.17 (d, J = 9.4 Hz, 0.5H), 3.86 (d, J = 9.2 Hz, 0.5H), 3.71 (d, J = 9.4 Hz, 0.5H), 2.00 (s, 3H), 1.73–1.69 (m, 4H), 1.63–1.60 (m, 2H), 1.51–1.44 (m, 5H), 1.44 (s, 9H), 1.42 (t, J = 7.2 Hz, 3H), 1.33–1.25 (m, 3H), 1.21–1.08 (m, 3H), 1.00–0.93 (m, 3H), 0.92 (s, 3H), 0.84 (s, 3H), 0.80 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.4, 171.3, 81.5, 74.6, 74.5, 70.5, 70.5, 66.5, 56.7, 56.4, 55.0, 42.4, 41.5, 41.5, 40.7, 40.7, 39.2, 37.5, 37.3, 37.3, 35.7, 35.6, 34.4, 30.0 27.4, 27.3, 24.9, 21.2, 20.1, 20.0, 17.8, 17.7, 15.6, 15.5, 13.6; IR (neat) ν 2923 (w), 2848 (w), 1729 (m), 1455 (w), 1440 (w), 1381 (m), 1365 (m), 1272 (m), 1243 (m), 1186 (m), 1164 (m), 1115 (w), 1035 (m), 954 (m), 884 (m), 853 (m), 713 (w), 672 (w), 634 (w), 598 (m), 565 (w) cm–1; TLC Rf = 0.42 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C29H49NO6S3•Na 626.2614; Found 626.2615.
3β-acetoxydihydrobetulinic tert-butyl((ethoxycarbonthioyl)thio) sulfamate (1q).
Prepared from 3q (200 mg, 0.32 mmol) following general procedure E. The compound was obtained as a white foamy solid (57 mg, 24% yield) after silica gel column chromatography using hexanes:EtOAc (98:2). The NMR data listed below were obtained at 25 °C and are presumed to characterize a mixture of rotamers. 1H NMR (400 MHz, CDCl3) δ 4.80–4.69 (m, 2H), 4.50–4.47 (dd, J = 10.1, 6.0 Hz, 1H), 4.37 (d, J = 9.3 Hz, 0.5H), 4.21 (d, J = 9.1 Hz, 0.5H), 3.94 (d, J = 9.3 Hz, 0.5H), 3.80 (d, J = 9.1 Hz, 0.5H), 2.04 (s, 3H), 1.92–1.78 (m, 3H), 1.74–1.45 (m, 9H), 1.52 (s, 9H), 1.49 (t, J = 7.1 Hz, 3H), 1.43–1.20 (m, 12H) 1.03 (s, 3H), 0.94 (s, 3H), 0.86–0.82 (m, 12H), 0.76, (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.3, 171.0, 80.9, 70.5, 66.4, 55.3, 49.9, 48.2, 48.1, 46.9, 46.8, 44.5, 44.4, 42.8, 40.9, 40.9, 38.3, 37.87 37.3, 37.0, 34.3, 34.3, 34.1, 34.0, 30.3, 29.9, 29.4, 29.3, 29.3, 27.9, 26.7, 23.6, 22.8, 21.5, 21.4, 21.3, 20.7, 18.1, 16.5, 16.1, 16.0, 15.9, 14.8, 14.6, 13.6; IR (neat) ν 2945 (w), 2868 (w), 1730 (w), 1653 (w), 1559 (w), 1540 (w), 1507 (w), 1457 (w), 1364 (w), 1242 (w), 1186 (w), 1163 (w), 1112 (w), 1035 (w), 966 (w), 881 (w), 835 (w), 657 (w), 633 (w), 594 (w) cm–1; TLC Rf = 0.40 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C39H68NO6S3 742.4203; Found 742.4194.
Preparation and Characterization of Alkylxanthate Sulfamate Esters:
General Procedure F.
In a flame-dried microwave vial equipped with magnetic stir bar and sealed with a disposable crimp cap containing a Teflon-lined septum, N-xanthylsulfamate ester (0.28 mmol, 1.0 equiv) was dissolved in anhydrous MeCN (4.0 mL, 0.07 M) under a nitrogen atmosphere. The vial was then placed in between two 26W CFL bulbs (ca. 3 cm away from each light source) and irradiated until complete consumption of starting material was observed by TLC (generally 12–24 hours, see below for specific details). Upon consumption of starting material as judged by TLC, the reaction solution was transferred to a round-bottomed flask rinsing the microwave vial with CH2Cl2 (ca. 4 mL) to achieve quantitative transfer. Volatiles were removed under reduced pressure. The resulting crude material was purified by flash column chromatography on silica gel column or manual flash chromatography using Florisil, eluting with a hexanes:EtOAc solvent system as noted below.
3-((ethoxycarbonothioyl)thio)pentyl tert-butylsulfamate (2a).
Prepared from N-xanthylsulfamate ester 1a (96.2 mg, 0.28 mmol) following general procedure F. After irradiating for 12 h, the product was obtained as colorless oil (87.6 mg, 91% yield) following purification by silica gel flash chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.68–4.60 (m, 2H), 4.30 (s, 1H), 4.23 (t, J = 6.6 Hz, 2H), 3.83–3.76 (m, 1H), 2.20–2.11 (m, 1H), 2.08–1.99 (m, 1H), 1.81 (ddd, J = 14.6, 7.3, 5.7 Hz, 1H), 1.68–1.76 (m, 1H), 1.43 (t, J = 7.1 Hz, 3H), 1.36 (s, 9H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.4, 69.8, 67.4, 54.5, 48.7, 32.7, 29.4, 27.1, 13.6, 11.0; IR (neat) ν 3296 (br), 2968 (w), 1428 (w), 1393 (w), 1344 (m), 1291 (w), 1211 (m), 1159 (s), 1110 (m), 1042 (s), 1002 (m), 974 (s), 898 (m), 873 (m), 804 (m), 770 (m), 614 (m), 587 (m) cm−1; TLC Rf = 0.15 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C12H25NO4S3•Na 366.0838; Found 366.0836.
3-((ethoxycarbonothioyl)thio)pentyl ethylsulfamate (2b).
Prepared from N-xanthylsulfamate ester 1b (88.3 mg, 0.28 mmol) following general procedure F. After irradiating for 24 h, the product was obtained as colorless oil (69.1 mg, 78% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.65 (q, J = 7.1 Hz, 2H), 4.27 (s, 1H), 4.26 (dd, J = 7.1, 6.0 Hz, 3H), 3.85–3.78 (m, 1H), 3.20 (qd, J = 7.2, 5.7 Hz, 2H), 2.21–2.13 (m, 1H), 2.08–1.99 (m, 1H), 1.84–1.68 (m, 2H), 1.43 (t, J = 7.1 Hz, 3H), 1.23 (t, J = 7.3 Hz, 3H), 1.03 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.8, 70.0, 67.9, 49.0, 39.0, 32.9, 27.4, 15.1, 13.7, 11.1; IR (neat) ν 3299 (br), 2964 (w), 2923 (w), 1427 (w), 1344 (m), 1289 (w), 1212 (m), 1170 (s), 1109 (m), 1041 (s), 979 (m), 952 (m), 899 (m), 785 (m), 583 (m) cm–1; TLC Rf = 0.11 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H22NO4S3 316.0706; Found 316.0709.
3-((ethoxycarbonothioyl)thio)pentyl methylsulfamate (2c).
Prepared from N-xanthylsulfamate ester 1c (84.4 mg, 0.28 mmol) following general procedure F. After irradiating for 12 h, the product was obtained as colorless oil (65 mg, 77% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.65 (q, J = 7.1 Hz, 2H), 4.36 (br s, 1H), 4.26 (t, J = 6.5 Hz, 3H), 3.85–3.78 (m, 1H), 2.82 (d, J = 5.3 Hz, 3H), 2.22–2.14 (m, 1H), 2.09–2.00 (m, 1H), 1.85–1.67 (m, 2H), 1.43 (t, J = 7.2 Hz, 3H), 1.03 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.8, 70.0, 68.0, 49.0, 32.9, 29.8, 27.4, 13.7, 11.1; IR (neat) ν 3309 (br), 2964 (w), 2932 (w), 1459 (w), 1411 (w), 1343 (m), 1290 (w), 1212 (s), 1171 (s), 1145 (m), 1110 (m), 1040 (s), 974 (s), 899 (m), 853 (s), 756 (m), 580 (m) cm–1; TLC Rf = 0.10 (hexanes:EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C9H19NO4S3•NH4 319.0815; Found 319.0819.
3-((ethoxycarbonothioyl)thio)pentyl (2,2,2-trichloroethyl)sulfamate (2d).
Prepared from N-xanthylsulfamate ester 1d (90.8 mg, 0.28 mmol) following general procedure F. After irradiating for 20 h, the product was obtained as colorless oil (52 mg, 57% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.90 (br s, 1H), 4.65 (q, J = 7.1 Hz, 2H), 4.31 (dd, J = 7.1, 5.9 Hz, 2H), 3.84–3.72 (m, 3H), 2.22–2.14 (m, 1H), 2.09–2.00 (m, 1H), 1.83–1.68 (m, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.01 (t, J = 7.4 Hz, 3H).
13C{1H} NMR (126 MHz, CDCl3) 213.8, 70.2, 69.1, 49.0, 45.1 (q, J = 36.0 Hz), 32.8, 27.5 13.7, 11.1 [full quartet from CF3 group was not detected]; 19F NMR (326 MHz, CDCl3) δ – 72.72 (t, J = 8.4 Hz); IR (neat) ν 3298 (br), 2966 (w), 2923 (w), 2852 (w), 1641 (w), 1459 (w), 1363 (m), 1273 (m), 1218 (m), 1153 (s), 1112 (m), 1044 (m), 962 (s), 915 (m), 856 (m), 736 (m), 702 (m), 664 (m), 560 (m) cm–1; TLC Rf = 0.38 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C10H18F3NO4S3•NH4 387.0688; Found 387.0687.
