

Abstract
Seventy-one 7-oxycoumarins, 66 synthesized and 5 commercially sourced, were tested for their ability to inhibit growth in murine PAM212 keratinocytes. Forty-nine compounds from the library demonstrated light-induced lethality. None was toxic in the absence of UVA light. Structure-activity correlations indicate that the ability of the compounds to inhibit cell growth was dependent not only on their physiochemical characteristics, but also on their ability to absorb UVA light. Relative lipophilicity was an important factor as was electron density in the pyrone ring. Coumarins with electron withdrawing moieties – cyano and fluoro at C3 – were considerably less active while those with bromines or iodine at that location displayed enhanced activity. Coumarins that were found to inhibit keratinocyte growth were also tested for photo-induced DNA plasmid nicking. A concentration-dependent alteration in migration on neutral gels caused by nicking was observed.
Keywords: 7-hydroxycoumarins, 7-oxycoumarins, furocoumarins, psoralens, methoxsalen, 8-MOP, phototoxicity, PAM212 keratinocytes, DNA photo-damage
Graphical Abstract
Forty-nine members of a set of seventy-one structurally-diverse 7-oxycoumarins bearing alkyl, alkenyl, alkynyl, halo, nitro, cyano, trifluoromethyl, and amino residues were found to display photo-induced toxicity (UVA light) against murine PAM212 keratinocytes in culture.
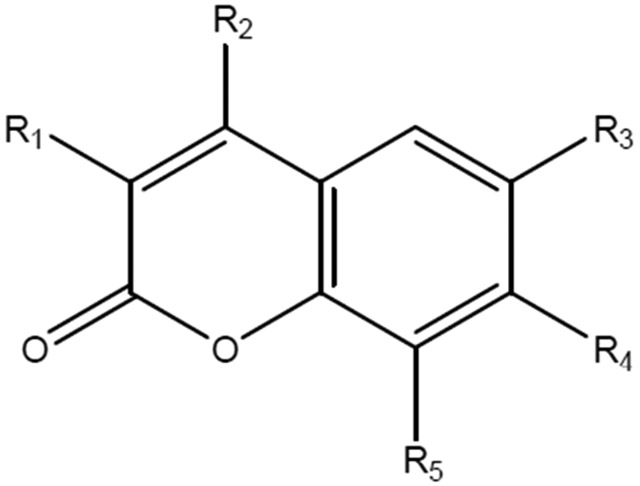
Structure-activity effects were evident and depended on halogen content and ring position, electron-withdrawing moieties at position R1, and relative lipophilicity of the alkenyl and alkynyl side chains at R3 and R4. A photo-induced DNA plasmid nicking was observed in phototoxic members of the set.
Introduction:
Simple monomeric 7-hydroxycoumarins (or their glycosides) as well as linear fused-ring furocoumarins, a.k.a, psoralens, are widely distributed in many dicotyledonous plant families, including the Apiaceae (Umbellifereae), Asteraceae, Fabiaceae, Moraceae, Rosaceae, Rubiaceae, Rutaceae and Solanaceae as well as in monocotyledonous plants, such as Gramineae, palms, lilies, and orchids(1) In traditional and in established medicine, these compounds have found broad pharmaceutical utility as anti-microbials, immune-modulators, anti-inflammatories, therapeutics for hyperproliferative skin diseases, anti-virals, and cancer chemotherapeutics.(2,3)
8-Methoxypsoralen, a.k.a., methoxsalen or 8-MOP, and many similar furocoumarins, have an especially long and extensive literature as phototoxin/phototherapeutics.(4-6) However, the story on phototoxicity of the 7-hydroxycoumarins is more limited and ambiguous. Several researchers have concluded that “simple coumarins (umbelliferone), show no phototoxic effects”(4, 7-9) But, nevertheless, a number of coumarins – substituted with electron donating groups – were reported to be photo-labile and undergoing a 2+2 cycloaddition (dimerization) at the C3-C4 double bond(10) Mousavi demonstrated a modest phototoxicity (IC50 476 μM) for umbelliferone (7-hydroxycoumarin) against the MCF-7 breast carcinoma cell line(11) In addition, Goard has shown that a conjugate containing 6-bromo-7-hydroxycoumarin, attached at C4 to the antibiotic anisomycin, served as a photo-activated inhibitor of protein biosynthesis better than did the parent antibiotic itself against CHO or HEK293 cells(12) Indeed, some coumarins can display phototoxic effects while others can display a structure-specific avidity for DNA binding although not by intercalation. Coumarin, esculetin, and a set of pyridyl-substituted coumarins formed reversible complexes with DNA; physical measurements supported their binding loci to be in the minor groove by a weak hydrogen-bonding association of the DNA with the coumarin carbonyl(13-15) Most psoralens, however, intercalate DNA and photo-crosslink at the pyrimidine bases(16) Thus, coumarins and psoralens exert their pharmacology by different mechanisms.
The psoralens and the 7-hydroxycoumarins are related in several ways. Virtually all the laboratory synthetic pathways to the linear furocoumarins pass through the functionalized 7-hydroxycoumarins as the penultimate precursors of the resulting three-ring psoralens(16) Nature also uses these same 7-hydroxycoumarins on the biosynthetic pathway to the natural product psoralens(16, 17) Besides sharing structural features and a similar synthetic pathway, the psoralens and the 7-hydroxycoumarins also share comparable absorption and qualitatively similar fluorescence behaviors. Methoxsalen. which absorbs at 220, 250, and 305 nM, can be excited at 250 nm, and thereafter gives a weak fluorescence emission at 505 nM with a quantum yield of 0.002 (18) Similarly, 7-hydroxycoumarin also has broad absorption bands at 220, 255, and 295 with a λmax of 330 nm and weak shoulder end-absorption at 370 nm. However, when excited at 330 nm it emits such intense blue fluorescence at 455 nm (with an impressive quantum yield of 0.81) that the compound finds wide use as a fluorescent whitener in laundry detergents(19-21) Because of their acidic phenolic –OH, 7-hydroxycoumarins show a range of pH-dependent excitations and emissions with quantum yields approaching unity above pH 9.4(21). The psoralens show no such pH-dependency.
Because the phenolic-capped O-acylated and the O-glycosylated 7-hydroxycoumarins – like the corresponding psoralens – display almost no fluorescence, they can serve as useful fluorogenic substrates for quantifying enzymatic activity (hydrolytic and glycolytic).(22-23) Thus, enzymatic liberation of the highly fluorescent free phenol provides a readily measured marker; molecular orbital calculations have been advanced to explain this phenomenon(24)
Intrigued by the structural commonalities, the spectral dissimilarities and the ambiguous reports on the photobiology of umbelliferones, a study of the phototoxic effects of seventy-one 7-hydroxycoumarins, their ethers and esters, (see Figure 1 and Table 1) was undertaken. It is well recognized that ultraviolet light irradiation in combination with chemical photosensitizers, such as psoralens, has considerable biological activity in the skin. In addition to causing erythema, sunburn, and tanning, the photoactivated psoralens cause alterations in keratinocyte growth and differentiation(25) Indeed, because of these biological responses, the psoralens have been used to treat many skin disorders, particularly, eczema and psoriasis(26) Many derivatives of psoralens have been evaluated in phototoxicity assays using keratinocyte growth inhibition to evaluate biological activity. In the present studies we used a mouse keratinocyte cell line to evaluate phototoxicity of a number of structural analogs of the 7-oxycoumarins. PAM212 cells in culture are a common mouse keratinocyte line often used to investigate mechanisms of drug action since results with these cells can lead directly to in vivo mouse models and subsequently to human keratinocytes as lead pharmaceuticals are advanced in preclinical development.
Figure 1.

Effects of coumarins on keratinocyte growth with and without UVA light. PAM212 cells were grown in tissue culture dishes and treated with increasing concentrations of the coumarins. After 30 min at 37°C, cells were treated with (closed symbols) or without (open symbols) UVA light. Cells were then refed with fresh growth medium. After 4-5 days, cells were removed from the dishes and counted on a Coulter Counter. Data are presented as percent inhibition of cell growth relative to controls. Data represent means ± SEM (n = 3). In a typical experiment, control plates contained 5.5 × 104 cells.
Table 1.
