

We identify CRTC2 as a key mediator of LKB1-mutant non–small cell lung cancer.
Abstract
The LKB1 tumor suppressor is often mutationally inactivated in non–small cell lung cancer (NSCLC). LKB1 phosphorylates and activates members of the AMPK family of Ser/Thr kinases. Within this family, the salt-inducible kinases (SIKs) modulate gene expression in part via the inhibitory phosphorylation of the CRTCs, coactivators for CREB (cAMP response element-binding protein). The loss of LKB1 causes SIK inactivation and the induction of the CRTCs, leading to the up-regulation of CREB target genes. We identified CRTC2 as a critical factor in LKB1-deficient NSCLC. CRTC2 is unphosphorylated and therefore constitutively activated in LKB1-mutant NSCLC, where it promotes tumor growth, in part via the induction of the inhibitor of DNA binding 1 (ID1), a bona fide CREB target gene. As ID1 expression is up-regulated and confers poor prognosis in LKB1-deficient NSCLC, our results suggest that small molecules that inhibit CRTC2 and ID1 activity may provide therapeutic benefit to individuals with NSCLC.
INTRODUCTION
The family of cAMP–regulated transcriptional coactivators (CRTCs) promotes cellular gene expression via an association with the transcription factor cAMP response element–binding protein (CREB) (1–3). The first evidence of an oncogenic role for CRTCs came from the identification of a t(11;19) translocation that generates a CRTC1/mastermind like transcriptional coactivator 2 (MAML2) chimeric protein as the major driver genetic alteration in up to 50% of mucoepidermoid carcinomas (MECTs) (4). The CRTC1/MAML2 chimera is thought to promote MECT oncogenesis by binding to CREB over relevant genes.
Consistent with its central position downstream of many growth factor signaling pathways, CREB influences the survival, growth, and differentiation of both normal and transformed cells; increases in CREB activity are associated with tumor progression, chemotherapy resistance, and reduced patient survival (5–9). The oncogenic response to CREB has prompted the design of therapies that block its activity. Several inhibitors have been used in preclinical trials (10–12), although the molecular mechanisms underlying CREB, as well as CRTC-mediated tumor development, remain unclear.
In the basal state, CRTCs (CRTC1, CRTC2, and CRTC3) are predominantly phosphorylated by a family of three salt-inducible kinases (SIKs)—namely, SIK1, SIK2, and SIK3—and sequestered in the cytoplasm through phosphorylation-dependent interactions with 14-3-3 proteins. Exposure to cAMP triggers the protein kinase A (PKA)–mediated phosphorylation of CREB at Ser133 and promotes its association with the histone acetyltransferase paralogs CBP (CREB-binding protein) and P300. In parallel, PKA directly phosphorylates and inhibits each of the SIK kinases, leading to dephosphorylation of the CRTCs, which migrate to the nucleus, bind to CREB, and stimulate recruitment of the transcriptional machinery (3).
The LKB1 tumor suppressor is the major upstream kinase that activates SIK kinases and other members of the AMP-activated protein kinase (AMPK) family (13). Loss of LKB1 inactivates the SIKs, leading to shuttling of the CRTCs into the nucleus and to the up-regulation of CREB target genes (3, 14, 15). LKB1 is mutationally inactivated in ∼20% of non–small cell lung cancer (NSCLC) (16, 17). Previous work in mouse models has uncovered an essential role for LKB1 loss in promoting lung cancer (18, 19). However, the molecular mechanisms underlying LKB1 tumor suppression and, specifically, the role of CRTCs in NSCLC, remain unclear. Although targeted therapeutics have made substantial advances in subsets of NSCLC, therapeutic options for LKB1-mutant tumors remain limited. Because LKB1 functional loss can be caused by multiple mechanisms, including somatic gene mutations, epigenetic silencing, or posttranslational modifications, the identification of more robust biomarkers appears critical for the early detection of LKB1-deficient lung cancers.
Here, we identify CRTC2 as a key regulator of LKB1-deficient NSCLC. We demonstrate that CRTC2 promotes NSCLC by stimulating the expression of the oncogene ID1 (inhibitor of DNA binding 1), a novel CRTC2/CREB target gene, which we show here to be up-regulated in LKB1-deficient NSCLC. Together, these studies point to the importance of the CREB/CRTC2 pathway in linking the loss of LKB1 activity in NSCLC to tumor development.
RESULTS
CRTC2 is activated and confers poor prognosis in NSCLC
We evaluated the potential role of CRTCs in NSCLC by bioinformatics analysis of NSCLC cell lines and The Cancer Genome Atlas (TCGA). CRTC2 is the most highly expressed CRTC family member in NSCLC (Fig. 1A and fig. S1A). The CRTC2 gene is genomically amplified in up to 13% of tumors from 230 patients (fig. S1B). Moreover, high levels of CRTC2, but not CRTC1 or CRTC3, expression appear to confer poor prognosis (Fig. 1B), prompting us to further evaluate the role of this coactivator in NSCLC.
Fig. 1. CRTC expression in NSCLC.
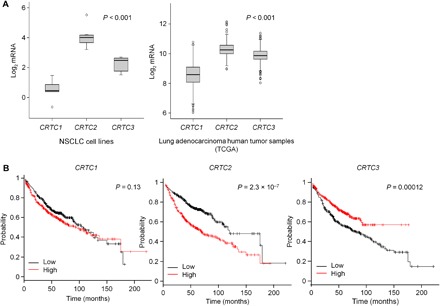
(A) Expression of CRTC1, CRTC2, and CRTC3 mRNA in a panel of NSCLC cell lines (n = 8) and a collection of lung adenocarcinoma patient samples (n = 517) from the TCGA. Statistical test: Analysis of variance (ANOVA) with Tukey’s method for multiple comparisons. (B) Association of CRTC1, CRTC2, and CRTC3 mRNA levels with overall survival (OS) within lung adenocarcinoma datasets from the KM plotter (n = 720). Statistical test: Log-rank test.
LKB1 has been shown to inhibit CRTC2 activity in different cellular contexts where it otherwise modulates metabolism and physiology (14). Because 20% of NSCLC harbors inactivating mutations in LKB1 (16–18), we hypothesized that CRTC2 activity may be up-regulated in LKB1-deficient tumors. To test this notion, we analyzed CRTC2 in lung adenocarcinoma cell lines where LKB1 is inactive (A549 and A427). We observed that CRTC2 is indeed unphosphorylated and therefore active in LKB1-deficient cell lines. Expression of wild-type but not kinase-dead LKB1 rescued CRTC2 phosphorylation (Fig. 2A). When CRTC2 is unphosphorylated, it shuttles to the nucleus (fig. S2A), where it binds to CREB over cAMP response elements (CREs) (3). Consistent with this scenario, the expression of wild-type but not kinase-dead LKB1 repressed a CRE-responsive luciferase reporter (Fig. 2B), and it down-regulated the expression of endogenous CREB target genes (RASD1, RGS2, IRS2, FOS, and NR4A1) (Fig. 2D and fig. S2B). Overexpression of phosphorylation-defective constitutively active CRTC2 (CRTC2CA) rescued the expression of endogenous CREB target genes (FOS and RASD1) in wild-type LKB1-expressing cells (fig. S2C). Basal CREB activity was elevated in LKB1-deficient NSCLC cells and decreased following LKB1 expression (Fig. 2, B and D). Accordingly, exposure to the adenylyl cyclase activator Forskolin (FSK) had no effect on CRE reporter activity in A549 cells, but it stimulated this reporter in LKB1-reconstituted cells, where basal CREB activity is correspondingly lower (Fig. 2C).