3-((ethoxycarbonothioyl)thio)pentyl 2-(pyridine-2-yl)propan-2-yl) sulfamate (2e).
Prepared from N-xanthylsulfamate ester 1e (114 mg, 0.28 mmol) following general procedure F. After irradiating for 16 h, the product was obtained as colorless oil (84 mg, 74% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 8.52 (dd, J = 4.8, 1.6 Hz, 1H), 7.74 (td, J = 7.7, 1.7 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.25–7.19 (m, 1H), 4.62 (q, J = 7.0 Hz, 2H), 4.24–4.15 (m, 2H), 3.78–3.71 (m, 1H), 2.12–2.04 (m, 1H), 2.02–1.93 (m, 1H), 1.80–1.64 (m, 2H), 1.73 (s, 6H), 1.41 (t, J = 7.1 Hz, 3H), 0.98 (t, J = 7.4 Hz); 13C{1H} NMR (126 MHz, CDCl3) δ 213.5, 162.9, 147.7, 137.4, 122.3, 118.8, 69.9, 67.4, 58.9, 48.8, 32.9, 28.3, 28.3, 27.2, 13.7, 11.0; IR (neat) ν 3260 (br), 2967 (w), 2933 (w), 1592 (w), 1573 (w), 1462 (w), (1433 (m), 1401 (w), 1380 (m), 1341 (m), 1293 (w), 1212 (m), 1171 (s), 1111 (m), 1042 (s), 978 (s), 918 (m), 825 (m), 785 (m), 748 (m), 655 (m), 622 (m), 591 (m), 559 (s) cm–1; TLC Rf = 0.20 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H27N2O4S3 407.1128; Found 407.1123.
3-((ethoxycarbonothioyl)thio)pentyl cyclohexylsulfamate (2f).
Prepared from N-xanthylsulfamate ester 1f (103 mg, 0.28 mmol) following general procedure F. After irradiating for 12 h, the product was obtained as colorless oil (85 mg, 82% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.28 (d, J = 7.0 Hz, 1H), 4.24 (t, J = 6.5 Hz, 2H), 3.84–3.77 (m, 1H), 3.35–3.26 (m, 1H), 2.20–2.12 (m, 1H), 2.08–1.99 (m, 3H), 1.84–1.68 (m, 4H), 1.62–1.57 (m, 2H), 1.43 (t, J = 7.1 Hz, 3H), 1.36–1.15 (m, 5H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.7, 70.0, 67.7, 53.5, 49.0, 33.6, 32.9, 27.3, 25.1, 24.6, 13.7, 11.1; IR (neat) ν 3294 (br), 2930 (m), 2854 (w), 1450 (m), 1342 (m), 1299 (w), 1211 (m), 1171 (s), 1147 (m), 1110 (m), 1043 (s), 977 (s), 914 (m), 887 (m), 842 (m), 803 (m), 782 (m), 757 (m), 678 (w), 579 (m) cm–1; TLC Rf = 0.20 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C14H27NO4S3•Na 392.0994; Found 392.0999.
3-((ethoxycarbonothioyl)thio)pentyl pentylsulfamate (4).
Prepared from N-xanthylsulfamate ester 1g (100 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (85.2 mg, 85% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.40 (br s, 1H), 4.13 (t, J = 6.6 Hz, 2H), 3.75 (q, J = 6.8 Hz, 1H), 3.17 (app q, J = 6.3 Hz, 2H), 1.78–1.68 (m, 6H), 1.42 (t, J = 7.1 Hz, 3H), 1.39 (d, J = 6.9 Hz, 3H), 1.37 –1.32 (m, 4H), 0.91 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 214.2, 70.8, 69.7, 45.2, 43.3, 32.8, 28.5, 27.6, 26.9, 22.1, 20.3, 13.8, 13.7; IR (neat) ν 3300 (br), 2956 (w), 2931 (w), 2870 (w), 1427 (w), 1345 (m), 1291 (w), 1209 (m), 1169 (s), 1109 (m), 1043 (s), 962 (m), 911 (m), 830 (m), 758 (m), 724 (m), 580 (m) cm–1; TLC Rf = 0.19 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H27NO4S3•Na 380.0994; Found 380.0998.
5-((tert-butyldimethylsilyl)oxy)-3-((ethoxycarbonothioyl)thio)pentyl tert-butylsulfamate (2h).
Prepared from N-xanthylsulfamate ester 1h (133 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (98 mg, 74% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.30 (br s, 1H), 4.24 (t, J = 6.7 Hz, 2H), 4.01–3.94 (m, 1H), 3.74 (t, J = 6.1 Hz, 2H), 2.28–2.19 (m, 1H), 2.15–2.06 (m, 1H), 1.96–1.87 (m, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.35 (s, 9H), 0.88 (s, 9H), 0.04 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.3, 70.0, 67.7, 60.1, 54.8, 44.4, 36.6, 33.7, 29.7, 25.9, 18.2, 13.8, 5.4; IR (neat) ν 3295 (br), 2953 (w), 2927 (w), 2855 (w), 1470 (w), 1392 (w), 1348 (m), 1291 (w), 1250 (m), 1211 (m), 1160 (s), 1108 (m), 1043 (s), 1004 (m), 977 (m), 932 (m), 876 (m), 833 (s), 809 (m), 773 (s), 662 (w), 615 (m), 587 (m) cm–1; TLC Rf = 0.44 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H39NO5S3Si•Na 496.1652; Found 496.1652.
Methyl 4-((ethoxycarbonothioyl)thio)-6-hydroxyhexanoate) tert-butylsulfamate (2i).
Prepared from N-xanthylsulfamate ester 1i (112 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (93.4 mg, 83% yield) after silica gel column chromatography using hexanes:EtOAc (80:20). 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.37 (s, 1H), 4.25 (t, J = 6.2 Hz, 2H), 3.92–3.85 (m, 1H), 3.68 (s, 3H), 2.51–2.47 (m, 2H), 2.20–2.04 (m, 3H), 2.02–1.91 (m, 1H), 1.43 (t, J = 7.1 Hz, 3H), 1.36 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3) δ 212.8, 173.1, 70.2, 67.2, 54.7, 51.7, 46.9, 33.5, 31.1, 29.6, 29.5, 29.4, 13.7; IR (neat) ν 3293 (br), 2975 (w), 1734 (m), 1435 (m), 1393 (w), 1346 (m), 1290 (w), 1211 (s), 1158 (s), 1110 (m), 1043 (s), 974 (s), 873 (m), 773 (m), 734 (m), 613 (m), 586 (m) cm–1; TLC Rf = 0.22 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C14H27NO6S3•NH4 419.1339; Found 419.1332.
5-phenyl-3-((ethoxycarbonothioyl)thio)pentyl tert-butylsulfamate (2j).
Prepared from N-xanthylsulfamate ester 1j (118 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (94 mg, 80% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 2H), 7.22–7.17 (m, 3H), 4.64 (q, J = 7.1 Hz, 2H), 4.25–4.22 (m, 3H), 3.90–3.83 (m, 1H), 2.81–2.73 (m, 2H), 2.23–2.17 (m, 1H), 2.13–1.99 (m, 3H), 1.42 (t, J = 7.1 Hz, 3H), 1.35 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.2, 140.9, 128.5, 128.4, 126.1, 70.1, 67.5, 54.8, 47.1, 36.5, 33.5, 33.0, 29.6, 13.7; IR (neat) ν 3294 (br), 2975 (w), 2934 (w), 1495 (w), 1453 (w), 1428 (w), 1392 (m), 1344 (m), 1290 (w), 1212 (w), 1159 (s), 1110 (m), 1043 (s), 973 (m), 906 (m), 875 (m), 809 (m), 779 (m), 747 (m), 698 (m), 613 (m), 584 (m) cm–1; TLC Rf = 0.53 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C18H28NO4S3 418.1186; Found 418.1178.
3-((ethoxycarbonothioyl)thio)-3-phenylpropyl tert-butylsulfamate (2k).
Prepared from N-xanthylsulfamate ester 1k (109 mg, 0.28 mmol) following general procedure F. After irradiating for 26 h, the product was obtained as colorless oil (83 mg, 76% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 4.3 Hz, 2H), 7.32–7.27 (m, 3H), 4.89 (dd, J = 9.1, 6.6 Hz, 1H), 4.60 (q, J = 7.1 Hz, 2H), 4.23 (s, 1H), 4.16–4.10 (m, 1H), 4.04–3.98 (m, 1H), 2.55–2.47 (m, 1H), 2.37–2.28 (m, 1H), 1.39 (t, J = 7.1 Hz, 3H), 1.33 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 212.0, 139.2, 128.8, 127.9, 127.9, 70.1, 67.2, 54.8, 50.2, 34.9, 29.6, 13.7; IR (neat) ν 3296 (br), 2976 (w), 1492 (w), 1468 (w), 1452 (w), 1427 (w), 1392 (w), 1345 (m), 1290 (w), 1265 (w), 1219 (m), 1159 (s), 1110 (m), 1089 (w), 1041 (s), 981 (s), 908 (m), 812 (w), 782 (m), 734 (s), 697 (s), 613 (m), 584 (m) cm–1; TLC Rf = 0.32 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H25NO4S3•Na 414.0838; Found 414.0834.
3-((ethoxycarbonothioyl)thio)-3,7-dimethyloctyl tert-butylsulfamate (2l).
