

Research reveals the structure of the TRPC5 ion channel, a potential drug target of kidney disease, depression and anxiety.
Abstract
The transient receptor potential canonical subfamily member 5 (TRPC5), one of seven mammalian TRPC members, is a nonselective calcium-permeant cation channel. TRPC5 is of considerable interest as a drug target in the treatment of progressive kidney disease, depression, and anxiety. Here, we present the 2.8-Å resolution cryo–electron microscopy (cryo-EM) structure of the mouse TRPC5 (mTRPC5) homotetramer. Comparison of the TRPC5 structure to previously determined structures of other TRPC and TRP channels reveals differences in the extracellular pore domain and in the length of the S3 helix. The disulfide bond at the extracellular side of the pore and a preceding small loop are essential elements for its proper function. This high-resolution structure of mTRPC5, combined with electrophysiology and mutagenesis, provides insight into the lipid modulation and gating mechanisms of the TRPC family of ion channels.
INTRODUCTION
The transient receptor potential canonical (TRPC1 to TRPC7) channels are Ca2+-permeant cation channels that depolarize cells and increase intracellular calcium levels. These channels are potentiated by G protein–coupled receptors (GPCRs) or tyrosine kinase receptor–mediated activation of phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol trisphosphate (1, 2). TRPC5 is closely related to TRPC4, with 65% sequence identity. TRPC4 and TRPC5 exist as homomers, as well as heteromers with TRPC1 (3, 4). TRPC2, TRPC3, TRPC6, and TRPC7 are potentiated by DAG, a cleavage product of PIP2 (5), whereas TRPC4 and TRPC5 become sensitive to DAG upon dissociation of the Na+/H+ exchanger regulatory factor adapter proteins from the C terminus (6).
TRPC5 is expressed in both excitable and nonexcitable cells, primarily in the brain and kidney (7), and TRPC5 knockout mice exhibit deficits in anxiety and innate fear behavior (8, 9). TRPC5 is a potential therapeutic target for progressive kidney disease, depression, anxiety, and other disorders (10–12). TRPC4’s function in the brain may overlap with that of TRPC5 (13), but TRPC4 appears to have unique functions in the vasculature (14, 15). TRPC5’s constitutive activity is higher in overexpression systems and native cells than that of TRPC4. Gαq and Gαi/o pathways have been reported to potentiate both TRPC4 and TRPC5 (16, 17), although the action of Gαi/o pathways on TRPC5 would benefit from studies with native agonists at physiological concentrations. TRPC5 is also potentiated by cold (18), which has not been reported for TRPC4.
To date, the cryo–electron microscopy (cryo-EM) structures of TRPC3, TRPC4, and TRPC6 have been determined, which reveal new structural features of these channels in apo- or ligand-occupied states (19–22). However, the physiological regulators and gating mechanism of TRPC5 and other TRPC channels are still being investigated. A detailed analysis of TRPC5 based on high-resolution structure will provide deeper insight into lipid-protein interactions and protein-protein interactions. Here, we report the cryo-EM structure of mouse TRPC5 to an overall resolution at 2.8 Å. Together with mutagenesis studies, this structure reveals critical determinants for ion permeation, channel activity, and lipid-protein interactions. Comparison of the TRPC5 structure with other transient receptor potential (TRP) channels provides essential information to help understand the diverse functional and physiological roles of this ion channel family.
RESULTS
Overall structure of the mouse TRPC5 tetrameric ion channel
A mouse TRPC5 construct lacking the C-terminal 210 residues (amino acids 1 to 765, excluding amino acids 766 to 975) yielded more protein after purification than that of the full-length construct. The baculovirus construct, consisting of a maltose binding protein tag at the N terminus, was stably purified to homogeneity in n-dodecyl β-d -maltoside and reconstituted in the amphipol, poly(maleic anhydride-alt-1-decene) substituted with 3-(dimethylamino) propylamine (PMAL-C8; fig. S1A). The amphipol-reconstituted detergent-free protein was then negatively stained and analyzed by single-particle cryo-EM.
Single-particle cryo-EM analyses of TRPC5 at an overall resolution of 2.8 Å were sufficient for de novo model building (fig. S2 and table S1). Disordered regions led to poor densities for 7 residues in the S1-S2 loop, 28 residues in the distal N terminus, and 3 residues in the truncated distal C terminus. Similar to other solved TRP channel structures, TRPC5 forms a fourfold symmetric homotetramer (Fig. 1, A and B) with dimensions of 100 Å by 100 Å by 120 Å. Each of the four monomers can be divided into a compact cytosolic domain and a transmembrane domain (TMD) (Fig. 1). The cytosolic domain is composed of the N-terminal region with an ankyrin repeats domain (ARD) and an HLH (helix-loop-helix) region of seven α helices; the C-terminal subdomain contains a connecting helix and a coiled-coil domain. The TMD is composed of six α helices (S1 to S6), a TRP domain, and several small helices, including a pore helix, pre-S1 elbow, pre-S1 helix, and an S2-S3 linker helix with connecting loops (Fig. 1, C and D).
Fig. 1. Overall structure of mouse TRPC5.
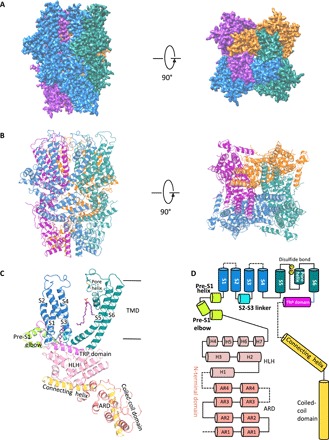
(A) Cryo-EM density map of mTRPC5 at 2.8 Å overall resolution with each monomer represented in different colors (left, side view; right, top view). (B) Ribbon diagrams of the mouse TRPC5 model with the channel dimensions indicated. (C) Ribbon diagrams depicting structural details of a single subunit. (D) Linear diagram depicting the major structural domains of the TRPC5 monomer, color-coded to match the ribbon diagram in (C). ARD, ankkyrin repeat domain.