Fig. 2. LKB1 regulates CRTC2 activity and CREB signaling in NSCLC.
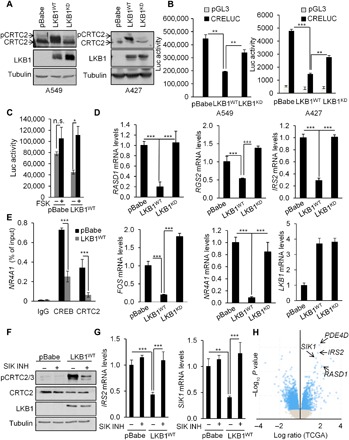
(A) Immunoblot analysis in A549 and A427 NSCLC cells expressing the pBabe vector, full-length LKB1 (LKB1WT), or kinase-dead LKB1 (LKB1KD) showing the effect of LKB1 expression on CRTC2 dephosphorylation and associated mobility shift. (B) Luciferase activity in A549 and A427 NSCLC cells expressing the pBabe vector, full-length LKB1, or kinase-dead LKB1 and transfected with a CRE-luciferace (CRE-luc) reporter. All values are expressed as means ± SD. **P < 0.01, and ***P < 0.001 determined by ANOVA with Tukey’s method. (C) Luciferase activity in A549 cells expressing the pBabe vector or full-length LKB1 and transfected with a CRE-luc reporter and treated with FSK (10 μM) for 4 hours. All values are expressed as means ± SD. *P < 0.05 determined by ANOVA with Tukey’s method. n.s., not significant. (D) RASD1, RGS2, IRS2, FOS, NR4A1, and LKB1 mRNA levels in A549 cells expressing the pBabe vector, full-length LKB1, or kinase-dead LKB1. All values are expressed as means ± SD. ***P < 0.001 determined by ANOVA with Tukey’s method. (E) Chromatin immunoprecipitation (ChIP) assay in A549 cells expressing the pBabe vector or full-length LKB1 showing the amounts of CREB and CRTC2 recruited to the NR4A1 promoter. All values are expressed as means ± SD. ***P < 0.001 determined by ANOVA with Tukey’s method. IgG, immunoglobulin G. (F) Immunoblot analysis in A549 cells expressing the pBabe vector or full-length LKB1 and treated with vehicle dimethyl sulfoxide (DMSO) or the selective SIK inhibitor YKL-05-096 (1 μM) for 3 hours showing the effect of the SIK inhibitor on CRTC2 phosphorylation at S274. (G) IRS2 and SIK1 mRNA levels in A549 cells expressing the pBabe vector or full-length LKB1 and treated with the SIK inhibitor YKL-05-096 (1 μM) for 3 hours. All values are expressed as means ± SD. **P < 0.01 and ***P < 0.001 determined by ANOVA with Tukey’s method. (H) Volcano plot that shows mRNA enrichment in LKB1-mutant (n = 40) versus LKB1 wild-type (n = 190) lung adenocarcinoma specimens obtained from the TCGA.
We examined the effects of LKB1 on the binding of CRTC2 and CREB to the proximal promoter of a well-established core CREB target gene (NR4A1). Consistent with the constitutive activation of this pathway, CREB and CRTC2 occupancy over the NR4A1 promoter was elevated in A549 cells by chromatin immunoprecipitation (ChIP) analysis, and re-expression of LKB1 decreased the binding of both factors to this site (Fig. 2E). Because LKB1 stimulates the kinase activity of all 13 AMPK family members, we evaluated the extent to which SIKs mediate the phosphorylation and inactivation of CRTC2. Exposure of A549 cells to the selective SIK inhibitor YKL-05-096 (20) blocked the effects of LKB1 expression on the phosphorylation of CRTC2 at S274 and the down-regulation of CREB target genes (IRS2 and SIK1) (Fig. 2, F and G). Using data from 40 LKB1-mutant and 190 LKB1 wild-type human lung adenocarcinoma specimens (TCGA), we observed that a number of the most significantly up-regulated genes in LKB1-mutant lung adenocarcinoma are bona fide CREB target genes (SIK1, PDE4D, IRS2, and RASD1) (Fig. 2H). Together, these results indicate that the loss of LKB1 in NSCLC leads to CRTC2 dephosphorylation and to the constitutive activation of the CREB pathway.
CRTC2 regulates oncogenesis and promotes expression of ID1 in LKB1-deficient NSCLC
On the basis of these results, we tested the potential role of CRTC2 in LKB1-deficient lung cancer. Depletion of CRTC2, by short hairpin RNA (shRNA)–mediated knockdown or CRISPR-Cas9–mediated gene knockout, modestly decreased cell proliferation (fig. S3A), but it substantially decreased anchorage-independent growth (Fig. 3A). Overexpression of phosphorylation-defective CRTC2CA was sufficient to rescue anchorage-independent growth in LKB1-expressing cells (fig. S3B). We also examined the impact of CRTC2 loss in lung tumor formation by intravenously injecting luciferase-expressing A549 cells into severe combined immunodeficient (SCID)/Beige mice to induce lung colonization. Control A549 cells formed lung tumors that grew rapidly over the 5-week experiment; these effects were disrupted in A549 cells depleted of CRTC2, demonstrating the importance of this coactivator in NSCLC (Fig. 3, B to D).
Fig. 3. Depletion of CRTC2 inhibits tumor growth in NSCLC.