Prepared from N-xanthylsulfamate ester 1l (116 mg, 0.28 mmol) following general procedure F. After irradiating for 12 h, the product was obtained as colorless oil (71 mg, 61% yield) after Florosil column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.66 (q, J = 7.1 Hz, 2H), 4.29 (br s, 1H), 4.24 (t, J = 7.4 Hz, 2H), 2.38 (dt, J = 14.6, 7.3 Hz, 1H), 2.29 (dt, J = 14.5, 7.4 Hz, 1H), 1.80 (ddd, J = 13.9, 11.2, 5.7, 1H), 1.67–1.61 (m, 1H), 1.57–1.51 (m, 2H), 1.46 (t, J = 7.2 Hz, 3H), 1.43 (s, 3H), 1.35 (s, 9H), 1.18–1.15 (m, 3H), 0.87 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 212.8, 69.6, 67.2, 56.8, 54.7, 40.2, 39.0, 37.9, 29.6, 27.7, 25.2, 22.5, 22.5, 21.8, 13.7; IR (neat) ν 3297 (br), 2954 (w), 2929 (w), 2868 (w), 1465 (m), 1393 (w), 1348 (m), 1226 (m), 1160 (s), 11128 (m), 1040 (m), 972 (m), 881 (m), 776 (m), 737 (m), 703 (w), 675 (w), 614 (m) cm–1; TLC Rf = 0.15 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C17H35NO4S3•NH4 431.2067; Found 431.2063.
(1R, 2R, 5R)-2-(2-(ethoxycarbonothioyl)thio)propan-2-yl)-5-methylcyclohexyl tert-butylsulfamate (2m).
Prepared from N-xanthylsulfamate ester 1m (115 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (73.8 mg, 64% yield) after silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.73–4.65 (m, 2H), 4.57 (td, J = 10.8, 4.4 Hz, 1H), 4.34 (s, 1H), 2.52–2.49 (m, 1H), 2.37–2.31 (m, 1H), 2.12–2.08 (m, 1H), 1.74–1.71 (m, 1H), 1.67 (s, 3H), 1.47 (s, 3H), 1.47 (t, J = 7.0 Hz, 3H), 1.38 (s, 9H), 1.23 – 1.10 (m, 4H), 0.94 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.9, 82.3, 69.3, 58.4, 55.3, 48.6, 42.3, 34.3, 31.4, 29.9,27.8, 26.7, 25.2, 21.6, 13.9; IR (neat) ν 3301 (br), 2956 (w), 2925 (w), 2870 (w), 1458 (w), 1391 (w), 1335 (m), 1264 (m), 1225 (m), 1159 (m), 1040 (s), 1002 (m), 972 (w), 946 (m), 872 (m), 809 (m), 724 (s), 702 (m), 641 (w), 620 (w), 572 (m) cm–1; TLC Rf = 0.32 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H33NO4S3•Na 434.1464; Found 434.1462.
3-((ethoxycarbonothioyl)thio)propyl tert-butylsulfamate (2n).
Prepared from N-xanthylsulfamate ester 1n (88.3 mg, 0.28 mmol) following general procedure F. After irradiating for 15 h, the product was obtained as white solid (42.6 mg, 48% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). Note: 2-((Ethoxycarbonothioyl)thio)propyl tert-butylsulfamate was formed as a byproduct (16.1 mg, 18% yield) from the xanthyl transfer of N-xanthyl sulfamate ester 1l. The characterization data is listed below. 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.49 (s, 1H), 4.20 (t, J = 6.0 Hz, 2H), 3.23 (t, J = 7.1 Hz, 2H), 2.14 (tt, J = 7.1, 6.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.36 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 214.1, 70.1, 68.4, 54.8, 31.8, 29.6, 27.9, 13.7; IR (neat) ν 3302 (br), 2968 (w), 2924 (w), 2899 (w), 1470 (w), 1428 (w), 1392 (w), 1369 (w), 1351 (m), 1292 (w), 1172 (m), 1160 (m), 1110 (m), 1046 (m), 1034 (s), 1013 (s), 989 (m), 921 (m), 868 (m), 774 (m) 774 (m), 653 (m), 617 (m), 584 (m) cm–1; TLC Rf = 0.16 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H22NO4S3 316.0706; Found 316.0706.
2-((Ethoxycarbonothioyl)thio)propyl tert-butylsulfamate (S4a).
The compound was formed as a minor byproduct from the xanthyl transfer of N-xanthyl sulfamate ester 1n. The compound was isolated as an colorless oil (16.1 mg, 18% yield) after silica gel column chromatography using hexanes:EtOAc. 1H NMR (400 MHz, CDCl3) δ 4.65 (q, J = 7.0 Hz, 2H), 4.37 (dd, J = 9.3, 4.0 Hz, 1H), 4.30 (br s, 1H), 4.12–4.07 (m, 1H), 4.07–4.00 (m, 1H), 1.45 (d, J = 7.0 Hz, 3H), 1.43 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 212.6, 71.8, 70.2, 54.9, 43.4, 29.6, 16.3, 13.7; IR (neat) ν 3297 (br), 292975 (w), 2929 (w), 1442 (w), 1394 (m), 1348 (m), 1212 (m), 1161 (s), 1111 (m), 1051 (m), 1016 (m), 966 (s), 873 (m), 813 (m), 709 (w), 614 (m), 578 (m) cm–1; TLC Rf = 0.2 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H22NO4S3 316.0706; Found 316.0701.
(1S, 2S)-1-(((ethoxycarbonothioyl)thio)methyl)-7,7-dimethylbicyclo[2.2.1]heptan-2-yl tert-butylsulfamate (2o).
Prepared from N-xanthylsulfamate ester 1o (115 mg, 0.28 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as colorless oil (53 mg, 46% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). Note: (1R, 2R, 3R)-3-((ethoxycarbonothioyl)thio)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl tert-butylsulfamate was formed as a byproduct (33 mg, 29% yield) from the xanthyl transfer of N-xanthyl sulfamate ester 1m. The characterization data is listed below. 1H NMR (400 MHz, CDCl3) δ 4.91 (dt, J = 9.9, 2.7 Hz, 1H), 4.65 (q, J = 7.1 Hz, 2H), 4.34 (s, 1H), 3.39 (d, J = 13.6 Hz, 1H), 3.28 (d, J = 13.6 Hz, 1H), 2.38 (ddd, J = 14.1, 9.8, 4.3 Hz, 1H), 2.00 (ddd, J = 13.5, 9.7 Hz, 4.4, 1H), 1.86–1.78 (m, 1H), 1.72 (t, J = 4.5 Hz, 1H), 1.59–1.49 (m, 3H), 1.42 (t, J =7.1 Hz, 3H), 1.38 (s, 9H), 1.00 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 214.9, 85.1, 70.0, 55.1, 52.2, 48.9, 45.4, 36.5, 36.1, 29.8, 27.7, 25.0, 20.4, 19.5, 13.8; IR (neat) ν 3292 (br), 2957 (w), 1428 (w), 1392 (m), 1368 (w), 1340 (m), 1305 (w), 1266 (w), 1161 (m), 1110 (m), 1046 (s), 985 (m), 964 (m), 938 (m), 890 (m), 844 (m), 800 (m), 784 (m), 731 (m), 616 (m), 566 (m) cm–1; TLC Rf = 0.37 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H31NO4S3•Na 432.1307; Found 432.1311.
(1R, 2R, 3R)-3-((ethoxycarbonothioyl)thio)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl tert-butylsulfamate (S4b).
The compound was formed as a byproduct from the xanthyl transfer of N-xanthyl sulfamate ester 1o. The compound was isolated as a colorless oil (33 mg, 29% yield) after silica gel column chromatography using hexanes:EtOAc (95:5).1H NMR (400 MHz, CDCl3) δ 4.69 (d, J = 4.5 Hz, 1H), 4.66–4.59 (m, 2H), 4.36 (s, 1H), 3.84 (d, J = 4.6 Hz, 1H), 1.97–1.89 (m, 3H), 1.61–1.54 (m, 2H), 1.41 (td, J = 7.1, 1.3 Hz, 4H), 1.34 (s 9H), 0.97 (s, 6H), 0.89 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.5, 89.4, 70.1, 56.7, 55.0, 52.6, 50.8, 47.1, 29.6, 29.1, 25.9, 20.5, 19.5, 13.8, 13.0; IR (neat) ν 3302 (br), 2957 (w), 2923 (w), 2853 (w), 1434 (w), 1393 (w), 1345 (m), 1304 (w), 1291 (w), 1209 (m), 1159 (m), 1111 (m), 1092 (w), 1052 (s), 1010 (m), 985 (m), 968 (m), 917 (m), 877 (m), 852 (m), 788 (m), 724 (w), 668 (m), 616 (m), 589 (w), 567 (m) cm–1; TLC Rf = 0.34 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C17H31NO4S3•NH4 427.1754; Found 427.1754.
(1R,3aS,5aS,5bS,7aS,9R,11aS,11bS,13aS,13bS)-3a-(((N-(tert-butyl)sulfamoyl)oxy)methyl)-3-((ethoxycarbonothioyl)thio)-1-isopropyl-5a,5b,8,8,11a-pentamethylicosahydro-1H-cyclopenta[a]chrysen-9-yl acetate (2p).