Whole-cell patch clamp recordings confirmed that the truncated TRPC5 construct used for cryo-EM analyses retained the basic electrophysiological properties of full-length TRPC5 but lost the receptor-operated activation response (fig. S1, B and C). Both the truncated and full-length constructs were activated by englerin A (EA; a guaiane sesquiterpenoid or cinnamate and glycolate ester) and inhibited by ML204, a TRPC4/5 blocker, but no detectable current was observed in cells transfected with empty vector (fig. S1B). The truncated construct failed to respond to the extracellular application of adenosine 5′-triphosphate (ATP), which robustly activated full-length TRPC5 via stimulating the purinergic receptors (P2Y2/P2Y11) endogenously expressed in human embryonic kidney–293 (HEK293) cells (fig. S1C).
The ion conduction pore
TRPC4 and TRPC5 are the most closely related TRPC proteins, with 65% amino acid identity at full length and 100% identity in S5 (amino acids 502 to 542), the pore helix (amino acids 569 to 580), S6 (amino acids 596 to 651, including the lower gate), and the selectivity filter (amino acids 580 to 584). The architecture of the ion conduction pathway is well conserved between these two channels (Fig. 2, A and B). TRPC5’s motif “TRAIDEPNN” (amino acids 544 to 552) forms a small loop in the middle of the extracellular loop connecting S5 and the pore helix [Fig. 2C (left), red arrowheads, and fig. S3]. This unique loop, close to the disulfide bond and on the top of the pore helix, is distinct from the corresponding motif “ETKGLS” of TRPC4 [Fig. 2C (right), red arrowhead, and fig. S3]. The TRPC5 motif contains three more amino acids, including one more negatively charged residue (Asp548) and the hydrophobic residue (Ile547) (Fig. 2D, red arrowhead), which may potentially attract more cations around the pore to increase conduction.
Fig. 2. TRPC5 ion conduction pathway compared with TRPC4 and other TRPCs.
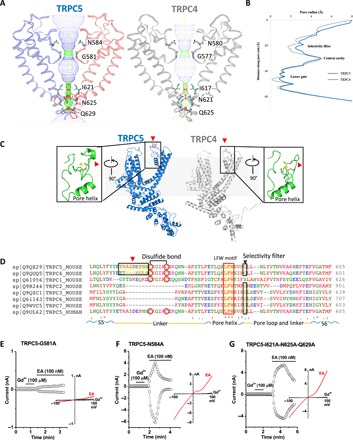
(A) Side view of TRPC5’s pore region with chains A and C compared with TRPC4 (gray). The ion conduction pathway is shown as dots and mapped using HOLE with key amino acid residues labeled. (B) Pore radius along the central axis. The side chains of glycine form a narrow constriction at the selectivity filter. “INQ” motif forms the lower gate. (C) Side view of TRPC5 (blue) monomer subunit compared with TRPC4 (gray). Differences in the organizations of a linker (red arrowheads) of S5 and the small loop above the pore helix of the extracellular pore domain are enlarged. (D) Sequence of the mouse TRPC5 aligned to TRPC4 and other TRPC subfamily members (Clustal Omega) between S5 and S6 including the linker, pore helix, and pore loop. Regions corresponding to different extracellular pore domains are indicated by the red arrowheads. The two cysteines forming disulfide bonds, a conserved “LFW” motif, and the selectivity filter are highlighted. (E to G) Patch clamp recordings of TRPC5 mutants in response to channel activators Gd3+ and EA.
Along with the pore, the filter/gate-forming residues of TRPC5 are identical to those of TRPC4. Gly581 marks a restriction point of 8.2 Å between diagonally opposed residues at the selectivity filter. INQ (Ile621, Asn625, and Gln629) defines a lower gate with a 5.0-Å constriction formed by the side chains of Asn625 (fig. S4); in TRPC4, the most restricted point is 3.6 Å between opposing Asn621 residues.
Structural comparisons between TRPC5 and other TRPC channels
We compared the architectural differences between the TRPC1/4/5 and TRPC3/6/7 channel subfamilies that reveal similarities and unique features (Fig. 3, A to D). An overlay of TRPC5 with the previously solved TRPC3, TRPC4, and TRPC6 channel structures shows a relatively high spatial conservation of the six-transmembrane bundle. Superimposition of the TRPC channels also reveals notable differences in the arrangement of S3 (Fig. 3D); the S3 helix is longer in TRPC3 and TRPC6 and protrudes into the extracellular space. In both TRPC4 and TRPC5 channels, the extracellular S3 region is shorter by four helical turns, limiting potential extracellular interactions. Several intracellular features of TRPC channels are also preserved, including the ankyrin repeats, the pre-S1 elbow in the N-terminal domain, and the connecting helix running parallel to the membrane bilayer. A key feature of TRPC channels is the conserved LFW motif inside the pore domain (Fig. 2D). In the TRPC5 structure, a π-π interaction between Phe576 and Trp577 stabilizes the key pore region, as is also seen in the available TRPC3/4/6 channel structures (Fig. 3B). Mutagenesis of the LFW motif in TRPC5 results in nonfunctional channels (4).
Fig. 3. Comparison of the TRPC5 structure with other TRP channel structures.
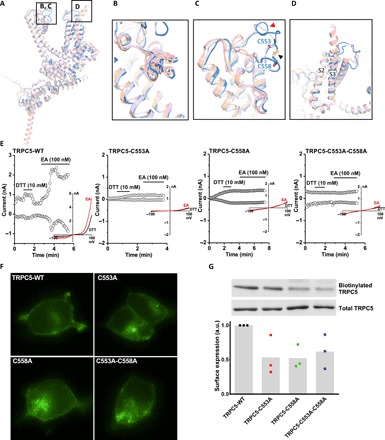
(A) Superimposed side views of mouse TRPC5 subunit (blue) compared with other TRPC family members, including mouse TRPC4 [Protein Data Bank (PDB) ID code 5Z96, gray] (19), human TRPC3 (PDB ID 5ZBG, pink) (20), and human TRPC6 (PDB ID 5YX9, yellow) (21). (B) The conserved LFW motif in the pore helix from a π-π interaction that stabilizes the pore region. (C) Key pore loop disulfide bond between Cys553 and Cys548 in TRPC5 (black arrowhead) and the corresponding pore loop disulfide bond in TRPC4 that close to a linker (red arrowheads) of S5 and loop above the pore helix. This disulfide bond is not present in TRPC3 or TRPC6. (D) Differences in the organization of the S3 helix between the TRPC4/5 and TRPC3/6. The S3 helices of TRPC3 and TRPC6 are longer than those of TRPC5 and TRPC4. (E) Patch clamp recordings of wild-type (WT) TRPC5 and cysteine mutants in response to the reducing agent [dithiothreitol (DTT)] and channel activator (EA). (F) Localization of enhanced yellow fluorescent protein–tagged WT TRPC5 and cysteine mutants in HEK293 cells. (G) Quantification of cell surface expression of TRPC5 cysteine mutants (n = 3). Mean values are shown as gray bars. By analysis of variance (ANOVA), there is no statistically significant difference. a.u., arbitrary units.
