

Abstract
A reproducible synthetic strategy was developed for facile large-scale (200 mg) synthesis of surface silanized magnetite (Fe3O4) nanoparticles (NPs) for biological applications. After further coupling a phosphate-specific affinity ligand, these functionalized magnetic NPs were used for the highly specific enrichment of phosphoproteins from a complex biological mixture. Moreover, correlating the surface silane density of the silanized magnetite NPs to their resultant enrichment performance established a simple and reliable quality assurance control to ensure reproducible synthesis of these NPs routinely in large scale and optimal phosphoprotein enrichment performance from batch-to-batch. Furthermore, by successful exploitation of a top-down phosphoproteomics strategy that integrates this high throughput nanoproteomics platform with online liquid chromatography (LC) and tandem mass spectrometry (MS/MS), we were able to specifically enrich, identify, and characterize endogenous phosphoproteins from highly complex human cardiac tissue homogenate. This nanoproteomics platform possesses a unique combination of scalability, specificity, reproducibility, and efficiency for the capture and enrichment of low abundance proteins in general, thereby enabling downstream proteomics applications.
Keywords: nanoparticles, nanoproteomics, surface functionalization, large-scale, phosphoprotein enrichment, top-down proteomics, mass spectrometry
Graphical abstract
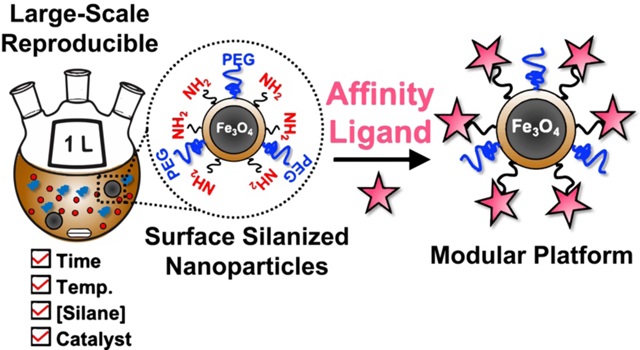
A reproducible and scalable nanomaterials platform for enabling proteomics applications. When coupled with a suitable affinity ligand, this nanomaterials platform allows for the capture, enrichment, and analysis of low abundance proteins in general.
1. Introduction
The design and applications of functionalized nanoparticles (NPs) for biological purposes attract enormous attention from researchers across diverse fields of chemistry, biology, and materials science, fostering the emerging research field of nanobiotechnology [1,2]. Various classes of NPs including semiconductors [3], magnetic materials [4], and coinage metals such as gold [5], have been extensively investigated for their biological applications. In particular, magnetic iron-oxide NPs of magnetite (Fe3O4), or maghemite (γ-Fe2O3), have been utilized in clinical diagnostics [6], in vivo chemical and biological sensing [7], and therapeutic targeting [8,9]. The most common synthetic method to obtaining scalable and high-quality magnetite NPs involves the high-temperature decomposition of an organic iron precursor, such as iron (III) oleate, in the presence of a stabilizing surfactant, such as oleic acid (OA) [10]. However, the NPs produced are rendered hydrophobic by the surface ligand and thus require an additional surface modification step to make them biocompatible. NP surface identity can be modified by ligand exchange and many reports have demonstrated the utilization of various carboxylates [11], phosphates [12], alcohols [13], and catechols [14] as exchange ligands to impart biocompatibility.
However, these popular methods yield ligands noncovalently bonded to the NP surface and are thus labile under biologically relevant conditions or in the presence of a complex biological milieu [15]. To address this issue, one general method is to use alkoxysilane derivatives to perform ligand exchange reactions, which would yield a covalently attached siloxane network on the surface of the Fe3O4 NPs [16]. Although capable of offering highly chemically stable covalent modifications and additional designable functionality, this surface silanization process is kinetically challenging to control [17,18] and thus rather irreproducible. Moreover, large-scale production of these silanized magnetite NPs with batch-to-batch reproducibility, properly designed surface modification, and high efficacy for downstream biological applications has so far been elusive, with only small-scale (~ 10s of mg) synthesis being reported [16,19–21]. Therefore, herein we aim to develop the synthetic route to reproducibly preparing highly effective surface silanized superparamagnetic magnetite NPs with size < 10 nm and strong magnetic response in a large-scale (Scheme 1).
Scheme 1.
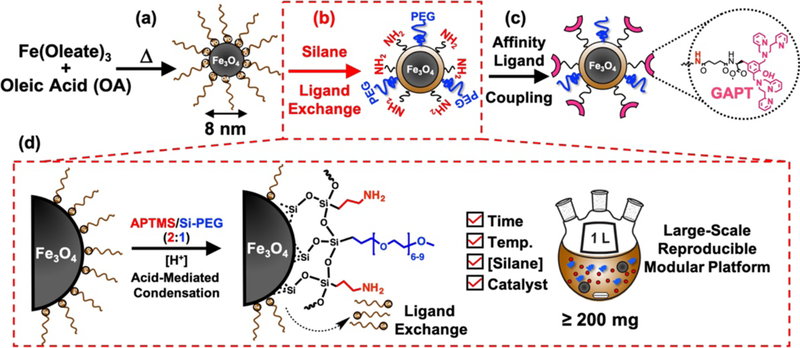
Schematic illustration of the large-scale reproducible synthesis of surface functionalized Fe3O4 NPs as an enabling nanoproteomics platform. (a) Synthesis of starting Fe3O4-OA NPs via thermal decomposition of iron-oleate in the presence of stabilizing ligand, oleic acid (OA). (b) Silane ligand exchange of the hydrophobic Fe3O4-OA NPs to impart a hydrophilic, biocompatible coating. (c) Affinity ligand coupling to free amines imparted by the silanization step enables various downstream biological applications. Specifically, the coupling of a phosphate binding GAPT ligand is highlighted for the current study. (d) Illustration of the key details of the optimized silanization chemistry allowing for robust and reproducible large-scale synthesis of surface functionalized NPs for biological applications.
One important application of such a reliable large-scale production of surface silanzed magnetite NPs is the establishment of a nanoproteomics platform (Scheme S1) for the specific enrichment of low-abundance proteins with post-translational modifications (PTMs) from complex biological samples. Proteomics (the large-scale analysis of proteins and their interactions) is essential for deciphering how biomolecules interact as a system and for understanding the function of cellular systems in human diseases [22–24]. A major challenge in proteomics is the high dynamic range of the proteome (108-1012) [25] which makes detection of low-abundance proteins and their PTMs extremely difficult [26,27] and thus requires a specific enrichment using affinity reagents prior to MS analysis [28]. In particular, protein phosphorylation is a critically important PTM that plays a central role in many biological processes [29]. We have previously developed functionalized magnetic NPs that utilize a dinuclear Zn(II)-dipicolylamine (Zn-DPA) complex as an affinity ligand to specifically enrich intact phosphoproteins [30], and further coupled this NP enrichment with a top-down phosphoproteomics strategy to characterize phosphoproteins on a chromatographic time scale [31]. However, the previous NP synthesis lacked scalability, reproducibility, and high throughput. Critical to the effective interrogation of the phosphoproteome with the developed strategy, a highly robust and scalable nanomaterial platform is necessary.
In this work, we develop the reproducible large-scale (200 mg) synthesis of silanized magnetite NPs for downstream biological applications, also provide a simple quality assurance control (QAC) method for ensuring such reproducible synthesis and subsequent performance, and then specifically demonstrate the successful exploitation of this nanoproteomics platform for enriching and characterizing the human cardiac phosphoproteome. By further integrating with a top-down LC-MS/MS workflow suitable for intact protein analysis, we have demonstrated the great promise of this highly scalable nanoproteomics platform for the global enrichment, identification, and characterization of endogenous phosphoproteins from the highly complex human cardiac system.
2. Experimental
Additional details of the experimental methods including chemicals and reagents, synthesis of the 8 nm Fe3O4 NPs, preparation of cardiac protein extracts and standard proteins for protein enrichment screening, and phosphoprotein enrichment procedure can be found in the electronic supplementary material (ESM).
