

This report focuses on the safety and effectiveness of regorafenib in real‐world outcomes in patients with metastatic colorectal cancer.
Keywords: Regorafenib, Metastatic colorectal cancer, Safety, Observational study, Postmarketing
Abstract
Background.
Regorafenib improved the overall survival (OS) of patients with metastatic colorectal cancer (mCRC) who progress after standard therapies in two phase III trials. The present large‐scale prospective observational study evaluated the safety and effectiveness of regorafenib administered to Japanese patients with mCRC in real‐life setting.
Materials and Methods.
Patients with mCRC were prospectively registered and initially received ≤160 mg oral regorafenib daily, at the investigator's discretion, for weeks 1–3 of each 4‐week cycle. The study's primary aim was to assess safety, particularly unexpected clinically significant adverse drug reactions (ADRs). A Cox's proportional hazards model was used to evaluate the association between OS, hand‐foot skin reaction (HFSR), and baseline characteristics.
Results.
We evaluated 1,227 of 1,301 patients (enrolled from March 2013 to May 2015). ADRs occurred in 89.3% of patients (mostly within the first 4 weeks) and were a major reason for discontinuing treatment. The most frequent ADRs were HFSR, liver injury, and hypertension. The cumulative incidence of HFSR and liver injury was higher in patients who initially received 160 mg than in those who received ≤120 mg. The incidence of hypertension and fatigue was similar between groups. Median OS was 6.9 months (95% confidential interval, 6.4–7.4). OS was associated with early onset of HFSR and good performance status (PS) but not with the initial dose.
Conclusion.
The outcomes of this study were consistent with those of clinical trials. There were no new safety concerns. Regorafenib treatment would not be recommended for patients with higher PS.
Implications for Practice.
Previous clinical trials demonstrated regorafenib improved overall survival in patients with metastatic colorectal cancer who progress after standard chemotherapies. Because the eligibility criteria of the trials were restricted compared with a real‐world setting, the data from the trials may not fully represent the profiles of regorafenib in clinical practice. This large‐scale observational study showed that the safety and effectiveness of regorafenib in clinical practice were generally consistent with previous trials. The majority of patients reported adverse drug reactions within the first 4 weeks, most commonly hand‐foot skin reaction. Regorafenib treatment would not be recommended for patients with higher performance status.
摘要
背景。在 2 个 III 期试验中,瑞戈非尼提高了经标准治疗后出现疾病进展的转移性结直肠癌 (mCRC) 患者的总生存期 (OS)。当前的大规模、前瞻性观察研究评估了在真实世界环境中将瑞戈非尼用于治疗日本 mCRC 患者的安全性和有效性。
材料和方法。mCRC 患者进行预先登记,并且,根据研究者的酌情决定,最初在每个周期(每 4 周为一个周期)的第 1–3 周内每天口服 ≤160 mg 瑞戈非尼。研究的主要目的为评估安全性,特别是意料之外的在临床上显著的药物不良反应 (ADR)。使用 Cox 比例风险模型来评估 OS、手足皮肤反应 (HFSR) 以及基线特征。
结果。我们对 1 301 名患者中的 1 227 名患者(自 2013 年 3 月至 2015 年 5 月入组)进行评估。89.3% 的患者(大多数在前 4 周)出现 ADR,这是患者中断治疗的一个重要原因。最常见的 ADR 为 HFSR、肝损伤和高血压。最初采用 160 mg 用药剂量的患者的 HFSR 和肝损伤的累积发生率要高于用药剂量 ≤120 mg 的患者。各组之间的高血压和疲劳发生率相似。中位 OS 为 6.9 个月(95% 置信区间,6.4–7.4)。OS 与 HFSR 的早发和良好体力状态(PS)相关,但与初始剂量无关。
结论。本研究的结果与临床试验的结果一致。没有新的安全问题。我们不建议 PS 较高的患者接受瑞戈非尼治疗。
实践意义:既往的临床试验已证明瑞戈非尼可以提高经标准化疗后出现疾病进展的转移性结直肠癌患者的总生存期。由于此类试验的资格标准与真实世界环境相比较为有限,这些试验数据可能无法充分反映瑞戈非尼在临床实践中的特征。本次大规模观察研究证明,瑞戈非尼在临床实践中的安全性和有效性与既往试验的结果大体一致。大部分患者在前 4 周内报告了药物不良反应,其中,最常见的是手足皮肤反应。我们不建议体力状态评分较高的患者接受瑞戈非尼治疗。
Introduction
Colorectal cancer (CRC) was the third leading cause of cancer death worldwide in 2015 and the second in Japan in 2016 [1], [2]. Chemotherapy is used to treat unresectable metastatic CRC (mCRC). The standard drugs for mCRC involve fluoropyrimidine/leucovorin plus oxaliplatin or irinotecan. Biological agents such as bevacizumab, ziv‐aflibercept, and ramucirumab are used in combination with cytotoxic agents. Cetuximab and panitumumab (anti‐epidermal growth factor receptor [EGFR] monoclonal antibody [MAb]) are used to treat patients with mCRC with wild‐type RAS [3], [4], [5], [6]. Regorafenib and TAS‐102 are standard drugs for third‐line or subsequent treatment of mCRC.
