

Extracranial malignant rhabdoid tumor (MRT) is a rare and aggressive tumor that occurs mainly in infants and young children. This review reports the clinical characteristics, treatment schedules, and outcomes of patients with MRT from a single institution in China.
Keywords: Extracranial malignant rhabdoid tumor, Clinical characteristics, Treatment schedules, Prognosis
Abstract
Background.
The aim of this study is to add to the current knowledge regarding extracranial malignant rhabdoid tumor (MRT), a rare and highly aggressive tumor that occurs most commonly in infants and young children.
Patients and Methods.
A retrospective medical record review was conducted on 53 patients with pathologically confirmed MRT in Beijing Children's Hospital between January 2007 and October 2017.
Results.
Fifty‐three patients were diagnosed with MRT at a median age of 16 months, including 32 cases of malignant rhabdoid tumor of the kidney (MRTK) and 21 cases of extrarenal extracranial rhabdoid tumor (EERT). Fourteen (14/32, 43.75%) patients with MRTK and five (5/21, 23.81%) patients with EERT had metastases at diagnosis, and quite a few number of cases occurred tumor rupture (26.42%). Among the 53 patients, 40 (75.47%) patients died, 10 (18.87%) patients survived, and 3 patients (5.66%) were lost to follow‐up. Among the 40 dead patients, 38 patients died from rapid progression of the disease or tumor recurrence, and 2 patients died of severe postoperative complications. Most of the recurrent or relapsed cases (94.11%) occurred within 8 months, with a median time of 76 days after diagnosis. The overall survival rates of 3 years and 5 years for the entire cohort were 23.71% and 18.44%, respectively. After survival analysis, it was clear that a younger age at diagnosis and distant stage patients had relatively poor outcomes. The effect of treatment was the most difficult to analyze because patients were not treated uniformly. Statistically significant differences in survival were noted among patients treated with standard chemotherapy, total resection, and radiotherapy.
Conclusion.
Extracranial MRT is still a highly aggressive tumor in children. Younger patients and those suffering from metastatic disease were most likely to have a poor outcome because of rapid progression or recurrence of the tumor.
Implications for Practice.
This is the largest single‐institutional report that investigates the clinical characteristics and outcomes of extracranial malignant rhabdoid tumor (MRT) in China. Our study showed that gross hematuria and tumor rupture were typical characteristics of malignant rhabdoid tumor of the kidney. After survival analysis, it was found that the advanced stage of the tumor and an age ≤12 months at diagnosis were significantly associated with poorer survival. Although extracranial MRT is still a highly aggressive tumor in children, multimodal treatment approach, including chemotherapy, surgery, and radiotherapy, should be employed for this disease.
Introduction
Malignant rhabdoid tumor (MRT) is a rare, highly aggressive malignant tumor that occurs most commonly in infants and young children [1]. Recent studies reported that MRT occurred in nearly every anatomical site [1]. Nomenclature for the three recognized forms of MRT reflect their anatomic localization and include malignant rhabdoid tumor of the kidney (MRTK), atypical teratoid rhabdoid tumor, involving the central nervous system, and extrarenal extracranial rhabdoid tumor (EERT). Regardless of the anatomic origin, MRT is genetically characterized by mutation of the tumor suppressor gene SMARCB1/INI1 on chromosome 22q [2].
Given the rarity of MRT, standardized treatment protocols have not yet been popularized worldwide. Generally, treatment protocols are based on a multimodal approach, combining surgery, chemotherapy, and radiotherapy. Despite many attempts to improve these various regimens, MRT is still described as lethal; little evidence of improved survival has been reported. According to the National Wilms’ Tumor Study (NWTS) from a series of 142 MRTK cases collected between 1969 and 2002, the overall survival (OS) at 4 years was 23.2% [3]. Of the 106 children diagnosed with extracranial MRT, in the U.K. from 1993 to 2010, the 1‐year survival was only 31% [4]. Furthermore, regarding the 100 patients suffering from extracranial MRT, from 12 countries between 2005 and 2014, from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) Non‐Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study, the 3‐year OS was 38.4% [5].