2.1. Large-scale synthesis of functionalized magnetite NPs
2.1.1. Fe3O4-APTMS/Si-PEG NPs
In a typical optimized large-scale synthesis of silane functionalized NPs, Fe3O4 NPs (10 mL from a 20 mg/mL stock) were added to anhydrous n-hexane (490 mL) in a 1 L round bottom flask equipped with a Teflon-coated egg-shaped magnetic stir bar (1–1/4” × 5”) to achieve a total NP concentration of 0.4 mg/mL. After the reaction mixture was heated to 60 °C with stirring (1100 rpm), APTMS (1.05 mL) and Si-PEG (1.45 mL), in a 2:1 molar ratio, were added dropwise to the flask for a 0.5% (v/v) total concentration of trialkoxysilane reagents, followed by dropwise addition of a small amount of acetic acid (50 μL) for an acidic catalyst concentration of 0.01% (v/v). After reaction under stirring for 48 h, the precipitate was collected via centrifugation (5000 rpm, 20 min) and washed three times (5000 rpm, 20 min) with n-hexane to remove excess silane molecules. The NPs were then dried under vacuum for later use.
2.1.2. Fe3O4-APTMS/Si-PEG-GAPT NPs
For preparing a typical large-scale batch of functionalized NPs, 200 mg Fe3O4-APTMS/Si-PEG NPs were first dispersed in anhydrous DMF (15 mL), and then N, N-diisopropylethylamine (DIPEA, 0.57 mmol) was added. The resulting mixture was sonicated for 5 min to ensure a homogenous dispersion and then was stirred vigorously at room temperature. A Zn-DPA phosphate binding ligand coupled to glutaric acid (GAPT) was synthesized following our previous published protocol [30], with minor modifications. In a separate vial, a GAPT (1.78 mmol) and O-(7-azabenzotriazole-1-yl)-N,N,N’N’-tetramethyluronium hexafluorophosphate (HATU, 1.78 mmol) were dissolved in anhydrous DMF (5 mL) and sonicated. DIPEA (5.33 mmol) was then added to this GAPT solution and vortexed for 3 min to preactivate the GAPT molecule. This mixture containing GAPT, HATU, and DIPEA was then added to the NP solution to achieve a NP concentration of 10 mg/mL, and the whole mixture was stirred vigorously at room temperature for 24 h. The resulting black-brown precipitate was collected and washed one time with DMF, two times with DMF/MeOH (v/v, 1:1), and one time with EtOH via centrifugation (13000 rcf, 10 min) and subsequently isolated magnetically with a DynaMag to remove unreacted GAPT, HATU, and DIPEA. The resulting GAPT functionalized NPs were redispersed in EtOH at a concentration of 8 mg/mL.
2.1.3. Fe3O4-APTMS/Si-PEG-GAPT-Zn NPs
For each NP phosphoprotein enrichment trial, a 750 μL aliquot of the Fe3O4-APTMS/Si-PEG-GAPT NP stock solution suspended in EtOH (at 8 mg/mL) was transferred to a 2 mL Eppendorf tube. After the NPs (6.0 mg) were washed with nanopure water three times to remove EtOH, 1.5 mL of 10 mM ZnCl2 (aq) was added to the NPs, and the mixture was shaken at room temperature for at least 3.5 h. The Fe3O4-APTMS/Si-PEG-GAPT-Zn NPs were collected and washed three times with nanopure water via centrifugation (13000 rcf, 5 min) and then isolated with the DynaMag.
2.2. Material characterization
Transmission electron microscopy (TEM) samples were prepared by pipetting a 10 μL drop of as-synthesized NPs at a concentration of 0.125 mg/mL onto a copper TEM grid with lacey carbon film. TEM was conducted on a FEI T12 microscope operated at 120 kV, equipped with a Gatan CCD image system with digital micrograph software program. Powder X-ray diffraction (PXRD) patterns were collected on as-synthesized Fe3O4 NPs deposited on a glass substrate using a Bruker D8 Advance using Cu Kα radiation (λ= 1.5418 Å), with the background from the glass substrate subtracted. Transmission Fourier transform infrared (FTIR) spectroscopy measurements were recorded on a Bruker Equinox 55 FT-IR spectrometer in the range of 4000 cm−1 to 400 cm−1 at 2 cm−1 resolution on NPs in a potassium bromide (KBr) pellet, at a sample mass loading of 0.33 wt.%. Thermogravimetric analysis (TGA) was carried out using a TA Instruments Q500 thermal analysis system under a N2 atmosphere and at a constant heating rate of 10 °C/min from 100 °C to 600 °C. All samples were first heated to 100 °C and held at that temperature for 3 min to remove adsorbed water. Dynamic light scattering (DLS) and zeta-potential (ζ-potential) measurements was carried out using a Malvern Zetasizer Nano and three independent measurements were made for each sample.
2.3. Reverse phase chromatography (RPC)
Reverse phase chromatography was performed with a nanoACQUITY UPLC system (Waters; Milford, MA, USA). Mobile phase A (MPA) contained 0.2 % formic acid in nanopure water, and mobile phase B (MPB) contained 0.2% formic acid in 50:50 acetonitrile and isopropanol. Prior to injection, protein samples were desalted by washing through 10 kDa MWCO filters using water six times. For each injection, 5 μL of desalted protein sample (1 μg/μL) was loaded on a home-packed 200 mm × 250 μm PLRP-S (5 μm, 1000Å; Agilent Technology, Santa Clara, CA, USA) capillary column. The column was placed in a column heater set at 50 °C with a constant 6 μL/min flow rate. The RPC gradient consisted of the following concentrations of MPB: 10% MPB at 0 min, 20% at 5 min, 65% at 65 min, 90% at 70 min, held at 90% until 75 min, adjusted back to 10% at 75.1 min, and held at 10% until 80 min. Each run was 80 min long.
2.4. Top-down MS analysis
Samples eluted from RPC separation were electrosprayed into an Impact II Q-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) for online LC/MS and LC/MS/MS experiments. End plate offset and capillary voltage were set at 500 and 4000 V, respectively. The nebulizer was set to 0.3 bar, and the dry gas flow rate was 4.0 L/min at 200 °C. The quadruple low mass was set to 500 m/z. Mass range was set to 200–3000 m/z and spectra were acquired at 1 Hz for LC/MS runs. For the top 3 data-dependent LC/MS/MS CID runs, spectra were acquired across 500–3000 m/z at 2–6 Hz with active exclusion after 4 spectra. All data were collected with OtofControl 3.4 (Bruker Daltonics) and analyzed and processed in DataAnalysis 4.3 (Bruker Daltonics). Maximum Entropy algorithm (Bruker Daltonics) was used to deconvolute all mass spectra with the resolution set to 50000. SNAP algorithm was applied to determine the monoisotopic mass of all detected ions. Fragmentation ion lists consisting of monoisotopic mass, intensity and charge were generated from DataAnalysis 4.3, and were subsequently converted to MSAlign files. TopPIC was used to search against the Uniprot-Swissprot human database which was released on November 09th, 2018 and contains 20395 protein sequences. Fragment mass tolerance was set to 15 ppm. All identifications were validated with statistically significant P and E values (<0.01) and satisfactory numbers of assigned fragment (>10). Tandem mass spectra were output from the DataAnalysis software and analyzed using MASH Suite Pro software. The spectra were deconvoluted with a signal-to-noise ratio of 3 and a cutoff fit score of 60%. All the program-processed data were manually validated to obtain accurate sequence and PTM information.
3. Results and discussion
3.1. Synthesis and characterization of surface functionalized Fe3O4 NPs
Superparamagnetic iron oxide (magnetite, Fe3O4) NPs with a diameter of about 8 nm were prepared by thermal decomposition of iron(III)-oleate following a modified procedure reported by Hyeon et al. [10]. These NPs are comparable in size to typical proteins to enhance the protein capture and enrichment [30,31] because they can: (1) penetrate better in complex protein mixtures, leading to a higher interaction rate; (2) reduce the probability that proteins are denatured; and (3) have good colloidal stability [32–34]. Transmission electron microscopy (TEM) revealed the uniformity and monodispersity of the as-synthesized Fe3O4 NPs with an average diameter of 8.0 ± 0.5 nm (Fig. 1a,b). Powder X-ray diffraction (PXRD) confirmed the magnetite phase (Fig. 1c). Although the synthesis yields large quantities of monodisperse nanocrystals (Fig. 1a,b), they are coated with oleic acid (OA) rendering them hydrophobic. To make the NPs biocompatible and allow for further functionalization, we performed a silane ligand exchange step (Scheme 1b) to displace the OA ligands on the NP surface with trialkoxysilanes (3-aminopropyl)trimethoxysilane (APTMS) and 2-[methoxy(polyethyleneoxy)6–9propyl]trimethoxysilane (Si-PEG) at a 2:1 molar ratio, granting the NPs hydrophilic character [35,36]. The APTMS provides terminal amines for further chemical modification, and the neutral PEG ligands can act as an antifouling polymer to reduce non-specific binding [37–39].