Despite the relatively high structural conservation, TRPC5’s TMD has several distinct features. Unlike TRPC3/6, TRPC4/5’s extracellular domains and long pore loops form intricate structures stabilized by a disulfide bond between Cys553 and Cys558 (Fig. 3C). As the two cysteines in the pore region are well conserved in TRPC1/4/5 subfamily (Fig. 2D), the disulfide bond may be important to gating. The wild-type (WT) TRPC5 can be activated by reducing agent dithiothreitol (DTT) and EA, while mutations of a single or both cysteine residues to alanine all led to inactive channels (Fig. 3E and fig. S1D). This differs from what we observed for TRPC4; the single-cysteine mutants were inactive, but the double-cysteine mutant was functional (19). Although mutation of cysteine residues resulted in more intracellular retention of both TRPC4 and TRPC5, mutant proteins still trafficked to the plasma membrane, as determined by fluorescence microscopy and biotinylation accessibility (Fig. 3, F and G, and fig. S5, A and B).
Potentiation by EA and the trivalent cations Gd3+ and La3+
An unusual feature of TRPC4 and TRPC5 is potentiation by micromolar concentrations of the trivalent metal cations lanthanum and gadolinium. Neutralization of the negatively charged amino acids Glu543 and Glu595/Glu598 resulted in a loss of potentiation (23). We found that the TRPC5 construct mutated at the selectivity filter (G581A) could not be activated by Gd3+ and is only weakly potentiated by EA (Fig. 2E and fig. S1D), although its trafficking to the plasma membrane was not significantly affected (fig. S1E). Mutation of an upper residue at the selectivity filter (N584A) or mutation of all three restriction residues at the lower gate (I621A-N625A-Q629A) resulted in loss of potentiation by Gd3+ but did not significantly affect channel activation by EA (Fig. 2, F and G). Electrophysiological analyses of additional TRPC5 mutants revealed a considerable number of amino acids required for direct channel potentiation by Gd3+ (fig. S6A). However, it is unlikely that all these residues, distributed from the intracellular to extracellular regions, are involved in Gd3+ binding or gating. A careful examination on the basal activities of all mutants before application of Gd3+ suggests that potentiation by Gd3+ is dependent on the partially open state of the TRPC5 channel, as Q629N is the only mutant that retained constitutive activity and exhibited sensitivity to Gd3+ (fig. S6, A and B). All other mutants of TRPC5, as well as TRPC4, were inactive at the basal condition and not stimulated by Gd3+ (fig. S6, A and C). Application of Gd3+ after EA had no effects for all constructs (fig. S6, B and C), suggesting that the unliganded state of TRPC5 is required for its potentiation by Gd3+.
As discussed above, the loop preceding TRPC5’s disulfide bond is distinct from that of TRPC4, with three extra residues (amino acids 546 to 548) in TRPC5 (red arrowheads in Fig. 2, C and D, and Fig. 3C). Mutagenesis analyses demonstrated that neutralization of the two negatively charged residues (D548N-E549Q) or replacement of Asp548 or Glu549 by alanine (D548A or E549A) in TRPC5 slowed the activation and inactivation kinetics by EA in some, but not all, transfected cells (Fig. 4, A to C, G, and H). Mutation of both residues into alanine (D548A-E549A) had a similar effect on channel activation but caused complete loss of inactivation in all cells (Fig. 4, D, G, and H), suggesting that both of the side chains of Asp548 and Glu549 may interact with other residues in the extracellular pore domain to facilitate the opening and closing of the channel. We also exchanged the motifs corresponding to the small loops in TRPC5 and TRPC4 to determine whether they would be functionally interchangeable. The activation and inactivation kinetics of the TRPC5 mutant carrying the TRPC4 motif varied substantially from cell to cell, similar to that observed for D548N-E549Q, D548A, and E549A mutants (Fig. 4, E, G, and H). In contrast, the TRPC4 mutant with the TRPC5 motif recapitulated the typical responses of WT TRPC5 and TRPC4 (Fig. 4, F to H). Because of the proximity between this small loop and the disulfide bond, we also tested whether it serves as a structural element determining the functionality of the double-cysteine mutants. However, we found that the TRPC5 mutant carrying the TRPC4 motif and C553A-C558A mutations was inactive (Fig. 4I).
Fig. 4. Mutations in the loop preceding the disulfide bond of TRPC5 alter channel inactivation kinetics.
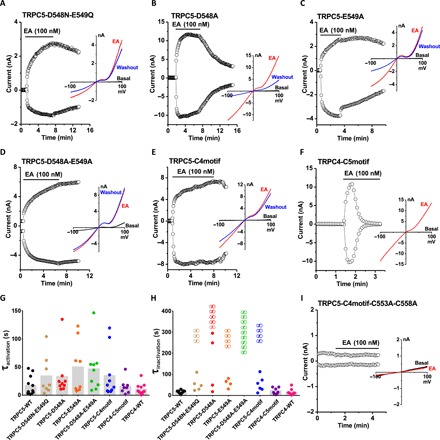
(A to F and I) The time course of whole-cell currents measured at +80 and −80 mV and representative I-V relationships in different conditions as labeled. TRPC5-C4motif, TRPC5 mutant carrying the TRPC4 motif ETKGLS; TRPC4-C5motif, TRPC4 mutant with the TRPC5 motif TRAIDEPNN; TRPC5-C4motif-C553A-C558A, TRPC5 mutant bearing the TRPC4 motif and double-cysteine mutations. (G and H) Time constants (τ) of channel activation and inactivation by 100 nM EA. τ values from each recording are plotted as dots. “∞” indicates that the current did not decay in the recording time frame. Mean τ values of each mutant are shown as gray bars. There is no significant difference by ANOVA in (G) (n = 8 to 11).