Prepared from N-xanthylsulfamate ester 1p (60.4 mg, 0.1 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as white foamy solid (46 mg, 76% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). Note: The characterization data listed below is of major diastereomer.1H NMR (700 MHz, CDCl3) δ 4.72 (dd, J = 9.9, 5.4 Hz, 1H), 4.64 (q, J = 7.1 Hz, 2H), 4.42 (d, J=9.6 Hz, 1H), 4.40 (br s, 1H), 4.28 (m, 1H), 3.87 (d, J= 9.6 Hz, 1H), 2.17–2.11 (m, 1H), 2.05 (s, 3H), 1.82–1.76 (m, 3H), 1.57–1.52 (m, 5H), 1.42 (t, J = 7.2 Hz), 1.37 (s, 9H), 1.35–1.32 (m, 1H), 1.29–1.18 (m, 5H), 1.14 (s, 3H), 1.12–1.04 (m, 3H), 0.90 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.7, 171.3, 81.4, 72.6, 70.0, 56.1, 54.9, 54.5, 42.0, 41.4, 41.4, 41.1, 40.6, 37.4, 34.8, 34.3, 29.8, 25.2, 24.8, 23.8, 21.2, 19.9, 19.6, 15.8, 13.7; IR (neat) ν 3285 (br), 2929 (w), 2847 (w), 1735 (w), 1713 (w), 14.39 (w), 1391 (w), 1351 (w), 1238 (w), 1207 (m), 1161 (m), 1110 (w), 1045 (m), 964 (m), 873 (w), 832 (w), 811 (w), 749 (w), 727 (w), 672 (w), 616 (w) cm–1; TLC Rf = 0.16 (hexanes:EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H50NO6S3 604.2795; Found 604.2796.
(3S,4R,4aS,6aS,8R,9R,11aR,11bS)-4-(((N-(tert-butyl)sulfamoyl)oxy)methyl)-3-((ethoxycarbonothioyl)thio)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalen-8-yl acetate (2q).
Prepared from N-xanthylsulfamate ester 1q (74.2 mg, 0.1 mmol) following general procedure F. After irradiating for 18 h, the product was obtained as white foamy solid (50.4 mg, 68% yield) after silica gel column chromatography using hexanes:EtOAc (95:5). Note: The characterization data listed below is of major diastereomer only. 1H NMR (400 MHz, CDCl3) δ 4.67–4.57 (m, 2H), 4.47 (dd, J = 10.9, 5.2 Hz, 1H), 4.36 (d, J = 9.8 Hz, 1H), 4.30 (s, 1H), 3.97–3.90 (m, 2H), 2.04 (s, 3H), 1.95–1.81 (m, 4H), 1.70–1.45 (m, 15H), 1.41 (t, J = 7.2 Hz, 3H), 1.40 (s, 9H), 1.33–1.23 (m, 6H), 0.86 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H), 0.84 (s, 3H), 0.83 (s, 3H), 0.81 (d, J = 6.6 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 216.1, 171.0, 80.8, 69.6, 67.9, 55.7, 55.3, 55.0, 51.0, 50.3, 49.8, 45.0, 44.8, 41.2, 38.3, 37.7, 37.0, 37.0, 35.8, 34.0, 33.5, 29.8, 29.7, 29.4, 27.9, 26.4, 23.6, 22.8, 21.3, 21.2, 20.6, 18.0, 16.4, 16.1, 14.9, 13.8; IR (neat) ν 3290 (br), 2952 (w), 2869 (w), 1732 (w), 1448 (w), 1392 (w), 1367 (m), 1243 (m), 1209 (m), 1162 (m), 1111 (m), 1047 (m), 1011 (m), 973 (m), 916 (w), 875 (m), 814 (m), 732 (m), 647 (w), 614 (m), 591 (w) cm–1; TLC Rf = 0.20 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C39H68NO6S3 742.4203; Found 742.4198. Crystals suitable for single crystal X-Ray diffraction were grown by dissolving in CHCl3 and layering pentane (ca. 1:3 CHCl3:pentane).
Further Transformation of Alkyl Xanthates.
3-mercapto pentyl tert-butylsulfamate (7).
Thiolation was performed using modified literature procedure.10aA flame-dried microwave vial with magnetic stir bar was charged with 3-((ethoxycarbonothioyl)thio)pentyl tert-butyl sulfamate (34.4 mg, 0.1 mmol, 1.0 equiv). The vial was capped with a crimp cap and evacuated and backfilled with N2 three times. Absolute ethanol (0.5 mL, 0.2 M) and 4-methylpiperidine (0.05 mL, 0.4 mmol, 4.0 equiv) were then added sequentially. The reaction was left to stir at 21 °C for 24 h. After 24 h, the yellow solution was transferred to a scintillation vial rinsing the reaction vial with CH2Cl2 (5 mL) to ensure quantitative transfer. The reaction solution was then concentrated under reduced pressure. The product was obtained as a colorless oil (20.7 mg, 81% yield) following silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.37–4.26 (m, 3H), 2.90–2.81 (m, 1H), 2.19–2.11 (m, 1H), 1.79–1.70 (m, 2H), 1.61–1.50 (m, 2H), 1.37 (s, 9H), 1.03 (t, J = 7.3 Hz); 13C{1H} NMR (126 MHz, CDCl3) δ 68.0, 54.8, 38.8, 37.6, 32.1, 29.7, 11.4; IR (neat) ν 3296 (br), 2967 (w), 2923 (w), 2875 (w), 2853 (w), 2572 (br)1462 (w), 1429 (w), 1393 (w), 1342 (m), 1231 (w), 1210 (w), 1158 (s), 1086 (w), 1041 (w), 976 (s), 916 (m), 874 (m), 771 (m), 750 (w), 616 (m), 587 (m) cm–1; TLC Rf = 0.52 (hexanes/EtOAc, 7:3); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H22NO3S2 256.1036; Found 256.1041.
3-azido pentyl tert-butylsulfamate (8).
Azidation was performed using modified literature procedure.10a A flame-dried microwave vial with stir bar was taken into the glovebox and charged with 3-((ethoxycarbonothioyl)thio)pentyl tert-butyl sulfamate (44.6 mg, 0.13 mmol, 1.0 equiv), ethyl sulfonyl azide11b (55 mg, 0.4 mmol, 3.0 equiv), and dilauroyl peroxide (5.4 mg, 0.013 mmol, 0.1 equiv). Anhydrous chlorobenzene (0.5 mL, 0.26 M) was added. The vial was sealed with a rubber septum and removed from the glovebox. The reaction was then heated in a pie block set at 100 °C. After stirring for 0.5 h, an additional 0.1 equiv of dilauroyl peroxide was added by briefly removing the septum and adding the dilauroyl peroxide in a single portion. An additional 0.1 equiv of dilauroyl peroxide was added every 0.5 h until a total of 0.6 equiv had been added. At the time when the next dilauroyl peroxide addition would have occurred, the reaction was removed from the heating block and allowed to cool to room temperature. Once cool, the reaction solution was transferred to a scintillation vial rinsing the reaction vial with CH2Cl2 (5 mL) to ensure quantitative transfer. The reaction solution was then concentrated under reduced pressure. The product was obtained as a colorless oil (27.1 mg, 79% yield) following silica gel column chromatography using gradient from hexanes (100%) to hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.39 (s, 1H), 4.22 (dd, J = 7.6, 5.1 Hz, 2H), 3.48–3.42 (m, 1H), 1.96 (dtd, J = 14.7, 7.4, 3.8, 1H), 1.75 (ddt, J = 14.7, 9.9, 5.1 Hz, 1H), 1.68–1.59 (m, 2H), 1.37 (s, 9H), 1.02 (t, J = 7.4, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 66.9, 60.4, 54.8, 33.3, 29.6, 27.6, 10.3; IR (neat) ν 3298 (br), 2971 (w), 2930 (w), 2095 (m), 1465 (w), 1429 (w), 1394 (w), 1341 (m), 1232 (m), 1159 (s), 1043 (w), 979 (s), 915 (m), 872 (m), 814 (w), 771 (m), 746 (w), 615 (m), 589 (m) cm–1; TLC Rf = 0.38 hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C9H19N4O3S 263.1183; Found 263.1179.
3-((trifluoromethyl)thio) pentyl tert-butylsulfamate (9).
Trifluoromethylthiolation was performed using modified literature procedure.10a A flame-dried microwave vial with stir bar was taken into the glovebox and charged with 3-((ethoxycarbonothioyl)thio)pentyl tert-butyl sulfamate (46.4 mg, 0.14 mmol, 1.0 equiv), ((2-phenylpropan-2-yl)oxy)(trifluoromethyl)sulfane44 (95 mg, 0.4 mmol, 3.0 equiv), and dilauroyl peroxide (25 mg, 0.07 mmol, 0.5 equiv). Anhydrous chlorobenzene (6.8 mL, 0.02 M) was added. The vial was sealed with a rubber septum and removed from the glovebox. The reaction was then heated in a pie block set at 100 °C. After stirring for 0.5 h, an additional 0.5 equiv of dilauroyl peroxide was added by briefly removing the septum and adding the dilauroyl peroxide in a single portion. An additional 0.5 equiv of dilauroyl peroxide was added every 0.5 h until a total of 4.0 equiv had been added. At the time when the next dilauroyl peroxide addition would have occurred, the reaction was removed from the heating block and allowed to cool to room temperature. Once cool, the reaction solution was transferred to a scintillation vial rinsing the reaction vial with CH2Cl2 (5 mL) to ensure quantitative transfer. The reaction solution was then concentrated under reduced pressure. The product was obtained as a colorless oil (33.2 mg, 76% yield) following silica gel column chromatography using using gradient from hexanes (100%) to hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.39 (s, 1H), 4.28 (t, J = 7.0 Hz, 2H), 3.27–3.21 (m, 1H), 2.19–2.11 (m, 1H), 2.04–1.95 (m, 1H), 1.82 (dt, J = 14.4, 7.3 Hz, 1H), 1.72 (dt, J = 14.6, 7.4 Hz, 1H), 1.36 (s, 9H), 1.02 (dd, J = 7.3, 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 130.9 (q, J = 306.5 Hz), 67.0, 54.9, 44.5, 34.0, 29.6, 28.3, 10.7; 19F NMR (376 MHz, CD3CN) δ – 38.71; IR (neat) ν 3298 (br), 2972 (w), 2924 (w), 1465 (w), 1431 (w), 1394 (w), 1345 (m), 1232 (w), 1157 (s), 1106 (s), 1041 (w), 978 (m), 901 (m), 771 (m), 755 (m), 616 (m), 588 (w) cm–1; TLC Rf = 0.32 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C10H19F3NO3S2 322.0764; Found 322.0757.