Figure 1.
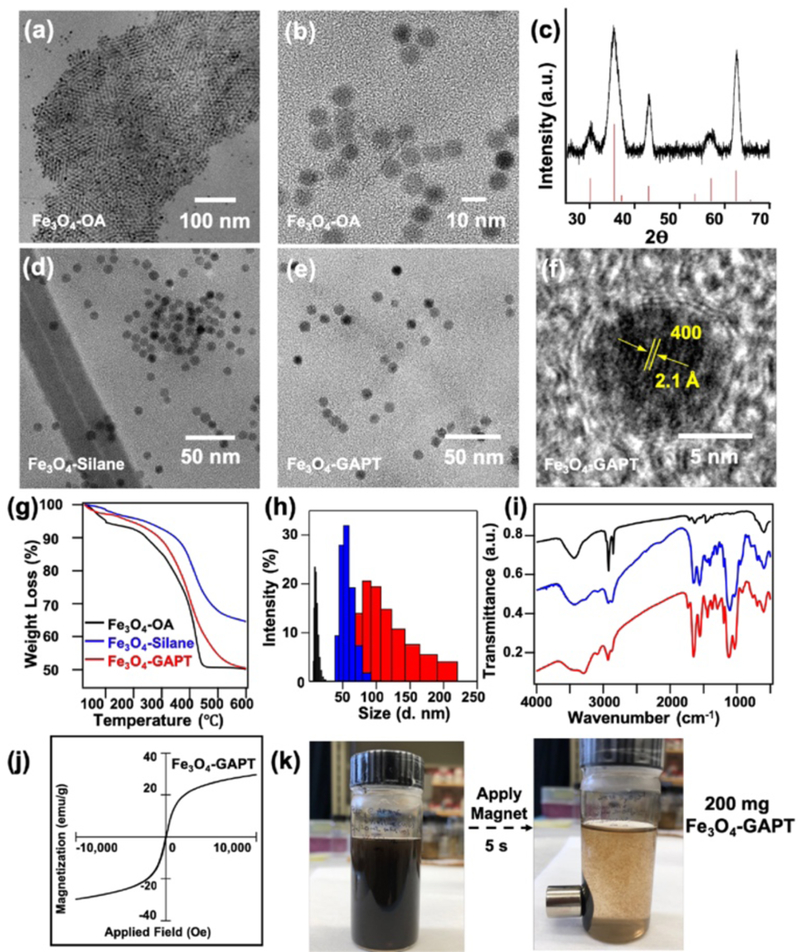
Structural characterization of large-scale surface functionalized Fe3O4 NPs. (a-b) TEM images of Fe3O4-OA. (c) PXRD pattern of the as-synthesized Fe3O4-OA NPs (black) in comparison with magnetite standard (red; JCPDS:19–0629). TEM images of (d) Fe3O4-APTMS/Si-PEG, and (e) Fe3O4-APTMS/Si-PEG-GAPT (Fe3O4-GAPT) NPs. (f) HRTEM of a representative Fe3O4-GAPT NP showing the crystalline core of the NP after functionalization. (g) TGA, (h) DLS size distribution, and (i) FTIR analysis of as-synthesized Fe3O4-OA (black), Fe3O4-APTMS/Si-PEG (blue), and Fe3O4-GAPT (red) NPs. (j) Field dependence of magnetization measured at 298 K for the final Fe3O4-GAPT NPs showing the superparamagnetic nature of the NPs. (k) Photographs of Fe3O4-GAPT dispersed in aqueous solution before (left) and after (right) a permanent magnet was placed next to the vial demonstrating the strong magnetic response of the functionalized NPs.
The careful control and optimization of this silanization step is the most crucial for the reproducible and large-scale synthesis of these functionalized Fe3O4 NPs to enable them as an effective nanoproteomics platform for effective biological applications, which will be discussed in detail later. When performed successfully, this silane ligand exchange yields a thin, stable, covalently linked silica coating on the surface of the Fe3O4 NPs with terminal functional groups (-NH2) allowing for further surface modification [16,19]. TEM analysis confirmed the absence of significant morphological changes to the functionalized Fe3O4-APTMS/Si-PEG NPs (Fig. 1d). Furthermore, a Zn-DPA ligand coupled to glutaric acid (hereafter referred to as GAPT), synthesized using our previously reported method [30] with minor modifications, was used as an affinity ligand to bind to phosphate groups for the specific capture, enrichment, and release of phosphoproteins. The GAPT ligands were coupled to reactive amine groups imparted by the silane coating on the Fe3O4-APTMS/Si-PEG NPs, yielding Fe3O4-APTMS/Si-PEG-GAPT (Fe3O4-GAPT) NPs. TEM revealed the uniform size distribution of the final Fe3O4-GAPT NPs (Fig. 1e), and HRTEM further showed that the surface coating was sufficiently robust to achieve a thin uniform coating of GAPT, while maintaining the crystalline core (Fig. 1f). Magnetic measurement confirmed the superparamagnetic nature and strong magnetic response (~30 emu/g) of the final Fe3O4-GAPT NPs (Fig. 1j). The superparamagnetism of the NPs prevents them from aggregating by themselves due to mutual magnetic attraction, but enables them to aggregate and precipitate readily under applied magnetic fields (Fig. 1k), making them attractive for biological applications [40,41].
Physiochemical properties of the surface functionalized NPs were also measured at each reaction step to both confirm the proper functionalization and elucidate the surface identity of the NPs. Thermogravimetric analysis (TGA) of the Fe3O4-OA NPs revealed a 44% weight loss, accounting for the relatively large amount of OA coating on as-synthesized NPs (Fig. 1g). The Fe3O4-APTMS/Si-PEG NPs show decreased weight loss (34%), because the silane ligands have lower molecular mass than the OA ligand they replaced. Finally, the Fe3O4-GAPT NPs showed an increased weight loss (47%) suggesting the successful attachment of the GAPT ligand. From the difference in weight loss (~13%) between the silane coated NPs and the final Fe3O4-GAPT NPs, a surface density of ~8 ligands/nm2 per particle corresponding to ~5 GAPT/nm2 per particle was inferred.
Dynamic light scattering (DLS) measurements showed that the hydrodynamic size of the NPs increased from the ~ 9 nm for OA coated NPs to ~ 50 nm for the APTMS/Si-PEG coated NPs (Fig. 1h). This increase in hydrodynamic size is expected and consistent with results reported for similar silane chemistry [19,42]. Additionally, upon coupling of GAPT, the hydrodynamic size further increased to ~ 97 nm illustrating successful coupling of the GAPT ligand. The broadening in DLS size intensity distribution and the slight increase in PDI (~ 0.1) of the Fe3O4-GAPT NPs is consistent with the increase in average hydrodynamic size of the Fe3O4-GAPT NPs (~ 97 nm) compared to the Fe3O4-silane NPs (~ 40 nm) and together imply partial aggregation of the Fe3O4-GAPT NPs, as seen in other reports [42]. At physiological pH, the positively charged, bulky pyridine groups of the GAPT ligand likely hydrogen bond with other GAPT ligands on nearby NPs, leading to the DLS size increase [19,43]. However, to demonstrate the colloidal stability of the final Fe3O4-GAPT NPs, we measured the zeta potential (ζ-potential), which measures the potential difference between the bulk material and the interfacial double layer situated some distance from the particle surface [44]. The ζ-potential for the Fe3O4-GAPT NPs in PBS buffer (pH 7.4) was ~ 33 mV, which has been previously shown to be sufficient for electrostatic repulsive forces to dominant over the van der Waals force, such that agglomeration is suppressed, leading to high colloidal stability [44]. The results obtained from both TGA and DLS measurements are summarized in Table 1.
Table 1.
Summary of materials characterization and surface functionalization analysis of various functionalized Fe3O4 NPs synthesized at a large-scale.
| Nanoparticle Sample | TEM Diameter (nm) |
DLS Diameter (nm)a |
PDIb | ζ-Potential pH 7.4 (mV)c |
TGA Weight Loss (%) |
Surface Density (ligands/nm2) |
|---|---|---|---|---|---|---|
| Fe3O4-OA | 8.0 ± 0.5 | 9.1 ± 0.5 | 0.106 ± 0.003 | - | 44 | 14 |
| Fe3O4-APTMS/Si-PEG | 8.0 ± 1.4 | 50 ± 2 | 0.15 ± 0.01 | 10.4 ± 0.3 | 34 | 7 |
| Fe3O4-GAPT | 8.2 ± 1.2 | 97 ± 8 | 0.24 ± 0.03 | 32.7 ± 0.7 | 47 | 8 |
DLS measurements were performed in n-hexane for the Fe3O4-OA, and in aqueous media for the Fe3O4-APTMS/Si-PEG and Fe3O4-GAPT NPs.