Regorafenib is an orally administered multikinase inhibitor (MKI) that inhibits the activities of protein kinases associated with angiogenesis, oncogenesis, and the tumor microenvironment [7]. In the international phase III CORRECT trial, 760 patients were randomized to receive regorafenib or placebo at a 2:1 ratio. Treatment with regorafenib significantly improved overall survival (OS) of patients with mCRC compared with placebo (hazard ratio [HR], 0.77; 95% confidence interval [CI], 0.64–0.94; one‐sided p = .0052). Most common regorafenib‐related adverse events (AEs) were fatigue and hand‐foot skin reaction (HFSR) [8]. The CORRECT trial included 100 Japanese patients, 67 of whom were randomly selected to receive regorafenib. The Japanese subpopulation in the trial (CORRECT‐J) achieved comparable efficacy and a manageable safety profile compared with non‐Japanese patients, although certain regorafenib‐related AEs such as HFSR were observed more frequently, and there was one case of fatal liver injury [9]. According to the CORRECT trial, regorafenib is recommended by the international guidelines as one of the standard drugs for treating mCRC [3], [4], [5], [6].
In clinical practice, patient populations are more heterogeneous compared with those treated in clinical trials. Therefore, the data from 67 Japanese patients included in the CORRECT may not fully represent the efficacy and safety profiles of regorafenib in real‐world settings. Consequently, large‐scale, real‐world studies may detect unexpected, clinically significant adverse drug reactions (ADRs). Therefore, post‐marketing surveillance (PMS) is a component of a risk management plan for new drugs required by the Pharmaceutical and Medical Devices Agency for Pharmacovigilance in Japan.
Here we report a large‐scale, prospective, multicenter, observational PMS study to investigate the safety and effectiveness of regorafenib under real‐world conditions.
Those data will be applied to mCRC patients treated with regorafenib in clinical practice, not only in Japan but also worldwide.
Materials and Methods
This study was a prospective observational study involving more than 500 centers in Japan. Patients with mCRC, who progressed after standard chemotherapies and for whom regorafenib administration was planned, were centrally registered. The exclusion criteria strongly recommend not treating patients with severe liver injury (aspartate aminotransferase [AST] or alanine aminotransferase [ALT] >5 times the upper limit of normal [ULN], bilirubin >2.0 times the ULN), uncontrolled hypertension, or Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≥2.
This study (NCT01843400) was conducted in compliance with the Good Post‐Marketing Study Practice (GPSP) and Good Vigilance Practice (GVP) of the Ministry of Health, Labor, and Welfare in Japan. Approval by the ethics committee of each institution was not mandatory because GPSP and GVP do not require such approval for a PMS. The recommended dose of regorafenib was 160 mg per day for 3 weeks and 1 week off as described in the CORRECT trial [8]. The dose of regorafenib could be modified according to the product label [10] and at the investigators’ discretion.
The main study objectives were to assess the safety, including occurrence of unknown and clinically significant ADRs, and the effectiveness of regorafenib in real‐world clinical practice. An ADR was defined as an adverse event for which a causal relationship with regorafenib could not be excluded. Data collected at 2 and 6 months after treatment began included demographic and disease characteristics, prophylaxis for HFSR, concomitant medications, dose of regorafenib, AEs, and laboratory values. The severity of AEs was graded using Common Terminology Criteria for Adverse Events version 4.0 [11]. Patients were observed for 6 months, which was based on the progression‐free survival and OS results of the CORRECT‐J trial [9]. Survival data were collected for all patients 1 year after treatment began. However, the observation period was 30 days after early discontinuation and immediately ended when it was confirmed that the patient was lost to follow‐up.