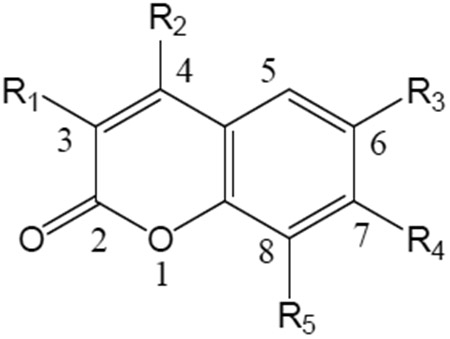
Effects of 7-Oxycoumarins plus UVA light on the growth of PAM212 keratinocytes
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | Source | Melting point (°C) |
R1 | R2 | R3 | R4 | R5 | IC50 (μM) |
| 1 | A | 228-230(*) | H | H | H | -OH | H | >100 |
| 2 | S | 145-146 | H | Me | H | -OC(Me)2C≡CH | Me | 4 |
| 3 | S | 93-94 | H | Me | H | -OCH2CH=CMe2 | Me | 10 |
| 4 | S | 83-84 | H | Me | H | -O(CH2)5Me | Me | 10 |
| 5 | S | 106-108 | H | Me | H | -O(CH2)3Me | Me | 10 |
| 6 | S | 103-104 | H | Me | H | -O(CH2)2Me | Me | 15 |
| 7 | S | 114-116 | H | Me | H | -OCH2CH=CHMe | Me | 33 |
| 8 | S | 166-167(31) | H | Me | H | -OMe | Me | 40 |
| 9 | S | 139-141(32) | H | Me | H | -OCH2C≡CH | Me | 41 |
| 10 | S | 87-88.5 | H | Me | H | -OCH2C≡C(CH2)4Me | Me | 60 |
| 11 | S | 80-82 | H | Me | H | -O(CH2)7Me | Me | 60 |
| 12 | B | 260.5-262 | H | Me | H | -OH | Me | 100 |
| 13 | S | 158-159.5(33) | H | Me | I | -OCH2CH=CH2 | Me | 1 |
| 14 | S | 146-147 | H | Me | I | -OCH2CH=C(Me)2 | Me | 5 |
| 15 | S | 124.5-126 | H | Me | I | -O(CH2)2Me | Me | 10 |
| 16 | S | 219-220(33) | H | Me | I | -OH | Me | 18 |
| 17 | S | 145.5-147.5 | H | Me | NH2 | -OCH2CH=CH2 | Me | 5 |
| 18 | S | 229.5-231.5 | H | Me | NO2 | -OH | Me | >100 |
| 19 | S | 199-200 | -CH2CH=C(Me)2 | Me | H | -OH | Me | 12 |
| 20 | S | 165-167 | H | Me | -CH2CH=C(Me)2 | -OH | Me | 1 |
| 21 | S | 168-170(34) | H | Me | -CH2CH=CH2 | -OH | Me | 10 |
| 22 | S | 175-176 | H | Me | -CH(Me)CH=CH2 | -OH | Me | 14 |
| 23 | S | 158-160 | H | Me | -CH2CH=CHMe | -OH | Me | 20 |
| 24 | S | 149.5-150(35) | H | Me | -CH2CH=CH2 | -OAc | Me | 21 |
| 25 | C | 159-161 | H | Me | H | -OMe | H | 60 |
| 26 | S | 101-102(36) | H | Me | H | -OCH2CH=CH2 | H | 10 |
| 27 | S | 254-256(32) | H | Me | H | -OH | I | >100 |
| 28 | S | 151.5-153 | H | Me | H | -OCH2-CH=C(CH3)2 | I | 22 |
| 29 | S | 129-130 | H | Me | H | -O(CH2)2Me | I | 50 |
| 30 | S | 194-195 | H | Me | H | -OMe | I | 70 |
| 31 | D | 119-120 | H | H | H | -OMe | OMe | 33 |
| 32 | C | 118-121 | H | H | H | -OMe | H | 50 |
| 33 | S | 182-183 | H | CF3 | -CH2CCI=CH2 | -OH | Me | 20 |
| 34 | S | 143-144.5 | H | CF3 | H | -OCH2(CO)Me | Me | 40 |
| 35 | S | 112-113 | H | CF3 | H | -O(CH2)2Me | Me | 100 |
| 36 | S | 76-77 | H | CF3 | H | -OCH2CH=CH2 | H | >200 |
| 37 | S | 197-198 | H | CF3 | H | -OH | Me | >200 |
| 38 | S | 160.5-161.5 | H | CF3 | H | -OCH2C≡CH | Me | >300 |
| 39 | S | 97-98 | H | CF3 | H | -OCH2CH2CH3 | H | >100 |
| 40 | S | 151-152 | H | CF3 | -CH2CH=CH2 | -OAc | Me | >100 |
| 41 | S | 112.5-113.5(37) | H | CF3 | H | -OMe | H | >100 |
| 42 | S | 110.5-112 | H | CF3 | H | -OCH2C≡CH | H | >100 |
| 43 | S | 174-175 | H | CF3 | -CH2CH=CH2 | -OH | Me | >300 |
| 44 | S | 114.5-115.5 | H | CF3 | H | -OCH2CH=CH2 | Me | >300 |
| 45 | S | 109.5-111(38) | H | CF3 | H | -OMe | Me | >300 |
| 46 | S | 158-159 | H | CF3 | H | -OCH2(CO)Me | H | >300 |
| 47 | S | 186-188(39) | H | CF3 | H | -OH | H | >300 |
| 48 | S | 149.5-150(35) | H | Me | -CH2CH=CH2 | -OAc | Me | 21 |
| 49 | S | 198-198.5(d)(40,41) | Br | Me | -CH2(CHBr)CH2Br | -OH | Me | 1 |
| 50 | S | 158-160(40,41) | Br | Me | -CH2CH=CH2 | -OH | Me | 5 |
| 51 | S | 269-271 (42) | Br | Me | H | -OH | Me | 10 |
| 52 | S | 143-143.5 | Br | H | H | -OCH2CH=CH2 | Me | 2 |
| 53 | S | 131.5-133 | Br | H | H | -OCH2CH=CMe2 | Me | 5 |
| 54 | S | 139.5-140 | Br | H | H | OCH2CH2CH3 | Me | 5 |
| 55 | S | 181-182 | Br | H | H | -OCH2C≡CH | Me | 10 |
| 56 | S | 154-155 | Br | H | H | -OMe | Me | 13 |
| 57 | S | 209-211(42) | Br | H | H | -OH | Me | 20 |
| 58 | S | 147-148 | Br | Me | H | -OCH2CH=CH2 | Me | 5 |
| 59 | S | 231-232 | Br | Me |  |
-OAc | Me | 20 |
| 60 | S | 158-160(41) | Br | Me | -CH2(CHBr)CH2Br | -OAc | Me | 48 |
| 61 | S | 160-161 (41) | Br | Me | -CH2CH=CH2 | -OAc | Me | 140 |
| 62 | S | 221-223(40,41) | CN | Me | -CH2(CHBr)CH2Br | -OH | Me | 3 |
| 63 | S | 170-171 (41) | CN | Me | -CH2(CHBr)CH2Br | -OAc | Me | 48 |
| 64 | S | 234-235(40,41) | CN | Me | -CH2CH=CH2 | -OH | Me | >300 |
| 65 | S | 120-124(d)(40,41) | CN | Me | -CH2CH=CH2 | -OAc | Me | >300 |
| 66 | S | 172-173(40,41) | CN | Me | H | -OCH2CH=CH2 | Me | >300 |
| 67 | S | 175-176(40,41) | F | Me | -CH2(CHBr)CH2Br | -OH | Me | 120 |
| 68 | S | 192-193(43) | F | Me | H | -OH | Me | >200 |
| 69 | S | 117-118(40,41) | F | Me | H | -OCH2CH=CH2 | Me | >300 |
| 70 | S | 163-164(40,41) | F | Me | -CH2CH=CH2 | -OH | Me | >300 |
| 71 | S | 228-230(d) | I | Me | H | -OH | Me | 61 |
: Compound 1 is umbelliferone also known as 7-hydroxycoumarin. An IC50 expressed as > (number) means no activity was observed up to that value, usually the solubility limit of the compound for the assay’s conditions. A = Sigma-Aldrich; B = Regis Chemical; C = TCI America Inc.; D = Elder (Valeant) Pharmaceuticals. S means that the compound was synthesized for these studies either following an existing procedure or a new one developed for that purpose, which is described in detail in the experimental part. Published methods for specific coumarins are indicated by references provided in the table and in the experimental part. Obvious decomposition upon melting is noted by (d).
Results and Discussion:
While coumarins including the 7-hydroxy compounds, their ethers and alkylated analogs, have shown broad promise as anti-inflammatories, anti-tumor, anti-HIV, anticoagulant, antimicrobial, antioxidant, hepatoprotective, anti-pyretic, anti-hypertensive, and anti-convulsant agents, they nevertheless display complex pharmacologies, which are truly difficult to correlate.(27) The problem is that for any given compound library, no validated quantitative structure-linked comparative principle has yet to be found although logP, electronic surface analysis, pKa, and size have all been considered. In coumarin papers, structure-activity relations are phrased in general terms and even then, any given set displays many exceptions. For example, “for TNF-α inhibitory effects...the C6 position must contain a halogen or methoxy…hydrophobic groups at C3 are favored…small aliphatic groups at C4 are optimal.”(27) Similar general statements are reported for anti-HIV coumarins(28) and for anti-fungal coumarins.(29) For instance, Kostova noted the anti-HIV activities for substituents on C4, were ”methyl>H>propyl>benzyl” but he similarly noted that “to describe structure-activity relationships on the potent anti-HIV coumarin derivatives is not an easy task.” The anti-fungal SARs for a series of 24 diverse coumarins were equally problematic. Guerra reported that all ethers of 6-hydroxycoumarin were inactive in his anti-fungal assay (the parent phenol did have modest activity) but the ethers of 7-hydroxycoumarin were active and with declining effect as the 7-alkoxy chain lengthened.(29) No other correlations were made.