For further insight into MRT, we reviewed all patients who had extracranial MRT diagnosed and treated in our institution for the past 10 years. In this report, we describe their clinical characteristics, treatment schedules, and outcomes. Moreover, we further compare the demographics and treatment variables to determine prognostic factors in our cohort.
Subjects, Materials, and Methods
Patients
A retrospective medical record review was conducted on 53 patients treated for extracranial MRT in Beijing Children's Hospital (BCH) between January 2007 and October 2017. All the patients were pathologically confirmed as MRT by the institutional pathologist based on morphological and immunohistochemical (IHC) evaluations.
The basic information of patients was collected by referring to their medical record: age, sex, clinical symptoms, tumor marker (lactate dehydrogenase [LDH]), imaging examination, primary tumor sites and metastatic sites, tumor rupture, treatment schedules, IHC staining of INI1, and prognosis characteristics. All patients were staged as localized, regional, distant according to the Surveillance, Epidemiology, and End Results (SEER) staging system [6]. All methods were carried out in accordance with relevant guidelines and regulations. The study has been approved by the Medical Ethics Committee of BCH (2018‐k‐28). A waiver of consent was granted to conduct analyses on the study.
Imaging Analysis
The radiology reports of ultrasound, computed tomography (CT), magnetic resonance imaging (MRI), and/or positron emission tomography‐CT were collected and analyzed retrospectively. According to the Response Evaluation Criteria in Solid Tumors (RECIST) guidance, published in 2000 and revised in 2009, tumor evaluations relied on the measurement of nonnodal target lesions based on the longest single diameter [7]. Maximum diameters of primary tumors in CT and/or MRI were analyzed for all patients.
Treatment
Patients with MRT were mainly treated with multimodal therapy based on surgery, chemotherapy, radiotherapy, and others. Surgical procedures included biopsy, total resection, subtotal resection, and debulking surgery (palliative surgery for relieving symptoms). Because of the lack of standardized treatment protocols for MRT, there were various combinations of chemotherapeutic drugs in this study. However, our institution referred to the UH‐1 protocol from the Children's Oncology Group (COG) (ClinicalTrails.gov NCT00335556) and performed chemotherapy with the use of vincristine, doxorubicin, cyclophosphamide, etoposide, and carboplatin on part of patients with extracranial MRT.
Statistical Analysis
Statistical analysis was performed by SAS 9.4 (SAS Institute, Cary, NC). Continuous variables were presented by mean with standard deviation, or median and interquartile range if normality hypothesis test rejected the null hypothesis of normal distribution. Receiver operating characteristic (ROC) curve analysis was performed to determine the most appropriate cutoff value for age. Student's t test or the Mann‐Whitney U test was used for comparing variables between different subgroups. Categorical variables were reported as counts and percentages and were studied using a chi‐square test. For the time‐to‐event analysis, we estimated event‐free survival (EFS) and OS with Kaplan‐Meier curves. P < .05 was considered as statistically significant.
Results
Patients and Clinical Characteristics
A total of 53 patients with extracranial MRT were studied, including 32 with MRTK (60.38%), and 21 with EERT (39.62%). The primary tumor sites are shown in Table 1. Twenty‐nine patients were male, and 24 were female, giving a male‐to‐female ratio of 1.21:1. The median age at presentation was 16 months (range, from birth to 105 months). Specimens of the 53 patients were studied for INI1 staining; 52 patients (98.11%) had loss of INI1 staining, and one patient (1.89%) had retained INI1 staining.
Table 1. Primary tumor sites of patients with extracranial MRT.
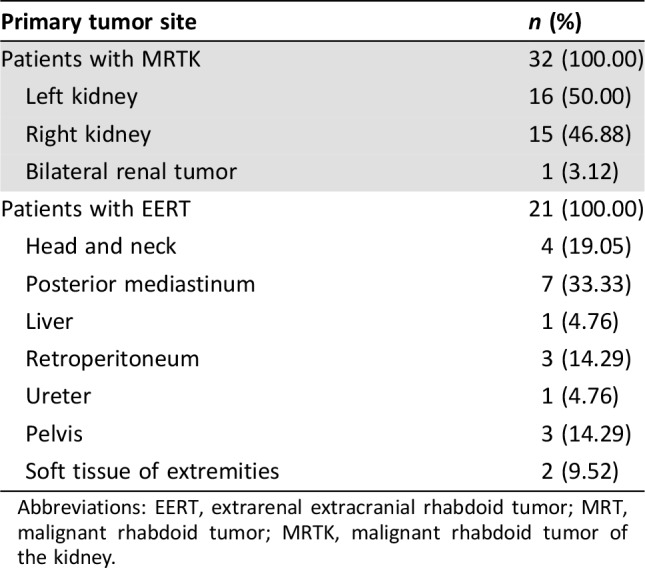
Abbreviations: EERT, extrarenal extracranial rhabdoid tumor; MRT, malignant rhabdoid tumor; MRTK, malignant rhabdoid tumor of the kidney.