All statistical analyses were performed using SAS version 9.2 (SAS Institute Inc., Cary, NC). Considering the exploratory nature of the study, all p values were nominal and considered descriptive. Demographic characteristics of patients are summarized descriptively. OS and time‐to‐treatment failure (TTF) were estimated using the Kaplan‐Meier method. OS was defined as the time from the date of first administration of regorafenib to the date of death (regardless of the cause) or censored on the last date of survival. TTF was defined as the time from the date of first administration of regorafenib to the date of treatment discontinuation for any reason.
Cox's proportional hazards model was used to evaluate the association between OS and baseline characteristics. Baseline variables with ≥10% missing data (including “not evaluable”) and with an extreme distribution (i.e., ≥95% in one category) were excluded from the analyses. Considering the correlation and hierarchy with respect to clinical importance among baseline variables, the latter were manually selected in advance. Baseline variables were included in the model as covariates and were finally selected using a stepwise method. Similar analyses were performed for TTF and the time to the first occurrence of HFSR.
A landmark analysis of HFSR occurrence up to the 28th day after treatment began was added to the Cox model as a covariate of the selected baseline variables. Patients who experienced an event of interest (OS or TTF) before day 28 were excluded from the landmark analysis. The time from day 28 to the event of interest was considered the event of interest.
Based on a lowest expected incidence of increased ALT in 0.4% of patients and increased AST in 0.4% as key events leading to either dose reduction or interruption, 1,186 patients were required for the analysis to detect this event in at least two patients, achieving a statistical power = 95%. Allowing for the exclusion of some patients from the safety analysis, the target sample size was 1,250 patients.
Results
Patients’ Characteristics
We enrolled 1,301 patients from March 2013 to May 2015. Twelve patients were excluded because they were not treated with regorafenib (n = 9) or were enrolled outside of the study period (n = 3). We excluded 62 patients whose case report forms could not be collected. Thus, we evaluated 1,227 patients for safety and effectiveness of treatment (supplemental online Fig. 1) by the 26 September 2016 cutoff. At baseline, 91.6% of patients had an ECOG PS = 0–1, and 8.3% had an ECOG PS ≥2 (Table 1). Prophylaxis for HFSR was administered to 83% of patients.
Table 1. Baseline demographics and disease characteristics.
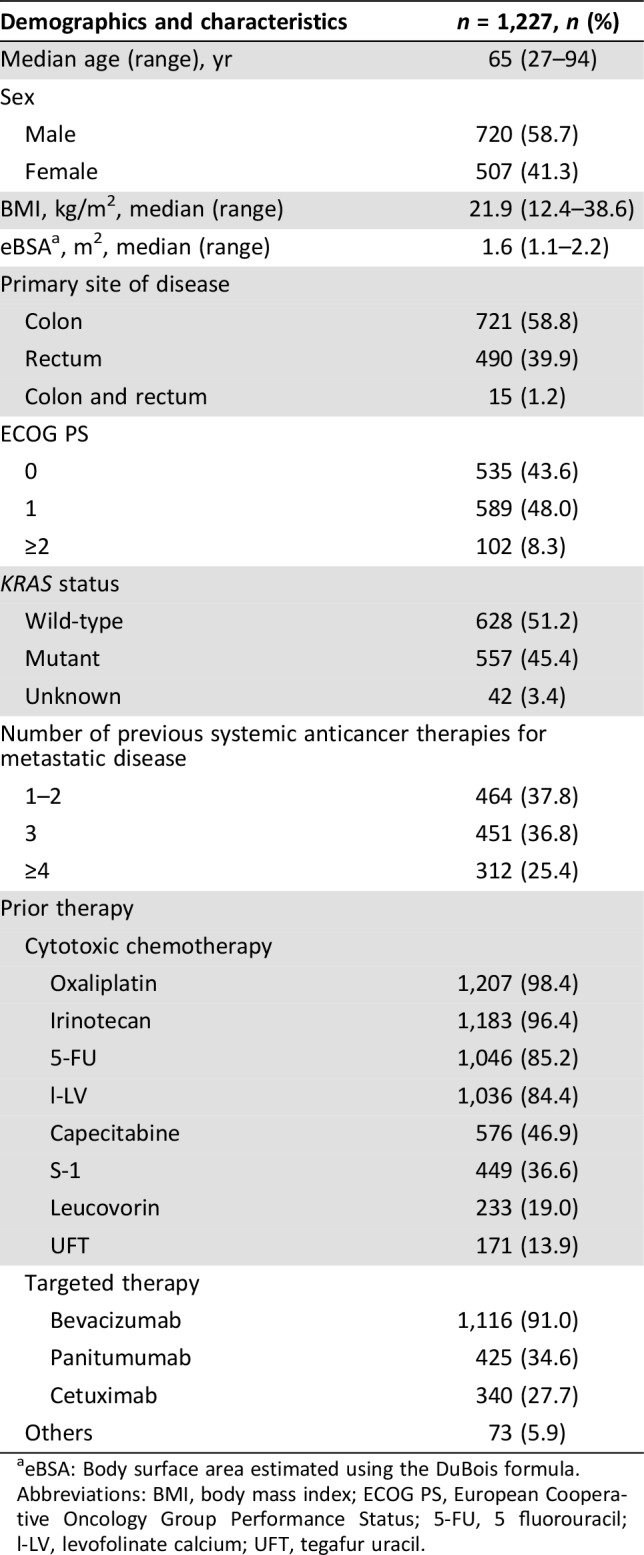
eBSA: Body surface area estimated using the DuBois formula.
