

Abstract
Antimicrobial drug resistance demands novel approaches for improving the efficacy of antibiotics, especially against Gram-negative bacteria. Here we report that conjugating a diglycine (GG) to a prodrug of antibiotics drastically accelerates intrabacterial hydrolysis of ester bond for regenerating the antibiotics against E. coli. Specifically, the attachment of GG to chloramphenicol succinate (CLsu) generates a novel conjugate (CLsuGG), which exhibits about an order of magnitude higher inhibitory efficacy than CLsu against E. coli. Further studies reveal that CLsuGG undergoes rapid hydrolysis catalyzed by intrabacterial esterases (e.g., BioH and YjfP) for generating chloramphenicol (CL) in E. coli. More importantly, the conjugate exhibits lower cytotoxicity to bone marrow stromal cells that CL does. The structural analogs of CLsuGG indicate that the conjugation of GG is an effective strategy for accelerating hydrolysis catalyzed by esterases and enhancing antibacterial efficacy of antibiotics. This work, for the first time, illustrates that dipeptide conjugation modulates intrabacterial hydrolysis for increasing antibiotic efficacy and reducing adverse drug effects.
Keywords: diglycine, intrabacterial esterase, hydrolysis, prodrug, antibiotics
Graphical Abstract
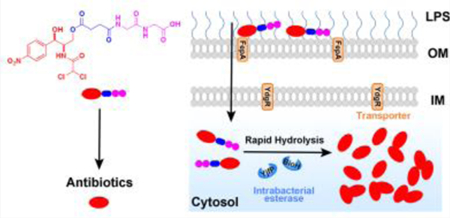
We report that conjugating a diglycine (GG) to a prodrug of antibiotics drastically accelerates intrabacterial hydrolysis of ester bond for regenerating the antibiotics against E. coli. This work, for the first time, illustrates that dipeptide conjugation modulates intrabacterial hydrolysis for increasing antibiotic efficacy and reducing adverse drug effects.
Infectious disease remains a major threat to public health.[1] One challenging problem in the treatment of bacterial infection is the emergence of multidrug resistant (MDR) bacteria, especially the MDR Gram-negative bacteria.[2] However, the development of alternatives to the existing strategies for killing pathogenic bacteria fails to keep pace with the outbreak of resistance, and the antibiotic supply in clinical pipeline remains limited.[2c] Thus, there is an urgent need for developing new antimicrobial approaches against MDR bacterial pathogens. Besides investigating novel agents against new targets in bacteria[3] via previously unexplored mechanisms, another strategy is to enhance the efficacy and safety of the existing antibiotics via drug combinations or increasing specificity. For example, Kahne et al. demonstrated that the use of novobiocin analogs greatly lower the dose of polymyxins against Gram-negative bacteria via inhibiting DNA gyrase, binding LptB, and disrupting outer membrane.[3a] Nolan et al. reported that the conjugate of a synthetic siderophore and ciprofloxacin affords excellent selectivity against a pathogenic E. coli strain.[4] Yan et al. reported nanoaggregates of ciprofloxacin for inhibiting E. coli.[5] Encouraged by these studies, as well as by our recent studies that show that esterases catalyze intracellular accumulation of simple D-dipeptides for inhibiting cancer cells,[6] we decided to explore the use of bacterial esterases to increase the retention of antibiotics inside Gram-negative bacteria for boosting the efficacy of antibiotics (Scheme 1).
Scheme 1.