Polydispersity index (PDI) obtained from DLS measurement.
Zeta-potential (ζ-potential) obtained in PBS buffer (pH 7.4). All errors indicate standard deviations from measurements on three independent batches of particles.
Moreover, Fourier transform infrared spectroscopy (FTIR) confirmed the proper functionalization of the NPs. The Fe3O4-OA NPs showed strong vibrations in the 3100–3500 cm−1 (-OH), 2800–2900 cm−1 (-CH), 1700 cm−1 and 1400–1550 cm−1 (carboxylate) regions characteristic of the OA coating on the NP surface, and a strong peak in the 520–700 cm−1 region characteristic of the ferrite stretches (Fig. 1i) [16,45]. After the APTMS/Si-PEG coating, strong peaks between 1000–1200 cm−1 characteristic to alkoxysilane species (Si-O-R, Si-O-Si, C-O-R) [16] were observed. After the GAPT functionalization, a sharp peak around 1690 cm−1 appeared indicating both the presence of an amide bond and the successful attachment of GAPT to the NP surface (Fig. 1i).
3.2. Evaluation of NP phosphoprotein enrichment performance using standard proteins
To evaluate the enrichment performance of the surface functionalized NPs, the NPs were further activated with Zn2+ and the resultant Fe3O4-GAPT-Zn NPs were added to a standard protein mixture consisting of 300 μg bovine serum album (BSA, nonphosphoprotein), 100 μg pepsin (monophosphorylated), and 100 μg β-casein (multiphosphorylated) in a HEPES buffer (50 mM, pH 7.4, 150 mM NaCl) as the loading mixture (LM). As illustrated in Figure 2a, after equilibration, the NPs were washed with the same HEPES buffer to yield the flow through (FT) solution, and the sodium phosphate (Na2HPO4) buffer (100 mM, pH 7.4, 50 mM NaCl) was used as a competitive binding agent for eluting the captured phosphoproteins off the NPs to generate the elution mixture (EM).
Figure 2.
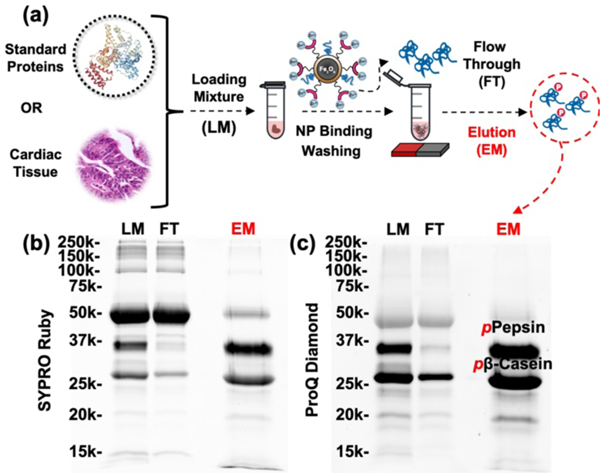
The process and performance of phosphoprotein enrichment using optimized large-scale functionalized NPs. (a) Workflow illustrating capture and enrichment process of phosphoproteins from a mixture of standard proteins or a biological sample using the Fe3O4-GAPT-Zn NPs. SDS-PAGE analysis with (b) SYPRO Ruby and (c) Pro-Q Diamond stains of a standard protein mixture containing 300 μg BSA, 100 μg β-casein, and 100 μg pepsin, respectively. Equal amount (10 μg) of the loading mixture (LM), flow-through (FT), and elution mixture after enrichment (EM) was loaded on the gel. Phosphorylated pepsin and β-casein are labeled in the Pro-Q EM showing the highly specific enrichment of phosphoproteins from the standard protein mixture.
To visualize the enrichment performance of these NPs, we performed SDS-PAGE analysis of the LM, FT, and EM fractions obtained from the enrichment step in equal loading of total protein amount (10 μg). The resultant gel was stained by SYPRO Ruby staining (Fig. 2b) to detect total proteins (both nonphosphoprotein and phosphoproteins) and ProQ-Diamond (Fig. 2c) to visualize phosphoproteins on the same gel. Clearly, the phosphoproteins (β-casein and pepsin) were specifically enriched from the protein mixture. The phosphoprotein specific stain further confirmed that the EM contained mostly phosphoproteins and the flow through (FT) contained very little phosphoproteins. Minimal nonspecific binding was observed. Moreover, the NPs unbiasedly captured and enriched the stringent monophosphorylated pepsin, which was nearly completely absent in the flow-through (FT).
To quantify the performance of these functionalized NPs and establish a quality control, we defined an enrichment performance metric based on SDS-PAGE analysis that uses image analysis software ImageJ [46] to determine the difference between the signal intensity of eluted phosphoproteins to that of eluted nonphosphoproteins, normalized to the total protein signal intensity by Equation(1) (see Fig. S1 and Table S1 in the ESM for example):
| (1) |
Such analysis of the total protein stained gel (Fig. 2a) revealed an enrichment performance index of ~ 0.72 (see Table S1 in the ESM), illustrating the highly specific enrichment of phosphoproteins and the resistance to nonspecific binding for the optimized large-scale functionalized NPs. Furthermore, by comparing the total amount of enriched phosphoproteins in the EM (143 μg) to the total amount of Fe3O4-GAPT NPs loaded (6 mg), we estimated a phosphoprotein capture efficiency of ~ 24 μg phosphoproteins/mg NP. (see details in Table S1).
3.3. Establishing a robust and reproducible surface silane functionalization method at large-scale
Initial experiments attempting to produce surface functionalized NPs at a large scale (> 50 mg per batch) suffered from inconsistencies in the final phosphoprotein enrichment performance from batch-to-batch. However, repeated enrichment trials using several parallel batches of Fe3O4-GAPT NPs derived from the same batch of Fe3O4-APTMS/Si-PEG NPs showed relatively constant enrichment performance (Fig. S2). We hypothesized that this silane ligand exchange step was likely the limiting factor in realizing high and reproducible enrichment efficacy. Therefore, the problem was reduced to finding a method to reliably synthesize high-quality large-scale silanized NPs to guarantee high performance for the entirety of the batch. We tested a series of silanization reaction conditions varying temperature (RT or 60 °C), reaction time (24 h or 48 h), and silane concentration (0.25 % or 0.5 % v/v), while fixing NP concentration (0.4 mg/mL) and adding a fixed amount of acetic acid as the reaction catalyst (0.01% v/v). It should be noted that in the case of Si alkoxides, the reactivity is sufficiently low, such that both hydrolysis and condensation reactions (see Eq. S1-S3 in the ESM) are kinetically separate [17].
Therefore, an acid catalyst (acetic acid) is used to expedite the hydrolysis reaction and promote NP surface silanization. After a total of about 20 silanization reaction runs and subsequent further functionalizing to the Fe3O4-GAPT-Zn NPs, the phosphoprotein enrichment performance using the same standard protein mixture was evaluated for each reaction condition, as illustrated in Fig. 2a. From the enrichment performance index (Eq. 1) determined from the enrichment results using each batch of NPs, we found the optimal silanization reaction condition to be a 60 °C reaction which proceeds for 48 h using a 0.5% (v/v) total silane concentration, a fixed 0.4 mg/mL NP concentration, and a 0.01% (v/v) acetic acid concentration.
3.4. Development of a quality assurance control (QAC) for silanized NPs
Because the silanization via trialkoxysilane species is kinetically challenging to control [17,18], the outcome of such reactions could still vary despite careful control. But, to our great benefit, the reaction condition screening revealed an interesting correlation between the NP enrichment performance and the relative FTIR intensity of vibrational modes associated with silane and ferrite character for the silanized Fe3O4-APTMS/Si-PEG NPs. As representative examples, three distinct batches of Fe3O4-APTMS/Si-PEG NPs prepared under different reaction conditions, denoted by (i), (ii), and (iii), displayed different the relative intensity and area under curve (AUC) for the respective FTIR peaks (Figure 3a) of silane stretches (marked with “arrow”) and ferrite stretches (marked with “*”). The relative changes in silane and ferrite peaks appeared to vary in direct correspondence to the eventual NP enrichment performance visualized by SDS-PAGE analysis using the same standard protein mixture of 300 μg BSA, 100 μg β-casein, and 100 μg pepsin (Fig. 3b). To quantify this potential correlation, we define a silane coverage density (SCD; see Fig. S3 for example) metric (Equation 2) which represents the relative silane surface coverage:
| (2) |
Figure 3.
