

This article reports the results of a study of the natural history of aggressive angiomyxoma, focusing on the disease's sensitivity to hormone therapy and on treatment outcomes.
Keywords: Sarcoma, Aggressive angiomyxoma, Hormone antagonists, Surgery
Abstract
Background.
Aggressive angiomyxoma (AA) is a rare, locally aggressive tumor usually arising from pelvis or perineum, with a high local‐recurrence rate after complete surgery. Anecdotal responses to hormone therapy have been reported. In the present study we aimed at studying surgical treatment outcomes and sensitivity to hormone therapy of AA.
Materials and Methods.
We conducted a multicenter, international retrospective effort including patients with AA treated at three European referral centers (Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy and the Italian Rare Cancer Network; Centre Léon Bérard, Lyon, France; and Hospital Universitario Virgen del Rocio, Seville, Spain).
Results.
A total of 36 patients were included. Median follow‐up was 51.3 months. Thirty‐three patients (92%) underwent complete (R0 + R1) surgery, with a local relapse rate of 50% and a median relapse‐free survival of 39 months (95% confidence interval [CI], 27–68.1). Thirteen patients received a first‐line systemic treatment with hormone therapy for locally advanced disease, with an overall response rate of 62% and a median progression‐free survival of 24.6 months (95% CI, 11.0–39.7). In two patients, adding an aromatase inhibitor (AI) on progression to first‐line GnRH agonist (GnRHa) resulted in a new tumor response.
Conclusion.
Our findings confirm that in AA, surgical local control may be challenging, with a significant rate of local relapse despite complete surgery. Hormone therapy is an active treatment option, with a potential of disease control and of being combined with surgery. The addition of an AI to first‐line GnRHa could be an effective second‐line systemic therapy in premenopausal female patients with AA.
Implications for Practice.
In this retrospective effort including 36 patients with aggressive angiomyxoma, local relapse rate after complete surgery was 50%, with a median relapse‐free survival of 39 months, confirming that local control is challenging. Overall response rate to first‐line hormone therapy was 62%, with a median progression‐free survival of 24.6 months. Thus, hormone therapy has a potential of disease control and of being combined with surgery.
Introduction
Aggressive angiomyxoma (AA) is a rare, locally aggressive tumor first described in 1983 [1], with several hundred cases reported in literature. It usually arises in perineum and pelvic region and presents as a deep‐seated lesion with indolent growth and high local‐recurrence rate. Distant metastases are exceptional, with only three cases observed [2], [3], [4]. AA predominantly affects women, especially in their reproductive age [5]. However, cases in men were reported [6]. Histologically, AAs exhibit a low to moderate cellularity and are composed by a population of uniform, spindled cells featuring a low mitotic count [7]. Neoplastic cells are immunoreactive for desmin, smooth muscle and muscle‐specific actin, and vimentin and usually also for estrogen receptors (ERs) and progesterone receptors (PgRs) [8]. Differential diagnosis includes angiomyofibroblastoma, myxoid smooth muscle tumors, lipomatous tumors, peripheral nerve sheath tumors, myxofibrosarcoma, and pelvic fibromatosis. HMGA2 chromosomal translocations have been reported [9]. Surgery is held as the treatment mainstay when feasible, but local recurrences are frequent and affect about half the patients, even after complete resections [10]. Responses to hormonal therapy have been anecdotally reported [11], [12], without any prospective study available.
The aim of this international, retrospective effort was to study the natural history of AA, its sensitivity to hormone therapy, and the treatment outcomes.