In the present studies, PAM212 mouse keratinocytes were used to evaluate the biological activity of the coumarins.(30) In this assay the PAM212 cells were treated with the coumarins and then exposed to UVA light. Control cells were treated with the coumarin without UVA light. The cells were then allowed to grow for 4-5 days and then analyzed for growth. Figure 1 shows typical keratinocyte growth inhibition experiments with several of the coumarins. Structure activity analysis for the photobiological activity of the coumarins is shown in Table 1. Without UVA light treatment, no significant growth inhibition by the coumarins is evident. However, following UVA light treatment, there is a marked inhibition of cell growth. Table 1 summarizes the concentration of the coumarins inhibiting growth by 50% (IC50) for the compounds in Figure 1 as well as other coumarins in the data set compounds (1–71).
Based on these data (Table 1), lipophilicity is an important factor. Compare for example the parent 7-hydroxycoumarins and their corresponding 7-alkoxy ethers. Dimethylumbelliferone, (12) logP = 2.1 displayed a modest IC50 of 100 μM while all its ethers (2 to 11) displayed substantially improved activity. The lower chain length ethers, compounds 2 to 9, (whose cLogP values fell between 3.2 and 3.9 were considerably more active than were the longer chain length ethers (10 and 11) for which cLogPs of > 5.8 reflected an enhanced – and apparently less optimal – lipophilicity.
Furthermore, when the alkenyl or alkyl moiety was rearranged from the 7-position (where it was an ether) onto either ring positions 3- or 6- (e.g., 19–24), all of these 7-hydroxycoumarins were more active (IC50’s 1–21 μM) than the parent dimethylumbelliferone (12) from which they were synthetically derived. In addition, all these 3- or 6- alkylated 7-hydroxycoumarins were more lipophilic than 12 with their cLogPs falling between 3.6 and 4.6.
Similarly, in the 6-iodo series, the parent phenol, 6-iododimethylumbelliferone (16 with clogP = 3.38) possessed an IC50 of 18 μM – much more photo-active than its nor-iodo, the dimethylumbelliferone (12, logP = 2.10) – but even here its more lipophilic ethers the 6-iodo-7-alkoxy derivatives (viz., 13–15) with clogPs of 4.3 to 5.4, were all more active. The 4,8-dimethyl-6-nitro-7-hydroxycoumarin (18) is a member of the family of acidic ortho-nitrophenols (in general pKa’s average 7.2 for o-nitrophenols and clogPs average 1.7), which are extensively ionized at physiological pH and thus might be expected to display poor membrane permeability. At a cLogP of 2.39 compound 18 possesses no photocytotoxicity up to its solubility maximum of 100 μM. On the other hand, the 4,8-dimethyl-6-amino-7-allyloxycoumarin (17), a member of the family of weakly basic aromatic ortho amino ethers, is unionized at physiological pH and in this case with clogP = 2.94 displays an IC50 of 5 μM.
In the 8-iodo series (27-30) the parent phenol, 4-methyl-8-iodo-7-hydroxycoumarin (27, cLogP = 2.93) was inactive while its more lipophilic 7-alkoxy variants (28, 29, and 30 with cLogPs of 4.81, 4.16, and 3.10 respectively) had IC50’s of 22, 50, and 70 μM respectively. Here the activity of the alkoxy compounds does increase with the increasing lipophilicity as provided by the carbon chain but since no analog has more than a five-carbon side chain a maximum lipophilicity on the Hansch curve has not been attained.
In keeping with the trend that the alkoxys are more photo-active than their parent phenols, as noted umbelliferone (7-hydroxycoumarin = 1) showed no activity in the screen; but its O-methyl ether (7-methoxycoumarin = 32) had an IC50 of 50 μM while its bis-methyl ether (7,8-dimethoxycoumarin = 31) had an IC50 of 33 μM.
The two families of fluoro-substituted coumarins constitute diverse classes. The first of these, those bearing a trifluoromethyl at R2, position 4 in traditional coumarin numbering, (e.g., 33-47), were largely inactive. The two parent phenols, compound 37 (4-trifluoromethyl-8-methyl-7-hydroxycoumarin) and the nor-methyl compound 47 (4-trifluoromethyl-7-hydroxycoumarin), were inactive at their solubility maxima. The traditional alkyl, alkenyl, or alkynyl ethers of 47 (e.g., 36, 39, 41, 42, and 46) which in the other 7-hydroxy structural families did impart significant activity over their less active parent phenols, were equally inactive in this particular 4-trifluoromethyl family (the one with no 8-methyl moiety). However, three analogs of phenol 37 (the 4-trifluoromethyl-8-methyl-7-hydroxy system), were active (viz., 33-35). Is this observed activity an effect of increased lipophilicity or is it derived from an electronic effect? The answer is probably both since two of the active derivatives (33 with cLogP = 4.09 and 35 with cLogP = 4.20) were indeed much more lipophilic than 37 (cLogP = 3.00), the inactive parent phenol, but the 7-O-acetonyl analog (34) displayed equivalent lipophilicity (clogP = 2.50) to the parent and yet a substantially enhanced 40 μM activity. There may be an explanation for the activity (IC50 = 20μM) seen in 33 with its pendant chloro-containing side chain. Platz has claimed an augmented photo-induced anti-virual activity for sensitizable heterocyclics with halogen-bearing side chains(44).
The second family of fluoro-substituted coumarins, compounds 67 to 70 bearing an F at R1, position 3 in the traditional coumarin numbering, was similarly an inactive class. Not only the parent phenol (3-fluoro-4,8-dimethyl-7-hydroxycoumarin, 68) but also the O-allyl and C-allyl analogs displayed no activity up to their solubility maxima. One exception, compound 67, with a 2,3-dibromopropyl sidechain, was modestly active at 120 μM probably by a free radical effect to be discussed later.
We are not the first to observed that fluoro-attachment can be a deactivating structural feature in otherwise promising umbelliferones and other coumarins as candidate therapeutics. A CF3 attachment at ring positions 3 or 4 (R1 and R2 in the generalized coumarin structure, see Figure on Table 1) was found to abolish otherwise significant anti-inflammatory activity in a study of more than 50 diverse coumarins(27) In addition, fluorine attachment to the ring carbons of 7-hydroxycoumarins has been found to impart a photostability, an inertness to photo-reactivity and a resistance to fluorescence quenching; these properties all being problematic to the successful use of non-fluorinated umbelliferones as fluorophors in biological systems(22) The photo-inactivity of the two types of fluoro coumarins in this study is therefore to be expected.
Not surprisingly, the 3-cyano coumarins (e.g., 62-66) show an activity profile similar to the trifluoromethyl and fluoro coumarins. The cyano, the trifluoromethyl, and the fluoro groups are all electronically-deactivating entities with closely related electron withdrawing sigmas of 0.56, 0.43, and 0,34(45) Very few members of these three electron withdrawing families display phototoxicity and those that do are the side-chain brominated analogs. As in the 3-fluoro class (see 67) so in the 3-cyano class only 62 and 63 (IC50’s 3 and 48 μM), with their 2,3-dibromopropyl side chains, demonstrated the photosensitization effect.
One unique structure-activity correlation is that all fourteen 3-bromo- and 3-iodo-coumarins (49-60 and 71) in this study displayed significant photosensitizing effects with eight of the set having IC50’s of 10 μM or less. The heavy-halogen class of coumarins is the most photo-active. The significant phototoxic activity observed in the 3-brominated /iodinated coumarins (viz. 49- 61 and 71) as well as in those derived from dibrominating the C6 (R3) allyl (viz. 49, 60, 62, 63, and 67), may arise from the well-known photo-homolytic cleavage of these labile carbon-halogen bonds(46) Not only is there a synthetically useful photo-radical cleavage which uses the 3-brominated/iodinated coumarins as starting materials for a coupling reaction(47) but also such 3-bromo/iodo species have demonstrated potent photo-viricidal properties(48) Park and colleagues advanced a theory in which 3-bromoumbelliferone – with a 7-alkoxy side chain to provide affinity to a viral nucleic acid strand – was photolyzed to a free radical at C3 which subsequently effected a cleavage of that strand(48) While other non-fluoro halogens (Cl, Br, and I) attached at C3 on the coumarin ring were claimed efficacious as photo-activated radical producers(48), Platz also noted that halogens on an aliphatic side chain off the coumarin ring were equally efficacious as light activated anti-viral agents(44) In the set of coumarins (see 49, 60, 62, 63 and 67), it was demonstrated that the 2,3-dibromopropyl side chain substantially augmented the phototoxicity of these dibromoalkyl compounds over that of close structural analogs lacking that moiety.
In summary, we established structural trends within the set of 71 coumarin derivatives presented herein. None of the compounds displayed growth inhibitory activity in the absence of light but 49 of them did in the presence of UVA light. Lipophilicity played a key role in the activity of those compounds. Within structural subsets, it was observed that the more lipophilic derivatives were the most active ones as long as the lipophilicity of said compounds did not exceed a value beyond which the activity started to diminish again. The addition of one or multiple halogens, such as bromo, iodo and chloro, resulted in enhanced activity; whether those substitutions occurred at the traditional ring positions (C3, C6 and C8) or on a more distal side chain. However, fluorine substitution, in the form of a single fluorine atom or a trifluoromethyl group, typically resulted in partial or complete loss of activity.