An intramembranous cation binding site
A strong nonprotein peak density is observed in the intracellular hydrophilic pocket between TRPC5’s S2 and S3 helices. Since Na+ was the only added cation in our purification buffer, this peak may represent a sodium ion, but we cannot rule out another cation, such as Ca2+. The cation is not in the putative ion conduction pathway; it is coordinated by hydrogen bonding with several highly conserved residues, including Glu418, Glu421, Asn436, and Asp439 (Fig. 5A). The geometry of the cation’s coordination in TRPC5 resembles that of the TRPC4 structure (19). Structural alignment of these residues from TRPC family members reveals a well-preserved cation binding site, similar to Ca2+ binding sites, in the TRPM4 (transient receptor protein cation channel subfamily M member 4) and TRPM2 structures (24, 25). Small deviations are seen in TRPM4 and TRPC4; Glu421 in the S2 helix’s cytosolic end is substituted by a glutamine (Fig. 5, B and C). A number of negatively charged residues— including Asp633, Asp636, Glu638, Asp652, and Glu653 in the TRP domain, as well as Asp424, Glu429, and Asp433 on S2-S3 linker—are aligned along a potential cation entry pathway (fig. S7). Asp652 and Glu653 in the TRP domain and negatively charged residues in the cytosolic region form a cation access pathway, which should facilitate cation entry to its binding site (fig. S7C). Substitution of the three negatively charged residues with structurally similar neutral residues (E418Q-E421Q-D439N) in TRPC5 did not affect channel activation by EA; however, replacing neutral Asn436 with positively charged arginine (N436R) resulted in loss of potentiation in most cases (Fig. 5D). We hypothesize that this site stabilizes the voltage sensing–like domain of the TRPs but may not be directly related to potentiation by trivalent cations or EA.
Fig. 5. The cation binding site in TRPC5.
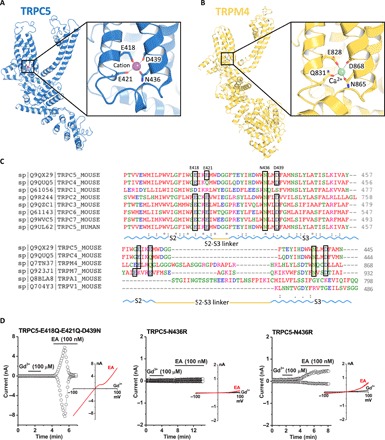
(A) A cation (purple sphere) on the cytosolic face is in the hydrophilic pocket of the S1-S4 domain, interacting with Glu418, Glu421, Asn436, and Asp439. Enlarged view of the cation binding site. (B) Comparable Ca2+ (green sphere) binding site and an enlarged view of TRPM4 as “E/Q/N/D” (TRPC5, “E/E/N/D”). (C) Sequence of the mouse TRPC5 aligned to mTRPC4 and other representative TRP members (Clustal Omega). The key residues are highlighted as E/E/N/D, which are conserved in TRPC2/3/5/6/7 and TRPM7 but as E/Q/N/D in TRPC4 and TRPM4. (D) Patch clamp recordings of TRPC5 mutants in response to the channel activators Gd3+ and EA. For the N436R mutant, 12 transfected cells showed no response to EA; weak activation was observed in three cells.
Lipid-binding sites
Eight densities corresponding to lipid molecules were resolved in the TRPC5 cryo-EM density and identified as four cholesteryl hemisuccinates (CHSs; a cholesterol-mimicking artificial detergent molecule; purple) and four phospholipids (PLs; identified as to be phosphatidic acids or ceramide-1-phosphates; yellow) (Fig. 6A). Each PL is embedded in the gap between the monomeric subunits. The PL interacts with the pore helix through its polar head and side-chain oxygen with amino acids Phe576 and Trp577 (Fig. 6, B and C). Mutation of both residues to alanine (F576A-W577A) resulted in an inactive channel (Fig. 6E). Four CHS heads face down at the interface with the plasma membrane, embedding into the space among the S4, S5, and S6 helices and the N-terminal domain (Fig. 6B). CHS stabilizes the domain through interactions with Asn500 on the S4/S5 linker and with N-terminal Trp315, Tyr316, and Trp322, which are spatially adjacent to the TRP domain (Fig. 6D). The polar heads of the identified lipids interact with positively charged regions of the channel (fig. S8). Mutation of Asn500 to alanine (N500A) did not affect channel activation by EA, while substitution of the N-terminal binding residues (W315A-W322A or Y316A) abolished channel function in most cases (weak activation for Y316A mutant was observed in 3 of 13 cells) (Fig. 6F). Despite their differential responses to EA, none of the four mutants described above were potentiated by ATP activation of G protein P2Y receptors (fig. S8F), suggesting that the two lipid binding sites may be required for receptor-operated activation of TRPC5.
Fig. 6. Lipid coordination in TRPC5.
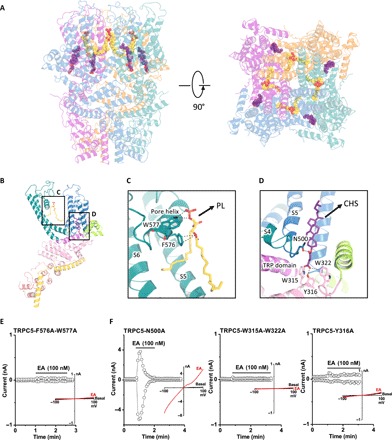
Lipid-channel interactions. (A) Side and top views of ribbon diagrams of the TRPC5 tetramer: four CHS molecules and four PLs (potentially ceramide-1-phosphate or phosphatidic acid) are shown as spheres with purple or yellow carbons, respectively. (B) Side views of each CHS and PL molecules per protomer. (C and D) Ribbon diagram of the TRPC5 lipid binding regions. (C) PL interacts with the pore helix through Trp577 and Phe576. (D) CHS, shown in purple, interacts with the S4/S5 linker at Asn500 and the N-terminal domains at Trp315, Tyr316, and Trp322. (E and F) Patch clamp recordings of lipid binding site mutants in response to EA.