Materials and Methods
Patients with histologically confirmed AA treated from 1999 to 2016 at three European reference centers (Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy and the Italian Rare Cancer Network; Centre Léon Bérard, Lyon, France; Hospital Universitario Virgen del Rocio, Seville, Spain) were retrospectively reviewed. Pathological diagnosis was reviewed at each center by a referral pathologist. Authorization from reviewing ethics committees was obtained according to local rules. Data regarding demographics, surgical and systemic therapy, and survival were collected. Response to hormone therapy was assessed according to Response Evaluation Criteria in Solid Tumors Guidelines (version 1.1) [13]. Fisher's exact test or χ2 test was used when appropriate to assess the association between relapse after complete surgery and investigate characteristics. Odds ratios (ORs) and 95% confidence intervals (CIs) were calculated. Statistical significance was set at p = .05. Relapse‐free survival (RFS) and progression‐free survival (PFS) were estimated by the Kaplan‐Meier method. Patients were censored at the last contact. Statistical analysis was performed with MedCalc Version_12.7.0.0 (Ostend, Belgium) and GraphPad Prism Version 5.02 (La Jolla, CA).
Results
A total of 36 patients with histologically confirmed AA were included. Patients and disease characteristics are summarized in Table 1. Median age at diagnosis was 43 years (range, 19–73). Female was the most prevalent gender in our series (78%). Median tumor diameter was 9.7 cm (range, 1–27), and positive immunostaining for ER and PgR was observed in 89% and 81% of patients, respectively. Tumor location was pelvis in 19 patients (53%), perineum in 16 patients (44%), and extrapelvic (fourth finger of the right hand) in 1 patient (35%).
Table 1. Patients and disease characteristics.
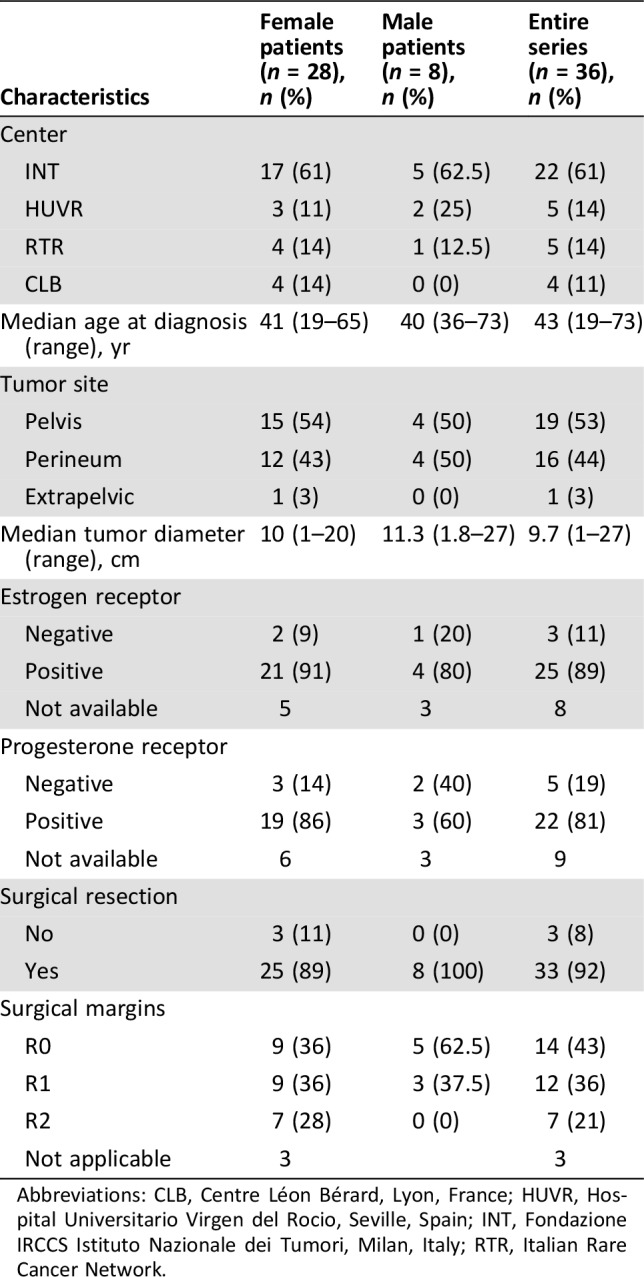
Abbreviations: CLB, Centre Léon Bérard, Lyon, France; HUVR, Hospital Universitario Virgen del Rocio, Seville, Spain; INT, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; RTR, Italian Rare Cancer Network.