As psoralens are known to intercalate into DNA, and a few psoralens are known to bind to the minor groove of DNA(13-15), we next determined if the photo-activated coumarins had the capacity to bind and damage DNA. For these studies, we used a plasmid DNA nicking and unwinding assay(30) Results obtained in this study demonstrate that there is a structure-specific photolytic interaction of coumarins and DNA. If the candidate coumarin is able to nick and linearize the plasmid upon light exposure, the change in electrophoretic migration of the altered DNA is readily evident, (see examples in Figure 2 and Table 2). A set of five structurally diverse coumarins was selected from a group that was found to strongly inhibit keratinocyte growth (see Figure 1) but which ranged from halogenated variants, to 7-hydroxyls to 7-allyloxys. There was little or no damage to the plasmid DNA by any of these compounds in the absence of UVA. However, with each of the coumarins (14, 48, 58, 62 and 71), UVA light treatment caused concentration-dependent alteration in migration in neutral gels caused by nicking, which resulted in a faster migrating supercoiled plasmid when compared to slower migrating linear or open circular forms of the plasmid. Generally similar results were evident when the plasmid was treated with the coumarins and UVA light and then analyzed in alkaline gels (14, 48 and 71). These data indicate that the photo-activated coumarins can modify the DNA plasmid. A direct structure activity correlation between the phototoxicity results (Table 1) and the plasmid nicking/unwinding results (Figure 2 and Table 2) would not be expected, nor was it observed. The phototoxicity study is a whole cell assay with attendant complications induced by membrane transport, the presence of growth media, and metabolism, while photo nicking is performed extracellularly in simple buffer solution. The C7 hydroxyl molecules 71 and 69 as well as the pre-hydroxyl (the readily hydrolyzed C7 acetoxy molecule) 48 all displayed nearly five times greater linearization of the plasmid compared to the 7-allyloxy ethers 14 and 58 although the latter were among the most phototoxic compounds of the set .
Figure 2.

Ability of coumarins and UVA light to damage plasmid DNA. Plasmid DNA was treated with increasing concentrations of the coumarins. Lane L shows products from the digestion of the plasmid with restriction endonuclease EcoRI to linearize the DNA (100 ng); in native gels, the linearized DNA migrates as one double-stranded band, whereas, in denaturing gels, it migrates as two well-separated single strands. Lane S shows untreated plasmid DNA (75 ng); in native gels, the intact plasmid DNA migrates mainly as one supercoiled double-stranded band, whereas in denaturing gels, it migrates as two closely migrating single strands. Plasmid was analyzed in neutral (panels A and B) or alkaline (panels C and D) agarose gels. Plasmid samples were treated with (panels B and D) or without (panels A and C) UVA light.
Table 2.
Percent linearization of plasmid DNA by coumarins plus UVA light
| Concentration (μM) |
Percentage of nicked plasmid | ||||
|---|---|---|---|---|---|
| Compd 14 |
Compd 48 |
Compd 58 |
Compd 62 |
Compd 71 |
|
| 0 | 0.2 | 0.2 | 1.0 | 1.2 | 2.2 |
| 1 | 1.0 | 4.0 | 3.2 | 3.2 | 6.8 |
| 10 | 8.0 | 60.5 | 12.0 | 55.4 | 80.2 |
| 100 | 20.5 | 90.8 | 20.2 | 96.6 | 98.6 |
Table 2. DNA nicking data from plasmids analyzed in neutral agarose gels (Figure 2, panel B). In these experiments, plasmids were treated with increasing concentrations of the coumarins followed by UVA light. Image analysis was used to calculate the percent of plasmid unwinding. Note that compounds 48, 62 and 71 were more effective in nicking DNA.
Experimental:
Materials, Methods and Instrumentation:
The 7-oxycoumarins were purchased or synthesized as indicated on Table 1, and in the following Syntheses. 1H NMR spectra were obtained on a Bruker 500MHz nuclear magnetic resonance spectrometer in CDC13 or DMSO-d6. All solvents and reagents were of the highest purity commercially available. Combustion analyses were within ± 0.4 of theoretical values (deviations are noted in the text) and NMR spectra (see below) were in accord with expected resonance shifts. The cLogPs reported herein were obtained from the Chemical Properties calculator in ChemDraw Professionnal version 17.0. If a measured logP is known, that value is used in the discussion.
Syntheses:
Coumarins from commercial sources or published syntheses are noted in Table 1 by the name of the supplier or by reference to a published article. The letter S means the indicated coumarin was synthesized for this study. In some cases, in which the published synthesis lacked experimental details or physical properties, we have repeated that synthesis and the newly measured properties are reported below. Compounds not present in the text below (8, 9, 31, 32, 50 and 61-70) were prepared according to methods described in the references displayed in Table 1 for those compounds. In addition, the synthetic methods for all previously unreported coumarins are described below.
4,8-Dimethyl-7-[(3-methyl-2-buten-1-yl)oxy]coumarin (3), 4,8-dimethyl-3-(3-methyl-2-buten-1-yl)-7-hydroxycoumarin (19), and 4,8-dimethyl-6-(3-methyl-2-buten-1-yl)-7-hydroxycoumarin (20) were all produced in the etherification of 4,8-dimethyl-7-hydroxycoumarin (12) when 1.9 g (10.0 mmol) of the latter compound were dissolved in 49 mL of aqueous sodium hydroxide (containing 0.40 g or 10.0 mmol) and reacted with 1-bromo-3-methyl-2-butene (1.5 g, 10.0 mmol) over 14 hr of vigorous stirring at ambient temperature. The precipitate which had formed was removed by filtration, washed on the filter with 20% aqueous sodium hydroxide, and the base-soluble phases combined. As expected the base insoluble compound (0.44 g, 17%) was the expected butenyl ether, 4,8-dimethyl-7-[(3-methyl-2-buten-1-yl)oxy]coumarin (3, mp = 93-94°C, recrystallized from MeOH). Elemental analysis and 1H NMR (DMSO-d6) supported the proposed structure: δ (ppm) 2.17 and 2.44 (two s, each 3H, ring methyls), 1.68 and 1.83 (two d by second order splitting, each 3H, =(CH3)2), 4.69 (m, 2H, -OCH2-), 5.40 (m, 1H, -CH=), 6.33 (s, 1H, C3-H), 7.43 (d, 1H, C5-H, J = 8.4 Hz), and 6.90 (d, 1H, C6-H, J = 8.4 Hz).
Neutralization with 6 N hydrochloric acid of the aqueous basic phase precipitated an orange oil which was separated by flash chromatography (silica gel, chloroform:ethyl acetate 95:5 elutant) into three phenols – unreacted starting material (not recovered) and two different C-alkylated phenols. The first eluted fraction (0.55 g, 21%, mp = 199-200°C, recrystallized from MeOH), elemental analysis within 0.4% of theoretical and 1H NMR (CDC13) supported the proposed structure of 4,8-dimethyl-3-(3-methyl-2-buten-1-yl)-7-hydroxycoumarin (19): δ (ppm) 2.32 and 2.36 (two s, each 3H, ring methyls), 1.68 (d, 3H, J = 1.2 Hz, =C(CH3)), 1.78 (s, 3H, =C(CH3)’), 3.35 (d, 2H, J = 6.5 Hz, -CH2-), 5.08 (dd, 1H, J = 6.5 Hz, J’ = 1.2 Hz, - CH=), 5.86 (br s, 1H, -OH, exchanges with D2O), 6.88 (d, 1H, J = 8.5 Hz, C6-H), and 7.27 ppm (d, 1H, J = 8.5 Hz, C5-H). The second component to elute was isolated as off-white short needles (0.31 g, 12%, mp = 165-167°C, recrystallized from MeOH), elemental analysis within 0.3% of theoretical and 1H NMR (CDC13) supported the proposed structure of 4,8-dimethyl-6-(3-methyl-2-buten-1-yl)-7-hydroxycoumarin (20): δ (ppm) 2.36 (s, 3H, C8-CH3), 2.39 (d, 3H, J = 1.2 Hz, C4-CH3 coupled to C3-H), 1.81 (two overlapping closely spaced s, 6H, =C(CH3)2), 3.42 (d, 2H, J = 6.8 Hz, -CH2-), 5.30 (m, 1H, J = 6.8 Hz, J’ = 1.2 Hz, -CH=), 5.95 (br s, 1H, phenolic -OH exchanges with D2O), 6.12 (q, 1H, J = 1.2 Hz, C3-H coupled to C4-CH3), and 7.18 ppm (s, 1H, C5-H).