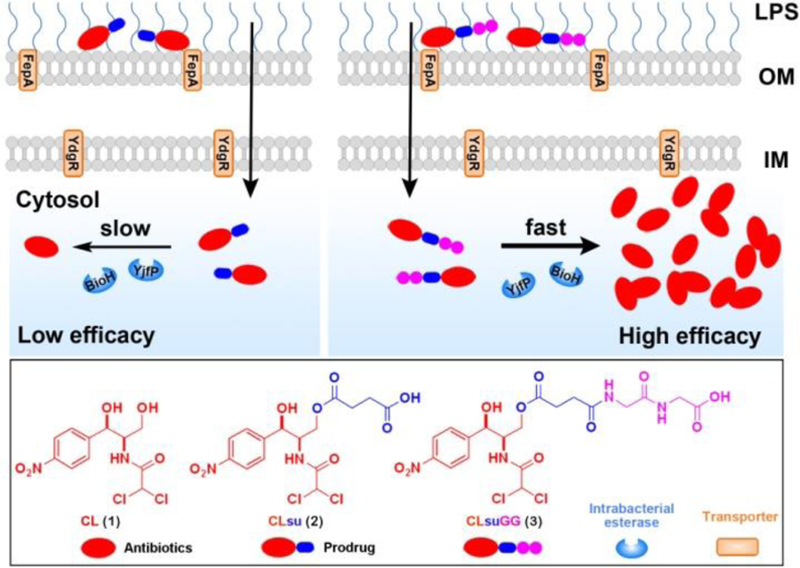
Illustration of diglycine speeding up the regeneration of chloramphenicol inside E. coli.
To demonstrate the concept, we chose to conjugate dipeptide with chloramphenicol succinate (CLsu), the soluble prodrug of chloramphenicol (CL). As a broad-spectrum antibiotic, CL inhibits proteins production in both of Gram-negative and Gram-positive bacteria.[7] Despite its potent activity against Gram-negative bacteria, clinical use of CL is rather limited because of its adverse effects such as bone marrow suppression.[8] Although short course of treatment should reduce the side effect,[9] CLsu, unfortunately, exits the body too fast to achieve adequate serum concentration when being administrated intravenously.[10] One strategy to address the problem is to enhance the uptake and increase the retention of CL inside the bacteria by conjugating it with dipeptide because Gram-negative bacteria express oligopeptide transporters for the uptake of di- and tripeptides.[11]
Based on the above rationale, we conjugated the simplest dipeptide, diglycine (GG), with CLsu to create a new conjugate CLsuGG. Besides exhibiting about 10 times higher inhibitory activity than CLsu against E. coli, CLsuGG shows lower cytotoxicity than CL towards bone marrow stromal cells. Mechanistic studies confirm that a proton-coupled and bacterial specific oligopeptide transporter, YdgR,[12] indeed, involves in the uptake of CLsuGG. More importantly, CLsuGG is rapidly hydrolyzed by intrabacterial esterases (e.g., BioH[13] and YjfP[14]) to regenerate CL (Scheme 1). In addition, the structural analogs of CLsuGG indicate that conjugating GG is an effective strategy for accelerating the hydrolysis catalyzed by intrabacterial esterases and for enhancing the efficacy of antibiotics. As the first example of increasing the rate of intrabacterial activation of antibiotic prodrugs, this work may lead to a new approach that increases intracellular accumulation of antibiotics for combating Gram-negative bacteria.
It is straightforward and easy to conjugate diglycine to CL (1) (Scheme S1). Simply acidifying the commercially available chloramphenicol succinate sodium generates CLsu (2), which is suitable for solid phase peptide synthesis (SPPS) to produce CLsuGG (3). To verify the generality and the roles of the conjugation of GG, we designed and synthesized seven analogs. Changing the number of the glycine residues gives CLsuG (4) and CLsuGGG (5) (Scheme S2); cyclohexane-1,2-dicarboxylic acid replaces the succinate in 2 and 3 to generate 6 and 7 (Scheme S3); ciprofloxacin substitutes CL to produce 8, 9, and 10 (Scheme S4).
We evaluated the antibacterial activities of the glycine-conjugates 2, 3, 4, and 5 against a wild-type E. coli (Figure 1A). Although these conjugates differ only in the number of glycine residues, they exhibit drastically different antibacterial activity. 3 shows high antibacterial activity, with the MIC value of 20 μM (10.7 μg/mL), which is comparable to that of 1 (20 μM (6.5 μg/mL), consistent with the reported result[15]). On the other hand, 4 exhibits the MIC of more than 200 μM (>95.8 μg/mL), which is similar with that of 2 (> 200 μM (>84.6 μg/mL)). The conjugate (5) that contains three glycine residues exhibits the same efficacy (20 μM (11.9 μg/mL)) with that of 3, indicating that GG is optimal for enhancing the antibacterial efficacy of the conjugates (Table S1).