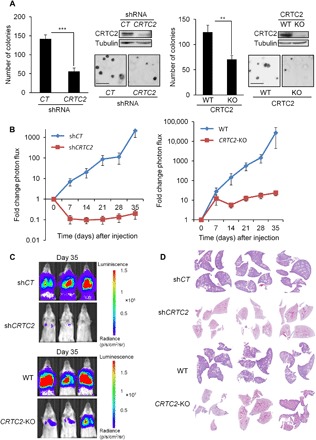
(A) Anchorage-independent growth assessed by soft agar assay in A549 shcontrol (shCT), A549 shCRTC2, A549 wild-type (WT), and A549 CRTC2 knockout (KO) cells. Representative images are shown. Lysates from A549 shcontrol, A549 shCRTC2, A549 wild-type, and A549 CRTC2 knockout cells were immunoblotted with the indicated antibodies. Scale bars, 200 μm. All values are expressed as means ± SD. **P < 0.01 and ***P < 0.001 determined by two-sided Student’s t test. (B) For lung colonization assay, A549 shcontrol, A549 shCRTC2, A549 wild-type, and A549 CRTC2 knockout cells were injected intravenously in the lateral tail vein of SCID/BEIGE mice. Tumor growth is represented as fold change in photon flux during each imaging time point relative to day 0 (n = 7 mice per condition). All values are expressed as means ± SEM. (C) Bioluminescence overlay images of lung growth in A549 shcontrol, A549 shCRTC2, A549 wild-type, and A549 CRTC2 knockout in nude mice. Images from day 35 were normalized to day 0. Images are representative for each condition. (D) Representative hematoxylin and eosin–stained sections of mouse lungs.
Realizing the importance of CRTC2 in LKB1-deficient NSCLC, we sought to identify relevant protein-coding genes that mediate CRTC2 oncogenic activity. Approximately 20% of genes whose expression is down-regulated by LKB1 also show LKB1-regulated binding of CRTC2 to their promoters, by RNA sequencing (RNA-seq) and ChIP sequencing (ChIP-seq) analyses of LKB1-mutant and LKB1-reconstituted NSCLC cells (Fig. 4A and fig. S4). Moreover, all CRTC2-occupied genes are co-bound with CREB (fig. S4), demonstrating the importance of this transcription factor in NSCLC. Thus, although CRTC can also bind to other transcription factors (21, 22), these results suggest that CRTC2 activity in NSCLC cells is mediated by CREB.
Fig. 4. ID1 is a direct CREB target.
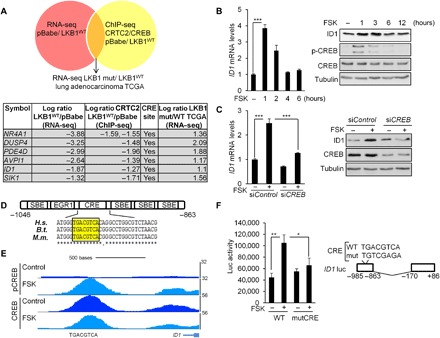
(A) Schematic representation of the strategy followed to find CRTC2/CREB transcriptional targets in LKB1-deficient NSCLC. A table describing the six genes identified is also shown. (B) Immunoblot and quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses in 293T cells treated with vehicle (DMSO) or FSK (10 μM). All values are expressed as means ± SD. ***P < 0.001 determined by two-sided Student’s t test. (C) Immunoblot and qRT-PCR analyses in 293T cells depleted of CREB by small interfering RNA (siRNA) transfection (20 nM) for 72 hours and treated with FSK for 1 hour before collection of cell lysates. All values are expressed as means ± SD. ***P < 0.001 determined by ANOVA with Tukey’s method. (D) Schematic representation of the ID1 promoter. The Smad binding elements (SBEs), EGR1, and CREB-binding site (CRE) are indicated relative to the transcription start site. ClustalW sequence alignment for three animal species [Homo sapiens (H.s.), Bos taurus (B.t.), and Mus musculus (M.m.)] shows the conservation of the CRE site. (E) Representative pCREB and CREB ChiP-seq profiles at the ID1 loci in 293T cells treated with FSK (10 μM) for 1 hour. (F) Luciferase activity in 293T cells transfected with the (−985/−863) wild-type or (−985/−863) mutCRE ID1 luciferase reporter constructs and treated with FSK (10 μM) for 4 hours. All values are expressed as means ± SD. *P < 0.05 and **P < 0.01 determined by ANOVA with Tukey’s method.
In an effort to identify genes that contribute to the growth of human NSCLC tumors, we compared the expression profiles of candidate target genes in A549 cells with a panel of 40 LKB1-mutant and 190 LKB1 wild-type human lung adenocarcinoma specimens from the TCGA (fig. S4). We found six genes (NR4A1, DUSP4, PDE4D, AVPI1, ID1, and SIK1) that are potentially regulated by the LKB1/CRTC2/CREB axis in lung adenocarcinoma (Fig. 4A). Within this list, the up-regulated gene ID1 is also overexpressed in many human tumors where it promotes both cancer initiation and progression (23, 24). Consistent with a potential role for the CREB pathway in this setting, exposure of human embryonic kidney (HEK) 293T cells to FSK increased ID1 mRNA and protein amounts (Fig. 4B), while RNA interference (RNAi)–mediated knockdown of CREB decreased them (Fig. 4C).
The ID1 promoter contains a proximal (−920) palindromic CRE, which is well conserved in mouse and other mammalian species (Fig. 4D). In ChIP-seq studies of HEK293T cells, we noted that CREB occupies the CRE on the ID1 promoter and undergoes Ser133 phosphorylation in response to FSK (Fig. 4E). In transient assays of HEK293T cells, exposure to FSK increased the activity of a wild-type reporter construct containing the proximal −985 and −863 regions of the ID1 promoter but not that of a reporter construct with inactivating mutations at the CRE site (Fig. 4F). Together, these data indicate that ID1 is indeed a canonical CREB target gene.
On the basis of the oncogenic role of ID1 in multiple tumor types (23, 24), we investigated whether ID1 is also relevant to the tumor progression in LKB1-deficient NSCLC. Supporting this idea, the expression of LKB1 in two different LKB1-deficient NSCLC cell lines (A549 and H157) decreased the mRNA and protein amounts for ID1 (Fig. 5A). Moreover, the inhibitory effect of LKB1 appears to be mediated by SIK kinases, as exposure to the selective SIK inhibitor YKL-05-096 blocked the down-regulation of ID1 in response to LKB1 expression (Fig. 5B). We tested components of the CREB pathway for their importance in supporting ID1 expression. RNAi-mediated depletion of CRTC2 or CREB decreased ID1 protein and mRNA amounts in LKB1-deficient cells (Fig. 5, C and D). Overexpression of CRTC2CA rescued the expression of ID1 in wild-type LKB1-expressing cells (fig. S5A). In keeping with these results, ChIP-seq analysis revealed that endogenous CRTC2 and CREB are recruited to the CREB binding site within the proximal ID1 promoter; LKB1 expression disrupted CRTC2 and, to a lesser extent, CREB occupancy over the ID1 gene (Fig. 5E). Consistent with an inability for FSK to up-regulate CREB reporter activity in LKB1-deficient cells (Fig. 2C), exposure to FSK increased ID1 expression in LKB1 wild-type but not LKB1-mutant NSCLC cells (Fig. 5F).
Fig. 5. The LKB1/CRTC2/CREB axis regulates ID1 expression in NSCLC cells.