Fourteen patients (14/32, 43.75%) with MRTK and five patients (5/21, 23.81%) with EERT had distant stage, according to the SEER staging system at diagnosis. The most common site of metastasis was the lung (10 patients). Other common sites included the liver, bones, lymph nodes, brain, and soft tissue (supplemental online Table 1).
The main characteristics of patients with MRTK were compared with those of patients with EERT (Table 2). Patients with MRTK were younger than those with EERT (median age of 11 months vs. 46 months, p = .0002), and the tumor marker of LDH (median level of 879 U/L vs. 311 U/L, p = .0056) in MRTK was higher than in EERT.
Table 2. Comparison of clinical characteristics between MRTK and EERT.

Results represent z value of Mann‐Whitney test and χ2 value of chi‐square test, respectively.
Normal range of serum LDH was 110–295 U/L.
Abbreviations: EERT, extrarenal extracranial rhabdoid tumor; IQR, interquartile range; LDH, lactate dehydrogenase; MRTK, malignant rhabdoid tumor of the kidney; SEER, Surveillance, Epidemiology, and End Results.
Treatment Schedules
There was no standardized treatment option for extracranial MRT, but multimodal therapy was recommended. Treatment schedules are listed in Table 3. Thirty‐five patients received standard chemotherapy (13 cases of UH‐1 protocol and 22 cases of other protocols). Patients received regimens composed of vincristine, dactinomycin, doxorubicin, cyclophosphamide, carboplatin, ifosfamide, cisplatin, topotecan, pirarubicin, and etoposide. Eighteen patients did not receive standard chemotherapy for the following reasons: early progressive disease (11 cases) between 28 days to 3 months, parents’ choice of treatment withdrawal (6 cases), and death from severe postoperative complications (1 case).
Table 3. Treatment schedules of patients with extracranial MRT.
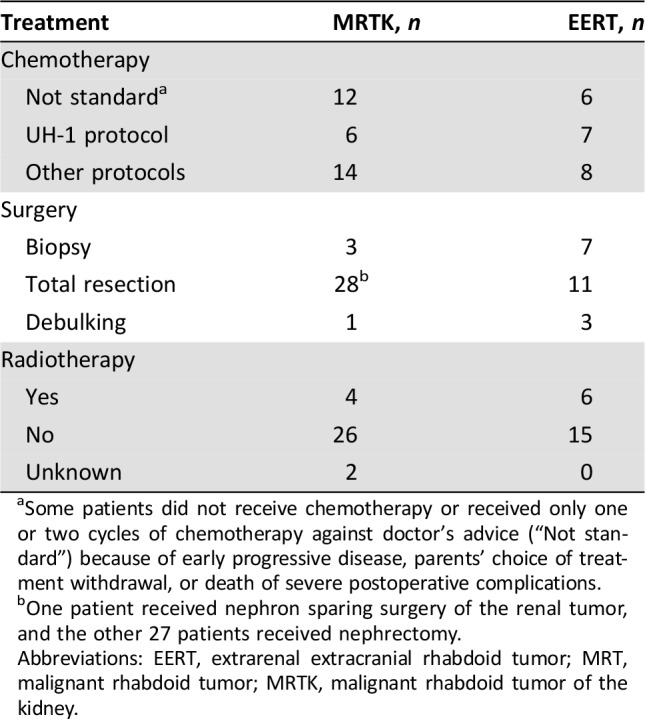
Some patients did not receive chemotherapy or received only one or two cycles of chemotherapy against doctor's advice (“Not standard”) because of early progressive disease, parents’ choice of treatment withdrawal, or death of severe postoperative complications.