Two lipid binding sites are also found in the TRPC3 and TRPC4 structures, while a clear lipid density, probably because of its relative resolution (3.8 Å), is not observed in the TRPC6 structure. CHS and PL are located in the same position in the structures of TRPC4 and TRPC5. In the TRPC3 structure, lipid1 located between S4/S5 linker and N-terminal domain has polar interactions with residues Asn548, Trp322, and Tyr323. Lipid2 is located near the pore helix, interacting with Lys607, Phe610, and Trp611 (fig. S8). It has been reported that a number of lipids can directly and indirectly activate some TRP channels and modulate channel activity (26). Endogenous phosphatidylinositides located near S4/S5 linker in TRPV1 (transient receptor protein cation channel subfamily V member 1) were shown to participate in the activation of TRPV1. The common features of lipid-protein interactions in the available TRPC channel structures, together with our mutagenesis study of the lipid binding sites in TRPC5, demonstrate that lipids can interact with TRP channels in diverse ways and participate in the activation mechanism of TRPC channels.
DISCUSSION
Here, we obtained and investigated the cryo-EM structure of TRPC5 at 2.8-Å resolution. The overall architecture of TRPC5 resembles that of other TRPC subfamily channels, but important differences in its pore may help explain its unique functional properties, particularly in contrast to its close homolog, TRPC4. The extracellular disulfide bond is a redox sensor that controls the opening and closing of the pore domain in both TRPC4 and TRPC5. However, our mutagenesis analyses suggest that it is dispensable for TRPC4 (19) but essential for the function of TRPC5. The small loop preceding the disulfide bond is the key structural element that differs in the pores of TRPC5 and TRPC4. Exchanging the corresponding sequences did not change the electrophysiological properties of TRPC4 but slowed or even abolished the inactivation of TRPC5. We observed a similar phenomenon for the TRPC5 mutants D548N-E549Q, D548A, E549A, and D548A-E549A, suggesting that the TRAIDEPNN motif tunes the activity of TRPC5. The side chains of Asp548 or Glu549 are coordinately involved in channel inactivation, as only the double mutant D548A-E549A exhibited nondecaying currents in all recordings. Mutation of a single residue (D548A or E549A) or minor modification of both residues (D548N-E549Q) produced largely variable inactivation kinetics but was not sufficient to cause loss of inactivation in all cells. We suggest that both Asp548 and Glu549 are required for TRPC5 channels to open and close in a consistent manner by coordinately interacting with other unrecognized residues. Mutation of a single residue may make this interaction unstable, while mutation of both residues would completely disrupt the interaction and result in complete loss of inactivation.
The significance of TRPC5’s disulfide bond has been disputed by two previous studies. An earlier study proposed that mutation of a single or both cysteines conferred constitutive activity on the mutant channel (27). A second study suggested that these mutants completely lost function (28), consistent with the current work. Moreover, we found that cotransfection of these cysteine mutants with WT TRPC5 did not suppress channel activation by EA and occasionally resulted in larger basal activity (fig. S5C). We suspect that the proposed constitutive activity (27) of the cysteine mutants may have been due to contamination from WT TRPC5.
Micromolar La3+ and Gd3+, which are usually inhibitors for other cation channels, reversibly potentiate TRPC5 and TRPC4. Mutagenesis studies suggest that the lanthanide-binding site is extracellular between residues Glu543 and Glu595 (23, 29). Mutation of glutamic acids Glu543 and Glu595/Glu598 in the extracellular mouth of the channel pore to glutamine resulted in loss of potentiation. However, Glu543 at the end of the S5 helix and Glu595 at the beginning of S6 are oriented such that cation binding is not possible in the state we captured (fig. S9A). Since our structure was obtained in a divalent-free buffer, we cannot rule out the possibility that Ca2+ binding may lead to a conformational change in this region. Here, we found that direct potentiation of TRPC5 by Gd3+ only occurred when the channel was unliganded and partially open, which may allow Gd3+ to access its potential binding sites and increase activity. Gd3+ can also increase TRPC5 activity in the presence of the GPCR ligand histamine or guanosine 5′-O-(3′-thiotriphosphate) (23, 29). Mutation of residues in the extracellular pore domain of TRPC5 often resulted in loss of potentiation, which confounds the interpretation of site mutagenesis. The Cryo-EM of Gd3+-bound TRPC5 will provide rational guidance for future investigation of the mechanism of Gd3+ potentiation.
PLC-dependent gating of TRPC5 was reported to require the pore loop residue Arg593 (29). In our structure, Arg593 has polar interactions with the Glu598 and Asn589 side chains (fig. S9A). Breaking this electrostatic interaction may lead to unfolding of this pore region. In addition, Arg593 is the only positively charged residue in the corresponding positions in TRPC channels (glycine in TRPC1, glutamine in TRPC4, asparagine in TRPC6, or negatively charged aspartic acid in TRPC7). We suggest that Arg593 serves as a molecular fulcrum, allowing the efficient transmission of gating force to TRPC5’s pore helix loop.
Another unique feature of TRPC4/5 channels is their unusual current-voltage (I-V) relation, particularly the flat segment in the 10 to 40 mV range attributed to Mg2+ block of outward current (2). TRPC5’s S6 terminal Asp633 residue is close to the intracellular mouth of channel pore and was proposed as the site for block of the ion conduction pathway upon binding Mg2+, whereas the downstream Asp636 did not affect Mg2+ block (30). In our structure, Asp633 is situated much closer to the channel pore than Asp636 (fig. S9B), which is consistent with it being a Mg2+ block site. However, because we used a divalent-free buffer for cryo-EM, direct evidence for Mg2+ binding on Asp633 and restriction of the channel pore should be studied in proteins purified in Mg2+-containing solution.
In TRPV channels, as in many other six transmembrane-spanning segment channels, the S4/S5 linker is crucial to channel gating (31–33). Consistent with this, a single point mutation in TRPC4/5’s S4/S5 linker, Gly504Ser, fully activates the channel. Introduction of a second mutation (Ser623Ala) into TRPC4 Gly503Ser suppressed the constitutive activation and partially rescued its function (34). These results indicate that the S4/S5 linker is a critical constituent of TRPC4/C5 channel gating and that disruption disinhibits gating control by the sensor domain.