3-ethylhex-5-en-1-yl tert-butylsulfamate (10).
Allylation was performed using modified literature procedure.10a A flame-dried microwave vial with stir bar was taken into the glovebox and charged with 3-((ethoxycarbonothioyl)thio)pentyl tert-butyl sulfamate (44.6 mg, 0.13 mmol, 1.0 equiv), allyl ethyl sulfone45 (52.3 mg, 0.4 mmol, 3.0 equiv), and dilauroyl peroxide (5.4 mg, 0.013 mmol, 0.1 equiv). Anhydrous chlorobenzene (0.5 mL, 0.26 M) was added. The vial was sealed with a rubber septum and removed from the glovebox. The reaction was then heated in a pie block set at 100 °C. After stirring for 0.5 h, an additional 0.1 equiv of dilauroyl peroxide was added by briefly removing the septum and adding the dilauroyl peroxide in a single portion. An additional 0.1 equiv of dilauroyl peroxide was added every 0.5 h until a total of 0.6 equiv had been added. At the time when the next dilauroyl peroxide addition would have occurred, the reaction was removed from the heating block and allowed to cool to room temperature. Once cool, the reaction solution was transferred to a scintillation vial rinsing the reaction vial with CH2Cl2 (5 mL) to ensure quantitative transfer. The reaction solution was then concentrated under reduced pressure. The product was obtained as a colorless oil (21.2 mg, 62% yield) following silica gel column chromatography using gradient from hexanes (100%) to hexanes:EtOAc (90:10).1H NMR (400 MHz, CDCl3) δ 5.79–5.69 (m, 1H), 5.06–5.01 (m, 2H), 4.30 (s, 1H), 4.14 (t, J = 7.0 Hz, 2H), 2.14–2.01 (m, 2H), 1.72–1.66 (m, 3H), 1.57–1.51 (m, 2H), 1.36 (s, 9H), 0.88 (t, J = 7.2, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 136.3, 116.5, 68.7, 54.6, 37.3, 35.4, 31.9, 29.7, 25.6, 10.7; IR (neat) ν 3297 (br), 2965 (w), 2923 (w), 2875 (w), 1466 (w), 1394 (w), 1346 (w), 1231 (w), 1161 (m), 1042 (w), 972 (m), 915 (m), 879 (w), 809 (w), 773 (w), 618 (w), 588 (w) cm–1; TLC Rf = 0.36 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C12H24NO3S 262.1482; Found 262.1486.
3-ethylhex-5-en-1-yl tert-butylsulfamate (11).
Vinylation was performed using modified literature procedure.10a A flame-dried microwave vial with stir bar was taken into the glovebox and charged with 3-((ethoxycarbonothioyl)thio)pentyl tert-butyl sulfamate (59.1 mg, 0.17 mmol, 1.0 equiv), styrl ethyl sulfone (S3, 102 mg, 0.51 mmol, 3.0 equiv. Anhydrous chlorobenzene (2.7 mL, 0.06 M) was added. The vial was sealed with a crimp cap and removed from the glovebox. Di-tert-butyl peroxide (0.02 mL, 0.017 mmol, 0.1 equiv) was then added via Hamilton syringe. The reaction was then heated in a pie block that had been preheated 130 °C. After stirring for 6 h, an additional 0.05 equiv of di-tert-butyl peroxide was added added via Hamilton syringe. An additional 0.05 equiv di-tert-butyl peroxide was added every 6 h until a total of 0.3 equiv had been added. Following the final di-tert-butyl peroxide addition, the reaction was left to stir at 130 °C for an additional 12 h. The reaction was then removed from the heating block and allowed to cool to room temperature. Once cool, the brown reaction solution was transferred to a scintillation vial rinsing the reaction vial with CH2Cl2 (5 mL) to ensure quantitative transfer. The reaction solution was then concentrated under reduced pressure. The product was obtained as a pale yellow oil (40.4 mg, 73% yield) following silica gel column chromatography using using gradient from hexanes (100%) to hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 7.36–7.20 (m, 5H), 6.40 (d, J = 15.8 Hz, 1H), 5.91 (dd, J = 15.8, 9.1 Hz, 1H), 4.19–4.06 (m, 3H), 2.29–2.20 (m, 1H), 1.99–1.91 (m, 1H), 1.73–1.69 (m, 1H), 1.57–1.49 (m, 1H), 1.47–1.34 (m, 1H), 1.34 (s, 9H), 0.91 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 137.3, 133.0, 131.3, 128.5, 127.2, 126.1, 68.7, 54.6, 41.4, 34.1, 29.7, 28.2, 11.7; IR (neat) ν 3296 (br), 3026 (w) 2963 (w), 2926 (w), 2874 (w), 1493 (w), 1450 (w), 1431 (w), 1393 (w), 1345 (m), 1231 (w, 1161 (m), 1045 (w), 967 (m), 907 (w), 876 (w), 812 (w), 747 (m), 694 (w), 617 (w), 589 (w) cm–1; TLC Rf = 0.40 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H27NO3S•Na 348.1604; Found 348.1602.
3-deuterio pentyl tert-butylsulfamate (d-3a).
Deuteration was performed using modified literature procedure.10c A flame-dried microwave vial with stirbar was rinsed with (3 × 2 mL) trimethylsilyl chloride (TMSCl) and then filled half-volume (5 mL) with TMSCl. The vial was sealed with rubber septum with a small needle for air flow and kept in the vacuum desiccator overnight. The vial was then taken inside glovebox where it was charged serially with alkyl xanthate (37.8 mg, 0.11 mmol, 1.0 equiv), BEt3 (1.0 M in THF) (0.56 mL, 0.5 mmol, 5.0 equiv), dodecanethiol (2.2 mg, 0.011 mmol, 0.1 equiv), d4–dichloroethane (0.4 mL), and D2O (0.2 mL). The vial was sealed with crimp cap and removed from the glovebox where the biphasic solution was sparged with oxygen for 30 seconds. The flask was then equipped with oxygen balloon and stirred at room temperature for 48 h. After 48 h, the mixture was diluted with dichloromethane (2 mL) and passed through a short silica plug, rinsing with 10% ethyl acetate in hexanes (2 × 5 mL) followed by ethyl acetate (2 × 5 mL). The crude was concentrated under reduced pressure and purified by silica gel flash chromatography using hexanes:EtoAc (95:5) to furnish the deuterated product as colorless oil (17.2 mg, 70% yield). HRMS analysis revealed 93% deuterium incorporation (average of two runs). HRMS analyses were performed using an Agilent 6224 TOF LC/MS with a Dual ESI Source. Samples were introduced by flow injection from an Agilent 1200 HPLC using a 50:50 water: acetonitrile mobile phase at a flow rate 0.35 μL/min. Mass spectra were collected in negative-ion mode from 110–1700 m/z using a capillary voltage of 3500 V, fragmentor voltage of 120 V, sheath gas temperature of 320 °C, sheath gas flow of 11 L/min, and nebulizer pressure of 33 psi. Isotopic purity was calculated based on %-area from extracted ion chromatograms for the labeled and unlabeled compounds. An Agilent isotope distribution calculator was used to correct for the contribution that the unlabeled compound made to the signal of the isotopically-labeled compound. 1H NMR (400 MHz, CDCl3) δ 4.24 (br s, 1H), 4.11 (t, J = 6.7 Hz, 2H), 1.74–1.68 (m, 2H), 1.39–1.31 (m, 3H), 1.36 (s, 9H), 0.91 (t, J = 7.1 Hz); 13C{1H} NMR (126 MHz, CDCl3) δ 70.4, 54.6, 29.6, 28.4, 27.3 (t, J = 19.5 Hz), 22.0, 13.8; IR (neat) ν 3297 (br), 2960 (w), 2932 (w), 2874 (w), 1467 (w), 1431 (w), 1394 (w), 1342 (m), 1230 (w), 1159 (s), 1043 (w), 970 (w), 908 (w), 874 (m), 804 (m), 616 (m), 584 (m) cm–1; TLC Rf = 0.12 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M – H]– Calcd for C9H19DNO3S 223.1232; Found 223.1236.
Nucleophilic Displacement of Sulfamate Anion.
3-((ethoxycarbonothioyl)thio)pentyl (tert-butoxycarbonyl)(ethyl)sulfamate (S5).