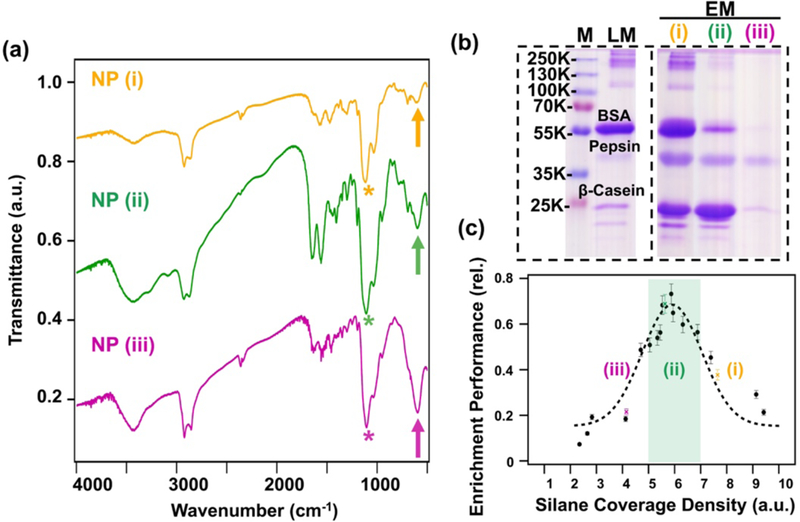
Quality check on the silane functionalization of NPs and the correlation to phosphoprotein enrichment performance. (a) FTIR spectra of three representative Fe3O4-APTMS/Si-PEG NPs synthesized under different conditions (i-iii) with vibrational modes corresponding to ferrite (“arrow”) and silane (“*”) denoted. Traces are offset along the y-axis for clarity. (b) SDS-PAGE analysis showing the enrichment performance of representative GAPT-functionalized NPs prepared from the same batches of NPs in (a) (i-iii). Phosphoprotein enrichment was screened using a standard protein mixture containing 300 μg bovine serum album (BSA), 100 μg β-casein, and 100 μg pepsin, respectively (loading mixture). The enrichment step was performed using 6 mg of NP, each lane was loaded to equal amount, and the gel was visualized using Coomassie blue. M: marker; LM: loading mixture: FT: flow through; EM: elution mixture. (c) Plot of enrichment performance against silane coverage density for all tested silane reaction conditions. The dashed line (---) represents a fitting for the experimental values. The green bar highlights samples that meet a minimum value of 0.5 enrichment performance, thereby illustrating the optimal silane functionalization condition to achieve highest enrichment performance.
Plotting the NP enrichment performance against the silane coverage density for all tested NP batches (Fig. 3c) revealed a striking correlation between the two metrics that illustrates the nature of the silanization step. We found that the best performing functionalized NPs (enrichment performance ≥ 0.5) had SCDs between 5–7. These results suggest: (1) the silane ligand exchange step has significant impact on the final enrichment performance of the functionalized NPs; (2) there exists an optimal SCD range corresponding to a particular reaction condition that achieves maximal NP enrichment performance (highlighted by the green bar in Fig. 3c). In particular, samples (i) and (iii) have “too-much” and “too-little” silane coating, respectively. Sample (i) suffers from too much nonspecific binding of nonphosphoproteins, and sample (iii) suffers from too little binding of phosphoproteins. On the other hand, sample (ii) represents the synthetic conditions required to achieve optimal silane coating to yield the highest enrichment performance. Obviously, insufficient silane functionalization will lead to poor overall NP functionalization and poor enrichment performance. But over silanization might lead to excess free amine groups after the GAPT functionalization step. From the previous studies on the use of APTMS or other silane reagents to provide free amines on the surface of NPs for further surface functionalization [19,20,35,47], it is known that terminal, uncapped amines can have noncovalent, nonspecific interactions with proteins that lead to nonspecific binding and NP aggregation [19,35,48,49]. Importantly, our optimized NP system shows little nonspecific binding and highly specific enrichment due to the additional PEG stealth layer coating on these NPs to reduce nonspecific interaction [50–52].
Hence, these results allowed for direct interrogation of the optimal surface silanization protocol (60 °C, 48 h reaction time, with a 0.5% silane v/v, 0.4 mg/mL NP loading, and 0.01% acetic acid v/v) to achieve maximal enrichment performance. Moreover, the synthesis could now be reliably scaled-up (~200 mg) using commensurate amounts of the described reagents without having to sacrifice enrichment performance. Furthermore, the correlation established between the SCD and the enrichment performance (Fig. 3c) serves as a simple but reliable metric for QAC from batch-to-batch. From now on, for any given large-scale batch of surface silanized Fe3O4 NPs, simply performing the FTIR measurement and checking the SCD against the plot in Fig. 3c could predict the NP enrichment performance a priori. Therefore, coupled with an optimized silanization protocol, this predictive capability over the final enrichment performance enables the robust nanoproteomics platform to guarantee reproducible and high phosphoprotein enrichment performance from batch-to-batch, without more tedious preliminary enrichment screening.
3.5. Top-down LC-MS/MS analysis of endogenous phosphoproteins enriched from human cardiac tissue
Following successful demonstration of the large-scale functionalized NP platform for the reproducible enrichment of phosphoproteins from a standard mixture, we applied them to directly interrogate the human heart phosphoproteome using a highly complex biological sample, an extraction of a human heart tissue homogenate (see ESM for details). SYPRO Ruby staining (Figure 4a) revealed total proteins, and ProQ-Diamond staining (Fig. 4b) revealed the phosphoproteins on the same gel. The abundant non-phosphoproteins in the LM were effectively depleted and washed out as FT, as shown by the nearly identical bands between LM and FT (Fig. 4b). The highly similar band patterns in the EM of the SYPRO Ruby and Pro-Q Diamond stains (Fig. 4a,b) show endogenous phosphoproteins are predominantly enriched from the complex human heart tissue. To further confirm the presence of phosphoproteins in EM, an additional dephosphorylation step using a shrimp alkaline phosphatase (rSAP) was performed on a portion of the EM fraction. The dephosphorylated elution fraction (dpEM) shows clearly the depletion of bands in the Pro-Q stain, while maintaining an identical band pattern as in the EM for the SYPRO Ruby stain (Fig. 4a,b). This further corroborates the results shown in the EM lane and together clearly confirm the successful enrichment of endogenous phosphoproteins from a highly complex human cardiac system. It should be noted that rSAP (54 kDa) also appears in the gel stain and has been denoted by an asterisk (“*”).
Figure 4.
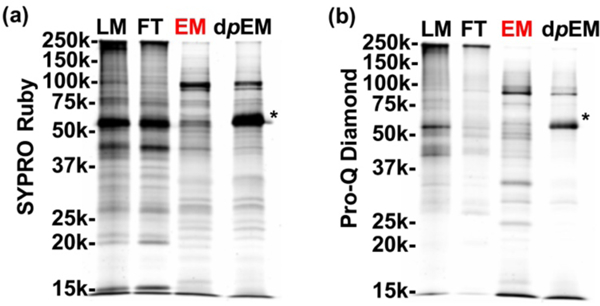
Specific enrichment of phosphoproteins from a complex human cardiac tissue homogenate. SDS PAGE analysis with (a) SYPRO Ruby and (b) Pro-Q Diamond staining of endogenous phosphoproteins from a HEPES buffer extraction of a human dilated cardiomyopathic (DCM) cardiac tissue homogenate. Equal amount (10 μg) of the loading mixture (LM), flow-through (FT), elution mixture after enrichment (EM), and elution mixture post enrichment with additional dephosphorylation step using alkaline phosphatase (dpEM) was loaded on the gel. The added alkaline phosphatase is a 54 kDa protein denoted by an asterisk (“*”).