In summary, our structure enables new comparisons between the different subgroups of the available TRPC channels. Mutagenesis of the lipid binding site revealed that binding events at both the extracellular and intracellular surfaces are required for channel activity and stabilization of the protein’s conformation. Our findings provide structural insights into conservation and divergence of TRPC channels but do not explain the mechanisms regulating GPCR-Gq-PLC activation of a receptor-operated channel. However, the findings have broad implications for understanding the structural basis underlying epitope antibody selectivity and can facilitate rational, structure-based approaches for the design of subtype-selective TRPC ligands as new therapeutic agents.
MATERIALS AND METHODS
Protein expression and purification
A synthetic gene fragment encoding residues 1 to 765 (excluding amino acids 766 to 975) of mouse TRPC5 was cloned into the pEGBacMam vector. The resulting protein contains a maltose-binding protein tag on its N terminus. P3 baculoviruses were produced in the Bac-to-Bac Baculovirus Expression System (Invitrogen). HEK293S GnTI− [from the American Type Culture Collection (ATCC)] cells were infected with 10% (v/v) P3 baculovirus at a density of 2.0 to 3.0 × 106 cells/ml for protein expression at 37°C. After 24 hours, 10 mM sodium butyrate was added, and the temperature was reduced to 30°C for 72 hours before harvesting. Cells were gently disrupted and resuspended in a solution containing 30 mM Hepes, 150 mM NaCl, and 1 mM DTT (pH 7.5) with EDTA-free protease inhibitor cocktail (Roche). Cells were solubilized for 2 to 3 hours in a solution containing 1.0% (w/v) N-dodecyl-β-d-maltopyranoside (Affymetrix), 0.1% (w/v) CHS (Sigma-Aldrich), 30 mM Hepes, 150 mM NaCl, and 1 mM DTT (pH 7.5) with EDTA-free protease inhibitor cocktail (Roche). After 30 min, the cell lysate was then centrifuged for 60 min at 100,000g, and the supernatant was incubated in amylose resin (New England Biolabs) at 4°C overnight. The resin was washed with 20 column volumes of wash buffer [25 mM Hepes, 150 mM NaCl, 0.1% (w/v) digitonin, 0.01% (w/v) CHS, and 1 mM DTT (pH 7.5) with EDTA-free protease inhibitor cocktail (Roche)]. The protein was eluted with four column volumes of wash buffer with 40 mM maltose. The protein was then concentrated to 0.5 ml and mixed with PMAL-C8 (Anatrace) at 1:4 (w/w) with gentle agitation for 4 hours. Bio-Beads SM-2 (50 mg/ml, reconstitution mixture; Bio-Rad) were added to remove additional detergent; a disposable Poly-Prep column was used to remove Bio-Beads. After incubation at 4°C overnight, the protein was further purified on a Superose 6 size exclusion column in 25 mM Hepes, 150 mM NaCl, and 1 mM DTT (pH 7.5). All purification procedures were carried out either on ice or at 4°C. The peak fractions corresponding to tetrameric TRPC5 was concentrated to 7.0 mg/ml and used for preparation of cryo-EM sample grids.
Electron microscopy data collection
Purified TRPC5 protein sample (3.5 μl) in PMAL-C8 at 7.0 mg/ml was applied to glow-discharged Quantifoil R1.2/1.3 holey carbon 400-mesh copper grids (Quantifoil). Grids were blotted for 7 s at 100% humidity and flash frozen by liquid nitrogen–cooled liquid ethane using a FEI Vitrobot Mark I (FEI). The grid was then loaded onto FEI TF30 Polara electron microscope operated at 300-kV accelerating voltage. Image stacks were recorded on a Gatan K2 Summit direct detector (Gatan) set in super-resolution counting mode using SerialEM (35), with a defocus range between 1.5 and 3.0 μm. The electron dose was set to 8 e−/physical pixel per s, and the subframe time was set to 0.2 s. A total exposure time of 8 s resulted in 40 subframes per image stack. The total electron dose was 42.3 e−/Å2 (~1.1 e−/Å2 per subframe).
Image processing and three-dimensional reconstruction
Image stacks were gain normalized and binned twice to a pixel size of 1.23 Å before drift and local movement correction using MotionCor2 (36), resulted in the sums of all frames of each image stack with and without dose weighting. The dose-weighting sum was binned by 8× and subjected to visual inspection; images of poor quality were removed before particle picking. Particle picking and subsequent bad particle elimination through two-dimensional (2D) classification were performed using Python scripts or programs (37) with minor modifications in the 8× binned images. The selected 2D class averages were used to build an initial model using the common lines approach implemented in SPIDER (38) through M. Liao’s Python scripts (37), which was applied to later 3D classification using RELION (39). Contrast transfer function (CTF) parameters were estimated using CTFFIND4 (40) using the sum of all frames without dose weighting. Quality particle images were then boxed out from the dose-weighted sum of all 40 frames and subjected to RELION 3D classification. RELION 3D refinements were then performed on selected classes for the final map. The resolution of this map was further improved by using the sum of subframes 1 to 14. The number of particles in each dataset and other details related to data processing are summarized in table S1.
Model building, refinement, and validation
At 2.8-Å resolution, the cryo-EM map was of sufficient quality for de novo atomic model building in Coot (41). Amino acid assignment was achieved mainly on the basis of the defined densities for bulky residues (Phe, Trp, Tyr, and Arg) and the absence of side-chain densities for glycine residues. The atomic model was further visualized in Coot; a few residues with side chains moving out of the density during the refinement were fixed manually, followed by further refinement. The TRPC5 model was then subjected to global refinement and minimization in real space using the module “phenix.real_space_refine” in PHENIX (42). The geometries of the model were assessed using MolProbity (43) in the “comprehensive model validation” section of PHENIX. The final model exhibited good geometry as indicated by the Ramachandran plot (preferred region, 98.25%; allowed region, 1.75%; outliers, 0%). The pore radius was calculated using HOLE (44). Local resolution estimation was from ResMap (45).