Following the procedure reported by Du Bois, Zare and coworkers,25b a flame-dried round bottom flask with stir bar was charged with 3-((ethoxycarbonothioyl)thio)pentyl ethylsulfamate (416 mg, 1.3 mmol, 1.0 equiv). The flask sealed with a rubber septum and evacuated and backfilled with N2 three times. Anhydrous CH2Cl2 (14 mL, 0.09 M) was added. Di-tert-butyl dicarbonate (403 mg, 1.85 mmol, 1.4 equiv) and 4-(dimethylamino)pyridine (177 mg, 1.45 mmol, 1.1 equiv) were added then sequentially by briefly removing the septum and adding each reagent in a single portion. The resulting pale yellow solution was stirred at 21 °C for 1 h. After 1 h, the solution was concentrated under reduced pressure. The product was obtained as a colorless oil (462 mg, 84% yield) following silica gel column chromatography using hexanes:EtOAc (90:10). 1H NMR (400 MHz, CDCl3) δ 4.64 (q, J = 7.1 Hz, 2H), 4.39 (t, J = 6.6 Hz, 2H), 3.77 (app q, J = 7.0 Hz, 3H), 2.22–2.13 (m, 1H), 2.11–2.04 (m, 1H), 1.85–1.68 (m, 2H), 1.53 (s, 9H), 1.43 (t, J = 7.1 Hz, 3H), 1.27 (t, J = 7.0 Hz, 3H), 1.03 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.3, 150.3, 84.3, 70.4, 69.9, 48.8, 44.4, 33.0, 27.8, 27.0, 14.6, 13.6, 11.1; IR (neat) ν 2976 (w), 2935 (w), 1731 (m), 1458 (w), 1369 (m), 1332 (w), 1284 (m), 1253 (m), 1215 (m), 1194 (s), 1111 (m), 1044 (s), 1002 (m), 953 (m), 917 (m), 843 (m), 769 (m), 727 (m), 648 (m), 620 (m), 571 (m) cm–1; TLC Rf = 0.64 (hexanes/EtOAc, 8:2); HRMS (ESI-TOF) m/z: [M + NH4]+ Calcd for C15H29NO6S3•NH4 433.1495; Found 433.1502.
O-ethyl (1-iodopentan-3-yl) carbonodithionate (12).
Following the procedure reported by Du Bois, Burns, Zare and coworkers,25b a flame-dried microwave vial with stir bar was charged with 3-((ethoxycarbonothioyl)thio)pentyl (tert-butoxycarbonyl)(ethyl)sulfamate (S5, 100 mg, 0.24 mmol, 1.0 equiv) and sodium iodide (120 mg, 0.79 mmol, 3.3 equiv) and taken into the glovebox. Anhydrous acetone (2.0 mL, 0.12 M) was added and the vial was capped with a disposable crimp cap containing a Teflon-lined septum and removed from the glovebox. The reaction was then heated in a pie block set at 35 °C for 6 h. After 6 h, the bright orange solution was transferred to a separtory funnel, rinsing the reaction vial with diethyl ether (ca. 5 mL). An additional 15 mL of diethyl ether was added and the organic phase was washed with DI water (20 mL). The organic phase was dried with MgSO4, filtered, and concentrated under reduced pressure. The product was obtained as a colorless oil (55 mg, 72% yield) following purification by silica gel column chromatography using 100% hexanes.1H NMR (400 MHz, CDCl3) δ 4.65 (q, J = 7.1 Hz, 2H), 3.77 (tt, J = 7.8, 5.6 Hz, 1H), 3.30–3.23 (m, 2H), 2.27–2.13 (m, 2H), 1.81–1.65 (m, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 213.8, 70.0, 53.3, 37.9, 27.0, 13.8, 11.1, 2.3; IR (neat) ν 2963 (w), 2930 (w), 1613 (w), 1441 (w), 1381 (w), 1361 (w), 1289 (w), 1206 (s), 1145 (m), 1108 (w), 1040 (s), 928 (m), 854 (w), 798 (m), 678 (w), 598 (w) cm–1; TLC Rf = 0.54 (hexanes:EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C8H16IOS2 318.9682 Found 318.9675.
(1-azidopentan-3-yl) O-ethyl carbondithioate (13).
Following the procedure reported by Du Bois, Burns, Zare and coworkers,25b a flame-dried microwave vial with stir bar was charged with 3-((ethoxycarbonothioyl)thio)pentyl (tert-butoxycarbonyl)(ethyl)sulfamate (S5, 100 mg, 0.24 mmol, 1.0 equiv) and sodium azide (51.8 mg, 0.79 mmol, 3.3 equiv) and taken into the glovebox. Anhydrous dimethyl sulfoxide (2.0 mL, 0.12 M) was added and the vial was capped with a disposable crimp cap containing a Teflon-lined septum and removed from the glovebox. The reaction was then heated in a pie block set at 35 °C for 6 h. After 6 h, the yellow solution was transferred to a separatory funnel, rinsing the reaction vial with diethyl ether (ca. 5 mL). An additional 45 mL of diethyl ether was added along with DI water (50 mL). The organic phase was separated and then washed with DI water (2 × 25 mL). The organic phase was dried with MgSO4, filtered, and concentrated under reduced pressure. The product was obtained as a yellowish oil (36.2 mg, 65% yield) following purification by silica gel column chromatography using gradient from hexanes (100%) to hexanes:EtOAc (99:1). 1H NMR (400 MHz, CDCl3) δ 4.65 (q, J = 7.2 Hz, 2H), 3.82–3.75 (m, 1H) 3.46–3.4 (m, 2H), 2.02–1.86 (m, 2H), 1.83–1.66 (m, 2H), 1.43 (t, J = 7.1 Hz, 3H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 214.0, 70.0, 50.0, 49.0, 33.0, 27.4, 13.8, 11.2; IR (neat) ν 2965 (w), 2930 (w), 2874 (w), 2091 (m), 1456 (w), 1365 (w), 1207 (s), 1145 (m), 1109 (m), 1041 (s), 854 (w), 811 (w), 675 (w), 555 (w) cm–1; TLC Rf = 0.52 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C8H16N3OS2 234.0729; Found 234.0727.
3-((ethoxycarbonothioyl)thio)pentyl acetate (14).
A flame-dried microwave vial with stir bar was charged with 3-((ethoxycarbonothioyl)thio)pentyl (tert-butoxycarbonyl)(ethyl)sulfamate (S5, 50 mg, 0.12 mmol, 1.0 equiv) and lithium acetate (26.5 mg, 0.4 mmol, 3.3 equiv) and taken into the glovebox. Anhydrous dimethyl sulfoxide (1.0 mL, 0.12 M) was added and the vial was capped with a disposable crimp cap containing a Teflon-lined septum and removed from the glovebox. The reaction was then heated in a pie block set at 40 °C for 18 h. After 18 h, the yellow solution was transferred to a separtory funnel, rinsing the reaction vial with diethyl ether (ca. 5 mL). An additional 20 mL of diethyl ether was added along with DI water (25 mL). The organic phase was separated and then washed with DI water (2 × 25 mL). The organic phase was dried with MgSO4, filtered, and concentrated under reduced pressure. The product was obtained as a yellowish oil (23.5 mg, 78% yield) following purification by silica gel column chromatography using gradient from hexanes (100%) to hexanes:EtOAc (95:5). 1H NMR (400 MHz, CDCl3) δ 4.62 (q, J = 7.1 Hz, 2H), 4.16 (t, J = 6.6 Hz, 2H), 3.80–3.74 (m, 1H), 2.04 (s, 3H), 2.05–1.91 (m, 2H), 1.82–1.65 (m, 2H), 1.41 (t, J = 7.1 Hz, 3H), 1.00 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 214.3, 171.0, 69.9, 62.0, 49.6, 32.7, 27.3, 21.0, 13.8, 11.2; IR (neat) ν 2964 (w), 2930 (w), 2874 (w), 1738 (m), 1456 (w), 1365 (m), 1223 (s), 1111 (m), 1040 (s), 853 (w), 809 (w), 731 (w), 676 (w), 635 (w), 605 (w) cm–1; TLC Rf = 0.42 (hexanes/EtOAc, 9:1); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H19O3S2 251.0770; Found 251.0770.
Supplementary Material
ACKNOWLEDGEMENTS
Funding was provided by the National Institutes of Health (R35GM128741–01) and Duke University. S.K.A was partially supported by C.R Hauser Memorial Fellowship. Characterization data were obtained on instrumentation secured by funding from the NSF (CHE-0923097, ESI-MS, George Dubay, the Duke Dept. of Chemistry Instrument Center), or the NSF, the NIH, HHMI, the North Carolina Biotechnology Center and Duke (Duke Magnetic Resonance Spectroscopy Center). All Bruker-Nonius X8 Kappa APEXII (CCD) instrument using MoKα radiation with an Oxford Cryostream 700 cold stream was used for crystallographic analyses and measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University (Roger Sommer). We thank Dr. Peter Silinski (Duke University) for high-resolution mass spectrometry, and Dr. Ben Bobay (Duke University) for NMR assistance. We thank Melanie A. Short (Duke University) for providing samples of precursors to compounds 1i and 1j and Melanie A. Short and J. Miles Blackburn (Duke University) for contributions to late-stage data compilation, formatting, and editing.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supporting Information Placeholder
The Supporting Information is available free of charge on the ACS Publications website.