Abbreviations: BMI, body mass index; ECOG PS, European Cooperative Oncology Group Performance Status; 5‐FU, 5 fluorouracil; l‐LV, levofolinate calcium; UFT, tegafur uracil.
Regorafenib Treatment
Two thirds of patients initiated regorafenib treatment at the standard daily dose of 160 mg (65.4%); the others started at a daily dose of 120 mg (21.6%) or lower (13.0%). By the fifth week of treatment, the proportion of patients receiving 160 mg decreased to 26.4%. The largest proportion of patients subsequently received 120 mg or ≤80 mg (supplemental online Fig. 2).
The median duration of treatment, including dose interruptions, was 7.6 weeks (range, 0.1–86.3). A dose interruption or reduction was required for treating 49.3% and 42.1% of patients, respectively. At the analysis cutoff date, 90.5% of patients had discontinued treatment, of which 33.3% discontinued treatment because of an ADR. An ADR was the most common reason for discontinuation during the first 4 weeks of treatment, and progressive disease (PD) was the leading reason that patients subsequently discontinued treatment (supplemental online Fig. 3).
Safety
ADRs were experienced by 89.3% of patients. Grade ≥3 ADRs were observed in 51.8% of patients (Table 2). The most common ADR was HFSR (58.2%). Twelve patients (1.0%) experienced grade 5 ADRs, including seven with a liver injury, and four of these patients reported other simultaneous ADRs (primary disease, four patients; gastrointestinal perforation, one patient).
Table 2. Regorafenib‐related adverse events reported in ≥5% of patients (n = 1,227).

HFSR, hand‐foot skin reaction; equivalent to hand‐and‐foot syndrome in MedDRA ver 19.0.
Includes all liver‐relevant events and significant changes in hepatic laboratory values.
ADRs experienced by 431 patients (35%) led to treatment discontinuation, including liver injury (11.1%) and HFSR (10.8%). ADRs led to interruption of treatment of 605 patients (49%) and dose reduction of 516 (42%), most frequently for HFSR (27.6% and 23.1%, respectively) and liver injury (11.6% and 7.3%, respectively).
Major ADRs leading to dose modification or discontinuation were observed mainly within the first 4 weeks after treatment began. The incidence of HFSR was the highest during the second week and subsequently decreased to relatively constant lower rates (Fig. 1).
Figure 1.

Proportion of patients experiencing the onset of major adverse events (AEs). Proportion of patients experiencing the first appearance of HFSR, liver injury, hypertension, fatigue, thrombocytopenia, or fever. Most AEs were observed during the early cycles of treatment.
Abbreviation: HFSR, hand‐foot skin reaction.
There was a higher, dose‐dependent cumulative incidence of HFSR and liver injury in patients initially treated with 160 mg compared with that of patients treated with ≤120 mg (Fig. 2A, 2B), although there was no difference in the incidence of hypertension or fatigue between the two initial dosing groups (Fig. 2C, 2D).
Figure 2.

Time to the first onset of adverse events (AEs) ≥ grade 3. Cumulative incidence rates for HFSR (A), liver injury (B), hypertension (C), and fatigue (D), with the first onset of those AEs plotted for patients grouped as having received an initial dose = 160 mg or ≤120 mg.
Abbreviation: HFSR, hand‐foot skin reaction.