Figure 1.
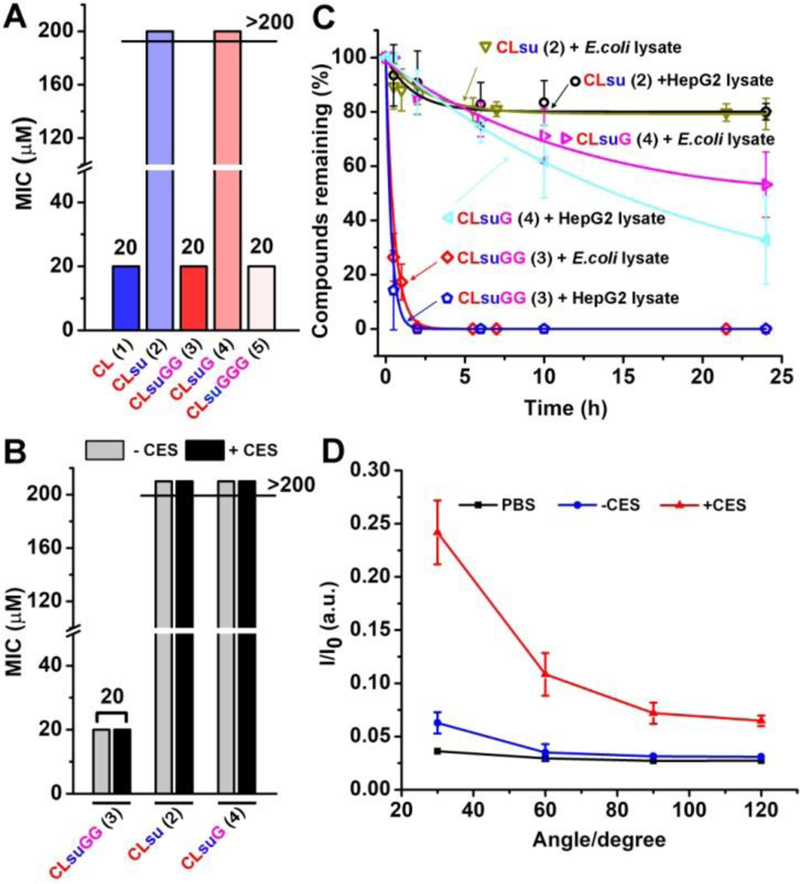
(A) The minimum inhibitory concentrations (MIC) of 1, 2, 3, 4, and 5 against a wild type E. coli strain (K-12). (B) The antibacterial activity of 2, 3 and 4 before and after the treatment of CES. (C) The hydrolysis curve of 2, 3 and 4 with the addition of the lysate of HepG2 cells or the lysate of E. coli (K-12); [2] = [3] = [4] = 200 M, [HepG2 lysate] = [E. coli lysate] = 0.1 U/mL. (D) The static light scattering signals of the solution of 3 before and after the addition of CES; [3] = 200 M, [CES] = 1 U/mL, t = 24 h.