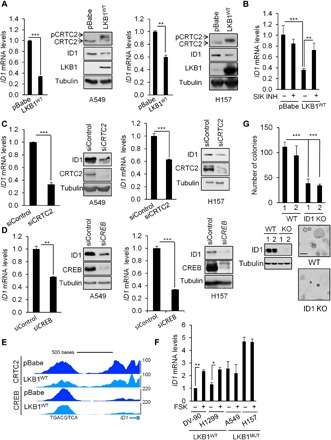
(A) Immunoblot and qRT-PCR analyses in A549 and H157 NSCLC cells expressing the pBabe vector or full-length LKB1 (LKB1WT). All values are expressed as means ± SD. **P < 0.01 and ***P < 0.001 determined by two-sided Student’s t test. Immunoblot shows the effect of LKB1 on ID1 expression, CRTC2 dephosphorylation, and associated mobility shift. (B) ID1 mRNA levels in A549 cells expressing the pBabe vector or full-length LKB1 and treated with the SIK inhibitor YKL-05-096 (1 μM) for 3 hours. All values are expressed as means ± SD. **P < 0.01 and ***P < 0.001 determined by ANOVA with Tukey’s method. (C) Immunoblot and qRT-PCR analyses in A549 and H157 NSCLC cells transfected with siRNA targeting CRTC2 (20 nM) for 72 hours. All values are expressed as means ± SD. ***P < 0.001 determined by two-sided Student’s t test. (D) Immunoblot and qRT-PCR analyses in A549 and H157 NSCLC cells transfected with siRNA targeting CREB (20 nM) for 72 hours. All values are expressed as means ± SD. **P < 0.01 and ***P < 0.001 determined by two-sided Student’s t test. (E) Representative CRTC2 and CREB ChiP-seq profiles in A549 NSCLC cells expressing the pBabe vector or full-length LKB1 at ID1 loci. (F) ID1 mRNA expression in a panel of LKB1 wild-type or LKB1-mutant (MUT) NSCLC cell lines treated with FSK (10 μM) for 1 hour. All values are expressed as means ± SD. *P < 0.05 and **P < 0.01 determined by two-sided Student’s t test. (G) Anchorage-independent growth assessed by soft agar assay in A549 clones deleted for ID1 by CRISPR-Cas9. Representative images are shown. Lysates were immunoblotted with the indicated antibodies. Scale bar, 100 μm. All values are expressed as means ± SD. ***P < 0.001 determined by two-sided Student’s t test.
On the basis of these results, we tested the potential role of ID1 in LKB1-deficient lung cancer. The depletion of ID1 in LKB1-deficient A549 cells, by CRISPR-Cas9–mediated gene knockout, decreased anchorage-independent growth (Fig. 5G), while the expression of ID1 in LKB1 wild-type DV-90 cells increased the number and size of colonies growing in soft agar (fig. S5B), demonstrating the ability of this factor to affect the oncogenic potential of NSCLC cells. These results are consistent with previous studies showing that ID1 functions as an oncogenic factor in other cancers (23–25). To further dissect how CRTC2-ID1 signaling affects anchorage-independent growth and lung colonization in LKB1-deficient NSCLC, we compared the transcriptomes of wild-type and ID1–CRISPR-Cas9 knockout A549 cells by RNA-seq. Pathway enrichment and Gene Ontology analyses of differentially expressed genes revealed that the top biological functions affected by ID1 loss included collagen formation, degradation and organization of the extracellular matrix (ECM), regulation of membrane-ECM interactions, cell adhesion, and substrate adhesion–dependent cell spreading (fig. S6). Together, these data suggest that ID1 regulates a genetic program that modulates the cytoskeleton, as well as cell-cell interactions and cellular binding to the ECM.
ID1 expression is induced in LKB1-deficient lung adenocarcinoma and confers poor prognosis
We wondered whether ID1 expression is commonly up-regulated in NSCLC. Using data from a collection of 48 human LKB1-mutant and 187 LKB1 wild-type lung adenocarcinoma specimens, we found that ID1 expression is indeed increased in LKB1-deficient lung adenocarcinoma; these results were corroborated in two additional datasets. By contrast with ID1, other family members (ID2, ID3, and ID4) appear to be comparably expressed in LKB1 wild-type and LKB1-mutant tumors (Fig. 6A).
Fig. 6. ID1 is highly expressed in LKB1-mutant lung adenocarcinoma tumors and confers poor prognosis.
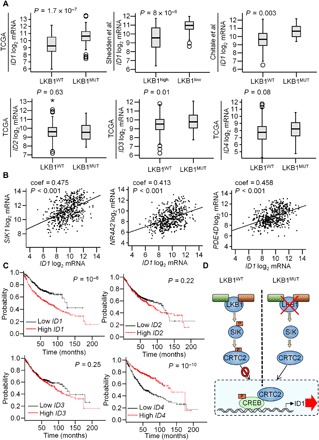
(A) ID1 mRNA expression in three different lung adenocarcinoma datasets. Sample sizes: n = 230 (TCGA), n = 193 (Shedden et al.), and n = 102 (Chitale et al.). ID2, ID3, and ID4 mRNA levels in the TCGA lung adenocarcinoma dataset are shown. Statistical test: Two-sided Student’s t test. (B) Graphs showing the correlation between ID1 and the CREB target genes SIK1, NR4A2, and PDE4D mRNA levels in a collection of lung adenocarcinoma patient samples (n = 517). Data obtained from the TCGA. A Spearman’s test was used, and the correlation coefficient (coef) and the two-tailed P values are shown. (C) Association of ID1, ID2, ID3, and ID4 mRNA levels with OS within lung adenocarcinoma datasets from the KM plotter (n = 720). Statistical test: Log-rank test. (D) Model: LKB1 phosphorylates SIK proteins which, in turn, phosphorylate CRTC2. pCRTC2 is sequestered in the cytoplasm through phosphorylation-dependent interactions with 14-3-3 proteins. The loss of LKB1 in NSCLC leads to the dephosphorylation of CRTC2. Dephosphorylated CRTC2 migrates to the nucleus, where it binds to CREB over the ID1 promoter and stimulates ID1 expression and tumor progression.
Consistent with the role of ID1 as a CREB target, we noted a positive correspondence between mRNA amounts for ID1 and other CREB target genes (SIK1, NR4A2, and PDE4D) in a collection of 517 lung adenocarcinoma tumors obtained from the TCGA (Fig. 6B). Moreover, overall survival was reduced in lung adenocarcinoma patients with tumors expressing high levels of ID1 but not other family members (ID2, ID3, or ID4) (Fig. 6C). Collectively, these studies demonstrate the importance of CRTC2 in mediating the oncogenic effects of LKB1 mutations in NSCLC through the CREB-dependent induction of ID1.
DISCUSSION
A growing number of studies indicate that CRTCs and the CREB pathway have an important role in the development of different types of cancers such as acute myeloid leukemia, MECT, and glioma (5–9, 26). Several CREB blocking agents have been developed and appear to have promising effects in preclinical assays (10–12).