Cox regression analysis suggested that HFSR was associated with previous bevacizumab treatment (yes/no, HR = 1.38; 95% CI, 1.02–1.85; p = .0340), ECOG PS (≥2/0–1, HR = 0.61, 95% CI, 0.44–0.85, p = .0037) and initial dose of regorafenib (<160 mg vs. 160 mg, HR = 0.77; 95% CI, 0.65–0.91, p = .0016). In contrast, HFSR was not associated with previous treatment with anti‐EGFR MAbs or capecitabine (Table 3).
Table 3. Landmark analysis of baseline variables that influenced overall survival, time‐to‐treatment failure, and hand‐foot skin reaction.

eBSA: Body surface area estimated using DuBois formula.
Abbreviations: CI, confidence interval; ECOG PS, European Cooperative Oncology Group Performance Status; HFSR, hand‐foot skin reaction; HR, hazard ratio.
Effectiveness
The median OS was 6.9 months (95% CI: 6.4–7.4; Fig. 3A). The estimated 6‐month and 1‐year OS rates were 54.9% (95% CI, 52.1–57.7) and 28.3% (95% CI, 25.6–31.0), respectively. The estimated median TTF was 2.2 months (95% CI, 2.1–2.3; Fig. 3B). The relationships between OS/TTF and baseline ECOG PS are shown in supplemental online Figures 4 and 5.
Figure 3.

Kaplan‐Meier analysis of OS (A) and TTF (B).
Abbreviations: CI, confidence interval; OS, overall survival; TTF, time‐to‐treatment failure.
Stepwise Cox multivariate regression analysis suggested that certain clinical features influenced OS and TTF in the landmark analysis (Table 3). Baseline ECOG PS had a significant effect on OS (HR, 1.41; ECOG PS ≥2 vs. 0–1; 95% CI, 1.09–1.83; p = .0092) as well as on TTF (HR = 1.38 95% CI, 1.03–1.83; p = .0293). The occurrence of HFSR within the first 28 days was associated with better OS (HR = 0.80; 95% CI, 0.70–0.91; p = .0010; supplemental online Fig. 6). However, the initial dose of regorafenib did not affect TTF or OS.
Discussion
To our knowledge, we present here the first large‐scale, prospective observational study to investigate the safety and effectiveness of regorafenib for Japanese patients with mCRC in routine clinical practice. Patients’ characteristics in the current study were mostly similar to those in the CORRECT‐J population [9], except that in this real‐world study, 8.3% of patients treated with regorafenib had a baseline ECOG PS ≥2. Approximately two thirds of patients started treatment with 160 mg of regorafenib, which was gradually decreased in subsequent cycles, likely to manage AEs. Treatment duration was similar to that of the CORRECT‐J trial; however, the rate of treatment discontinuation because of ADRs was higher compared with that caused by PD, particularly in the first 4 weeks. Major ADRs were observed during the early cycles of treatment and subsequently remained at relatively low rates. These findings support monitoring AEs early during treatment and promptly initiating appropriate dose modification or interruption to minimize discontinuing treatment.
HFSR was the most common ADR, which is consistent with the findings of other trials. HFSR is a known AE associated with MKIs such as sorafenib [12], [13], [14], and Asian patients seem to be more susceptible [9], [15]. However, it is unknown whether specific genetic polymorphisms are associated with the development of HFSR. An exploratory Cox regression analysis conducted here suggests that baseline ECOG PS, initial dose, and previous bevacizumab treatment influenced the occurrence of HFSR.
There is an association between better ECOG PS and MKI‐related HFSR [16], [17], possibly explained by the generally more active lifestyles (e.g., walking) of patients with a better ECOG PS [18] and longer treatment compared with patients with a poor ECOG PS. Therefore, it is important that health care professionals educate patients on managing their physical activity during treatment.
Molecularly targeted agents such as inhibitors of vascular endothelial growth factor (VEGF) or EGFR induce dermatological toxicity, including HFSR. Our data suggest that prior bevacizumab treatment was a risk factor for regorafenib‐related HFSR, but anti‐EGFR agents were not. Inhibition of the VEGF signal transduction pathway may represent one of the factors associated with the development of MKI‐associated HFSR [19]. Although regorafenib was administered as a single agent here, treatment with bevacizumab followed by regorafenib may contribute to long‐term inhibition of VEGF, which may explain the observed higher incidence of HFSR in patients previously treated with bevacizumab.