To understand the origin of the antibacterial activity of 3, we first examined whether esterase is able to activate the prodrugs. We added the commercially available mammalian carboxylesterase (CES)[16] (1 U/mL) into the solution of 2, 3 and 4, and compared the antibacterial activity of 2, 3 and 4 before and after the treatment of CES. Surprisingly, the addition of exogenous CES changes little on the inhibitory efficacies of 2, 3 and 4, implying that 3 likely is hydrolyzed by the catalysis of intrabacterial esterases. To confirm this assumption, we measured the rate of the ester bond hydrolysis of 2, 3 and 4 by the mammalian CES[16] and the lysates of E. coli and HepG2 (a hepatocyte cell which overexpress mammalian esterases[6c]) (Figure 1C and S1), after normalizing their activities. Figure 1C shows the intrabacterial esterases (0.1 U/mL, which is normalized by the fluorescence intensity of 5-carboxyfluorescein diacetate, see in SI) hydrolyze 3 completely within 2 h (with the apparent first-order rate constant of 1.868 h−1). Less than 20% 2 and only 55% of 4 are hydrolyzed after 24 h with the addition of the E. coli lysate. Being catalyzed by HepG2 lysates, 3 hydrolyzes completely within 0.5 h, but over 80% of 2 remains after 24 h, and more than 50% of 4 remains after 10 h. These results confirm that rapid intrabacterial hydrolysis of 3 contributes to its high activity against E. coli in cell assay. Further comparison of the rate of ester bond hydrolysis of di-, mono- and tri-glycine conjugates (i.e., 3, 4 and 5) by CES, the lysates of E. coli and HepG2 (Figure S1) supports this inference. Though 3 and 5 hydrolyze in almost same rate, the increased molecular weight of 5 leads to the higher dosage, so diglycine is optimal for enhancing the antibacterial efficacy of the conjugates. In addition, these results also provide an explanation of the clinical failure of CLsu.[10]
LC-MS analysis confirms that hydrolysis of 3 regenerates 1 (Figure S2). Moreover, the intensities of the static light scattering signals increase drastically after the addition of CES to the solutions of 3 for 24 h (Figure 1D), agreeing with that the transmission electron microscopy (TEM) shows increased amount of nanoparticles after the addition of CES (Figure S3). Besides confirming that 3 becomes 1 after the hydrolysis catalyzed by esterases, the above results, collectively, indicate that the soluble 3 is able to undergo rapid intrabacterial hydrolysis to form 1. Because of the poor solubility of 1, such a conversion favors the retention of 1 inside E. coli.
To verify whether bacterial oligopeptide transporters involve in the uptake of 3, we compared the activities of 3 against the YdgR mutant and wild-type E. coli (Figure S4). The deletion of YdgR, an inner membrane (IM) proton-coupled transporter for uptaking di- and tripeptides,[12] only rescues the bacteria slightly (i.e., increasing cell viability for about 10%), suggesting that YdgR unlikely is the major contributor for the precursors entering the bacteria. Further examination reveals that the deletion of siderophore transporter, FepA,[17] substantially increase the viability of the bacteria (Figure S5), indicating 3 enters the bacteria via multiple paths, including diffusion.
To examine the roles of different intrabacterial esterases on the hydrolysis of 3, we measured the activities of 3 against eight E. coli mutants that have one of bacterial esterase genes (i.e., BioH, YjfP, FrsA, YbfF, YeiG, YfbB, YpfH or YqiA) deleted. As shown in Figure 2A, the single gene deletion of the cytoplasmic esterase of E. coli reduces the antibacterial activities of 3 slightly at different magnitudes, but the differences among individual esterase knockout are statistically insignificant. Thus, mutation one esterase unlikely would lead to the drug resistance to 3. To confirm the role of intrabacterial esterases, we generated double mutant E. coli, with the deletion of BioH and YjfP genes (Figure S6). The double-mutant strains exhibit comparable growth rate with those of wild type and single mutant E. coli (Figure S7), suggesting that the increase of the viability of the double-mutant unlikely is resulted from its overall fitness. As shown in Figure 2B, the double deletion of BioH and YjfP significantly attenuates the antibacterial activity of 3, down to 50% of the activity against the wild type E. coli. This result indicates that BioH and YjfP play the considerable role in the hydrolysis of 3. We also generated E. coli mutants that overexpress BioH or YjfP, and found that the overexpression of BioH or YjfP is able to increase the susceptibility of the mutant (Figure S8). These studies confirm that, after 3 entering E. coli, various esterases in bacterial cytoplasm rapidly convert 3 to the active antibiotic agent (i.e., CL (1)).
Figure 2.