Electrophysiology
The full-length, truncated TRPC5 constructs or empty vector was transfected into HEK293T cells (from the ATCC), together with an mCherry plasmid. Cells with red fluorescence were selected for whole-cell patch recordings (HEKA EPC 10 USB amplifier, PATCHMASTER 2.90 software). A 1-s ramp protocol from −100 to +100 mV was applied at a frequency of 0.2 Hz. Signals were sampled at 10 kHz and filtered at 3 kHz. The pipette solution contained 130 mM CsCl, 1 mM MgCl2, 5.7 mM CaCl2, 10 mM EGTA, and 10 mM Hepes (calculated free Ca2+, 200 nM), and the pH was titrated to 7.2 using CsOH. The standard bath solution contained 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, and 10 mM d-glucose, and the pH was adjusted to 7.4 with NaOH. The recording chamber (150 μl) was perfused at a rate of ~2 ml/min. All recordings were performed at room temperature.
Quantification of proteins at the plasma membrane
TRPC5-tranfected 293T cells were incubated with Sulfo-NHS-Biotin [0.5 mg/ml in phosphate-buffered saline (PBS), Aladdin] for 30 min and washed thrice with 50 mM glycine in PBS. Cells were lysed and aliquoted for extraction of plasma membrane proteins and total proteins separately. Biotinylated plasma membrane proteins were captured with streptavidin magnetic beads (Invitrogen). Western blotting was then performed to detect TRPC5 in the plasma membrane and total proteins. The ratio between biotinylated and total TRPC5 proteins was used to represent the level of TRPC5 proteins expressed at the cell surface. TRPC4 constructs were analyzed in the same way. Primary antibodies against TRPC5 and TRPC4 were purchased from Sangon Biotech (Shanghai, China) and Proteintech (Wuhan, China), respectively.
Supplementary Material
Acknowledgments
We thank S. Harrison and the Cryo-EM Facility (Harvard Medical School) for use of their microscopes. We thank M. Liao for providing the Python scripts and help in image processing. Funding: J.Zhang was supported by the Thousand Young Talents Program of China, the National Natural Science Foundation of China (grant no. 31770795), and the Jiangxi Province Natural Science Foundation (grant no. 20181ACB20014). J.Li was supported by the National Natural Science Foundation of China (grant no. 81402850). Functional studies in this project were supported by the National Natural Science Foundation of China (grant nos. 31300949 to B.Z. and 31300965 to G.-L.C.). Author contributions: J.Z., J.D., and D.E.C. designed the project. J.Z. and J.D. designed and made constructs for BacMam expression and determined the conditions used to enhance protein stability. J.Z. purified the protein. B.L. assisted with initial constructs screening for cryo-EM. Z.L. carried out detailed cryo-EM experiments, including data acquisition and processing. J.L. and J.Z. built the atomic model on the basis of cryo-EM maps. G.-L.C., Y.G., J.L., K.X., and B.Z. performed mutagenesis and functional studies. X.P., J.Z., Y.Z., J.X., C.X., L.Z., W.L., X.-L.T., and J.W. assisted with protein purification and the mutation of TRPC5 constructs for functional studies. J.D., B.Z., and J.Z. drafted the manuscript. All authors contributed to structure analysis/interpretation and manuscript revision. J.Z., D.E.C., and Z.L. initiated the project, planned and analyzed the experiments, and supervised the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Cryo-EM electron density map of the mouse TRPC5 has been deposited in the Electron Microscopy Data Bank (accession no. EMD-9615; www.ebi.ac.uk/pdbe/emdb/), and the fitted coordinate has been deposited in the Protein Data Bank (PDB ID code 6AEI; www.pdb.org). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw7935/DC1
Fig. S1. Biochemical and functional characterization of the TRPC5 constructs.
Fig. S2. Flow chart for TRPC5 cryo-EM data processing.
Fig. S3. Pore loop structures of known TRP channels.
Fig. S4. Comparison of ion conducting pathways between TRPC5 and TRPC4.
Fig. S5. Cell surface expression levels of TRPC4 cysteine mutants and function of TRPC5 cysteine mutants.
Fig. S6. Gd3+ potentiation is dependent on the unliganded and partially open state of TRPC5.
Fig. S7. The cation binding sites.
Fig. S8. Lipid-channel interactions.
Fig. S9. Pore loop interactions and intracellular mouth of the channel pore.
Table S1. Cryo-EM data collection, refinement, and validation statistics.
REFERENCES AND NOTES
- 1.Clapham D. E., TRP channels as cellular sensors. Nature 426, 517–524 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Schaefer M., Plant T. D., Obukhov A. G., Hofmann T., Gudermann T., Schultz G., Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 275, 17517–17526 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Strübing C., Krapivinsky G., Krapivinsky L., Clapham D. E., TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29, 645–655 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Strübing C., Krapivinsky G., Krapivinsky L., Clapham D. E., Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 278, 39014–39019 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Albert A. P., Gating mechanisms of canonical transient receptor potential channel proteins: Role of phosphoinositols and diacylglycerol. Adv. Exp. Med. Biol. 704, 391–411 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Storch U., Forst A.-L., Pardatscher F., Erdogmus S., Philipp M., Gregoritza M., y Schnitzler M. M., Gudermann T., Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. U.S.A. 114, E37–E46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofmann T., Schaefer M., Schultz G., Gudermann T., Transient receptor potential channels as molecular substrates of receptor-mediated cation entry. J. Mol. Med. 78, 14–25 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Riccio A., Li Y., Moon J., Kim K.-S., Smith K. S., Rudolph U., Gapon S., Yao G. L., Tsvetkov E., Rodig S. J., Van’t Veer A., Meloni E. G., Carlezon W. A. Jr., Bolshakov V. Y., Clapham D. E., Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 137, 761–772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puram S. V., Riccio A., Koirala S., Ikeuchi Y., Kim A. H., Corfas G., Bonni A., A TRPC5-regulated calcium signaling pathway controls dendrite patterning in the mammalian brain. Genes Dev. 25, 2659–2673 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naziroglu M., Demirdas A., Psychiatric disorders and TRP channels: Focus on psychotropic drugs. Curr. Neuropharmacol. 13, 248–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y., Castonguay P., Sidhom E.