Optimization table of xanthylation reaction, pictures of xanthylation reaction set up, description of quantum yield experiments, UV-Vis spectra, crystallographic analyses, and copies of NMR spectra (PDF)
X-ray crystallographic data for betulin-derived 2q (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).For select reviews, see: (a) Bergman RG C–H activation. Nature 2007, 446, 391–393. [DOI] [PubMed] [Google Scholar]; (b) Davies HML; Manning JR Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature. 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons TW; Sanford MS Palladium-catalyzed ligand-directed CH functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) White MC Adding Aliphatic C–H Bond Oxidations to Synthesis. Science, 2012, 335, 807–809. [DOI] [PubMed] [Google Scholar]; (e) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev. 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chu JCK; Rovis T Angew. Chem. Int. Ed. 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For a review, see: Yamaguchi J; Yamaguchi AD; Itami K C-H bonds functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- (3).Blackburn JM; Short MA; Castanheiro T; Ayer SK; Muellers TD; Roizen JL Synthesis of N-Substituted Sulfamate Esters from Sulfamic Acid Salts by Activation with Triphenylphosphine Ditriflate. Org. Lett. 2017, 19, 6012–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Short MA; Blackburn JM; Roizen JL Sulfamate Esters Guide Selective Radical-Mediated Chlorination of Aliphatic C–H Bonds. Angew. Chem. Int. Ed. 2018, 57, 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mo F; Tabor JR; Dong G Alcohols or masked alcohols as directing groups for CH bond functionalization. Chem. Lett. 2014, 43, 264–271. [Google Scholar]
- (6).For alcohol-guided γ-C(sp3)–H oxygenation, see: (a) Simmons EM; Hartwig JF Catalytic functionalization of unactivated primary C-H bonds directed by an alcohol. Nature 2012, 483, 70–73. For carbamate-guided oxidation that is selective for oxidation of benzylic or tertiary β-C(sp3)–H or γ-C(sp3)–H centers, see [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen K; Richter JM; Baran PS 1,3-Diol synthesis via controlled, radical-mediated C-H functionalization. J. Am. Chem. Soc. 2008, 130, 7247–7249. [DOI] [PubMed] [Google Scholar]
- (7).For select examples of sulfamate esters in C–H amination and aziridination reactions, see: (a) Espino CG; Wehn PM; Chow J; Du Bois J Synthesis of 1,3-Difunctionalized Amine Derivatives through Selective C–H bond Oxidation. J. Am. Chem. Soc. 2001, 123, 6935–6936. [Google Scholar]; (b) Duran F; Leman FL; Ghini A; Burton G; Dauban P; Dodd RH Intramolecular PhI=O Mediated Copper-Catalyzed Aziridination of Unsaturated Sulfamates: A New Direct Access to Polysubstituted Amines from Simple Homoallylic Alcohols. Org. Lett. 2002, 4, 2481–2483. [DOI] [PubMed] [Google Scholar]; (c) Milczek E; Boudet N; Blakey S Enantioselective C-H amination using cationic ruthenium(II)-pybox catalysts. Angew. Chem. Int. Ed. 2008, 47, 6825–6828. [DOI] [PubMed] [Google Scholar]; (d) Liu Y; Guan X; Lai-Ming Wong E.; Liu P; Huang J-S; Che C-M Nonheme Iron-Mediated Amination of C(Sp3)–H Bonds. Quinquepyridine-Supported Iron-Imide/Nitrene Intermediates by Experimental Studies and DFT calculations. J. Am. Chem. Soc. 2013, 135, 7194–7204. [DOI] [PubMed] [Google Scholar]; (e) Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM Ligand-Controlled, Tunable Silver-Catalyzed C–H Amination. J. Am. Chem. Soc. 2014, 136, 16720–16723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Subbarayan V; Jin L-M; Cui X; Zhang XP Room Temperature Activation of Aryloxysulfonyl Azides by [Co(II)(TPP)] for Selective Radical Aziridination of Alkenes via Metalloradical Catalysis. Tetrahedron Lett. 2015, 56, 3431–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Paradine SM; Griffin JR; Zhao J; Petronico AL; Miller SM; White MC A Manganese Catalyst for Highly Reactive Yet Chemoselective Intramolecular C(sp3)–H Amination. Nature Chem. 2015, 7, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For pioneering reports, see: (a) Barton DHR; McCombie SW A new method for the deoxygenation of secondary alcohols. J. Chem. Soc., Perkin Trans. 1 1975, 0, 1574–1585. [Google Scholar]; (b) Barrett AGM; Prokopiou PA; Barton DHR The deoxygenation of NN-dialkylaminothiocarbonyloxyalkanes. J. Chem. Soc., Perkin Trans. 1 1981, 0, 1510–1515. [Google Scholar]; (c) Barton DHR; Crich D; Löbberding A; Zard SZ On the mechanism of the deoxygenation of secondary alcohols by the reduction of their methyl xanthates by tin hydrides. Tetrahedron 1986, 42, 2329–2338. [Google Scholar]
- (9).For select reviews of xanthate ester reactivity, see: (a) Crich D; Quintero L Radical chemistry associated with the thiocarbonyl group. Chem. Rev. 1989, 89, 1413–1432. [Google Scholar]; (b) Zard SZ The genesis of the reversible Radical Addition-Fragmentation-Transfer of Thiocarbonylthio Derivatives From the Barton-McCombie Deoxygenation: A Brief Account and Some Mechanistic Observations. Aust. J. Chem. 2006, 59, 663–668. [Google Scholar]
- (10).(a) Czaplyski WL; Na CG; Alexanian EJ A General Strategy for Aliphatic C–H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 13854. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Spiegel DA; Wilberg KB; Schacherer LN; Medeiros MR; Wood JL Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source. J. Am. Chem. Soc. 2005, 127, 12513–12515. [DOI] [PubMed] [Google Scholar]; (c) Soulard V; Villa G; Vollmar DP; Renaud P Radical Deuteration with D2O: Catalysis and Mechanistic Insights. J. Am. Chem. Soc. 2018, 140, 155–158. [DOI] [PubMed] [Google Scholar]
- (11).(a) Ollivier C; Renaud P Formation of Carbon–Nitrogen Bonds via a Novel Radical Azidation Process. J. Am. Chem. Soc. 2000, 122, 6496–6497. [DOI] [PubMed] [Google Scholar]; (b) Ollivier C; Renaud P A Novel Approach for the Formation of Carbon–Nitrogen Bonds: Azidation of Alkyl Radicals with Sulfonyl Azides. J. Am. Chem. Soc. 2001, 123, 4717–4727. [DOI] [PubMed] [Google Scholar]
- (12).Bertrand F; Quiclet-Sire B; Zard SZ A New Radical Vinylation Reaction of Iodides and Dithiocarbonates. Angew. Chem. Int. Ed. 1999, 38, 1943–1946. [DOI] [PubMed] [Google Scholar]
- (13).Sire B; Seguin S; Zard SZ A new Radical Allylation Reaction of Dithiocarbonates. Angew. Chem. Int. Ed. 1998, 37, 2864–2866. [DOI] [PubMed] [Google Scholar]
- (14).Sato A; Yorimitsu H; Oshima K O-alkyl S-3,3-dimethyl-2-oxobutyl dithiocarbonates as versatile sulfur-transfer agents in radical C(sp3)-H functionalization. Chem. Asian. J. 2007, 2, 1568–1573. [DOI] [PubMed] [Google Scholar]
- (15).For select resources describing site-selectivity in C–H functionalization, see: (a) Chen K; Eschenmoser A; Baran PS Strain-Release in C–H Bond Activation? Angew. Chem. Int. Ed. 2009, 48, 9705–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brückl T; Baxter RD; Ishihara Y; Baran PS Innate and guided C–H functionalization logic. Acc. Chem. Res. 2012, 45, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cernak T; Dykstra KD; Tyagarajan S; Vachal P Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (16).Na CG, Alexanian EJ, A General Approach to Site-Specific, Intramolecular C–H Functionalization Using Dithiocarbamates. Angew. Chem. Int. Ed. 2018, 57, 13106–13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Zard SZ Recent progress in the generation and use of nitrogen-centered radicals. Chem. Soc. Rev. 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (b) Xiong T; Zhang Q New amination strategies based on nitrogen-centered radical chemistry. Chem. Soc. Rev. 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]; (c) Kärkäs MD Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS Catal. 2017, 7, 4999–5022. [Google Scholar]
- (18).For select examples of distal C(sp3)–H functionalization via 1,5-HAT-mediated processes, see: (a) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic alkylation of remote C-H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chu JCK; Rovis T Amide-directed photoredox-catalysed C-C bond formation at unactivated sp3 C-H bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shu W; Nevado C Visible-Light-Mediated Remote Aliphatic C-H Functionalizations through a 1,5-Hydrogen Transfer Cascade. Angew. Chem. Int. Ed. 2017, 56, 1881–1884. [DOI] [PubMed] [Google Scholar]; (d) Jiang H; Studer A α-Aminoxy-Acid-Auxiliary-Enabled Intermolecular Radical γ-C(sp3)–H Functionalization of Ketones. Angew. Chem. Int. Ed. 2018, 57, 1692–1696. [DOI] [PubMed] [Google Scholar]; (e) Wappes EA; Nakafuka KM; Nagib DA Directed β C–H Amination of Alcohols via Radical Relay Chaperons. J. Am. Chem. Soc. 2017, 139, 10204–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen D-F; Chu JCK; Rovis T Directed γ-C(sp3)-H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu T; Myers CM; Yu JQ Copper-Catalyzed Bromination of C(sp3)–H Bonds Distal to Functional Groups. Angew. Chem. Int. Ed. 2018, 56, 306–309. [DOI] [PubMed] [Google Scholar]; (h) Liu T; Mei ST; Yu JQ γ,δ,ε-C(sp3)–H Functionalization through Directed Radical H-Abstraction. J. Am. Chem. Soc, 2015, 137, 5871–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Morcillo SP; Daunchey EM; Kin JH; Douglas JJ; Sheikh NS; Leonori D Angew. Chem. Int. Ed. 2018, 57, 12945–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Dauncey EM; Morcillo SP; Douglas JJ; Sheikh NS; Leonori D Angew. Chem. Int. Ed. 2018, 57, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Davies J; Morcillo SO; Douglas JJ; Leonori D Chem. Eur. J. 24, 12154–12163. [DOI] [PubMed] [Google Scholar]; (l) Bergès J; Garcial B; Muñiz K Angew. Chem. Int. Ed. 2018, 57, 15891–15895. [DOI] [PubMed] [Google Scholar]; (m) Becker P; Duhamel T; Martínex C; Muñiz K Angew. Chem. Int. Ed. 2018, 57, 5166–5170. [DOI] [PubMed] [Google Scholar]