One patient received nephron sparing surgery of the renal tumor, and the other 27 patients received nephrectomy.
Abbreviations: EERT, extrarenal extracranial rhabdoid tumor; MRT, malignant rhabdoid tumor; MRTK, malignant rhabdoid tumor of the kidney.
Of the 32 cases of MRTK, 28 patients received total resection, which included one case of nephron‐sparing surgery and 27 cases of nephrectomy (20 patients underwent upfront resection, and 8 patients underwent delayed surgery after chemotherapy). Two patients declined therapy after the biopsy or debulking surgery, and two primary tumors were unresectable even after chemotherapy. For the 21 cases of EERT, 11 patients underwent total resection, including 4 cases of upfront resection and 7 cases of delayed surgery. Five patients declined therapy, whereas another four patients did not receive surgery because of tumor progression after chemotherapy, and the other one patient with unresectable head and neck tumor only received biopsy. Ten of the 53 patients received radiation (four patients with MRTK and six with EERT), and the dose and extent of radiation varied upon the primary site and patients’ age.
Outcome
The EFS rates of 1 year and 3 years for the entire cohort were 21.80% and 14.53%, respectively (Fig. 1A). The OS rates of 3 years and 5 years were 23.71% and 18.44%, respectively (Fig. 1B). Among the 53 patients in this study, 40 (75.47%) patients died, 10 (18.87%) patients survived, and 3 patients (5.66%) were lost to follow‐up. Among the 40 dead patients, 38 patients died of disease progression or tumor recurrence, and 2 patients died of severe postoperative complications (one case of severe pneumonia and respiratory failure and one case of hemorrhage after biopsy). Thirty‐eight patients died within 10 months after the diagnosis (10 months’ OS was only 23.71%), and the other two patients died after 39 months and 41 months, respectively (supplemental online Table 2 shows the characteristics of these two patients). Nine patients were long‐term survivors (range, 32 to 107 months), and one patient has survived up to now for 7 months without tumor progression or recurrence. Table 4 summarizes the details of the clinical characteristics, treatment schedules, and outcomes of the ten long‐term survivors.
Figure 1.

Kaplan‐Meier curves for event‐free survival of the cohort (A), overall survival of the cohort (B), patients according to different primary sites (C), patients according to SEER (Surveillance, Epidemiology, and End Results) stage (D), patients according to age at diagnosis (E), and patients according to tumor rupture (F).
Abbreviations: EERT, extrarenal extracranial rhabdoid tumor; MRTK, malignant rhabdoid tumor of the kidney.
Table 4. Clinical characteristics and treatment schedules of 10 survivors.

Nephron‐sparing surgery of the renal tumor.
Tumor recurrence occurred in primary site in 8 months after diagnosis, and pathological result of surgery confirmed as relapsed MRT (negative of INI1).
Tumor rupture occurred spontaneously, and transcatheter arterial embolization of tumor and nephrectomy were performed.
Abbreviations: ADR, doxorubicin; CBP, carboplatin; CDDP, cisplatin; CTX, cyclophosphamide; EERT, extrarenal extracranial rhabdoid tumor; F, female; IFO, ifosfamide; IHC, immunohistochemical; M, male; MRTK, malignant rhabdoid tumor of the kidney; SEER, Surveillance, Epidemiology, and End Results; THP: pirarubicin; TOPO, topotecan; VCR, vincristine; VP16, etoposide.
Prognostic Characteristics of Different Primary Tumor Sites.
The influence of primary tumor sites on the outcome was further evaluated. As shown in Figure 1C, the prognosis of patients with EERT was better than that of those with MRTK; however, the difference did not reach statistical significance (p = .2005). For the 32 patients with MRTK, 3‐year OS was 14.13%, one (1/32, 3.13%) patient died of severe postoperative complications (severe pneumonia and respiratory failure), 24 (24/32, 75.00%) patients died of tumor recurrence or progression, 4 (4/32, 12.50%) patients survived without disease, and 3 (3/32, 9.38%) patients were lost to follow‐up. For the 21 patients with EERT, the 3‐year and 5‐year OS were 37.50% and 25.00% respectively, 14 (14/21, 66.67%) patients died of tumor recurrence or progression, 1 (1/21, 4.76%) patient (1 month of age at diagnosis) with soft tissue giant tumor of the left lower extremity (8.0 cm × 6.5 cm × 7.3 cm) died of hemorrhage after biopsy, 5 (5/21, 23.81%) patients survived without disease, and 1 (1/21, 4.76%) patient survived after tumor recurrence (Patient 4 in Table 4).