Using human data mining analysis, xenograft models, and cellular systems, we found that CRTC2 is associated with NSCLC. CRTC2 is dephosphorylated in LKB1-deficient NSCLC, which accounts for 20% of NSCLC, because of the loss of LKB1 activity and the subsequent inactivation of SIK kinases. Dephosphorylated CRTC2 shuttles to the nucleus where it binds to CREB over relevant promoters and stimulates their expression. Thus, CREB/CRTC2 signaling is constitutively up-regulated in LKB1-deficient NSCLC.
Notably, the CRTC2 gene is amplified in NSCLC (TCGA and Tumorscape), and high concentrations of CRTC2 confer poor prognosis in individuals with NSCLC, suggesting that gene amplification may provide a separate mechanism by which CRTC2 regulates NSCLC tumor growth. NSCLC cells with lower CRTC2 expression show a reduced anchorage-independent growth and a decreased capacity to generate tumors in the lungs.
We found that the CREB/CRTC2 pathway stimulates oncogenesis in part by up-regulating ID1 expression. ID1 is a transcriptional regulator that heterodimerizes with and antagonizes the DNA binding capacity of basic helix-loop-helix transcription factors (23). During development, ID1 inhibits the differentiation of embryonic stem cells (27, 28). In cancer, ID1 functions as a central hub for the coordination of multiple cancer hallmarks. The expression of ID1 has a prognostic value and is considered a therapeutic target and biomarker in different types of human tumors, including breast cancer, colon cancer, and glioma (23, 24). The expression of ID1 has been shown to be essential for lung colonization by breast cancer cells (29, 30); ID1 is 1 of the 18 genes that constitute a lung metastasis signature for breast cancer (31). In NSCLC, previous work has shown that epidermal growth factor receptor and α-7-nicotine-acetylcholine receptor induce ID1 expression through bone morphogenetic protein signaling and MYC (25) and that the expression of ID1 causes resistance to chemotherapy (24, 32). SMAD sites flank the CRE site on the ID1 promoter (33, 34), suggesting a potential cross-talk between these two pathways in promoting oncogenesis.
We found that ID1 is a canonical CREB target gene whose expression is up-regulated by CREB/CRTC2 signaling (Fig. 6D). ID1 is overexpressed and is a poor prognostic factor in LKB1-mutant lung adenocarcinoma. High ID4 expression, on the other hand, correlated with good prognosis, in line with previous studies indicating that ID4 acts as a tumor suppressor (35).
In NSCLC, ID1 appears to regulate genes that modulate the ECM, cytoskeleton, cell-cell interactions, and cell-ECM adhesion, perhaps explaining why CRTC2 and ID1 induce anchorage-independent growth and lung colonization. These results are consistent with previous studies showing that ID1 promotes tumor progression via regulation of the ECM and cellular adhesion (23). Future studies will be aimed at further defining how the LKB1/CRTC2/ID1 signaling regulates these cellular mechanisms.
In addition to CRTC2, CRTC1 also appears to be hypophosphorylated in LKB1-deficient NSCLC cell lines, where it appears to promote the expression of certain CREB targets that were proposed to induce cell growth and survival (15, 36–38). The extent to which CRTC1 contributes to NSCLC, however, remains unclear. We found that the expression of CRTC2, but not CRTC1 or CRTC3, correlates with poor survival in NSCLC. Although none of the CREB target genes described in those reports reached our cutoffs, we imagine that these genes may further enhance the effects of ID1.
Small-molecule and peptide-based inhibitors that directly target ID proteins have been developed (39, 40); these may be useful for the treatment of LKB1-deficient NSCLC. Future studies should provide insight into the utility of ID1 inhibitors in this setting. Our results suggest that small-molecule inhibitors of CRTC2 and ID1 may have beneficial effects in LKB1-deficient NSCLC.
MATERIALS AND METHODS
Cell culture
A549, A427, H157, 293 T, and H1299 cells were obtained from the American Type Culture Collection, and DV-90 cells were purchased from DSMZ. Cells were cultured in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cell lines with stable expression of wild-type LKB1 complementary DNA (cDNA) or kinase-inactive LKB1 cDNA were described previously (41). Stable expression of green fluorescent protein (GFP)–CRTC2CA (S171A, S274A, and S306A) was performed using retrovirus-mediated transduction of GFP-CRTC2CA cloned into pBabeD vector (MRC PPU Reagents and Services). Stable expression of ID1 in DV-90 cells was performed using retrovirus-mediated transduction of the ID1 cDNA cloned into pBabe vector. Briefly, for retroviral infection, the constructs were transfected along with the ampho packaging plasmid into growing HEK293T cells. Viral supernatants were collected 48 hours after transfection and filtered, and target cells were infected in the presence of polybrene (8 μg/ml). Twenty-four hours later, cells were selected with puromycin. Stable knockdown of CRTC2 in A549 cells was performed using lentivirus-mediated transduction of an shRNA targeting CRTC2 (MISSION shRNA TRCN0000229832, Sigma-Aldrich) cloned into TRC2-pLKO vector. The shRNA-containing vectors were transfected into HEK293T cells with lentiviral packaging plasmids VSVG (vesicular stomatitis virus glycoprotein), GAG/pol, and REV. Viruses were collected 48 hours after transfection, and target cells were infected in the presence of polybrene (8 μg/ml). Twenty-four hours later, cells were selected with puromycin. For CRISPR-Cas9 knockout of CRTC2 in A549 cells, cells were transfected with the guide RNA (gRNA) cloned into pX459, and cells were selected with puromycin after 48 hours. The gRNA targets nucleotides 199 to 218 (ATGGGTATGGGGGTAACCGC) in the second exon of CRTC2 and was a gift from M. Green (42). For CRISPRCas9 knockout of ID1 in A549 cells, cells were transfected with the gRNA (TGGATCTCACCTCGGCCGTC) cloned into pX458, and cells were selected by fluorescence-activated cell sorting to generate single knockouts after 48 hours. RNAi transfection was carried out by transfection of small interfering RNA (siRNA) oligos using RNAiMAX (Invitrogen) according to the manufacturer’s protocol. siRNA duplexes were used in pairs at 20 nM. Cells were harvested at 72 hours after siRNA transfection. Invitrogen siRNAs used were the BLOCK-IT Fluorescent Control (no. 460926) and the Stealth CREB1 siRNA (HSS102264 and HSS102262). Ambion siRNAs used were the Silencer Select Negative Control No. 1 (no. 4390843) and the siCRTC2 (s47209 and s47208).
For soft agar assay, experiments were carried out in six-well plates coated with a base layer of DMEM containing 0.5% agar. Cells were seeded at a density of 30,000 cells per well in DMEM containing 0.35% agar and 10% FBS for 14 days.