- (19).Wolff ME Cyclization of N-Halogenated Amines (The Hofmann-Löffler Reaction). Chem. Rev. 1963, 63, 55–64. [Google Scholar]
- (20).(a) Hofmann AW Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine. Chem. Ber. 1883, 16, 558–560. [Google Scholar]; (b) Löffler K; Freytag C Über aine neue Bildungsweise von N-alkylierten Pyrrolidinen. Chem. Ber. 1909, 42, 3427–3431. [Google Scholar]
- (21).For select recent reviews focusing on intramolecular 1,5-HAT, see: (a) Robertson J; Pillai J; Lush RK Radical translocation reactions in synthesis. Chem. Soc. Rev. 2001, 30, 94–103. [Google Scholar]; (b) Čeković Ž Reactions of carbon radicals generated by 1,5-transposition of reactive centers. J. Serb. Chem. Soc. 2005, 70, 287–318. [Google Scholar]; (c) Chiba S, Chen H. sp3 C–H oxidation by remote H-radical shift with oxygen- and nitrogen-radicals: a recent update. Org. Biomol. Chem. 2014, 12, 4051–4060. [DOI] [PubMed] [Google Scholar]
- (22).(a) Löffler K Über eine neue Bildungsweise N‐alkylierter Pyrrolidine. Ber. Dtsch. Chem. Ges. 1910, 43, 2035–2048. [Google Scholar]; (b) Corey EJ; Hertler WRA Study of the Formation of Haloamines and Cyclic Amines by the Free Radical Chain Decomposition of N-Haloammonium Ions (Hofmann-Löffler Reaction). J. Org. Chem. 1960, 82, 1657–1668. [Google Scholar]; (c) Yates BF; Radom L Intramolecular hydrogen migration in ionized amines: a theoretical study of the gas-phase analogs of the Hofmann-Loeffler and related rearrangements. J. Am. Chem. Soc. 1987, 109, 2910–2915. [Google Scholar]; (d) Janovsky I; Knolle W; Naumov S; Williams F EPR Studies of Amine Radical Cations, Part 1: Thermal and Photoinduced Rearrangements of n‐Alkylamine Radical Cations to their Distonic Forms in Low‐Temperature Freon Matrices. Chem. Eur. J. 2004, 10, 5524–5534. [DOI] [PubMed] [Google Scholar]
- (23).Seven membered transition states are often kinetically disfavoured, see: a) Dorigo AE; Houk KN Transition structures for intramolecular hydrogen-atom transfers: the energetic advantage of seven-membered over six-membered transition structures. J. Am. Chem. Soc. 1987, 109, 2195–2197. [Google Scholar]; (b) Dorigo AE; Houk KN The relationship between proximity and reactivity. An ab initio study of the flexibility of the OH.buI. + CH4 hydrogen abstraction transition state and a force-field model for the transition states of intramolecular hydrogen abstractions. J. Org. Chem. 1988, 53, 1650–1664. [Google Scholar]; (c) Dorigo AE; McCarrick MA; Loncharich RJ; Houk KN Transition structures for hydrogen atom transfers to oxygen. Comparisions of intermolecular and intramolecular processes, and open- and closed-shell systems. J. Am. Chem. Soc. 1989, 112, 7508–7514. [Google Scholar]
- (24).Nechab M; Mondal S; Bertrand MP 1,n-Hydrogen-Atom Transfer (HAT) Reactions in which n≠5: An Updated Inventory. Chem. Eur. J. 2014, 20, 16034. [DOI] [PubMed] [Google Scholar]
- (25).(a) Zalatan DN; Du Bois J Oxidative Cyclization of Sulfamate Esters Using NaOCl – A Metal-Mediated Hoffman-Löffler Freytag Reaction. Synlett 2009, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sathyamoothi S; Banerjee S; Du Bois J; Burns NZ; Zare RN Site-selective bromination of Sp3 C–H bonds. Chem. Sci. 2018, 9, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Ruppel JV; Kamble RM; Zhang XP Cobalt–catalyzed intramolecular C-H amination with arylsulfonyl azides. Org. Lett 2007, 9, 4889–4892. [DOI] [PubMed] [Google Scholar]; (b) Lu H; Lang K; Jiang H; Wojtas L; Zhang XP Intramolecular 1,5-C(sp3)–H radical amination via Co(II)-based metalloradical catalysis for five-membered cyclic sulfamides. Chem. Sci. 2016, 7, 6934–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rumble John R., Ed., CRC Handbook of Chemistry and Physics; 98th Edition (Internet Version 2018) CRC Press/Taylor & Francis, Boca Raton, FL. [Google Scholar]
- (28).(a) Hilal SH; Karickhoff SW A Rigorous Test for SPARC’s Chemical Reactivity Models: Estimation of More Than 4300 Ionization pKas. Quant. Struct.-Act. Relat. 1995, 14, 348. [Google Scholar]; (b) http://www.archemcalc.com/sparc.html See the Supporting Information for additional details on how calculations were performed. [Google Scholar]
- (29).Tertiary alkyl xanthates have been accessed by conjugate addition into α,β-unsaturated carbonyl compounds: Binot G; Quiclet-Sire B; Saleh T; Zard SZA Convergent Construction of Quaternary Centers and Polycyclic Structures. Synlett 2003, 3, 382–386. [Google Scholar]
- (30).Tertiary alkyl xanthates have been accessed from acyl xanthates and from S-alkoxycarbonyl xanthates. For select pioneering examples, see: (a) Barton DHR; George MV; Tomoeda M Photochemical Transformations. Part XIII. A New Method for the Production of Acyl Radicals. J. Chem. Soc. 1962, 0, 1967–1974. [Google Scholar]; (b) Delduc P; Tailhan C; Zard SZ A convenient source of alkyl and acyl radicals. J. Chem. Soc., Chem. Commun. 1988, 0, 308–310. [Google Scholar]; (c) Forbes JE; Zard SZ New radical chain reaction of S-alkoxycarbonyl dithiocarbonates: a useful source of alkyl radicals from alcohols. J. Am. Chem. Soc. 1990, 112, 2034–2036. [Google Scholar]
- (31).Tertiary acyl xanthates have been used as surrogates for tertiary xanthates: Jenkins EN; Czaplyski WL; Alexanian EJA General Approach to Quaternary Center Construction from Couplings of Unactivated Alkenes and Acyl Xanthates. Org. Lett. 2017, 19, 2350–2353. [DOI] [PubMed] [Google Scholar]
- (32).For an early example where a 3,7-dimethyloctanol derivative engages in selective C(7) oxidation, see: Gal C; Ben-Shoshan G; Rozen S Selective fluorination on tertiary carbon-hydrogen single bonds in the aliphatic series. Tetrahedron Lett. 1980, 21, 5067–5070. [Google Scholar]
- (33).Chen MS; White MC A predictably selective aliphatic C-H oxidation reaction for complex molecules synthesis. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]
- (34).McNeill E; Du Bois J Ruthenium-Catalyzed Hydroxylation of Unactivated Tertiary C–H bonds. J. Am. Chem. Soc. 2010, 132, 10202–10204. [DOI] [PubMed] [Google Scholar]
- (35).Cismesia MA; Yoon TP Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hatchard CG; Parker CA A new sensitive chemical actinometer – II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. London Ser. A 1956, 235, 518–536. [Google Scholar]
- (37).Noble A; MacMillan DWC Photoredox α-vinylation of α-amino acids and N-aryl amines. J. Am. Chem. Soc. 2014, 136, 11602–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gregerson CE; Trentadu KN; Phipps EJ; Kirsch JK; Reed KM; Dyke GD; Jansen JH; Otteman CB; Stachowski JL; Johnson JB Oxidative coupling of Michael acceptros with aryl nucleophiles produced through rhodium-catalyzed C-C bond activation. Org. Biomol. Chem. 2017, 15, 5944–5948. [DOI] [PubMed] [Google Scholar]
- (39).Frankowski KJ; Golden EJ; Zeng Y; Lei Y; Aube J Syntheses of the stemona Alkaloids (±)-Stenine, (±)-Neostenine, and (±)-13-Epineostenine Using a Stereodivergent Diels-Alder/Azido-Schmidt Reaction. J. Am. Chem. Soc. 2008, 130, 6018–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Farina P; Guazzi G; Innocenti S; Marotta V; Valcavi U; Bosone E New Synthesis of Methyl 7-Oxoheptanoate: An Useful Intermediate for the preparation of 2-(6-Methoxycarbonylhexyl)-cyclopent-2-en-1-one. Synthesis 1983, 11, 942–944. [Google Scholar]
- (41).Kelly CB; Lambert KM; Mercadante MA; Ovian J M; Bailey, W. F.; Leadbeater, N. E. Access to Nitriles from Aldehydes Mediated by an Oxoammonium Salt. Angew. Chem. Int. Ed. 2015, 54, 4241–4245. [DOI] [PubMed] [Google Scholar]
- (42).Samoshina NF; Denisenko MV; Denisenko VA; Uvarova NI Synthesis of Glycosides of Lupane-Type Triterpene Acids. Chemistry of Natural Compounds 2003, 39, 575–582. [Google Scholar]
- (43).Mintz MJ; Walling C t-BUTYL HYPOCHLORITE. Org. Synth. 1969, 49, 9–10. [Google Scholar]
- (44).Shao X; Xu C; Lu L; Shen Q Structure–Reactivity Relationship of Trifluoromethanesulfenates: Discovery of an Electrophilic Trifluoromethylthiolating Reagent. J. Org. Chem. 2015, 80, 3012–3021. [DOI] [PubMed] [Google Scholar]
- (45).Palmer RJ; Stirling CJM Elimination and addition reactions. 36. Acceleration of nucleophilic eliminative ring fission by bond strain. J. Am. Chem. Soc. 1980, 102, 7888–7892. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.