4,8-dimethyl-6-allyl-7-hydroxycoumarin (21, mp = 168-170°C) and 4,8-dimethyl-6-allyl-7-acetoxycoumarin (24, mp = 149.5-150°C) were prepared as reported.(34)
Preparation of 4,8-dimethyl-7-[(2-buten-1-yl)oxy]coumarin (7) and 4,8-dimethyl-6-(2-buten-1-yl)-7-hydroxycoumarin (23). To a solution of 0.95 g (5.0 mmol) of 4,8-dimethyl-7-hydroxycoumarin (12), in 30 mL of aqueous sodium hydroxide containing 0.2 g (5 mmol) of the base, was added 1-chloro-2-butene (0.68 g, 5 mmol). The resulting solution was stirred at room temperature for 18 h, acidified with 10% hydrochloric acid, chilled, and filtered. The dried crude product was subjected to flash chromatography (silica gel, 99:1 methylene chloride:2-propanol elutant) with the first product fraction identified as 4,8-dimethyl-7-[(2-buten-1-yl)oxy]coumarin (7, 0.44 g, 36%, mp = 114-116°C, recrystallized from methanol/ethyl acetate) and the second product fraction identified as 4,8-dimethyl-6-(2-buten-1-yl)-7-hydroxycoumarin (23, 0.47 g, 39%, mp = 158-160°C). Alternatively, 7 can be prepared without contamination by 23 if the acetone, potassium carbonate phenolic alkylation of 12 is used.(49)
4,8-Dimethyl-6-(3-buten-2-yl)-7-hydroxycoumarin (22) was prepared by the Claisen rearrangement of 0.40 g (16 mmol) of 4,8-dimethyl-7-[(2-buten-1-yl)oxy]coumarin (7) refluxed for eight hours under a dry nitrogen stream in 30 mL of N,N-diethylaniline. The cooled reaction contents were poured into 40 mL of chloroform and extracted 3X with 30 mL each of 25% hydrochloric acid. The organic layer was washed with 30 mL of water, dried over anhydrous sodium sulfate, and evaporated in vacuo to a yellow-brown solid, which was crystallized from methanol to a fluffy beige solid (0.31 g, 78% yield, mp = 175-176°C). Elemental analysis was within ± 0.1% of theoretical and 1H NMR confirmed the structure as 4,8-dimethyl-6-(3-buten-2-yl)-7-hydroxycoumarin (22). 1H NMR (CDC13): δ (ppm) 1.41 (d, 3H, J = 6.8 Hz, C-CH3), 2.32 (s, 3H, C8-CH3), 2.42 (d, 3H, J = 0.98 Hz, C4-CH3), 3.80 (m, 1H, -CHCH3), 5.15-5.35 (m, 2H, =CH2), 6.12 (q, 1H, J = 0.98 Hz, C3-H), 7.24 (3, 1H, C5-H).
These 4,8-dimethyl-7-alkoxycoumarins (2, 4, 5, 6, 10 and 11) were prepared by alkylation of 4,8-dimethyl-7-hydroxycoumarin (12) (Regis Chemical Company) by the general method for phenolic etherification.(49) The requisite 7-hydroxycoumarin (1.0 mmol), the corresponding alkyl/alkenyl/alkynyl bromide (1.5 mmol), 50 mL of anhydrous acetone, and 3 g of potassium carbonate were stirred vigorously at reflux for 16 hr, chilled, filtered, and the solid residue washed on the filter with a minimum of boiling methanol. The organic phase (acetone, methanol and coumarin ether product) was evaporated and the crude solid triturated with 20% aqueous sodium hydroxide to remove any unreacted 7-hydroxycoumarin. The dried organic solid was crystallized from methanol to yield the pure ethers in 45-79% yields with elemental analyses (± 0.4% of theory) and 1H NMR (CDC13) in accord with the expected: (2) mp = 145 -146°C, (4) mp 83-84°C, (5) mp 106-108°C, (6) mp 103-104°C, (10) mp = 87.0-88.5°C, and (11) mp = 80-82°C.
Dimethyl-7-allyloxycoumarin (26) was prepared as described by Kaufman.(36)
4,8-Dimethyl-3-bromo-6-[3-(1,3-dioxoisoindolin-2-yl)prop-1-en-1-yl]-7-acetoxycoumarin (59) was prepared by the heating with vigorous stirring at 100°C of 1.85 g (10 mmol) of potassium phthalimide in 50 mL of dimethylacetamide with 2.55 g (5 mmol) of 4,8-dimethyl-3-bromo-6-(2,3-dibromopropyl)-7-acetoxycoumarin (60). The latter was prepared by our published method(41) At the end of five hours the reaction medium was chilled to room temperature in an ice-water bath and diluted with 300 mL of cold water to precipitate the organic materials. These were filtered, dried, taken up in methylene chloride and flash chromatographed. 59 was isolated (0.64 g, 26% yield, mp = 231-232 °C, recrystallized from CH2Cl2/hexane). 1H NMR (CDCl3) confirmed the structure: δ (ppm) 2.22 and 2.37 (two s, each 3H, C4–CH3 and C8-CH3), 2.59 (s, 3H, –OAc), 4.44 (dd, 2H,-CH2-N), 6.22 and 6.63 (two m, each 1H, side chain -CH=CH-), 7.53 (s, 1H, C5-H), 7.73 and 7.85 ppm (two m, each 2H, phthalimido Ar-H).
4,8-dimethyl-7-hydroxy-6-iodocoumarin (16): 4,8-dimethyl-7-hydroxycoumarin (12, 10.0 g, 52.0 mmol) was dissolved in 80 mL of dioxane with 200 mL of concentrated aqueous ammonia subsequently added. A solution prepared from 13.6 g (53.5 mmol) of iodine in 500 mL of aqueous 5% KI was added dropwise with vigorous stirring over a two-hour period. The greenish color of the solution gradually disappeared and a yellow precipitate formed. The pH was adjusted to 3.0 with hydrochloric acid, cooled, and filtered to remove the product, which was taken up in a minimum of ethyl acetate and washed with water (3 × 50 ml). Evaporation of the ethyl acetate gave 15.7 g (94%) of the title compound which was recrystallized from MeOH to analytical purity: elemental analysis (C,H), mp = 219-220°C.(33) 1H NMR (DMSO-d6) confirmed that iodination had taken place on C6: δ (ppm) 2.25 and 2.36 (two s, each 3H, CH3’s), 6.13 (s, 1H, C3-H), 7.84 (s, 1H, C5-H), 10.11 (br S, 1H, OH, exchangeable with D2O).
4-Methyl-8-iodo-7-hydroxycoumarin (27) was prepared as described in 35% yield, mp as reported 254-256°C.(32)
4-Methyl-7-alkoxy-8-iodocoumarins, (28-30). A solution of 27 (3.87 g, 12.8 mmol), 2 g of 1-chloro-3-methyl-2-butene (19.1 mmol), 8.0 g potassium carbonate (58 mmol), and 200 mL of acetone was stirred at reflux for four hrs. After filtration the potassium carbonate was washed on the filter three times with hot acetone. Combined filtrates and washings were evaporated and the product, 4-methyl-7-[(3-methyl-2-buten-1-yl)oxy]-8-iodocoumarin (28), isolated as white microneedles (2.60 g, 55% yield, mp = 151.5-153°C, recrystallized from ethyl acetate). Elemental analysis (C,H,I within ± 0.2% of theory) and 1H NMR (CDC13) confirmed the structure: δ (ppm) 1.77 and 1.79 (two s, each 3H, =C(CH3)2), 2.42 (d, 3H, J = 1.2 Hz, C4-CH3), 4.65 (d, 2H, J = 6.8, -OCH2), 5.56 (dt, 1H, J = 6.8, J’ = 1.1, -CH=), 6.13 (d, 1H, J = 1.2, C3-H), 6.83 (d, 1H, J = 8.8, C6-H), 7.51 (d, 1H, J = 8.8, C5-H). Similarly, with n-propyl bromide as the alkylating agent 4-methyl-7-n-propoxy-8-iodocoumarin (29) was obtained (49% yield, mp =129-130°C, recrystallized from ethanol). Elemental analysis (C,H,I within ± 0.15% of theoretical) and 1H NMR (CDC13) confirmed the assigned structure: δ (ppm) 1.13 (t, 3H, J = 7.33, -CH3), 1.85 (m, 2H, -CH2-), 2.41 (s, 3H, C4-CH3), 4.10 (t, 2H, J = 6.36, -OCH2-), 6.14 (s, 1H, C3-H), 6.77 (d, 1H, J =8.8, C6-H), 7.54 (d, 1H, J = 8.8, C5-H).
Similarly, by the above method using methyl iodide as the alkylating agent, a 54% yield of 4-methyl-7-methoxy-8-iodocoumarin (30) was obtained (mp = 194-195°C, recrystallized from ethanol). Elemental analysis (C,H,I within ± 0.1% of theoretical) and 1H NMR (CDC13) confirming of the assigned structure: δ (ppm) 2.42 (s, 3H, C4-CH3), 4.00 (s, 3H, -OCH3), 6.15 (s, 1H, C3-H), 6.82 (d, 1H, J = 8.8, C6-H), 7.57 (d, 1H, J = 8.8, C5-H).
Syntheses of (13-15). 4,8-dimethyl-6-iodo-7-alkoxycoumarins The allyl, 3-methyl-2-buten-1-yl, and n-propyl ethers were prepared by the general method of Vyas(49) from condensation of 4,8-dimethyl-6-iodo-7-hydroxycoumarin (16) and the corresponding bromides (viz. allyl bromide, 3-methyl-2-butenyl bromide, and n-propyl bromide) in dry acetone over potassium carbonate in yields of 60-85% (recrystallization EtOH). Elemental analyses (C,H) and 1H NMR were in accord with theoretical values and respective mp’s of the purified ethers were (13) 158-159.5°C, (14) 146-147°C, and (15) 124.5-126°C.