Reagents
Specific antibodies against LKB1 (no. 3047, Cell Signaling Technology), tubulin (no. 05-829, Millipore), ID1 (BCH1/no. 195-14, BioCheck), HSP90 (sc-13119, Santa Cruz Biotechnology), and Lamin A/C (sc-7293, Santa Cruz Biotechnology) were used for immunoblot. For CREB, pCREB (pSer133), pCRTC2/3 (pS273) (43), and CRTC2 detection, rabbit polyclonal antibodies were raised against their respective antigens. FSK (Sigma-Aldrich) and the selective SIK inhibitor YKL-05-096 (a gift from N. S. Grey) were used at the indicated concentration. Nuclear and cytoplasmic fractions were isolated using an NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific) following the manufacturer’s conditions.
Luciferase assays
Cells were transfected with the CRE-luciferase (pGL3 vector) or the ID1-Iuciferase reporter (pGL2 vector), and luciferase activity was measured in the GloMax-Multi Microplate Reader (Promega). Luciferase activity was normalized to β-galactosidase activity. When indicated, cells were treated with FSK. The ID1 promoter reporter construct was a gift from F. Ventura (34).
ChIP and ChIP-seq
ChIP was performed as described previously (44). For ChIP-seq experiments, libraries were prepared using NEBNext ChIP-seq reagents from Illumina. Samples were run on MiSeq using v2 chemistry with 25–base pair (bp) paired-end reads. FASTQ sequencing files were mapped to the genome using bowtie2, and peaks were identified. Differential binding and annotation to the genome were performed using HOMER.
Real-time qRT-PCR and RNA-seq
Cultured cells were disrupted in lysis buffer from the RNeasy Mini Kit (Qiagen), and mRNA was purified following the manufacturer’s instructions. cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche). Quantitative polymerase chain reaction real-time qRT-PCR was carried out with diluted cDNA, appropriate primers, and LightCycler 480 SYBR Green I Master (Roche) on a Lightcycler 480 instrument (Roche). Relative mRNA levels were calculated using the 2−ΔΔCt method, with L32 serving as the internal control. RNA-seq libraries were prepared using the mRNA isolation protocol and NEBNext Ultra kits from New England BioLabs following the manufacturer’s protocols. Libraries were quantitated by Qubit (Invitrogen) and run on a MiSeq instrument with 75-bp paired-end reads using v3 chemistry (Illumina). Data were analyzed by TopHat2 and Cuffdiff against the human hg19 genome build.
Mouse studies
All procedures using animals were approved by the Salk Institute’s Institutional Animal Care and Use Committee. Eight-week-old female SCID/Beige (PrkdcscidLystbg-J/Crl; no. 250, Fox Chase) were intravenously injected with 1 × 106 luciferase-expressing A549 cells (n = 7 mice per condition). All mice were imaged via bioluminescence imaging (BLI) 20 min after intravenous injection. Mice were monitored weekly for lung tumor growth by BLI. For histopathology studies, lungs were inflated with 10% formalin, fixed in 10% formalin for 18 hours, and then paraffin-embedded.
Bioinformatics analysis
For mRNA expression analysis, data from the lung adenocarcinoma patients with RNA-seq and/or sequencing information in the TCGA dataset were extracted from cbioportal.org. Expression levels of the selected genes were also obtained from the dataset reported by Shedden et al. (45) and Chitale et al. (46). Correlation of mRNA expression levels to patient survival in lung adenocarcinoma was done using the KM plotter [kmplot.com; (47)]. Patients were split by median mRNA expression, and analysis was performed using the lung adenocarcinoma dataset irrespective of grade, stage, or previous treatment regimen. For enrichment analysis, data from lung adenocarcinoma patients with sequencing and RNA-seq information in the TCGA dataset were extracted from cbioportal.org. For correlation analysis, data from lung adenocarcinoma patients with RNA-seq information in the TCGA were extracted from cbioportal.org, and Spearman correlation test was applied to analyze the relationship between paired genes.
To identify genes regulated by LKB1 and CRTC2, datasets from RNA-seq and ChIP-seq experiments in A549-LKB1WT and A549-pBabe cells were cross-referenced. Forty-nine protein-coding genes were identified with mRNA expression and CRTC2 binding log2 fold change < −1. Among those 49 genes, we further analyzed for the presence of canonical CRE sites and mRNA expression in a panel of LKB1-mutant and LKB1 wild-type lung adenocarcinoma tumors (TCGA). Six genes were identified as putative LKB1/CRTC2/CREB targets in the lung adenocarcinoma. Pathway enrichment and Gene Ontology analyses of differentially expressed genes in A549 ID1 knockout and ID1 wild-type cells were done using CANCERTOOL (48).
Statistical analysis
Statistical analyses are described in each figure legend. For statistical test selection, distribution, fitting, and variance testing were determined to justify test selection. Most of the data had similar variance, and all data met the assumptions of the statistical test. For all statistical analyses, significance was accepted at the 95% confidence level. Data in all graphs are represented as means ± SD of biologic triplicates.
Supplementary Material
Acknowledgments
We are grateful to T. Sonntag for technical help during the revision of the manuscript. We thank M. Green for the CRISPR-Cas9 CRTC2 construct, F. Ventura for the ID1 reporter construct, and N. S. Grey for the YKL-05-096 SIK inhibitor. Funding: This work was supported by grants to M.M. from the National Institutes of Health (R01 DK083834), The Leona M. and Harry B. Helmsley Charitable Trust, the Clayton Foundation for Medical Research, and the J. W. Kieckhefer Foundation. This study was supported by grants to R.J.S. from the National Institutes of Health (R35CA220538 and P01CA120964) and by The Leona M. and Harry B. Helmsley Charitable Trust grant no. 2012-PG-MED002. L.R. was supported by a postdoctoral fellowship from the Tobacco-Related Disease Research Program (25FT-0006). R.U.S. was supported by a postdoctoral fellowship from the American Cancer Society (ACS no. 124183-PF-13-023-01- CSM). L.J.E. was supported by a postdoctoral fellowship from the American Cancer Society (PF-15-037-01-DMC). The Salk CCSG P30 CA014195 and The Leona M. and Harry B. Helmsley Charitable Trust supported the Functional Genomics Core and the Bioinformatics Core and provide partial support for K. McIntyre and the histology core of the Salk Institute. Author contributions: L.R. conceived the study, designed and performed the experiments, analyzed the data, and wrote the manuscript. M.M. conceived the study, provided suggestions for the experiments, and reviewed and edited the manuscript. R.J.S. provided suggestions for the experiments and feedback on the manuscript. L.R. performed all the experiments except as noted. R.U.S. performed the lung colonization experiments shown in Fig. 3, B to D. E.W. assisted with the RNA-seq and ChIP-seq experiments. M.G.H.C. generated the NSCLC cell lines with and without LKB1 add-back, and M.G.H.C. and L.J.E. performed the RNA-seq analysis in fig. S1A. W.-W.T. performed the ChIP-seq analysis in Fig. 4E. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The accession number for the ChIP-seq and RNA-seq data reported here is GSE128873. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The materials used in this research can be provided by M. Montminy and R. J. Shaw pending scientific review and a completed material transfer agreement. Requests for the materials should be submitted to M. Montminy or R. J. Shaw. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw6455/DC1
Fig. S1. mRNA expression and incidence of CRTC alterations in NSCLC.