In real‐world clinical practice, some physicians select a starting dose lower than the approved dose but without the support of clinical evidence or consideration of patients’ characteristics. In the present study, the initial dose of regorafenib did not affect TTF or OS. Two small phase II trials recently presented the efficacy and safety of a lower initial dose of regorafenib. These results did not reduce the efficacy of regorafenib and were associated with a slightly, but not drastically, lower incidence of HFSR compared with the previous clinical trial data [20], [21]. Interestingly, the ReDOS study, a randomized phase II trial comparing a dose escalation arm and a standard arm, suggested that the rapid dose escalation strategy showed significantly higher proportion of initiation of Cycle 3 as the primary endpoint and a better survival trend [21]. Although this strategy might be an option for some patients depending on their condition, more robust data are required to consolidate the dose escalation strategy in clinical practice.
Our study showed that OS was associated with the early onset of HFSR, which is consistent with findings for regorafenib and other MKIs [22], [23], [24]. Post hoc analysis of the CORRECT and RESORCE phase III trials of regorafenib for CRC and hepatocellular carcinoma also showed an association between HFSR and OS, as in our study [25], [26]. Onset of HFSR could indicate high sensitivity for patients treated with regorafenib. Considering those results, even though these were a post hoc analysis, the early onset of HFSR might be a surrogate marker of better prognosis by regorafenib treatment. However, HFSR is the most common reason for discontinuing treatment, and it should therefore be well managed. In the present study, 17.0% of patients did not receive prophylaxis for HFSR. Health care professionals must be well informed about the risks for and management of HFSR during long‐term treatment with regorafenib.
Liver injury is a critical ADR caused by regorafenib. Although the incidence of serious liver injury in clinical practice was not higher in our study compared with the CORRECT‐J trial [9], the risk of liver injury requires careful monitoring and minimization because of the risk of death. A recent review of regorafenib‐induced liver injury in Japanese patients considered this effect idiosyncratic, that predictive biomarkers have not been identified, and that the second cycle was the most frequently observed time to onset. These findings highlight the importance of conducting weekly tests for liver function, particularly during the first two cycles, to identify hepatic events early and discontinue treatment, which are the most important strategies for risk mitigation [27].
The benefit conferred upon OS by regorafenib treatment of patients with mCRC in a real‐world setting is consistent with the experience of randomized clinical trials [8], [15]. Here, an exploratory Cox regression analysis suggests that worse ECOG PS (≥2) was significantly associated with poorer OS and TTF compared with better ECOG PS (0–1). The current study included patients with ECOG PS ≥2 (8.3 %), whereas the CORRECT and CONCUR trials only included patients with ECOG PS 0–1 [8], [15]. The REBECCA study of a cohort of patients in the French compassionate program, including patients with ECOG PS ≥2 (10.6%), showed that worse ECOG PS was associated with poorer OS [22], consistent with our results. The possible benefits of regorafenib in patients with ECOG PS ≥2 cannot be excluded because changes in ECOG PS after treatment initiation were not investigated here. Importantly, the available data indicated that treatment with regorafenib would not be recommended for patients with poor ECOG PS (≥2).
This study has several limitations. Certain baseline data, including those for tumor markers and laboratory values, were not analyzed because of the large number of missing data. These may be important predictors of the effectiveness and safety of regorafenib treatment that require further investigation. Although safety and effectiveness were not prospectively analyzed, the Cox regression analysis was exploratory and retrospective, and the data require careful interpretation. Nevertheless, the information obtained from this cohort of 1,227 patients in real‐world settings will be useful for making clinical decisions to use regorafenib to treat patients with mCRC.
Conclusion
Our data suggest that the safety and effectiveness profiles of regorafenib of Japanese patients with mCRC treated in clinical practice were consistent with those of clinical trials. No new safety concerns were observed. To prevent early treatment discontinuation because of ADRs, weekly monitoring for the first 2 months is strongly recommended. Treatment with regorafenib would not be recommended for patients with poor ECOG PS.
See http://www.TheOncologist.com for supplemental material available online.
Acknowledgments
The study was supported by Bayer Yakuhin Ltd. Bayer collaborated with the authors to design the study. Bayer worked with the investigators on the collection, analysis, and interpretation of the data, and on the preparation of this report. The authors made the final decision to submit the article for publication. We thank the people who were involved in this study, including the patients and their families as well as the personnel at all institutions. Medical writing and editorial assistance were provided by ASCA Corporation.