Furthermore, we identified and characterized the enriched phosphoproteins by top-down mass spectrometry. An equal amount of proteins from LM, FT, EM, and dpEM that were acquired from the same human cardiac tissue extract (and used for the SDS-PAGE analysis) was subjected to reversed-phase chromatography (RPC) LC-MS and LC-MS/MS analyses using a Waters nanoACUITY UPLC and a Bruker Impact II Q-TOF mass spectrometer. Consistent with the SDS-PAGE analysis (Fig. 4a,b), the overlapping separation pattern in the total ion chromatograms (TIC) between the LM and flow through (FT) (Fig. S4) demonstrates that the majority of the proteins from LM were washed out to the FT fraction. Subsequent MS analysis revealed that most of the proteins in the LM and FT fractions are highly abundant non-phosphoproteins (Fig. S5). Notably, blood proteins such as myoglobin and hemoglobin (α and β) were significantly depleted after the enrichment step along with other highly abundant non-phosphoproteins (Fig. S5 and S6). This significant depletion of highly abundant nonphosphoproteins from the cardiac tissue loading mixture enabled us to detect the low abundance phosphoproteins in the elution mixture. The deconvoluted mass spectra of representative phosphoproteins specifically enriched from the highly complex cardiac tissue homogenate are illustrated in Figure 5. Enrichment of the endogenous phosphoproteins is apparent by comparing the intensity of the phosphorylated proteoforms across the same chromatographic elution time frame of LM, FT, EM, and dpEM. Comparison of the mass spectra between EM and dpEM directly revealed both the presence of a phosphorylated site and the total number of phosphorylated sites on each of the captured phosphoproteins by tracking the appearance/disappearance of a phosphate group (80 Da mass increase; labeled as +HPO3). Based on the directly overlapped TIC-MS of the EM and dpEM fractions, the dephosphorylation treatment did not significantly alter the protein population (Fig. S7).
Figure 5.
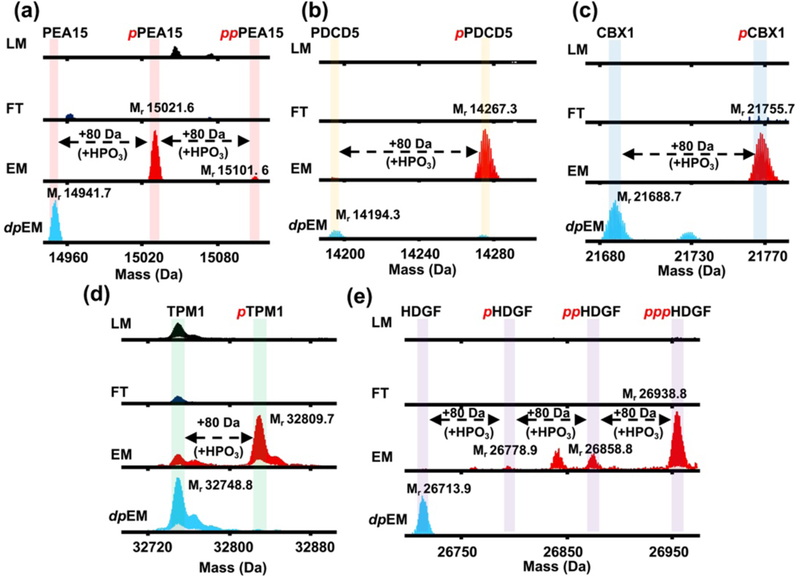
Top-down MS analysis of representative endogenous phosphoproteins specifically enriched from a highly complex human cardiac tissue homogenate. Deconvoluted spectra of (a) astrocytic phosphoprotein 15 (PEA15), (b) programmed cell death protein 5 (PDCD5), (c) chromobox protein homolog 1 (CBX1), (d) tropomyosin alpha (TPM1), and (e) hepatoma derived growth factor (HDGF). Equal total protein amount (5 μg) of the loading mixture (LM), flow-through (FT), elution mixture after enrichment (EM), and elution mixture post enrichment with additional dephosphorylation step using alkaline phosphatase (dpEM) was used for the MS analysis. Phosphorylation is indicated by a red “p”. Mr: monoisotopic mass. Mass difference of 80 Da corresponds to covalent addition of a phosphate group (+HPO3). All deconvoluted spectra corresponding to a particular protein were normalized.
This optimized large-scale nanoproteomics platform allowed us to specifically enrich endogenous phosphoproteins that were otherwise undetectable in the LM by top-down MS. We were able to identify and characterize two previously undetected low abundance apoptosis related proteins. One of them is the phosphorylated astrocytic phosphoprotein 15 (PEA15; monoisotopic mass, Mr 14267.3; Fig. 5a and S8), which is an important apoptosis regulatory protein that interacts with extracellular signal-regulated kinase 1 and 2 (ERK½) in the mitogen activated protein kinase (MAPK) pathway and has recently been reported as a potential therapeutic target for cardiovascular disease, breast cancer, and ovarian cancer [53–55]. Further analysis of the collision-induced dissociation (CID) MS/MS data using MASH Suite Pro [56] allowed us to map the sequence of the enriched PEA15 and determine the most probable phosphorylation site as Ser90 (Fig. S8a,c). Top-down MS/MS not only revealed an N-terminal methionine excision, but also a coexisting acetylation in addition to the phosphorylation. We also selectively enriched both phosphorylated (Mr 15021.6) and bis-phosphorylated (Mr 15101.6) programmed cell death protein 5 (PDCD5; Fig. 5b and S8). PDCD5 is an important modulator of the apoptotic pathway required for normal cardiovascular development, and serum PDCD5 has been recently shown to reflect vascular endothelial status, which is significantly correlated with cardiovascular risk [57–59]. Along with a N-terminal methionine excision and a coexisting acetylation, the most probable phosphorylation site was determined to be Ser119 (Fig. S8b,d). Furthermore, using this integrated top-down phosphoproteomics strategy, we have enriched, detected and examined multiple known cardiac phosphoproteins that were previously undetected or in extremely low abundance in the HEPES cardiac tissue extraction [31,60,61]: phosphorylated chromobox protein homolog 1 (CBX1; Mr 21755.7; Fig. 5c), phosphorylated tropomyosin alpha (TPM1; Mr 32809.7; Fig. 5d), and triply-phosphorylated hepatoma derived growth factor (HDGF; Mr 26938.l8; Fig. 5e). Due to scalability and reproducible high enrichment performance, this robust nanoproteomics platform can be utilized for the interrogation of the human heart phosphoproteome. The identification and characterization of phosphorylated proteoforms of intrinsically low abundance cell death signaling proteins illustrates the high specificity and robustness of our nanoproteomics platform for phosphoprotein enrichment.
Moreover, the utilization of a cost-effective and high performance phospho-specific affinity ligand provides distinct advantages [62] over common phosphoprotein enrichment strategies that rely on phospho-specific anti-phosphotyrosine monoclonal antibodies (mAbs) [63] or immobilized metal ion affinity chromatography (IMAC) [64]. IMAC suffers from low specificity, poor reproducibility, and mandates pH conditions outside the physiological condition of endogenous proteins [65]. mAbs selected against phosphoserine/threonine containing proteins (which represent > 99% of all phosphoproteins) [66] suffer from high costs, low specificity, and batch-to-batch variability [62]. Importantly, because affinity ligands are generally designed to select for broader protein families (proteoforms) and are not necessarily limited to single analytes or epitopes such as mAbs, the use of a high quality phospho-specific affinity ligand allows for a global and deep selection of phosphoproteins without bias [30,31,62,67], as demonstrated above in the enrichment of multiple phosphoproteins. Furthermore, due to the easily modifiable amine-coated surface, this nanoproteomics platform can be easily furnished with other affinity ligands, thereby enabling a broad scope of enrichment strategies for other low abundance proteins in general and other biological applications.
4. Conclusions
By refining and optimizing the NP surface silane ligand exchange reaction, we have developed a reproducible large-scale (200 mg) synthesis of surface silanzed Fe3O4 NPs and further surface functionalization for the reliable and highly effective enrichment of phosphoproteins from a complex biological mixture. Moreover, we developed a simple and reliable quality assurance control to both ensure the proper surface composition necessary to achieve optimal phosphoprotein enrichment performance, and to enable the routine synthesis of these functionalized NPs in a large scale with minimal batch-to-batch variation of enrichment performance. We also demonstrated the successful exploitation of a top-down phosphoproteomics strategy that integrates this scalable and high throughput NP-based phosphoprotein enrichment platform with online LC-MS/MS to enrich, identify, and characterize endogenous phosphoproteins from highly complex human cardiac tissue. SDS-PAGE analysis clearly showed the specific enrichment of phosphoproteins, and top-down LC-MS results revealed a variety of low-abundance endogenous phosphoproteins that were otherwise undetectable without enrichment. These results highlight the great promise of these robust and scalable surface silanized Fe3O4 NPs as an enabling nanoproteomics platform. Although the current study specifically focuses on phosphoprotein enrichment, this nanoproteomics platform can be adapted for capturing, enriching, and analysis of other low abundance proteins in general when a suitable affinity binding can be developed. A combination of scalability, reproducibility, and high throughput screening are attractive features to this nanoproteomics platform that will enable new affinity capture strategies utilizing other robust affinity ligands.