Fig. S2. LKB1 down-regulates the expression of CREB target genes.
Fig. S3. CRTC2 promotes anchorage-independent growth in NSCLC cells.
Fig. S4. Genes regulated by LKB1 at the mRNA level that show LKB1-regulated binding of CRTC2 to their promoters in NSCLC.
Fig. S5. ID1 expression is regulated by CRTC2 and promotes anchorage-independent growth in NSCLC cells.
Fig. S6. Genes and molecular functions regulated by ID1 in NSCLC cells.
REFERENCES AND NOTES
- 1.Conkright M. D., Canettieri G., Screaton R., Guzman E., Miraglia L., Hogenesch J. B., Montminy M., TORCs: Transducers of regulated CREB activity. Mol. Cell 12, 413–423 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Iourgenko V., Zhang W., Mickanin C., Daly I., Jiang C., Hexham J. M., Orth A. P., Miraglia L., Meltzer J., Garza D., Chirn G.-W., McWhinnie E., Cohen D., Skelton J., Terry R., Yu Y., Bodian D., Buxton F. P., Zhu J., Song C., Labow M. A., Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 100, 12147–12152 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altarejos J. Y., Montminy M., CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonon G., Modi S., Wu L., Kubo A., Coxon A. B., Komiya T., O'Neil K., Stover K., El-Naggar A., Griffin J. D., Kirsch I. R., Kaye F. J., t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 33, 208–213 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Conkright M. D., Montminy M., CREB: The unindicted cancer co-conspirator. Trends Cell Biol. 15, 457–459 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Sakamoto K. M., Frank D. A., CREB in the pathophysiology of cancer: Implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 15, 2583–2587 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steven A., Seliger B., Control of CREB expression in tumors: From molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 7, 35454–35465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodón L., Gonzàlez-Juncà A., del Mar Inda M., Sala-Hojman A., Martinez-Sáez E., Seoane J., Active CREB1 promotes a malignant TGFβ2 autocrine loop in glioblastoma. Cancer Discov. 4, 1230–1241 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Johannessen C. M., Johnson L. A., Piccioni F., Townes A., Frederick D. T., Donahue M. K., Narayan R., Flaherty K. T., Wargo J. A., Root D. E., Garraway L. A., A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Best J. L., Amezcua C. A., Mayr B., Flechner L., Murawsky C. M., Emerson B., Zor T., Gardner K. H., Montminy M., Identification of small-molecule antagonists that inhibit an activator: Coactivator interaction. Proc. Natl. Acad. Sci. U.S.A. 101, 17622–17627 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie F., Li B. X., Kassenbrock A., Xue C., Wang X., Qian D. Z., Sears R. C., Xiao X., Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J. Med. Chem. 58, 5075–5087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B. X., Gardner R., Xue C., Qian D. Z., Xie F., Thomas G., Kazmierczak S. C., Habecker B. A., Xiao X., Systemic inhibition of CREB is well-tolerated in vivo. Sci. Rep. 6, 34513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkela T. P., Hardie D. G., Alessi D. R., LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw R. J., Lamia K. A., Vasquez D., Koo S.-H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C., The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komiya T., Coxon A., Park Y., Chen W.-D., Zajac-Kaye M., Meltzer P., Karpova T., Kaye F. J., Enhanced activity of the CREB co-activator Crtc1 in LKB1 null lung cancer. Oncogene 29, 1672–1680 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L., Getz G., Wheeler D. A., Mardis E. R., McLellan M. D., Cibulskis K., Sougnez C., Greulich H., Muzny D. M., Morgan M. B., Fulton L., Fulton R. S., Zhang Q., Wendl M. C., Lawrence M. S., Larson D. E., Chen K., Dooling D. J., Sabo A., Hawes A. C., Shen H., Jhangiani S. N., Lewis L. R., Hall O., Zhu Y., Mathew T., Ren Y., Yao J., Scherer S. E., Clerc K., Metcalf G. A., Ng B., Milosavljevic A., Gonzalez-Garay M. L., Osborne J. R., Meyer R., Shi X., Tang Y., Koboldt D. C., Lin L., Abbott R., Miner T. L., Pohl C., Fewell G., Haipek C., Schmidt H., Dunford-Shore B. H., Kraja A., Crosby S. D., Sawyer C. S., Vickery T., Sander S., Robinson J., Winckler W., Baldwin J., Chirieac L. R., Dutt A., Fennell T., Hanna M., Johnson B. E., Onofrio R. C., Thomas R. K., Tonon G., Weir B. A., Zhao X., Ziaugra L., Zody M. C., Giordano T., Orringer M. B., Roth J. A., Spitz M. R., Wistuba I. I., Ozenberger B., Good P. J., Chang A. C., Beer D. G., Watson M. A., Ladanyi M., Broderick S., Yoshizawa A., Travis W. D., Pao W., Province M. A., Weinstock G. M., Varmus H. E., Gabriel S. B., Lander E. S., Gibbs R. A., Meyerson M., Wilson R. K., Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill R. K., Yang S.-H., Meerzaman D., Mechanic L. E., Bowman E. D., Jeon H.-S., Roy Chowdhuri S., Shakoori A., Dracheva T., Hong K.-M., Fukuoka J., Zhang J.-H., Harris C. C., Jen J., Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene 30, 3784–3791 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji H., Ramsey M. R., Hayes D. N., Fan C., McNamara K., Kozlowski P., Torrice C., Wu M. C., Shimamura T., Perera S. A., Liang M.-C., Cai D., Naumov G. N., Bao L., Contreras C. M., Li D., Chen L., Krishnamurthy J., Koivunen J., Chirieac L. R., Padera R. F., Bronson R. T., Lindeman N. I., Christiani D. C., Lin X., Shapiro G. I., Jänne P. A., Johnson B. E., Meyerson M., Kwiatkowski D. J., Castrillon D. H., Bardeesy N., Sharpless N. E., Wong K. K., LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Shackelford D. B., Shaw R. J., The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sundberg T. B., Liang Y., Wu H., Choi H. G., Kim N. D., Sim T., Johannessen L., Petrone A., Khor B., Graham D. B., Latorre I. J., Phillips A. J., Schreiber S. L., Perez J., Shamji A. F., Gray N. S., Xavier R. J., Development of chemical probes for investigation of salt-inducible kinase function in vivo. ACS Chem. Biol. 11, 2105–2111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canettieri G., Coni S., Della Guardia M., Nocerino V., Antonucci L., Di Magno L., Screaton R., Screpanti I., Giannini G., Gulino A., The coactivator CRTC1 promotes cell proliferation and transformation via AP-1. Proc. Natl. Acad. Sci. U.S.A. 106, 1445–1450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Vera L., Fischer W. H., Montminy M., The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasorella A., Benezra R., Iavarone A., The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 14, 77–91 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Roschger C., Cabrele C., The Id-protein family in developmental and cancer-associated pathways. Cell Commun. Signal 15, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pillai S., Rizwani W., Li X., Rawal B., Nair S., Schell M. J., Bepler G., Haura E., Coppola D., Chellappan S., ID1 facilitates the growth and metastasis of non-small cell lung cancer in response to nicotinic acetylcholine receptor and epidermal growth factor receptor signaling. Mol. Cell. Biol. 31, 3052–3067 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escoubas C. C., Silva-Garcia C. G., Mair W. B., Deregulation of CRTCs in aging and age-related disease risk. Trends Genet. 