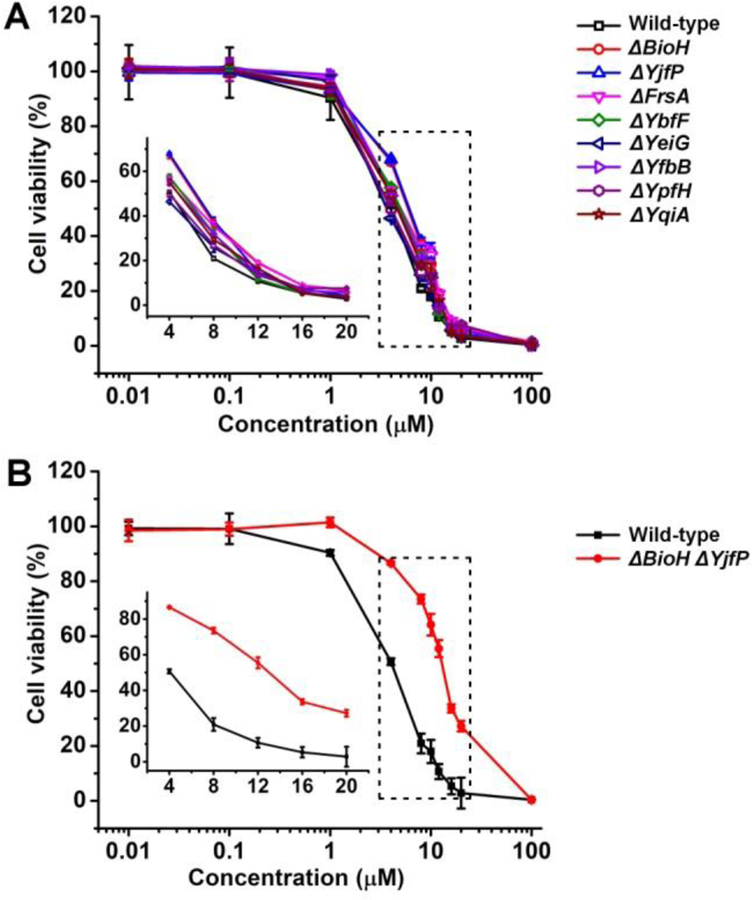
The antibacterial activity of 3 against (A) single esterase (BioH, YjfP, FrsA, YbfF, YfbB, YqiA, YeiG, or and YpfH) deletion mutants and (B) a double esterase (BioH and YjfP) deletion mutant of E. coli (inset: magnified image in the dashed square).
CL inhibits the peptidyl transferase activity of the bacterial ribosome to exhibit excellent antibacterial activity, but its ability to inhibit protein synthesis also results in adverse effects against mammalian cells, including bone marrow suppression.[18] To assess the side effects of the conjugates, we compared the cytotoxicities of 1, 2, and 3 towards HS-5,[19] a bone marrow stromal cell line. At the concentration of 20 μM (the MIC values of effective conjugates), while 1 reduces the viability of HS-5 cells, 2 and 3 are largely innocuous to HS-5 cells. We also compared the cytotoxicities of 1, 2, and 3 against a hepatocyte cell line (HepG2) and a kidney cell line (HEK293) and found that the conjugation of GG hardly alters the cytotoxicity of CL. Due to its resistance to esterases (Figure 1B), 2 also exhibits reduced cytotoxicity against HepG2. These results (Figure S9) indicate that the conjugation of GG likely would reduce the major adverse effect of CL (i.e., bone marrow suppression).
We next investigated whether the strategy shown in Scheme 1 is general for other type antibiotics. After synthesized the analogs (Figure 3A), we examined their activities against E. coli (Figure 3B). Although the replacement of succinate by cyclohexane-1,2-dicarboxylic acid produces a prodrug (6) with rather low activity (MIC > 200 μM), the conjugation of GG to 6 significantly increases the activity of the prodrug (7, MIC = 50 μM). Because the ciprofloxacin derivative, with the diglycine conjugated at the carboxylic acid of ciprofloxacin, hardly shows activity against E. coli (Scheme S5 and Figure S10), we made a ciprofloxacin derivative (8), which has 2-hydroxyacetic acid attach to the piperazine end of ciprofloxacin, for conjugating GG to ciprofloxacin. To further demonstrate the design that conjugating GG to antibiotic succinate accelerates intrabacterial hydrolysis of ester bond compared with the antibiotic succinate, for regenerating the active antibiotic against E. coli, we genereated 9, the succinate derivative of 8, which exhibits lower antibacterial activity (MIC = 1.0 μM) than that of 8 (MIC = 0.5 μM). The conjugation of GG to 9 results in 10 (MIC = 0.5 μM), which is more potent than 9 against E. coli. In addition, the comparison of the cytotoxicity of 6, 7, 8, 9, and 10 indicates low adverse effects against HS-5, HepG2, and HEK293 cells after the conjugation of GG (Figure S11). These results further confirm that the conjugation of GG to the succinate ester is an effective strategy for activating prodrugs via rapid intrabacterial hydrolysis, thus boosting the efficacy of antibiotics and reducing side effects.