Tumor Progression and Recurrence.
Tumor progression or recurrence occurred in 40 patients of the entire cohort (supplemental online Table 3 showed the progression or recurrence sites). Most events (94.11%) occurred within 8 months (including 8 months), with a median time of 76 (30, 175) days after diagnosis of extracranial MRT. Thirty‐eight patients died of tumor progression or recurrence with a median period time between progression to death of 31 (18, 61) days, one patient died of severe postoperative complications after resection of relapsed tumor (severe pneumonia and respiratory failure), and only one patient with EERT survived after chemotherapy, surgical resection of relapsed tumor, and radiotherapy (Patient 4 in Table 4).
Tumor Rupture.
In our series, tumor rupture was a typical feature of MRT, and there were 14 (26.42%) cases of tumor rupture in 53 patients in this study, including 11 patients with MRTK and 3 patients with EERT (two cases of retroperitoneal tumor and one case of liver tumor). Twelve cases were detected by ultrasound or CT scan, and two cases reported obsolete tumor rupture in the process of the surgery. Spontaneous rupture of tumors occurred in nine cases; two patients with MRTK experienced rupture after four cycles induction of chemotherapy (dactinomycin and vincristine); one case of retroperitoneal tumor ruptured after core needle biopsy; one case of retroperitoneal tumor ruptured because of trauma; and one case of MRTK ruptured after violent activity. Nine MRTK patients received nephrectomy immediately after the diagnosis of tumor rupture, one patient with MRTK received transcatheter arterial embolization (TAE) of tumor and further nephrectomy, and three patients received induction chemotherapy and delayed resection. One case of retroperitoneal tumor progressed and was unresectable even after chemotherapy. There was only one patient with MRTK who has survived until now (32 months) event‐free after multimodal therapies, including TAE of the tumor, nephrectomy, chemotherapy, and local radiotherapy (Patient 6 in Table 4). Among the other 13 patients, 12 patients died of tumor recurrence or progression, and one case was lost to follow‐up.
Prognostic Factors.
After survival analysis, we found that survival was not impacted by the primary site (p = .2005, Fig. 1C) or if the tumor ruptured (p = .0890, Fig. 1F). The most appropriate cutoff value for age was 12 months, determined by ROC curve analysis. The stage of tumor (p = .0261, Fig. 1D) and the age (p = .0069, Fig. 1E) were associated with significant differences in the survival. The effect of treatment was the most difficult to analyze as patients were not treated uniformly. Statistically significant differences in survival were noted among patients treated with total resection (p = .0380, Fig. 2A), radiotherapy (p = .0017, Fig. 2C), and anthracyclines (p = .0107, Fig. 2D). Otherwise, patients who received UH‐1 or other protocols of chemotherapy had better prognosis than those without standard chemotherapy (p = .0120, Fig. 2B).
Figure 2.

Kaplan‐Meier curves for overall survival of patients according to surgical procedures (A), patients according to different chemotherapy protocols (B), patients according to radiotherapy (C), and patients according to treatment with or without anthracyclines (D). Note: “Not standard” in Figure 2B: some patients did not receive chemotherapy or received only one or two cycles of chemotherapy, against doctor's advice, because of early progressive disease, parents’ choice of treatment withdrawal, or death from severe postoperative complications.
Discussion
This is the largest single‐institutional report that investigates the clinical characteristics and outcomes of extracranial MRT in China. The overall survival rates of 3 years and 5 years for the entire cohort were 23.71% and 18.44%, which was equivalent to previous reports [3]. Most of the patients died of rapid tumor progression or recurrence within 8 months (including 8 months) after diagnosis of extracranial MRT. Our study showed that gross hematuria and tumor rupture were typical characteristics of MRTK. After survival analysis, it was found that the advanced stage of the tumor and an age ≤12 months at diagnosis were significantly associated with poorer survival, which was consistent with former reports [3], [6]. Otherwise, statistically significant differences in survival were noted among patients treated with standard chemotherapy, total resection, and radiotherapy.