33, 303–321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romero-Lanman E. E., Pavlovic S., Amlani B., Chin Y., Benezra R., Id1 maintains embryonic stem cell self-renewal by up-regulation of Nanog and repression of Brachyury expression. Stem Cells Dev. 21, 384–393 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Ying Q.-L., Nichols J., Chambers I., Smith A., BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Gupta G. P., Perk J., Acharyya S., de Candia P., Mittal V., Todorova-Manova K., Gerald W. L., Brogi E., Benezra R., Massagué J., ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. U.S.A. 104, 19506–19511 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stankic M., Pavlovic S., Chin Y., Brogi E., Padua D., Norton L., Massagué J., Benezra R., TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 5, 1228–1242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minn A. J., Gupta G. P., Siegel P. M., Bos P. D., Shu W., Giri D. D., Viale A., Olshen A. B., Gerald W. L., Massagué J., Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ponz-Sarvisé M., Nguewa P. A., Pajares M. J., Agorreta J., Lozano M. D., Redrado M., Pio R., Behrens C., Wistuba I. I., García-Franco C. E., García-Foncillas J., Montuenga L. M., Calvo A., Gil-Bazo I., Inhibitor of differentiation-1 as a novel prognostic factor in NSCLC patients with adenocarcinoma histology and its potential contribution to therapy resistance. Clin. Cancer Res. 17, 4155–4166 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Kang Y., Chen C.-R., Massagué J., A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 11, 915–926 (2003). [DOI] [PubMed] [Google Scholar]
- 34.López-Rovira T., Chalaux E., Massagué J., Rosa J. L., Ventura F., Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J. Biol. Chem. 277, 3176–3185 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Patel D., Morton D. J., Carey J., Havrda M. C., Chaudhary J., Inhibitor of differentiation 4 (ID4): From development to cancer. Biochim. Biophys. Acta 1855, 92–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao C., Gao R., Zhang M., Amelio A. L., Fallahi M., Chen Z., Gu Y., Hu C., Welsh E. A., Engel B. E., Haura E. B., Cress W. D., Wu L., Zajac-Kaye M., Kaye F. J., Role of LKB1-CRTC1 on glycosylated COX-2 and response to COX-2 inhibition in lung cancer. J. Natl. Cancer Inst. 107, 358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z., Li J.-L., Lin S., Cao C., Gimbrone N. T., Yang R., Fu D. A., Carper M. B., Haura E. B., Schabath M. B., Lu J., Amelio A. L., Cress W. D., Kaye F. J., Wu L., cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J. Clin. Invest. 126, 2267–2279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng Y., Wang Y., Wang Z., Fang Z., Li F., Gao Y., Liu H., Xiao T., Zhou Y., Zhai Q., Liu X., Sun Y., Bardeesy N., Wong K.-k., Chen H., Xiong Z.-q., Ji H., The CRTC1-NEDD9 signaling axis mediates lung cancer progression caused by LKB1 loss. Cancer Res. 72, 6502–6511 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhary J., William G., Salvador R., A novel small molecule inhibitor of Id proteins (AGX-51) blocks cell survival in vitro and diminishes angiogenesis and tumor growth in vivo. FASEB J. 23, 761.4 (2009). [Google Scholar]
- 40.Henke E., Perk J., Vider J., de Candia P., Chin Y., Solit D. B., Ponomarev V., Cartegni L., Manova K., Rosen N., Benezra R., Peptide-conjugated antisense oligonucleotides for targeted inhibition of a transcriptional regulator in vivo. Nat. Biotechnol. 26, 91–100 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Shackelford D. B., Abt E., Gerken L., Vasquez D. S., Seki A., Leblanc M., Wei L., Fishbein M. C., Czernin J., Mischel P. S., Shaw R. J., LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang M., Pak M. L., Chamberlain L., Xing W., Yu H., Green M. R., The CREB coactivator CRTC2 is a lymphoma tumor suppressor that preserves genome integrity through transcription of DNA mismatch repair genes. Cell Rep. 11, 1350–1357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sonntag T., Moresco J. J., Vaughan J. M., Matsumura S., Yates J. R. III, Montminy M., Analysis of a cAMP regulated coactivator family reveals an alternative phosphorylation motif for AMPK family members. PLOS ONE 12, e0173013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravnskjaer K., Kester H., Liu Y., Zhang X., Lee D., Yates J. R. III, Montminy M., Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 26, 2880–2889 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma , Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 14, 822–827 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chitale D., Gong Y., Taylor B. S., Broderick S., Brennan C., Somwar R., Golas B., Wang L., Motoi N., Szoke J., Reinersman J. M., Major J., Sander C., Seshan V. E., Zakowski M. F., Rusch V., Pao W., Gerald W., Ladanyi M., An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 28, 2773–2783 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Győrffy B., Surowiak P., Budczies J., Lánczky A., Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLOS ONE 8, e82241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cortazar A. R., Torrano V., Martin-Martin N., Caro-Maldonado A., Camacho L., Hermanova I., Guruceaga E., Lorenzo-Martín L. F., Caloto R., Gomis R. R., Apaolaza I., Quesada V., Trka J., Gomez-Muñoz A., Vincent S., Bustelo X. R., Planes F. J., Aransay A. M., Carracedo A., CANCERTOOL: A visualization and representation interface to exploit cancer datasets. Cancer Res. 78, 6320–6328 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw6455/DC1
Fig. S1. mRNA expression and incidence of CRTC alterations in NSCLC.
Fig. S2. LKB1 down-regulates the expression of CREB target genes.
Fig. S3. CRTC2 promotes anchorage-independent growth in NSCLC cells.
Fig. S4. Genes regulated by LKB1 at the mRNA level that show LKB1-regulated binding of CRTC2 to their promoters in NSCLC.
Fig. S5. ID1 expression is regulated by CRTC2 and promotes anchorage-independent growth in NSCLC cells.
Fig. S6. Genes and molecular functions regulated by ID1 in NSCLC cells.