Figure 3.
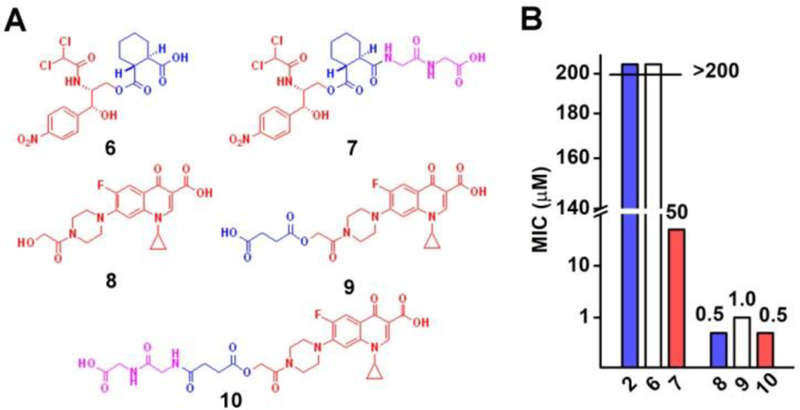
(A) The molecular structures and (B) the MIC values of the diglycine conjugated antibiotics (6, 7, 8, 9, and 10) against a wild type E. coli strain (K12).
In conclusion, this work demonstrates that conjugating GG to succinate prodrugs of antibiotics enhances the efficacy and improves the safety of the existing antibiotics. Although the original design aims to utilize the peptide transporters, rapid intrabacterial hydrolysis turns out to be the major factor for boosting the efficacy of the antibiotic prodrugs. This result implies that rapid intrabacterial hydrolysis may act as a useful approach for countering the efflux pumps[15] or other bacterial machineries,[20] which warrants further investigation. Besides underscore the versatile applications of intracellular enzymatic reactions,[21] this work suggests that judiciary conjugation of peptides to drugs is a powerful way for developing new therapeutics.[22]
Supplementary Material
Acknowledgements
This work is partially supported by NIH (R21AI130560) and NSF (DMR-1420382).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, Hayflick L, Butler RN, Allison DB, Ludwig DS, N. Engl. J. Med 2005, 352, 1138–1145. [DOI] [PubMed] [Google Scholar]
- [2].(a) Neu HC, Science 1992, 257, 1064–1073; [DOI] [PubMed] [Google Scholar]; (b) Ghafourian S, Sadeghifard N, Soheili S, Sekawi Z, Curr. Issues Mol. Biol 2015, 17, 11–21; [PubMed] [Google Scholar]; (c) Butler MS, Blaskovich MA, Cooper MA, J. Antibiot 2013, 66, 571–591; [DOI] [PubMed] [Google Scholar]; (d) Carattoli A, Int. J. Med. Microbiol 2013, 303, 298–304. [DOI] [PubMed] [Google Scholar]
- [3].(a) Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D, J. Am. Chem. Soc 2018, 140, 6749–6753; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schaefer K, Owens TW, Kahne D, Walker S, J. Am. Chem. Soc 2018, 140, 2442–2445; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, Crawford JJ, Durk MR, Higuchi RI, Kang J, Murray J, Paraselli P, Park S, Phung W, Quinn JG, Roberts TC, Rouge L, Schwarz JB, Skippington E, Wai J, Xu M, Yu Z, Zhang H, Tan MW, Heise CE, Nature 2018, 561, 189–194; [DOI] [PubMed] [Google Scholar]; (d) Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG, Science 2008, 321, 1673–1675. [DOI] [PubMed] [Google Scholar]
- [4].Neumann W, Sassone-Corsi M, Raffatellu M, Nolan EM, J. Am. Chem. Soc 2018, 140, 5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xie S, Manuguri S, Proietti G, Romson J, Fu Y, Inge AK, Wu B, Zhang Y, Häll D, Ramström O, Proc. Natl. Acad. Sci 2017, 114, 8464–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].