Syntheses of (52-57): 8-methyl-3-bromo-7-alkoxycoumarins. The allyl, 3-methyl-2-buten-1-yl, n-propyl, propargyl, and methyl ethers were prepared by the general method.(49) The parent coumarin, 8-methyl-3-bromo-7-hydroxycoumarin (57), mp = 209-211°C, was prepared as described(42) and subsequently condensed with these corresponding bromides (viz., allyl, 3-methyl-2-butenyl, n-propyl, propargyl, and methyl bromide) in dry acetone over potassium carbonate in yields of 68-90%. Elemental analyses (C,H) and 1H NMR were in accord with theoretical values and the mp’s of the respective purified ethers were (52) 143-143.5°C, (53) 131.5-133°C, (54) 139.5-140°C, (55) 181-182°C, and (56) 154 - 155°C.
(58) 4,8-dimethyl-3-bromo-7-allyloxycoumarin was prepared in 52% yield by allyl bromide alkylation in dry acetone/potassium carbonate of 4,8-dimethyl-3-bromo-7-hydroxycoumarin (51) by the general method.(49) The starting material (51) was prepared as reported, mp = 269-271°C.(42) The title compound (58) appeared from MeOH as tiny white needles (mp = 147-148°C). 1H NMR and elemental analysis were in accord with theoretical values.
(37) 4-trifluoromethyl-8-methyl-7-hydroxycoumarin and (47) 4-trifluoromethyl-7-hydroxycoumarin, were prepared as described(50) and had melting points (197-198°C and 186-188°C respectively) and 1H NMRs in accord with those expected.
Synthesis of (34, 35, 38, 44, and 45), 4-trifluoromethyl-8-methyl-7-alkoxycoumarins. The 2-ketopropyl, n-propyl, propargyl, allyl, and methyl ethers were prepared by the general method(49) from condensation of 4-trifluoromethyl-8-methyl-7-hydroxycoumarin (37) with chloroacetone, n-propyl bromide, propargyl bromide, allyl bromide and methyl iodide in yields of 31 to 56% (recrystallization from EtOH). Elemental analyses (C,H) and 1H NMR were in accord with theoretical values and respective mp’s of the purified ethers were (34) 143-144.5°C, (35) 112-113°C, (38) 160.5-161.5°C, (44) 114.5-115.5°C, and (45) 109.5-111°C.
Synthesis of (36, 39, 41, 42, and 46). These 4-trifluoromethyl-7-alkoxycoumarins were prepared by etherification of 4-trifluoromethyl-7-hydroxycoumarin (47) with allyl bromide, n-propyl bromide, methyl iodide, propargyl bromide, and chloroacetone in 28 to 58% yields (recrystallized from EtOH) with elemental analyses (C,H within ± 0.3% of theory) and 1H NMR in accord with theory. The respective mp’s of the purified ethers were (36) 76-77°C, (39) 97-98°C, (41) 112.5-113.5°C, (42) 110.5-112°C, and (46) 158-159°C.
Syntheses of (33, 43 and 40). These 6-allyl-substituted coumarins were prepared by the Claisen rearrangement of the corresponding 7-hydroxy ethers. The rearrangement procedure in N,N-diethylaniline as described above for preparation of compound (22) was followed. First, 4-trifluoromethyl-8-methyl-7-hydroxycoumarin (37) was etherified by Vyas’s method(49) with 2-chloroallyl chloride (also known as 2,3-dichloropropene) and with allyl chloride respectively. The crude ethers were evaporated to dryness from the acetone solvent used to prepare them and each was solubilized in N,N-diethylaniline (five times the weight of aniline per unit weight of allyl ether) and heated at reflux under nitrogen for 12 hours. Work-up and product isolation as described for (22) gave 24% yield of chloroallyl (33), light tan needles, mp 182-183°C, with elemental analysis (C,H,F,C1 within ± 0.3% of theory) and 1H NMR in accord with theory. The same procedure for the 7-allyloxy compound (44) gave 34% of the rearranged 6-allyl-7-hydroxy (43) as off-white crystals, mp 174-175°C. Elemental analysis (C,H,F) was within ± 0.3% and 1H NMR was in accord with theory. Acetylation of (43) with acetic anhydride/sodium acetate gave 58% of the O-acetyl coumarin, compound (40), mp 151-152°C (recrystallization from MeOH). Analytical data confirmed structure and purity.
(18) 4,8-dimethyl-6-nitro-7-hydroxycoumarin. 4,8-Dimethyl-7-hydroxycoumarin (12, 7.50 g, 39.4 mmol) was dissolved in 75 mL concentrated sulfuric acid at room temperature, chilled to −20° C, and treated to the addition of a pre-chilled nitrating mixture (3 mL concentrated HNO3 added to 9 mL concentrated H2SO4). Stirring was continued for three hrs at −20°C with the mixture allowed to warm before pouring into ice. Bright yellow crystals were filtered, washed with water and dried to recover 7.50 g (81% yield) crude solid. The product (18) was recrystallized from ethanol to give yellow green crystals with mp = 229.5-231.5°C. C,H,N analysis was within ± 0.25% of theoretical. 1H NMR (DMSO-d6): δ (ppm) 2.29 and 2.41 (two s, 3H each, C4-CH3 and C8-CH3), 6.37 (s, 1H, C3H), 8.19 (s, 1H, C5H), 11.30 (br s, 1H, -OH). 13C NMR (DMSO-d6): δ (ppm) 8.75, 18.00, 112.45, 112.93, 115.12, 119.90, 132.80, 152.91, 154.18, 155.10, 158.96.
(17) 4,8-Dimethyl-6-amino-7-allyloxycoumarin. 4,8-Dimethyl-6-nitro-7-hydroxycoumarin (18, 4.70 g, 20 mmol) was added to 40 mL dry acetone with K2CO3 (2.76 g, 20 mmol) in an oven dried flask. Allyl bromide (2.90 g, 24 mmol) in 40 mL of acetone was added slowly with heating and stirring. After five hrs of vigorous agitation at reflux the mixture was filtered and the solid on the filter washed with boiling acetone. Evaporation of the filtrate gave 3.30 g (60%) of crude 4,8-dimethyl-6-nitro-7-allyloxycoumarin which was employed directly in the next step. To a round bottom flask was added finely ground 4,8-dimethyl-6-nitro-7-allyloxycoumarin (3.00 g, 11 mmol), tin (3.24 g, 27.3 mmol), SnCl2 (3.04 g, 16.0 mmol) and 2 mL concentrated HCl in 200 mL ethanol. The mixture was stirred overnight during which all tin dissolved. Most of the ethanol was removed in vacuo. As the solution was allowed to cool, a gel formed which was filtered to recover a small amount of pearlized crystals. Diethyl ether was added to the filtrate and a precipitate formed which was filtered, and rinsed with ether to remove SnCl2. The solids were dried and extracted with hot ethanol on the funnel of a filtration flask. Evaporation of the ethanol gave a crude product that was recrystallized from ethanol to provide 2.20 g (82%) of the 4,8-dimethyl-6-amino-7-allyloxycoumarin, (17). The bright mustard yellow crystals had a mp of 145.5-147.5°C. C,H,N analysis within ± 0.30% of theoretical. 1H NMR (DMSO-d6): δ (ppm) 2.28 and 2.49 (two s, each 3H, C4-CH3 and C8-CH3), 4.54 (d, 2H, -O-CH2-), 5.20 (br s, 2H, -NH2), 5.28 (d, 1H, =CH), 5.48 (d, 1H, =CH’), 6.16 (m, 1H, -CH=), 6.39 (s, 1H, C3-H), 7.58 (s, 1H, C5-H). 13C NMR (DMSO-d6): δ (ppm) 9.43, 18.14, 74.57, 113.64, 115.13, 116.00, 118.69, 120.09, 125.52, 133.48, 147.0, 151.49, 152.50, 159.62.
(71) 3-Iodo-4,8-dimethyl-7-hydroxycoumarin. Finely pulverized 4,8-dimethyl-7-hydroxycoumarin (12) (3.00 g, 15.7 mmol) was added to 150 mL concentrated acetic acid with subsequent dropwise addition of ICl (5.35 g, 33.0 mmol). This mixture was heated at 50°C with stirring overnight. The mixture was cooled and the crystals collected by filtration. The yield was 3.22 g (65%). The white crystals were purified by recrystallization from methanol, with mp = 228-230°C. C,H,N analysis was within + 0.10% of theoretical. 1H NMR (DMSO-d6): δ (ppm) 2.14 and 2.58 (two s, each 3H, C4-CH3 and C8-CH3), 6.84 (d, 1H, C6-H, J = 8.7 Hz), 7.55 (d, 1H, C5-H, J = 8.7 Hz), 10.53 (br s, 1H, -OH).