Author Contributions
Conception/design: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Toshirou Nishida, Naoya Yamazaki, Hajime Takikawa, Takashi Morimoto, Masayuki Chosa, Toshiyuki Sunaya, Yoko Hamada, Kei Muro, Kenichi Sugihara
Provision of study material or patients: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Kei Muro, Kenichi Sugihara
Collection and/or assembly of data: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Kei Muro, Kenichi Sugihara
Data analysis and interpretation: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Toshirou Nishida, Naoya Yamazaki, Hajime Takikawa, Takashi Morimoto, Masayuki Chosa, Toshiyuki Sunaya, Yoko Hamada, Kei Muro, Kenichi Sugihara
Manuscript writing: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Toshirou Nishida, Naoya Yamazaki, Hajime Takikawa, Takashi Morimoto, Masayuki Chosa, Toshiyuki Sunaya, Yoko Hamada, Kei Muro, Kenichi Sugihara
Final approval of manuscript: Kensei Yamaguchi, Yoshito Komatsu, Taroh Satoh, Hiroyuki Uetake, Takayuki Yoshino, Toshirou Nishida, Naoya Yamazaki, Hajime Takikawa, Takashi Morimoto, Masayuki Chosa, Toshiyuki Sunaya, Yoko Hamada, Kei Muro, Kenichi Sugihara
Disclosures
Kensei Yamaguchi: Bayer (H); Yoshito Komatsu: Bayer, Taiho, Lilly, Sanofi, Daiichi Sankyo, Chuugai, ONO Pharmaceutical Co. Ltd, Yakult, Shionogi, Bristol‐Myers Squibb, Kirin, Takeda (RF), Bayer, Taiho, Lilly, Sanofi, Daiichi Sankyo, Chuugai, ONO Pharmaceutical Co. Ltd, Yakult, Pfizer, Nipro, Takeda (H); Taroh Satoh: Eli Lilly & Co., Daiichi Sankyo (C/A), Chugai Pharmaceutical Co. Ltd, Merck Serono Co. Ltd, Bristol‐Myers Squibb, Taiho Pharmaceutical Co. Ltd, Baer Pharmaceutical, Takeda Pharmaceutical Co. Ltd (C/A, H), Chugai Pharmaceutical Co. Ltd, ONO Pharmaceutical Co. Ltd, Yakult Honsha Co. Ltd. (RF); Takayuki Yoshino: Chugai, Merck Sharp & Dohme, Sanofi, Sumitomo Dainippon, GlaxoSmithKline, Boehringer Ingelheim (RF), Sanofi, Chugai, Eli Lilly & Co., Merck Serono (H); Naoya Yamazaki: ONO, Bristol‐Myers Squibb, Novartis (RF), Bayer, ONO Pharmaceutical Co. Ltd, Bristol‐Myers Squibb, Novartis (SAB), H: ONO Pharmaceutical Co. Ltd, Bristol‐Myers Squibb, Novartis (H); Masayuki Chosa: Bayer Yakuhin Ltd. (E); Takashi Morimoto: Bayer Yakuhin Ltd. (E); Toshiyuki Sunaya: Bayer Yakuhin Ltd. (E); Yoko Hamada: Bayer Yakuhin (E); Kei Muro: Gilead Sciences, Merck Serono, Merck Sharp & Dohme, Daiichi Sankyo, Sanofi, ONO Pharmaceutical Co. Ltd, Shionogi, Medisience Planning, Pfizer, Kyowa Hakko Kirin (RF), Chugai, Eli Lilly & Co, Takeda, ONO Pharmaceutical Co. Ltd, Taiho, Bayer (H); Kenichi Sugihara: Chugai Pharmaceutical Co., Taiho Pharmaceutical Co., (RF), Chugai Pharmaceutical Co., Eli Lilly & Co., Novarutis Pharma K, Bayer Yakuhin Ltd., Sanofi Co. (H).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.World Health Organization . Cancer fact sheet. Available at http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed December 16, 2017.
- 2.Vital Statistics Japan (Ministry of Health, Labour and Welfare) . Available at http://ganjoho.jp/reg_stat/statistics/dl/index.html. Accessed December 16, 2017.