(a) Zhou J, Du X, Li J, Yamagata N, Xu B, J. Am. Chem. Soc 2015, 137, 10040–10043; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z, Liu J, Dinulescu DM, Xu B, Angew. Chem. Int. Ed 2015, 54, 13307–13311; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li J, Shi J, Medina JE, Zhou J, Du X, Wang H, Yang C, Liu J, Yang Z, Dinulescu DM, Xu B, Adv. Healthc. Mater 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F, Nature 2001, 413, 814; [DOI] [PubMed] [Google Scholar]; (b) Noller HF, Hoffarth V, Zimniak L, Science 1992, 256, 1416–1419. [DOI] [PubMed] [Google Scholar]
- [8].Yunis AA, Smith US, Restrepo A, Arch. Intern. Med 1970, 126, 272–275. [PubMed] [Google Scholar]
- [9].Choremis CB, Megas HA, Askouni AD, Acta Pediatr 1963, 52, 136–142. [DOI] [PubMed] [Google Scholar]
- [10].Yogev R, Kolling WM, Williams T, Pediatrics 1981, 67, 656. [PubMed] [Google Scholar]
- [11].Weitz D, Harder D, Casagrande F, Fotiadis D, Obrdlik P, Kelety B, Daniel H, J. Biol. Chem 2007, 282, 2832–2839. [DOI] [PubMed] [Google Scholar]
- [12].Prabhala BK, Aduri NG, Sharma N, Shaheen A, Sharma A, Iqbal M, Hansen PR, Brasen C, Gajhede M, Rahman M, Mirza O, J. Biol. Chem 2018, 293, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tomczyk NH, Nettleship JE, Baxter RL, Crichton HJ, Webster SP, Campopiano DJ, FEBS lett 2002, 513, 299–304. [DOI] [PubMed] [Google Scholar]
- [14].Kuznetsova E, Proudfoot M, Sanders SA, Reinking J, Savchenko A, Arrowsmith CH, Edwards AM, Yakunin AF, FEMS Microbiol. Rev 2005, 29, 263–279. [DOI] [PubMed] [Google Scholar]
- [15].Li XZ, Plesiat P, Nikaido H, Clin. Microbiol. Rev 2015, 28, 337–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Whitesides GM, Wong C-H, Angew. Chem. Int. Ed 1985, 24, 617–638. [Google Scholar]
- [17].Hollifield WC Jr, Neilands J, Biochemistry 1978, 17, 1922–1928. [DOI] [PubMed] [Google Scholar]
- [18].Morley A, Trainor K, Remes J, Br. J. Haematol 1976, 32, 525–532. [DOI] [PubMed] [Google Scholar]
- [19].Roecklein BA, Torokstorb B, Blood 1995, 85, 997–1005. [PubMed] [Google Scholar]
- [20].Salomon D, Orth K, Curr. Biol 2015, 25, R265–R266. [DOI] [PubMed] [Google Scholar]
- [21].(a) Ye D, Liang G, Ma ML, Rao J, Angew. Chem. Int. Ed 2011, 50, 2275–2279; [DOI] [PubMed] [Google Scholar]; (b) Takaoka Y, Nishikawa Y, Hashimoto Y, Sasaki K, Hamachi I, Chem. Sci 2015, 6, 3217–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].(a) Yang C, Ren C, Zhou J, Liu J, Zhang Y, Huang F, Ding D, Xu B, Liu J, Angew. Chem. Int. Ed 2017, 56, 2356–2360; [DOI] [PubMed] [Google Scholar]; (b) Cheetham AG, Zhang P, Lin Y.-a., Lock LL, Cui H, J. Am. Chem.Soc 2013, 135, 2907–2910; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cheetham AG, Chakroun RW, Ma W, Cui H, Chem. Soc. Rev 2017, 46, 6638–6663; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhao F, Ma ML, Xu B, Chem. Soc.Rev 2009, 38, 883–891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.