Biological Evaluation – Experimental Methods:
Growth inhibition assays:
Assays to measure keratinocyte growth inhibition were performed as previously described.(30) Briefly, PAM212 mouse keratinocytes (25,000 cells per well) were inoculated into 6-well Falcon plastic tissue culture dishes in Dulbecco's Modified Eagle's medium supplemented with 10% newborn calf serum in an incubator containing 5% carbon dioxide at 37°C. After 24 hr, the medium was changed to fresh growth medium supplemented with control medium or medium containing increasing concentrations of the coumarins. The medium contained 10% serum and 0.2% DMSO which facilitated the solubilization of the coumarins. After an additional 30 min, the uncovered culture plates were exposed to UVA light (320-400 nm) emitted from a bank of four BLB fluorescence light tubes (F40 BL, Sylvania) placed approximately 10 cm above the cell culture plates. In these experiments, the incident light on the culture plates was 2.4 mW/cm2 as measured with an International Light UV radiometer, Model IL442A. The cells were exposed to 1.28 J/cm2 of UVA. After exposing the cells to UVA light, the cell culture medium was drained and the cells refed with fresh growth medium. After incubating the cells for 4-5 days, cells on the plates were removed with trypsin and counted with a Coulter Counter. Results (see Table 1 and Figure 1) were presented as percent inhibition of cell growth when compared to controls.
Plasmid DNA nicking and unwinding assays:
Assays to evaluate the effects of coumarins on plasmid DNA were performed as previously described.(30) Briefly, 10 μL reaction mixtures were prepared that contained 75 ng of plasmid pZeoSV DNA, 0.1 μL coumarin deriviative solution (prepared as a 100 × concentrate in DMSO) and TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8). The reaction volume and components were scaled up several-fold so that multiple aliquots of 10 μL could be removed for analysis in agarose gels before and after UVA light treatment. Treatment with UVA light was performed in V-bottomed 96-well plates as described above for the cell cultures. Samples of the reaction mixtures containing the plasmids were then analyzed by electrophoresis using 1.2% agarose gels either in neutral gel buffer (40 mM Tris base, 20 mM acetic acid, 1 mM EDTA) or under denaturing conditions. To denature the DNA, samples were first mixed with 2 M NaOH supplemented with 0.1 M EDTA and heated in a water bath at 90°C for 1 min. After cooling on ice, DNA samples were loaded on 1.2% alkaline agarose gels prepared and electrophoresed in alkaline gel buffer (50 mM Tris base, 45 mM boric acid, 30 mM NaOH, 1 mM EDTA). After electrophoresis, gels were washed and stained with ethidium bromide (0.5 μg/mL). Gels were then photographed (see examples in Figure 2) with an Eagle Eye II digital documentation system (Stratagene, San Diego, CA). Bands shown in Figure 2 were then subjected to image analysis to calculate the percent of plasmid unwinding (see Table 2) which ranged from 0.2 to 99% in the set chosen.
Conclusions:
Forty-nine 7-oxycoumarins from a library of 71 structural variants demonstrated phototoxicity against PAM212 mouse keratinocytes. None of the compounds was phototoxic in the absence of UVA light. These data indicate that the ability of the compounds to inhibit cell growth was dependent not only on their physiochemical characteristics, but also on their ability to absorb UVA light. Based on the results, definite structure-activity correlations could be made. Relative lipophilicity was an important factor especially among structurally similar families of coumarins. In most substituted oxycoumarins, the 7-alkoxys were more active than the 7-hydroxy compounds. Coumarins with electron withdrawing moieties – cyano and fluoro at C3 – were considerably less active while those with bromines or iodine at that location displayed enhanced activity. The electron withdrawing trifluoromethyl at C4 was generally a deactivating functionality while brominated alkyl side chains substantially improved activity.
Of interest were the findings that photocytotoxic coumarins also possessed the ability to photo-damage plasmid DNA albeit not in the same rank order of their activity as inhibitors of keratinocyte growth. There is no evidence that the two-ring coumarins, unlike the three-ring furocoumarins (psoralens), intercalate DNA prior to manifesting phototoxicity, but outer groove DNA-affinity achieves the same proximity necessary for DNA binding. It thus appears that binding of the coumarins to DNA is sufficient to initiate photo-damage following exposure to UVA light. This includes both DNA strand nicking and unwinding. Taken together, our data indicates that the DNA in keratinocytes is likely to be an important target for the coumarins. Further studies are needed to characterize coumarin-induced DNA photo-damage and its role in regulating keratinocyte cell growth.
Highlights.
Polyfunctionalized non-toxic coumarins displayed a photo-induced toxicity against cultured keratinocytes. This toxicity may arise from a light-catalyzed nicking of DNA.
Acknowledgments:
This work was funded in part by the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH Grant number U54AR055073). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Weinmann I, (1997), History of the development and applications of coumarin and coumarin-related compounds In: O’Kennedy R & Thornes RD, eds. Coumarins – Biology, Applications and Mode of Action, John Wiley & Sons Ltd, Chichester, UK, pp. 1–22. [Google Scholar]
- (2).Ojala T, (2001), Biological screening of plant coumarins, Doctoral dissertation, University of Helsinki, Finland, 2 February 2001. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.407.8611&rep=rep1&type=pdf [Google Scholar]
- (3).Bruneton J, (1999), Pharmacognosy, Phytochemistry, Medicinal Plants, 2nd Ed. Intercept Ltd, Hampshire, UK, pp. 263–277. [Google Scholar]
- (4).Asif M, (2015), Pharmacological activities and phytochemistry of various plant containing coumarin derivatives. Curr. Sci. Perspectives, 1(3): 77–90. [Google Scholar]
- (5).Averbeck D, (1989), Recent advances in psoralen phototoxicity mechanism. Photochem. Photobio, 50(6): 859–882. DOI: 10.1111/j.1751-1097.1989.tb02917.x [DOI] [PubMed] [Google Scholar]
- (6).Laskin JD, Lee E, Yurkow EJ, Laskin DL, Gallo MA, (1985), A possible mechanism of psoralen phototoxicity not involving direct interaction with DNA. Proc. Natl. Acad. Sci. USA, 82(18): 6158–6162. DOI: 10.1073/pnas.82.18.6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stansbury J, (2011), Photosensitizing herbs: skin diseases and cancers. DDNR, Anti-aging, dermatology, oncology, 1 May 2011, pp. 1–11. http://ndnr.com/oncology/photosensitizing-coumarins-for-skin-diseases-and-cancers/ [Google Scholar]
- (8).Nigg HN, Nordby HE, Beier RC, Dillman A, Macias C, Hansen RC, (1993), Phototoxic coumarins in limes. FoodChem. Toxicol, 31(5): 331–335. DOI: 10.1016/0278-6915(93)90187-4 [DOI] [PubMed] [Google Scholar]
- (9).Ojala T, Vuorela P, Kiviranta J, Vuorela H, Hiltunen R, (1999), A bioassay using Artemia salina for detecting phototoxicity of plant coumarins. Planta Med., 65(8): 715–718. DOI: 10.1055/s-1999-14049 [DOI] [PubMed] [Google Scholar]
- (10).Jivaramonaikul W, Rashatasakhon P, Wanichwecharungruang S, (2010), UVA absorption and photostability of coumarins. Photochem, Photobiol, Sci, 9(8): 1120–1125. DOI: 10.1039/C0PP00057D [DOI] [PubMed] [Google Scholar]
- (11).Mousavi SH, Davari AS, Iranshahi M, et al. , (2015), Comparative analysis of the cytotoxic effects of 7-prenyloxycoumarin compounds and herniarin on MCF-7 cell line. Avicenna J. Phytomed, 5(6): 520–530. [PMC free article] [PubMed] [Google Scholar]
- (12).Goard M, Aakalu G, Fedorya OD, et al. , (2005), Light-mediated inhibition of protein synthesis. Chem. & Biol, 12(6): 685–693. DOI: 10.1016/i.chembiol.2005.04.018 [DOI] [PubMed] [Google Scholar]
- (13).Sarwar T, Rehman SU, Husain MA, Ishqi HM, Tabish M, (2015), Interaction of coumarin with calf thymus DNA: deciphering the mode of binding by in vitro studies. Int. J. Biol. Macromol, 73: 9–16. DOI: 10.1016/j.ijbiomac.2014.10.017 [DOI] [PubMed] [Google Scholar]
- (14).Sarwar T, Husain MA, Rehman SU, Ishqi HM, Tabish M, (2015), Multi-spectroscopic and molecular modelling studies on the interaction of esculetin with calf thymus DNA. Mol. Biosyst, 11(2): 522–531. DOI: 10.1039/C4MB00636D [DOI] [PubMed] [Google Scholar]
- (15).Pivetta T, Valletta E, Ferino G, Isaia F, Pani A, Vascellari S, Castellano C, Demartin F, Cabiddu MG, Cadoni E, (2017), Novel coumarins and related copper complexes with biological activity: DNA binding, molecular docking and in vitro antiproliferative activity. J. Inorg. Biochem, 177:101–109. DOI: 10.1016/j.jinorgbio.2017.09.013 [DOI] [PubMed] [Google Scholar]
- (16).Bisagni E, (1992), Syntheses of psoralens and analogues. J. Photochem. Photobio. B: Biol, 14: 23–46. DOI: 10.1016/1011-1344(92)85081-5 [DOI] [PubMed] [Google Scholar]
- (17).Matern U, Luer P and Kreusch D, (1999), Biosynthesis of coumarins In: Barton D, Nakanishi K, Meth-Cohn O and Sankawa U (eds.): Comprehensive Natural Products Chemistry, Vol. 1, Polyketides and Other Secondary Metabolites Including Fatty Acids and Their Derivatives. Elsevier Science Ltd, Oxford, UK: pp. 623–637. [Google Scholar]
- (18).Yeargers E, Augenstein L, (1965), Absorption and emission spectra of psoralen and 8- methoxypsoralen in powders and in solutions. J. Invest. Derm, 44(3): 181–186. DOI: 10.1038/jid.1965.32 [DOI] [PubMed] [Google Scholar]
- (19).Mattoo BN, (1956), Absorption and fluorescence spectra of coumarins. Trans. Faraday Soc, 52: 1184–1194. DOI: 10.1039/TF9565201184 [DOI] [Google Scholar]
- (20).Fink DW, Koehler WR, (1970), pH effects on fluorescence of umbelliferone. Anal. Chem, 42(9): 990–993. DOI: 10.1021/ac60291a034 [DOI] [Google Scholar]
- (21).Zhang J, Wei Y, Liu C, (2011), Fluorescence quantum yield and ionization constant of umbelliferone, Chem. Bull, 74: 957–960. [Google Scholar]
- (22).Gee KR, Haugland RP, Sun W-C, (1998), Derivatives of 6,8-difluoro-7-hydroxycoumarin. U. S. Patent 5,830,912 3 November 1998.