Thirty‐three patients (92%) underwent surgical resection; macroscopically complete surgery (R0 or R1) was performed in 26 patients, whereas incomplete surgery (R2) was performed in 7 patients. Notably, 32 patients received surgery as upfront treatment, whereas 1 out of 4 patients who started an upfront hormone therapy with a selective estrogen receptor modulator (SERM) for locally advanced disease underwent subsequent surgical resection after experiencing a tumor shrinkage. None of the patients with complete resection (R0 or R1) received immediate postoperative (adjuvant) hormonal therapy.
Median follow‐up of the entire series was 51.3 months (range, 2.0–165.5). Following complete surgery (R0 or R1), 13 out of 26 patients (50%) experienced a local relapse, with a median RFS of 39.0 months (95% CI, 27.0–68.1; Fig. 1, panel A). A trend toward a higher chance of recurrence for female compared with male patients (85% vs. 15%, respectively; OR, 4.71; 95% CI, 0.73–30.28; p = .10) and a lower chance of recurrence for R0 compared with to R1 surgery (38% vs. 62%, respectively; OR, 0.28; 95% CI, 0.05–1.41; p = .12) were observed (Table 2). No difference in recurrence‐free survival was observed between patients treated with R0 and R1 surgery (38,99 vs. 40,93 respectively, p = .31; supplemental online Fig. 1).
Figure 1.

Kaplan‐Meier curves for relapse‐free survival after complete surgery and progression‐free survival to any‐type, first‐line hormone therapy. (A): Relapse‐free survival. (B): Progression‐free survival.
Table 2. Patients and disease characteristics after complete (R0 or R1) surgery according to the presence of relapse.

Fisher's exact test or χ2 test as appropriate.
Abbreviations: CI, confidence interval; OR, odds ratio.
Thirteen patients (including one male patient) received first‐line systemic treatment with hormone therapy (Table 3): four patients upfront for locally advanced disease, three patients after R2 surgery, and six patients for a local relapse. Two complete responses (CRs), six partial responses (PRs), and five stable diseases (SDs) were achieved, for an overall response rate (ORR) of 62%. For patients experiencing PR or CR as best response, median time to response was 3 months. With a median follow‐up of 40.4 months (range, 3.7–94.1), 7 out of 13 patients (53.8%) experienced a progression of disease with a median PFS for any kind of first‐line hormone therapy of 24.6 months (95% CI, 11.0–39.7; Fig. 1, panel B).
Table 3. Clinico‐pathological characteristics and clinical outcomes of patients treated with hormone therapy.

Patient 20 discontinued hormone therapy 3 months after achieving complete response and restarted GnRH agonist at the time of disease progression (which occurred 2 months later), obtaining a new CR (still on therapy at her last follow‐up and free from progression after 43 months).
Patients free from progression at time of data cut‐off or at last contact.
Patient 32 withheld therapy after 1 month because of adverse events.
Following a partial response, patient 17 underwent surgical resection (after 57 months on therapy) and continued SERM as adjuvant treatment for a further 2 years.
Abbreviations: AI, aromatase inhibitor; CR, complete response; ER, estrogen receptor; GnRH, gonadotropin releasing hormone; NA, not available; PFS, progression‐free survival; PgR, progesterone receptor; PR, partial response; SD, stable disease; SERM, selective estrogen receptor modulator.
One of the two patients who achieved a CR discontinued hormone therapy (triptorelin) after 8 months on treatment and restarted the same GnRH agonist (GnRHa) at the time of disease progression (which occurred 2 months later), obtaining a new CR.
Three patients progressed to first‐line GnRHa and received a second‐line systemic therapy; two patients added an aromatase inhibitor (AI) to GnRHa and reached a PR and a CR, respectively. One patient discontinued GnRHa at progression and started an AI with SD as best response. All patients were alive at their last follow‐up.