- 3.Van Cutsem E, Cervantes A, Adam R et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386–1422. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Muro K, Ajioka Y et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2016 for the treatment of colorectal cancer. Int J Clin Oncol 2018;23:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Comprehensive Cancer Network (2017) NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Colon Cancer. Version 2 2017. [Google Scholar]
- 6.National Comprehensive Cancer Network (2017) NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Rectal Cancer. Version 3 2017. [Google Scholar]
- 7.Wilhelm SM, Dumas J, Adnane L et al. Regorafenib (BAY 73‐4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011;129:245–255. [DOI] [PubMed] [Google Scholar]
- 8.Grothey A, Van Cutsem E, Sobrero A et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:303–312. [DOI] [PubMed] [Google Scholar]
- 9.Yoshino T, Komatsu Y, Yamada Y et al. Randomized phase III trial of regorafenib in metastatic colorectal cancer: Analysis of the CORRECT Japanese and non‐Japanese subpopulations. Invest New Drugs 2015;33:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bayer Yakuhin, Ltd. Package insert of Stivarga tablets 40 mg [in Japanese]. Available at http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/630004_4291029F1028_1_08. Accessed December 16, 2017.
- 11.Zhang S, Chen Q, Wang Q. The use of and adherence to CTCAE v3.0 in cancer clinical trial publications. Oncotarget 2016;7:65577–65588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chanprapaph K, Rutnin S, Vachiramon V. Multikinase inhibitor‐induced hand‐foot skin reaction: A review of clinical presentation, pathogenesis, and management. Am J Clin Dermatol 2016;17:387–402. [DOI] [PubMed] [Google Scholar]
- 13.Llovet JM, Ricci S, Mazzaferro V et al.; SHARP Investigators Study Group . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 14.McLellan B, Ciardiello F, Lacouture ME et al. Regorafenib‐associated hand‐foot skin reaction: Practical advice on diagnosis, prevention, and management. Ann Oncol 2015;26:2017–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Qin S, Xu R et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2015;16:619–629. [DOI] [PubMed] [Google Scholar]
- 16.Kanbayashi Y, Hosokawa T, Yasui K et al. Predictive factors for sorafenib‐induced hand‐foot skin reaction using ordered logistic regression analysis. Am J Health Syst Pharm 2016;73:e18–23. [DOI] [PubMed] [Google Scholar]
- 17.Dranitsaris G, Vincent MD, Yu J et al. Development and validation of a prediction index for hand‐foot skin reaction in cancer patients receiving sorafenib. Ann Oncol 2012;23:2103–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iijima M, Fukino K, Adachi M et al. Sorafenib‐associated hand‐foot syndrome in Japanese patients. J Dermatol 2011;38(3):261–266. [DOI] [PubMed] [Google Scholar]
- 19.Azad NS, Aragon‐Ching JB, Dahut WL et al. Hand‐foot skin reaction increases with cumulative sorafenib dose and with combination anti‐vascular endothelial growth factor therapy. Clin Cancer Res 2009;15:1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kudo T, Kato T, Kagawa Y et al. Phase II dose titration study of regorafenib for patients with unresectable metastatic colorectal cancer that progressed after standard chemotherapy. J Clin Oncol 2018; 36(suppl 4):821. [Google Scholar]
- 21.Bekaii‐Saab T, Ou F, Anderson D et al. Regorafenib Dose Optimization Study (ReDOS): A randomized phase II trial to evaluate dosing strategy for regorafenib in refractory metastatic colorectal cancer (mCRC)—An ACCRU Network study. J Clin Oncol 2018; 36(suppl 4):611. [Google Scholar]
- 22.Adenis A, de la Fouchardiere C, Paule B et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: Results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer 2016;16:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reig M, Torres F, Rodriguez‐Lope C et al. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J Hepatol 2014;61:318–324. [DOI] [PubMed] [Google Scholar]
- 24.Branco F, Alencar RS, Volt F et al. The impact of early dermatologic events in the survival of patients with hepatocellular carcinoma treated with sorafenib. Ann Hepatol 2017;16:263–268. [DOI] [PubMed] [Google Scholar]
- 25.Grothey A, Huang L, Wagner A et al. Hand‐foot skin reaction (HFSR) and outcomes in the phase 3 CORRECT trial of regorafenib for metastatic colorectal cancer (mCRC). J Clin Oncol 2017;35(suppl 15):3551. [Google Scholar]
- 26.Bruix J, Merle P, Granito A et al. Hand‐foot skin reaction (HFSR) and overall survival (OS) in the phase 3 RESORCE trial of regorafenib for treatment of hepatocellular carcinoma (HCC) progressing on sorafenib. J Clin Oncol 2018;36(suppl 15):412. [Google Scholar]
- 27.Uetake H, Sugihara K, Muro K et al. Clinical features of regorafenib‐induced liver injury in Japanese patients from post‐marketing experience. Clin Colorectal Cancer 2018;17:e49–e58. [DOI] [PubMed] [Google Scholar]