Diagnosis of extracranial MRT as a distinct entity is a challenge, which shows histological heterogeneity with a diverse immunophenotypic profile. Thus, with the discovery that the loss of INI1 gene contributes to the oncogenesis of MRT, INI1 antibody IHC analysis became an important tool in its diagnosis [8]. In the current study, specimens of 53 patients were studied for INI1 staining; 52 patients (98.11%) had loss of INI1 staining, and one patient (1.89%) had retained INI1 staining despite pathologic confirmation as MRT based on morphological and other IHC studies (supplemental online Fig. 1). Previous studies have reported that up to 20% MRTs have no alterations in the INI1 gene at the DNA or RNA level, and that loss or mutation of the SMARCA4 (BRG1) gene is a much less frequent molecular alteration characteristic of MRT [8]. And the patient who had retained INI1 staining in our cohort showed loss of BRG1 staining (supplemental online Fig. 2). However, the impact that the loss of INI1, BRG1, and other genes has on the clinical characteristics and prognosis of MRT requires further evaluation.
The clinical manifestations of MRT varied depending on tumor location. The most common presentation of MRTK is hematuria [9]; Amar et al. reported gross hematuria was present in 59% of MRTK [10]. In our cohort, gross hematuria happened in 56.25% (18/32) of MRTK, compared with 18.2% previously reported in Wilms tumor [10]. MRTK is more medially located within the kidney, whereas Wilms tumor is found more peripherally within the kidney [10]. Thus, as a presenting symptom, hematuria is associated with tumor invasion of the renal pelvis. Based on this experience, urine routine tests are recommended as part of the initial evaluation for all children with tumors of the kidney at the first visit to the hospital.
Notably, tumor rupture, which is an uncommon finding in childhood tumors, was observed in 14 of 53 cases (26.4%) in our study (11 cases of MRTK and 3 cases of EERT). Tumor rupture had not been yet reported in patients with MRT except for some sporadic case reports [11], [12], [13]. Otherwise, according to our center analysis, from 2008 to 2017, about 45 out of 565 cases (45/565, 7.96%) of Wilms tumor occurred tumor rupture. Agrons et al. [14] found a peripheral crescent of fluid attenuation representing subcapsular renal hemorrhage or peripheral tumor necrosis in more than two thirds of MRTK. Conversely, only 12% of patients with other pediatric renal tumors had these fluid‐filled crescents. It supported that MRT had potential risk of rupture (especially MRTK) and a high degree of malignancy. For the patients with tumor rupture, open surgery and TAE should be the effective choices. Among them, some patients have unresectable tumors. For them, a conservative treatment is needed to stabilize vital signs, immediately followed with chemotherapy to control tumor growth and create conditions for delayed surgery. In this study, 12 patients with ruptured tumors died of tumor recurrence or progression, one case was lost to follow‐up, and there was only one patient with MRTK who has survived until now (32 months) event‐free after undertaking multimodal therapies including TAE, nephrectomy, chemotherapy, and local radiotherapy (Patient 6 in Table 4). Although the final diagnosis of MRTK still depends on pathological features, a distinct clinical presentation, hematuria, tumor rupture, a young age, and a high tumor stage at presentation suggest the diagnosis of MRTK [10].
Because of the scarcity of the disease and the diversity of primary tumor sites, there exists no uniform treatment guideline. MRT is treated with multimodality therapies, including surgery, chemotherapy, and radiotherapy. In this study, eight patients with MRTK were not pathologically confirmed as having MRT at initial diagnosis. They were clinically suspected as having Wilms tumors based on radiological imaging and received induction chemotherapy of dactinomycin and vincristine, and none of them showed a response to preoperative treatment. According to postoperative pathological results, patients in our series received multiagent chemotherapy. The agents mainly included vincristine, doxorubicin, carboplatin, etoposide, cyclophosphamide, pirarubicin, cisplatin, and ifosfamide. In general, anthracycline‐based therapy seemed to be favorable, which was consistent with our result (Fig. 2D) [15]. Thirteen patients were treated with the UH‐1 protocol (recommended by COG), which involved alternating courses of cyclophosphamide, carboplatin, and etoposide with vincristine, doxorubicin, and cyclophosphamide. Using this protocol, the EpSSG study showed that the 3‐year OS was 38.4% of children with extracranial MRT. It suggested that this protocol of intensive therapy could be delivered to patients with extracranial MRT, with a possible improvement in outcome [5]. However, our data did not appear to demonstrate this survival advantage (Fig. 2B). In recent years, high‐dose chemotherapy (HDCT) followed by autologous stem cell transplantation (aSCT) in patients with MRT has been used as a salvage strategy as well as intensive initial therapy [16]. However, no HDCT or aSCT was involved in our cohort, and the treatment effect should be assessed in the future.