Discussion
Our study reports on 36 patients with AA treated at three European reference institutions and within the Italian Rare Cancer Network for 17 years. In this series, 92% of patients underwent surgery, with a local relapse rate after complete surgery (R0 or R1) of 50% and a median RFS of 39 months. Patients undergoing R1 surgery had a higher chance of recurrence. Thirteen patients received a first‐line systemic treatment with hormone therapy reaching an ORR of 62% and a median PFS around 24 months.
The limitations of our study mainly consist of its retrospective nature and the relatively small number of cases. However, published series in this disease are scant in regard to surgical outcomes (supplemental online Table 1), with a number of patients between 5 and 18. From such reports, it is evident that a remarkable local‐recurrence rate is to be expected in this disease [14], and our series confirms this. With regard to hormone therapy, the disease is known to be sensitive (supplemental online Table 2), but available evidence is basically anecdotal. Our series provides further proofs that hormonal therapy is active in the disease. Thus, hormone therapy may well be an option when surgery is not feasible, or when surgery would be mutilating or challenging. Of note, one patient with a response to SERM became operable and is free from relapse after 74 months (she had continued SERM as an adjuvant for a further 2 years).
The other settings in which hormone therapy may be an option are patients with a rapidly progressing disease (acting as neoadjuvant therapy before surgery) and those with a relapse. Although the clinician may be confident that the probability of response to hormone therapy is high, the main clinical question may have to do with the duration of treatment. The rapid progression experienced by a patient in this series after GnRHa discontinuation would suggest that continuing hormone therapy is needed in these cases. However, in the same patient, GnRHa reintroduction resulted in a new maintained response.
We observed objective responses following first‐line systemic therapy with GnRHa, SERM, or a combination of the two class of agents, whereas the two patients treated with first‐line AI experienced disease stabilization. In the lack of any established criteria, gender, menopausal status, toxicity profile, and patient preferences could be used for treatment selection. Remarkably, in two patients, we saw that adding an AI on progression to first‐line GnRHa resulted in a new tumor response, suggesting this strategy as an effective second‐line systemic therapy in premenopausal female patients with AA. Clearly, to overcome acquired resistance to hormone therapy, a deep understanding of the biology of AA it is essential to explore new targeted strategies.
Conclusion
We confirm that local control in AA is challenging. Hormone therapy is an active treatment option, with a potential of disease control and of combination with surgery. The choices of hormone therapy as well as treatment duration are worth being explored in the future. How to study this exceedingly rare condition is challenging as well. This is a source of additional practical problems for patients; for example, as may happen with ultra‐rare cancers, effective medical therapy in AA (hormones) remains off‐label. An effort to collate data on these patients into prospective clinical registries would be worthwhile, following the collaborative intent which underlined this multi‐institutional, retrospective case series analysis.
See http://www.TheOncologist.com for supplemental material available online.
Contributed equally.