Supplementary Material
Acknowledgements
The financial support for this project is provided by NIH R01 GM117058 (to S.J. and Y.G.). Moreover, Y.G. would like to acknowledge the NIH R01 GM125085, and S10 0D018475. T.N.T. would like to acknowledge support from the NIH Chemistry-Biology Interface Training Program NIH T32GM008505.
Footnotes
Electronic Supplementary Material: Supplementary material (detailed experimental procedures, instrumentation and methods, example of enrichment performance calculation, workflow scheme, Si alkoxide silanization reaction steps, example of silane coverage density calculation, and additional figures) is available in the online version of this article at http://dx.doi.org/10.1007/s12274-***-****-* (automatically inserted by the publisher).
References
- [1].De M; Ghosh PS; Rotello VM Applications of nanoparticles in biology. Adv. Mater. 2008, 20, 4225–4241. [Google Scholar]
- [2].Mitragotri S; Anderson DG; Chen X; Chow EK; Ho D; Kabanov AV; Karp JM; Kataoka K; Mirkin CA; Petrosko SH; Shi J; Stevens MM; Sun S; Teoh S; Venkatraman SS; Xia Y; Wang S; Gu Z; Xu C Accelerating the Translation of Nanomaterials in Biomedicine. ACS Nano. 2015, 9, 6644–6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Michalet X; Pinaud FF; Bentolila LA; Tsay JM; Doose S; Li JJ; Sundaresan G; Wu AM; Gambhir SS; Weiss S Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005, 307, 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gao J; Gu H; Xu B Multifunctional magnetic nanoparticles: design, synthesis, and biomedical applications. Acc. Chem. Res. 2009, 42, 1097–1107. [DOI] [PubMed] [Google Scholar]
- [5].Giljohann DA; Seferos DS; Daniel WL; Massich MD; Patel PC; Mirkin CA Gold Nanoparticles for Biology and Medicine. Angew. Chemie Int. Ed. 2010, 49, 3280–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Assa F; Jafarizadeh-Malmiri H; Ajamein H; Anarjan N; Vaghari H; Sayyar Z; Berenjian A A biotechnological perspective on the application of iron oxide nanoparticles. Nano Res. 2016, 9, 2203–2225. [Google Scholar]
- [7].Xie J; Liu G; Eden HS; Ai H; Chen X Surface-engineered magnetic nanoparticle platforms for cancer imaging and therapy. Acc. Chem. Res. 2011, 44, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ho D; Sun X; Sun S Monodisperse magnetic nanoparticles for theranostic applications. Acc. Chem. Res. 2011, 44, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang W; Liu L; Chen H; Hu K; Delahunty I; Gao S; Xie J Surface impact on nanoparticle-based magnetic resonance imaging contrast agents. Theranostics. 2018, 8, 2521–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Park J; An K; Hwang Y; Park JEG; Noh HJ; Kim JY; Park JH; Hwang NM; Hyeon T Ultra-large-scale syntheses of monodisperse nanocrystals. Nat. Mater. 2004, 3, 891–895. [DOI] [PubMed] [Google Scholar]
- [11].White MA; Johnson JA; Koberstein JT; Turro NJ Toward the syntheses of universal ligands for metal oxide surfaces: Controlling surface functionality through click chemistry. J. Am. Chem. Soc. 2006, 128, 11356–11357. [DOI] [PubMed] [Google Scholar]
- [12].Grancharov SG; Zeng H; Sun S; Wang SX; O’Brien S; Murray CB; Kirtley JR; Held GA Bio-functionalization of monodisperse magnetic nanoparticles and their use as biomolecular labels in a magnetic tunnel junction based sensor. J. Phys. Chem. B. 2005, 109, 13030–13035. [DOI] [PubMed] [Google Scholar]
- [13].Hong R; Fischer NO; Emrick T; Rotello VM Surface PEGylation and ligand exchange chemistry of FePt nanoparticles for biological applications. Chem. Mater. 2005, 17, 4617–4621. [Google Scholar]
- [14].Xu C; Xu K; Gu H; Zheng R; Liu H; Zhang X; Guo Z; Xu B Dopamine as A Robust Anchor to Immobilize Functional Molecules on the Iron Oxide Shell of Magnetic Nanoparticles. J. Am. Chem. Soc. 2004. 126, 9938–9939. [DOI] [PubMed] [Google Scholar]
- [15].Lattuada M; Hatton TA Functionalization of monodisperse magnetic nanoparticles. Langmuir. 2007, 23, 2158–2168. [DOI] [PubMed] [Google Scholar]
- [16].De Palma R; Peeters S; Van Bael MJ; Van Den Rul H; Bonroy K; Laureyn W; Mullens J; Borghs G; Maes G Silane ligand exchange to make hydrophobic superparamagnetic nanoparticles water-dispersible. Chem. Mater. 2007, 19, 1821–1831. [Google Scholar]
- [17].Plueddemann EP Reminiscing on silane coupling agents. J. Adhes. Sci. Technol. 1991, 5, 261–277. [Google Scholar]
- [18].Arkles B; Steinmetz JR; Zazyczny J; Mehta P Factors contributing to the stability of alkoxysilanes in aqueous solution. J. Adhes. Sci. Technol. 1992, 6, 193–206. [Google Scholar]
- [19].Cano M; Núñez-Lozano R; Lumbreras R; González-Rodríguez V; Delgado-García A; Jiménez-Hoyuela JM; De La Cueva-Méndez G Partial PEGylation of superparamagnetic iron oxide nanoparticles thinly coated with amine-silane as a source of ultrastable tunable nanosystems for biomedical applications. Nanoscale. 2017, 9, 812–822. [DOI] [PubMed] [Google Scholar]
- [20].Jana NR; Earhart C; Ying JY Synthesis of water-soluble and functionalized nanoparticles by silica coating. Chem. Mater. 2007, 19, 5074–5082. [Google Scholar]
- [21].Smolensky ED; Park HYE; Berquo TS; Pierre VC Surface functionalization of magnetic iron oxide nanoparticles for MRI applications -effect of anchoring group and ligand exchange protocol. Contrast Media Mol. Imaging. 2011, 6, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aebersold R; Mann M Mass-spectrometric exploration of proteome structure and function. Nature. 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- [23].Cai W; Tucholski TM; Gregorich ZR; Ge Y Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev. Proteomics. 2016, 13, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen B; Brown KA; Lin Z; Ge Y Top-Down Proteomics: Ready for Prime Time? Anal. Chem. 2018, 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anderson NL; Anderson NG The Human Plasma Proteome. Mol. Cell. Proteomics. 2002, 1, 845–867. [DOI] [PubMed] [Google Scholar]
- [26].Siuti N; Kelleher NL Decoding protein modifications using top-down mass spectrometry. Nat. Methods. 2007, 4, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brown KA; Chen B; Guardado-Alvarez TM; Lin Z; Hwang L; Ayaz-Guner S; Jin S; Ge Y A photo-cleavable surfactant for top-down proteomics. Nat. Methods., in press, DOI: 10.1038/s41592-019-0391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xie S; Moya C; Bilgin B; Jayaraman A; Walton SP Emerging affinity-based techniques in proteomics. Expert Rev. Proteomics. 2009, 6, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hunter T Signaling—2000 and Beyond. Cell. 2000, 100, 113–127. [DOI] [PubMed] [Google Scholar]
- [30].Hwang L; Ayaz-Guner S; Gregorich ZR; Cai W; Valeja SG; Jin S; Ge Y Specific enrichment of phosphoproteins using functionalized multivalent nanoparticles. J. Am. Chem. Soc. 2015, 137, 2432–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen B; Hwang L; Ochowicz W; Lin Z; Guardado-Alvarez TM; Cai W; Xiu L; Dani K; Colah C; Jin S; Ge Y Coupling functionalized cobalt ferrite nanoparticle enrichment with online LC/MS/MS for top-down phosphoproteomics. Chem. Sci. 2017, 8, 4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rosi NL; Mirkin CA Nanostructures in biodiagnostics. Chem. Rev. 2005, 105, 1547–1562. [DOI] [PubMed] [Google Scholar]