Previous studies suggested that radical surgery seemed to benefit the prognosis in MRTK [1], but the prognosis of patients with delayed surgery after induction chemotherapy was worse than that of upfront operation [15]. In our series, total resection appears to be a favorable factor for survival (Fig. 2A). Moreover, nine patients with long‐term survival had total resection, with the exception of one (unresectable head and neck tumor), which further proved that surgical resection of the tumor was an important part of MRT treatment.
Most of the patients (41/53, 77.36%) in this study did not receive radiotherapy for various reasons (treatment withdrawal, death of postoperative complications, rapid tumor progression, young age, and distant stage disease). Only ten patients (10/53, 18.87%) received radiotherapy, and the role of radiotherapy as an important factor affecting outcome was shown in our series (Fig. 2C). Regarding the SEER database series, radiotherapy was often given to those with a higher clinical stage and to groups with older patients. Analysis showed that use of radiotherapy was significantly associated with improved survival [6]. In another multivariate analysis of 349 non‐Wilms renal tumors (including 40 with MRTK), radiotherapy improved outcome [17]. However, one might argue that radiation contributed to the improved survival of MRT in older patients who were irradiated to higher doses than younger patients [3]. Therefore, the role of radiotherapy still needs to be analyzed by randomized controlled trials in the future.
In 2010, Sultan et al. confirmed that age, stage, and radiotherapy, but not primary tumor site, affected the outcome of MRT patients from SEER data. Their univariate and multivariate analyses showed that age at diagnosis (> 2 years), localized stage, and use of radiotherapy were significantly associated with improved survival [6]. In 2005, NWTS reported that the age at diagnosis is a prognostic factor of MRTK [3]. Infant prognosis is very poor, whereas older children have a more favorable outcome. Former reports showed that approximately two thirds of patients with MRT presented with advanced‐stage disease [15]. MRTK and EERT tend to metastasize to the lung and brain. The presence of brain metastasis was always in conjunction with widespread disease involving other sites and was presumed to represent end‐stage disease [3]. Our results coincided with the above studies. Even though the analysis of the prognosis of different tumor sites in our cohort did not reach statistical significance (p = .2005), EERT might have a better prognostic tendency than MRTK.
Whether all tumors with SMARCB1 mutations belong to the same category remains controversial. MRTK and EERT show heterogeneity not only in terms of clinical features, age distribution, and treatment response but also in epigenetic changes and gene expression levels of mutation [18]. The low degree of differentiation in this category and malignant tumors of unknown origin cells require more in‐depth genomic research to reveal the answer. In terms of treatment improvement, clinical data of large samples obtained through cooperation of multiple centers, retrospective analysis of treatment and prognostic factors of the disease, and prospective randomized controlled clinical trials are best used to determine the treatment protocol. At the same time, targeted therapy based on the pathogenesis may be a promising tool for MRT and is expected to become a breakthrough point to improve the prognosis of the disease.
Conclusion
Extracranial MRT is still a highly aggressive tumor in children. In our study, gross hematuria and tumor rupture were typical characteristics of MRTK. Otherwise, the advanced stage of tumor and an age ≤12 months at diagnosis were significantly associated with poor survival. Multimodal treatment approach should be employed for this disease, which include chemotherapy, surgery, and radiotherapy.
See http://www.TheOncologist.com for supplemental material available online.
Contributed equally.