Author Contributions
Conception/design: Angelo Paolo Dei Tos, Alessandro Gronchi, Paolo Giovanni Casali, Roberta Sanfilippo
Provision of study material or patients: Isabelle Ray‐Coquard, Angelo Paolo Dei Tos, Javier Martin‐Broto, Domenica Lorusso, Francesco Raspagliesi, Maria Abbondanza Pantaleo, Bruno Vincenzi, Alessandro Gronchi, Paolo Giovanni Casali, Roberta Sanfilippo
Collection and/or assembly of data: Giovanni Fucà, Nadia Hindi, Vittoria Colia, Mehdi Brahmi, Maria Abbondanza Pantaleo, Bruno Vincenzi, Elena Fumagalli
Data analysis and interpretation: Giovanni Fucà, Nadia Hindi, Isabelle Ray‐Coquard, Angelo Paolo Dei Tos, Javier Martin‐Broto, Paola Collini, Domenica Lorusso, Francesco Raspagliesi, Alessandro Gronchi, Paolo Giovanni Casali, Roberta Sanfilippo
Manuscript writing: Giovanni Fucà, Nadia Hindi, Elena Fumagalli
Final approval of manuscript: Giovanni Fucà, Nadia Hindi, Isabelle Ray‐Coquard, Vittoria Colia, Angelo Paolo Dei Tos, Javier Martin‐Broto, Mehdi Brahmi, Paola Collini, Domenica Lorusso, Francesco Raspagliesi, Maria Abbondanza Pantaleo, Bruno Vincenzi, Elena Fumagalli, Alessandro Gronchi, Paolo Giovanni Casali, Roberta Sanfilippo
Disclosures
Paolo Giovanni Casali: Deciphera Pharmaceuticals, Eisai, Eli Lilly and Co., Nektar (C/A), Eli Lilly and Co., Pfizer, PharmaMar (H), Advenchen Laboratories, Amgen, AROG, Bayer, Blueprint Medicinces, Daiichi Sankyo, Deciphera, Eli Lilly and Co., Epizyme, Karyopharm Therapeutics, Nektar, PharmaMar (RF); Javier Martin‐Broto: PharmaMar, GlaxoSmithKline, Novartis, Amgen, Bayer, Lilly (C/A), Novartis, Eisai, PharmaMar (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Steeper TA, Rosai J. Aggressive angiomyxoma of the female pelvis and perineum. Report of nine cases of a distinctive type of gynecologic soft‐tissue neoplasm. Am J Surg Pathol 1983;7:463–475. [DOI] [PubMed] [Google Scholar]
- 2.Geng J, Cao B, Wang L. Aggressive angiomyxoma: an unusual presentation. Korean J Radiol 2012;13:90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siassi RM, Papadopoulos T, Matzel KE. Metastasizing aggressive angiomyxoma. N Engl J Med 1999;341:1772. [DOI] [PubMed] [Google Scholar]
- 4.Blandamura S, Cruz J, Faure Vergara L et al. Aggressive angiomyxoma: A second case of metastasis with patient's death. Hum Pathol 2003;34:1072–1074. [DOI] [PubMed] [Google Scholar]
- 5.Haldar K, Martinek IE, Kehoe S. Aggressive angiomyxoma: A case series and literature review. Eur J Surg Oncol 2010;36:335–339. [DOI] [PubMed] [Google Scholar]
- 6.Kidric DM, MacLennan GT. Aggressive angiomyxoma of the male genital region. J Urol 2008;180:1506. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher CDM, Bridge JA, Hogendoorn PCW et al. WHO Classification of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press, 2013. [Google Scholar]
- 8.Fetsch JF, Laskin WB, Lefkowitz M et al. Aggressive angiomyxoma: A clinicopathologic study of 29 female patients. Cancer 1996;78:79–90. [DOI] [PubMed] [Google Scholar]
- 9.Sutton BJ, Laudadio J. Aggressive angiomyxoma. Arch Pathol Lab Med 2012;136:217–221. [DOI] [PubMed] [Google Scholar]
- 10.Chan YM, Hon E, Ngai SW et al. Aggressive angiomyxoma in females: Is radical resection the only option? Acta Obstet Gynecol Scand 2000;79:216–220. [PubMed] [Google Scholar]
- 11.Fine BA, Munoz AK, Litz CE et al. Primary medical management of recurrent aggressive angiomyxoma of the vulva with a gonadotropin‐releasing hormone agonist. Gynecol Oncol 2001;81:120–122. [DOI] [PubMed] [Google Scholar]
- 12.McCluggage WG, Jamieson T, Dobbs SP et al. Aggressive angiomyxoma of the vulva: Dramatic response to gonadotropin‐releasing hormone agonist therapy. Gynecol Oncol 2006;100:623–625. [DOI] [PubMed] [Google Scholar]
- 13.Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 14.Coppola S, Desai A, Tzanis D et al. Conservative en bloc surgery for aggressive angiomyxoma achieves good local control: Analysis of 14 patients from a single institution. Int J Gynecol Cancer 2013;23:540–545. [DOI] [PubMed] [Google Scholar]