- (23).Invitrogen, (2010), Molecular probes handbook: a guide to fluorescent probes and labeling technologies. Chapter 10, 11th Edition (2010); Invitrogen publication by Thermo Fisher Scientific. [Google Scholar]
- (24).Abu-Eittah RH, El-Tawil BAH, (1985), The electronic absorption spectra of some coumarins. A molecular orbital treatment. Can. J. Chem, 63: 1173–1176. DOI: 10.1139/v85-200 [DOI] [Google Scholar]
- (25).Laskin JD, (1994), Cellular and molecular mechanisms in photochemical sensitization: studies on the mechanism of action of psoralens. Food Chem. Toxicol, 32(2): 1191–1127. DOI: 10.1016/0278-6915(94)90172-4 [DOI] [PubMed] [Google Scholar]
- (26).Racz E, Prens EP, (2015), Phototherapy and photochemotherapy for psoriasis. Dermatol. Clin, 33(1): 79–89. DOI: 10.1016/j.det.2014.09.007 [DOI] [PubMed] [Google Scholar]
- (27).Kayal G, Jain K, Malviya S, Kharia A, (2014), Comparative SAR of synthetic coumarin derivatives for their anti-inflammatory activity. Int. J. Pharm. Sci. Res, 5(9): 3577–3583. DOI: 10.13040/IJPSR.0975-8232.5(9).3577-84 [DOI] [Google Scholar]
- (28).Kostova I, Raleva S, Genova P, Argirova R, (2006), Structure-Activity Relationships of Synthetic Coumarins as HIV-1 Inhibitors. Bioinorg. Chem. Appl, 2006 (68274): 1–9. DOI: 10.1155/BCA/2006/68274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Guerra FQ, de Araujo RS, de Sousa JP, de Pereira FO, Mendoza-Junior FJ, Barbosa-Filho JM, de Oliveira Lima E, (2013), Evaluation of Antifungal Activity and Mode of Action of New Coumarin Derivative, 7-Hydroxy-6-nitro-2H-1-benzopyran-2-one, against Aspergillus spp. Evid Based Complement Alternat. Med 2015(925096): 8 pages. DOI: 10.1155/2015/925096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mariano TM, Vetrano AM, Gentile SL, Heck DE, Whittemore MS, Guillon CD, Jabin I, Rapp RD, Heindel ND, Laskin JD, (2002), Cell-impermeant pyridinium derivatives of psoralens as inhibitors of keratinocyte growth, Biochem. Pharmacol, 63(1): 31–39. DOI: 10.1016/S0006-2952(01)00855-3 [DOI] [PubMed] [Google Scholar]
- (31).Gupta VN, (1957), Nuclear methylation of coumarins. J. Scientific Industrial Res, 16B: 257–62. [Google Scholar]
- (32).Rodighiero P, Manzini P, Pastorini G, Bordin F, Guitto A, (1987), Synthesis of methyl derivatives of 8-desmethylxanthyletine and 8-desmethylseseline, potential antiproliferative agents. J. Heterocyclic Chem, 24: 485–488. DOI: 10.1002/jhet.5570240234 [DOI] [Google Scholar]
- (33).Yan H, Yin W, Liu P, Liu J, et al. , (2014), Palladium-catalyzed synthesis of 6-allylcoumarins using organotin reagents as multicoupling organometallic nucleophiles. Appl. Organometallic Chem, 28(10): 747–749. DOI: 10.1002/aoc.3188 [DOI] [Google Scholar]
- (34).Rangaswami S and Seshadri TR, (1938), Fixation of the aromatic double bonds in the coumarin ring system. Proc. - Indian Acad. Sci, 7A: 8–12. [Google Scholar]
- (35).Kaufman KD, (1961), New synthesis of methylsubstituted psoralens and isopsoralens. Journal of Organic Chemistry, 26, 117–121. DOI: 10.1021/jo01060a028 [DOI] [Google Scholar]
- (36).Kaufman KD, (1965), Preparation of alkylpsoralens. U.S. Patent P 17 August1965.
- (37).Bissell ER, Larson DK, Croudace MC, (1981), Some 7-substituted-4-(trifluoromethyl)coumarins. Journal of Chemical and Engineering Data. 1981, 26(3), 348–350. DOI: 10.1021/je00025a041 [DOI] [Google Scholar]
- (38).Goldfarb DS, (2009), Method using lifespan-altering compounds for altering the lifespan of eukaryoyic organisms and screening for such compounds. U. S. Patent (Appl. Pub.), 20090163545 A1.
- (39).Whalley WB, (1951), Organic fluoro compounds. VI. Some (trifluoromethyl)chromones and – coumarins. Journal of the Chemical Society, 3235–3238. DOI: 10.1039/JR9510003235 [DOI] [Google Scholar]
- (40).Heindel ND, Laskin JD, Heck DE, Rapp RD, Whittemore MS, McNeel TE, Jabin I (2001), Amino- and mercurio-substituted 4’,5’-dihydropsoralens and therapeutic uses thereof. U. S. Patent 6,255,324B1, 3 July 2001.
- (41).Jabin I, Heindel ND, Rapp RD, Laskin JD, (2000), Synthetic approaches to 3-substituted-5’- (N-pyridiniummethyl)-4’,5’-dihydropsoralens. J. Heterocyclic Chem, 37: 31–34. DOI: 10.1002/jhet.5570370106 [DOI] [Google Scholar]
- (42).Chilin A, Confente A, Pastorini G, Guiott A, (2002), Synthesis of furo[3.2 g][1,4]benzoxazin-3-ones, new psoralen isosters, Eur. J. Org. Chem, 2002: 1937–1940.DOI: [DOI] [Google Scholar]
- (43).Takaoka A, Ibrahim MK, Kagaruki SRF, Ishikawa N, (1985), Synthesis of monofluoro heterocycles using fluoroolefins as starting materials. Nippon Kagaku Kaishi. (11), 2169–2176. DOI: 10.1246/nikkashi.1985.2169 [DOI] [Google Scholar]
- (44).Platz MS, Goodrich RP Jr, Yerram N, (1995), Method of inactivation of viral and bacterial blood contaminants. U. S. Patent 5,418,130, 23 May 1995.
- (45).Leffler JE, Grunwald E, (1963), Rates and Equilibria of Organic Reactions. Wiley, New York, Table 7.1. [Google Scholar]
- (46).Reid ST, (1990), Photoelimination, Chapter 7 in Bryce-Smith D, Gilbert A. (eds), Photochemistry, Vol. 21, Royal Soc. of Chem. publishers, Cambridge, UK, p. 472. [Google Scholar]
- (47).Meng JB, Shen M-G, Fu D-C, Gao Z-H, et al. , (1990), A photochemical synthesis of 3-arylcoumarins. Synthesis, 1990(8): 719–721. DOI: 10.1055/s-1990-26993 [DOI] [Google Scholar]
- (48).Park SC, Goodrich RP Jr., Yerram N, et al. , (1996), Photoinactivation of viral and bacterial blood contaminants using halogenated coumarins. U. S. Patent 5,516,629, 14 May 1996.
- (49).Vyas GN, Shah NM, (1951), Quinacetophenone monomethyl ether. Organic Syntheses, 31, 90–92. DOI: DOI: 10.15227/orgsyn.031.0090 [DOI] [Google Scholar]
- (50).Bayer V, Pastor R, Cambon A, (1982), Synthesis and spectra study of 4-(perfluoroalkyl)-coumarins. J. Fluorine Chem, 20(2): 187–202. DOI: 10.1016/S0022-1139(00)82207-1 [DOI] [Google Scholar]