Author Contributions
Conception/design: Huanmin Wang
Provision of study material or patients: Xiaoli Ma, Hong Qin, Weiping Zhang
Collection and/or assembly of data: Haiyan Cheng, Shen Yang, Libing Fu, Qi Zeng, Mingjie Wen
Data analysis and interpretation: Haiyan Cheng, Shen Yang, Siyu Cai, Xiaoxia Peng
Manuscript writing: Haiyan Cheng, Shen Yang
Final approval of manuscript: Haiyan Cheng, Shen Yang, Siyu Cai, Xiaoli Ma, Hong Qin, Weiping Zhang, Libing Fu, Qi Zeng, Mingjie Wen, Xiaoxia Peng, Huanmin Wang
Disclosures
The authors indicated no financial relationships.
References
- 1.Madigan CE, Armenian SH, Malogolowkin MH et al. Extracranial malignant rhabdoid tumors in childhood: The Childrens Hospital Los Angeles experience. Cancer 2007;110:2061–2066. [DOI] [PubMed] [Google Scholar]
- 2.Versteege I, Sevenet N, Lange J et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203–206. [DOI] [PubMed] [Google Scholar]
- 3.Tomlinson GE, Breslow NE, Dome J et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: Age at diagnosis as a prognostic factor. J Clin Oncol 2005;23:7641–7645. [DOI] [PubMed] [Google Scholar]
- 4.Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: What we have learned so far and future directions. Lancet Oncol 2013;14:e329–e336. [DOI] [PubMed] [Google Scholar]
- 5.Brennan B, De Salvo GL, Orbach D et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non‐Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study‐EpSSG NRSTS 2005. Eur J Cancer 2016;60:69–82. [DOI] [PubMed] [Google Scholar]
- 6.Sultan I, Qaddoumi I, Rodriguez‐Galindo C et al. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 2010;54:35–40. [DOI] [PubMed] [Google Scholar]
- 7.Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 8.Hoot AC, Russo P, Judkins AR et al. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra‐renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 2004;28:1485–1491. [DOI] [PubMed] [Google Scholar]
- 9.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: Results from the First National Wilms’ Tumor Study. Cancer 1978;41:1937–1948. [DOI] [PubMed] [Google Scholar]
- 10.Amar AM, Tomlinson G, Green DM et al. Clinical presentation of rhabdoid tumors of the kidney. J Pediatr Hematol Oncol 2001;23:105–108. [DOI] [PubMed] [Google Scholar]
- 11.Chang CJ, Yeh ML, Chen CC. Rhabdoid tumor of the kidney with spontaneous rupture: Case report and review of literature. Pediatr Surg Int 2008;24:451–453. [DOI] [PubMed] [Google Scholar]
- 12.Kachanov D, Teleshova M, Kim E et al. Malignant rhabdoid tumor of the liver presented with initial tumor rupture. Cancer Genet 2014;207:412–414. [DOI] [PubMed] [Google Scholar]
- 13.Yuri T, Danbara N, Shikata N et al. Malignant rhabdoid tumor of the liver: Case report and literature review. Pathol Int 2004;54:623–629. [DOI] [PubMed] [Google Scholar]
- 14.Agrons GA, Kingsman KD, Wagner BJ et al. Rhabdoid tumor of the kidney in children: A comparative study of 21 cases. AJR Am J Roentgenol 1997;168:447–451. [DOI] [PubMed] [Google Scholar]
- 15.van den Heuvel‐Eibrink MM, van Tinteren H, Rehorst H et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: A report of the SIOP renal tumour study group. Pediatr Blood Cancer 2011;56:733–737. [DOI] [PubMed] [Google Scholar]
- 16.Koga Y, Matsuzaki A, Suminoe A et al. Long‐term survival after autologous peripheral blood stem cell transplantation in two patients with malignant rhabdoid tumor of the kidney. Pediatr Blood Cancer 2009;52:888–890. [DOI] [PubMed] [Google Scholar]
- 17.Zhuge Y, Cheung MC, Yang R et al. Pediatric non‐Wilms renal tumors: Subtypes, survival, and prognostic indicators. J Surg Res 2010;163:257–263. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Dagar V, Algar E et al. Rhabdoid tumour: A malignancy of early childhood with variable primary site, histology and clinical behaviour. Pathology 2008;40:664–670. [DOI] [PubMed] [Google Scholar]