- [33].Pan Y; Long MJC; Lin HC; Hedstrom L; Xu B Magnetic nanoparticles for direct protein sorting inside live cells. Chem. Sci. 2012, 3, 3495–3499. [Google Scholar]
- [34].Aubin-Tam ME; Hamad-Schifferli K Structure and function of nanoparticle-protein conjugates. Biomed. Mater. 2008, 3, 034001. [DOI] [PubMed] [Google Scholar]
- [35].Bagwe RP; Hilliard LR; Tan W Surface modification of silica nanoparticles to reduce aggregation and nonspecific binding. Langmuir. 2006, 22, 4357–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Palma R; Laureyn W; Frederix F; Bonroy K; Pireaux JJ; Borghs G; Maes G Formation of dense self-assembled monolayers of (n-decyl)trichlorosilanes on Ta/Ta2O5. Langmuir. 2007, 23, 443–451. [DOI] [PubMed] [Google Scholar]
- [37].Verma A; Stellacci F Effect of surface properties on nanoparticle-cell interactions. Small. 2010, 6, 12–21. [DOI] [PubMed] [Google Scholar]
- [38].Scott AW; Garimella V; Calabrese CM; Mirkin CA Universal Biotin-PEG-Linked Gold Nanoparticle Probes for the Simultaneous Detection of Nucleic Acids and Proteins. Bioconjug. Chem. 2017, 28, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li Y; Zhang Y; Wang W Phototriggered targeting of nanocarriers for drug delivery. Nano Res. 2018, 11, 5424–5438. [Google Scholar]
- [40].Ling D; Lee N; Hyeon T Chemical synthesis and assembly of uniformly sized iron oxide nanoparticles for medical applications. Acc. Chem. Res. 2015, 48, 1276–1285. [DOI] [PubMed] [Google Scholar]
- [41].Na H. Bin; Song IC; Hyeon T Inorganic nanoparticles for MRI contrast agents. Adv. Mater. 2009, 21, 2133–2148. [Google Scholar]
- [42].Rejeeth C; Pang X; Zhang R; Xu W; Sun X; Liu B; Lou J; Wan J; Gu H; Yan W; Qian K Extraction, detection, and profiling of serum biomarkers using designed Fe3O4@SiO2@HA core-shell particles. Nano Res. 2018, 11, 68–79. [Google Scholar]
- [43].Cano M; De La Cueva-Mendez G Self-assembly of a superparamagnetic raspberry-like silica/iron oxide nanocomposite using epoxy-amine coupling chemistry. Chem. Commun. 2015, 51, 3620–3622. [DOI] [PubMed] [Google Scholar]
- [44].Jiang J; Oberdorster G; Biswas P Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies. J. Nanoparticle Res. 2009, 11, 77–89. [Google Scholar]
- [45].Gao F; Cai Y; Zhou J; Xie X; Ouyang W; Zhang Y; Wang X; Zhang X; Wang X; Zhao L; Tang J Pullulan acetate coated magnetite nanoparticles for hyper-thermia: Preparation, characterization and in vitro experiments. Nano Res. 2010, 3, 23–31. [Google Scholar]
- [46].Xu L; Feng Y; Fan Z; Yun D Research into the grading method of kiwi fruit based on volume estimation and surface defect. INMATEH -Agric. Eng. 2014, 44, 93–102. [Google Scholar]
- [47].Li YC; Lin YS; Tsai PJ; Chen CT; Chen WY; Chen YC Nitrilotriacetic acid-coated magnetic nanoparticles as affinity probes for enrichment of histidine-tagged proteins and phosphorylated peptides. Anal. Chem. 2007, 79, 7519–7525. [DOI] [PubMed] [Google Scholar]
- [48].Panja P; Das P; Mandal K; Jana NR Hyperbranched Polyglycerol Grafting on the Surface of Silica-Coated Nanoparticles for High Colloidal Stability and Low Nonspecific Interaction. ACS Sustain. Chem. Eng. 2017, 5, 4879–4889. [Google Scholar]
- [49].Wang J; Shen H; Huang C; Ma Q; Tan Y; Jiang F; Ma C; Yuan Q Highly efficient and multidimensional extraction of targets from complex matrices using aptamer-driven recognition. Nano Res. 2017, 10, 145–156. [Google Scholar]
- [50].Liu Z; Cai W; He L; Nakayama N; Chen K; Sun X; Chen X; Dai H In vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in mice. Nat. Nanotechnol. 2007, 2, 47–52. [DOI] [PubMed] [Google Scholar]
- [51].Sun X; Liu Z; Welsher K; Robinson JT; Goodwin A; Zaric S; Dai H Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang C; Ye Y; Hu Q; Bellotti A; Gu Z Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv. Mater. 2017, 29, 1606036. [DOI] [PubMed] [Google Scholar]
- [53].Greig FH; Nixon GF Phosphoprotein enriched in astrocytes (PEA)-15: A potential therapeutic target in multiple disease states. Pharmacol. Ther. 2014, 143, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lee J; Bartholomeusz C; Krishnamurthy S; Liu P; Saso H; Lafortune TA; Hortobagyi GN; Ueno NT PEA-15 unphosphorylated at both serine 104 and serine 116 inhibits ovarian cancer cell tumorigenicity and progression through blocking P-catenin. Oncogenesis. 2012, 1, e22–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xie X; Tang H; Liu P; Kong Y; Wu M; Xiao X; Yang L; Gao J; Wei W; Lee J; Bartholomeusz C; Ueno NT; Xie X Development of PEA-15 using a potent non-viral vector for therapeutic application in breast cancer. Cancer Lett. 2015, 356, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cai W; Guner H; Gregorich ZR; Chen AJ; Ayaz-Guner S; Peng Y; Valeja SG; Liu X; Ge Y MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol. Cell. Proteomics. 2016, 15, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lee S-H; Seo J; Park S-Y; Jeong M-H; Choi H-K; Lee CJ; Kim MJ; Guk G; Lee S; Park H; Jeong J-W; Ha CH; Park S; Yoon H-G Programmed cell death 5 suppresses AKT-mediated cytoprotection of endothelium. Proc. Natl. Acad. Sci. 2018, 115, 201712918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Park SY; Seo J; Choi HK; Oh HJ; Guk G; Lee YH; Lee J; Jun WJ; Choi KC; Yoon HG Protein serine/threonine phosphatase PPEF-1 suppresses genotoxic stress response via dephosphorylation of PDCD5. Sci. Rep. 2017, 7, 39222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kwak S; Lee S-H; Han E-J; Park S-Y; Jeong M-H; Seo J; Park S-H; Sung G-J; Yoo J-Y; Yoon H-G; Choi K-C Serine/threonine kinase 31 promotes PDCD5-mediated apoptosis in p53-dependent human colon cancer cells. J. Cell. Physiol. 2019, 234, 2649–2658. [DOI] [PubMed] [Google Scholar]
- [60].Gregorich ZR; Cai W; Lin Z; Chen AJ; Peng Y; Kohmoto T; Ge Y Distinct sequences and post-translational modifications in cardiac atrial and ventricular myosin light chains revealed by top-down mass spectrometry. J. Mol. Cell. Cardiol. 2017, 107, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cai W; Tucholski T; Chen B; Alpert AJ; Mcllwain S; Kohmoto T; Jin S; Ge Y Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal. Chem. 2017, 89, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kinoshita E; Kinoshita-Kikuta E; Koike T Separation and detection of large phosphoproteins using phos-tag sds-page. Nat. Protoc. 2009, 4, 1513–1521. [DOI] [PubMed] [Google Scholar]
- [63].Schmidt SR; Schweikart F; Andersson ME Current methods for phosphoprotein isolation and enrichment. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 154–162. [DOI] [PubMed] [Google Scholar]
- [64].Porath J; Carlsson J; Olsson I; Belfrage G Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975, 258, 598–599. [DOI] [PubMed] [Google Scholar]
- [65].Kaur-Atwal G; Weston D; Bonner P; Crosland S; Green P; Creaser C Immobilised Metal Affinity Chromatography for the Analysis of Proteins and Peptides. Curr. Anal. Chem. 2008, 4, 127–135. [Google Scholar]
- [66].Wu H; Liao P Analysis of protein phosphorylation using mass spectrometry. Chang Gung Med. J. 2008, 20, 217–227. [PubMed] [Google Scholar]
- [67].Regnier FE; Kim J Proteins and Proteoforms: New Separation Challenges. Anal. Chem. 2018, 90, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.