-H., Clark A. R., Dvela-Levitt M., Kim S., Sieber J., Wieder N., Jung J. Y., Andreeva S., Reichardt J., Dubois F., Hoffmann S. C., Basgen J. M., Montesinos M. S., Weins A., Johnson A. C., Lander E. S., Garrett M. R., Hopkins C. R., Greka A., A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 358, 1332–1336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Just S., Chenard B. L., Ceci A., Strassmaier T., Chong J. A., Blair N. T., Gallaschun R. J., del Camino D., Cantin S., D’Amours M., Eickmeier C., Fanger C. M., Hecker C., Hessler D. P., Hengerer B., Kroker K. S., Malekiani S., Mihalek R., McLaughlin J., Rast G., Witek J., Sauer A., Pryce C. R., Moran M. M., Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLOS ONE 13, e0191225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riccio A., Li Y., Tsvetkov E., Gapon S., Yao G. L., Smith K. S., Engin E., Rudolph U., Bolshakov V. Y., Clapham D. E., Decreased anxiety-like behavior and Galphaq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J. Neurosci. 34, 3653–3667 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freichel M., Suh S. H., Pfeifer A., Schweig U., Trost C., Weißgerber P., Biel M., Philipp S., Freise D., Droogmans G., Hofmann F., Flockerzi V., Nilius B., Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 3, 121–127 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Makarewich C. A., Zhang H., Davis J., Correll R. N., Trappanese D. M., Hoffman N. E., Troupes C. D., Berretta R. M., Kubo H., Madesh M., Chen X., Gao E., Molkentin J. D., Houser S. R., Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 115, 567–580 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeon J.-P., Hong C., Park E. J., Jeon J. H., Cho N.-H., Kim I.-G., Choe H., Muallem S., Kim H. J., So I., Selective Gαi subunits as novel direct activators of transient receptor potential canonical (TRPC)4 and TRPC5 channels. J. Biol. Chem. 287, 17029–17039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thakur D. P., Tian J.-b., Jeon J., Xiong J., Huang Y., Flockerzi V., Zhu M. X., Critical roles of Gi/o proteins and phospholipase C-δ1 in the activation of receptor-operated TRPC4 channels. Proc. Natl. Acad. Sci. U.S.A. 113, 1092–1097 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimmermann K., Lennerz J. K., Hein A., Link A. S., Kaczmarek J. S., Delling M., Uysal S., Pfeifer J. D., Riccio A., Clapham D. E., Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 108, 18114–18119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan J., Li J., Zeng B., Chen G.-L., Peng X., Zhang Y., Wang J., Clapham D. E., Li Z., Zhang J., Structure of the mouse TRPC4 ion channel. Nat. Commun. 9, 3102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan C., Choi W., Sun W., Du J., Lu W., Structure of the human lipid-gated cation channel TRPC3. eLife 7, e36852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Q., Guo W., Zheng L., Wu J.-X., Liu M., Zhou X., Zhang X., Chen L., Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 28, 746–755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinayagam D., Mager T., Apelbaum A., Bothe A., Merino F., Hofnagel O., Gatsogiannis C., Raunser S., Electron cryo-microscopy structure of the canonical TRPC4 ion channel. eLife 7, e36615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung S., Mühle A., Schaefer M., Strotmann R., Schultz G., Plant T. D., Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J. Biol. Chem. 278, 3562–3571 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Autzen H. E., Myasnikov A. G., Campbell M. G., Asarnow D., Julius D., Cheng Y., Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z., Tóth B., Szollosi A., Chen J., Csanády L., Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 7, e36409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taberner F. J., Fernández-Ballester G., Fernández-Carvajal A., Ferrer-Montiel A., TRP channels interaction with lipids and its implications in disease. Biochim. Biophys. Acta 1848, 1818–1827 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Xu S.-Z., Sukumar P., Zeng F., Li J., Jairaman A., English A., Naylor J., Ciurtin C., Majeed Y., Milligan C. J., Bahnasi Y. M., al-Shawaf E., Porter K. E., Jiang L.-H., Emery P., Sivaprasadarao A., Beech D. J., TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong C., Kwak M., Myeong J., Ha K., Wie J., Jeon J.-H., So I., Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflugers Arch. 467, 703–712 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Chen X., Li W., Riley A. M., Soliman M., Chakraborty S., Stamatkin C. W., Obukhov A. G., Molecular determinants of the sensitivity to Gq/11-phospholipase C-dependent gating, Gd3+ potentiation, and Ca2+ permeability in the transient receptor potential canonical type 5 (TRPC5) channel. J. Biol. Chem. 292, 898–911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obukhov A. G., Nowycky M. C., A cytosolic residue mediates Mg2+ block and regulates inward current amplitude of a transient receptor potential channel. J. Neurosci. 25, 1234–1239 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao E., Liao M., Cheng Y., Julius D., TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao M., Cao E., Julius D., Cheng Y., Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y., Cao E., Julius D., Cheng Y., TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck A., Speicher T., Stoerger C., Sell T., Dettmer V., Jusoh S. A., Abdulmughni A., Cavalié A., Philipp S. E., Zhu M. X., Helms V., Wissenbach U., Flockerzi V., Conserved gating elements in TRPC4 and TRPC5 channels. J. Biol. Chem. 288, 19471–19483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ru H., Chambers M. G., Fu T.-M., Tong A. B., Liao M., Wu H., Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell 163, 1138–1152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A., SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Scheres S. H., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smart O. S., Neduvelil J. G., Wang X., Wallace B. A., Sansom M. S., HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw7935/DC1
Fig. S1. Biochemical and functional characterization of the TRPC5 constructs.
Fig. S2. Flow chart for TRPC5 cryo-EM data processing.
Fig. S3. Pore loop structures of known TRP channels.
Fig. S4. Comparison of ion conducting pathways between TRPC5 and TRPC4.
Fig. S5. Cell surface expression levels of TRPC4 cysteine mutants and function of TRPC5 cysteine mutants.
Fig. S6. Gd3+ potentiation is dependent on the unliganded and partially open state of TRPC5.
Fig. S7. The cation binding sites.
Fig. S8. Lipid-channel interactions.
Fig. S9. Pore loop interactions and intracellular mouth of the channel pore.
Table S1. Cryo-EM data collection, refinement, and validation statistics.