

Abstract
Dysregulation of nuclear and cytoplasmic O-linked β-N-acetylglucosamine (O-GlcNAc) cycling is implicated in a range of diseases including diabetes and cancer. This modification maintains cellular homeostasis by regulating several biological processes, such as cell signaling. This highly regulated cycle is governed by two sole essential enzymes, O-GlcNAc transferase and O-GlcNAcase that add O-GlcNAc and remove it from over a thousand substrates, respectively. Until recently due to lack of structural information, the mechanism of substrate recognition has eluted researchers. Here we review recent successes in structural characterization of these enzymes and how this information has illuminated key features essential for catalysis and substrate recognition. Additionally, we highlight recent studies which have used this information to expand our understanding of substrate specificity by each enzyme.
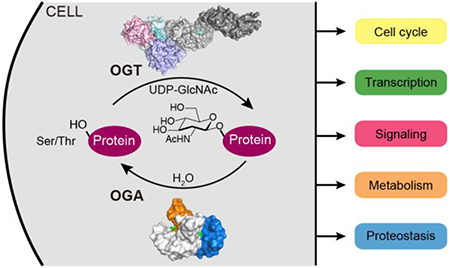
Graphical Abstract
Introduction
Historically, eukaryotic protein glycosylation was thought to occur exclusively in the endoplasmic reticulum and Golgi apparatus as part of the secretory pathway, which produces a vast array of diverse membrane glycoproteins. In the mid-1980’s, however, Hart and coworkers found O-linked β-N-acetylglucosamine (O-GlcNAc) on nuclear and cytoplasmic proteins (Figure 1a) [1]. The O-GlcNAc modification is dynamic, and its addition and removal are governed by a single pair of enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) (Figure 1b) [2–4]. Thousands of nucleocytoplasmic proteins are substrates of these O-GlcNAc-cycling enzymes. Because O-GlcNAc levels change substantially in response to nutrient availability and multiple forms of environmental stress (e.g., hypoxia, oxidative stress, thermal stress), it is thought that OGlcNAc cycling serves to maintain cell homeostasis by impacting cell signaling, gene expression, and proteostasis, among other processes [5,6]. Dysregulated O-GlcNAc abundance has been linked to several human diseases, including diabetes, cardiovascular disease, cancer, and neurodegenerative diseases, and it has been speculated that OGT and OGA may be therapeutic targets [7–10]. While the importance of O-GlcNAc cycling in metazoan physiology is by now indisputable, the functional significance of O-GlcNAc on individual substrates is extraordinarily challenging to decipher because there are so many O-GlcNAc substrates and the rules governing substrate selection are still unclear. Therefore, methods to selectively manipulate the cellular repertoire of O-GlcNAc are currently limiting. For OGT, the challenge is compounded by the recent discovery that this enzyme uses the same active site to attach O-GlcNAc and to effect another physiologically relevant modification, the cleavage of the essential cell cycle regulator, HCF-1 (Figure 1b) [11,12]. Progress in deconvoluting the functions of the O-GlcNAc cycling enzymes depends on having structural information to guide cellular experiments. A number of major advances have been made on this front in the past five years. This review will summarize key findings of structural studies on human OGT and OGA, with our apologies for the many omissions made due to space limitations. Information about structures mentioned in the text is provided in Figure 1
Figure 1: O-GlcNAc cycling is controlled by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA).

(a) Assembly of most cellular glycoproteins occur in the endoplasmic reticulum and Golgi apparatus as part of the secretory pathway. O-GlcNAc is uniquely found on nuclear and cytoplasmic proteins. (b) The O-GlcNAc cycling, the addition and removal of nuclear and cytoplasmic protein O-GlcNAc modifications, is controlled by two essential enzymes, OGT and OGA, and serves to maintain cellular homeostasis. OGT also catalyzes a second, physiological relevant protein modification, the cleavage of the transcriptional coactivator HCF-1. (c) Schematic of human OGT and constructs used for crystallography. (d) A partial list of human OGT crystal structures with different ligands. (e) Peptide sequences crystallized with OGT4.5. The TAB1 peptide was covalently fused to the N-terminus of OGT4.5. (f) Cartoon representation of human OGA isoforms and constructs for crystallography. (g) A partial list of reported human OGA crystal structures from different constructs and in complex with different ligands. The superscript numbers following PDB codes in Figure 1d and 1g represent the corresponding references.
O-GlcNAc Transferase
OGT belongs to the metal-independent GT-B superfamily of glycosyltransferases, which has been well-reviewed previously [13,14]. OGT is an essential gene encoded on the X-chromosome and it has two main regions: a long N-terminal tetratricopeptide repeat (TPR) region and a C-terminal catalytic region (Figure 1c) [15–17]. The two Rossmann-folded catalytic lobes that are characteristic of GT-B superfamily members are separated in primary sequence by a unique intervening domain of unknown function. Although there are two shorter splice variants, the TPR region of the primary, full-length OGT contains 13.5 TPRs. These are 34 amino acid helix-turn-helix motifs that typically mediate protein-protein interactions [18]. Consistent with this, removing TPRs from OGT abolishes protein glycosylation even when the active site is still functional; moreover, some cellular proteins have been shown to form stable interactions with the TPR region [17,19–22]. A crystal structure of the TPR region of OGT obtained in 2004 (PDB 1W3B; Figure 1c and 2d) shows an elongated, right-handed superhelix [23]. More recently, a structure of the TPR region containing a single point mutation distorts the superhelix, and it is speculated that this distortion alters the O-GlcNAc proteome or the OGT interactome, affecting brain development [24••].
Figure 2: OGT crystal structures provide clues to catalytic mechanisms and substrate recognition.

(a) Left panel: Cartoon representation of the structure of OGT complexes with CKII, a glycosylation substrate (yellow spheres), and UDP (magenta spheres). Middle panel: View of the active site of the OGT4.5-UDP-5SGlcNAc-CKII(A) complex showing key residues, selected diphosphate contacts and important bound water molecules. The β-phosphate of UDP-5SGlcNAc is anchored at the N-terminus of an α-helix and is stabilized by K842; a substrate-substrate interaction between the α-phosphate of UDP-5SGlcNAc and a backbone amide of CKII is proposed to assist in catalysis. The D554 side chain also plays an important role in catalysis. Right panel: Mechanism of O-GlcNAcylation highlighting the proposed role of D554 in acting as a terminal proton acceptor. (b) Left panel: Cartoon representation of OGT4.5-UDP-5SGlcNAcHCF-1rep complex shows that the cleavage region of HCF-1rep binds in the active site while the threonine-rich region binds in the TPR lumen. Middle panel: Composite structure of full-length OGT built in Pymol from PDB 4N3B and 1W3B. Right panel: The CKII(A) peptide (yellow sticks) and the cleavage region of HCF-1rep(Q) (cyan sticks) bind almost identically over UDP-5SGlcNAc (magnetic sticks) in the active site of OGT, providing structural evidence that glycosylation on glutamate is the first step in the mechanism of HCF-1 cleavage. (c) Substrates are anchored in the TPR lumen of OGT through bidentate contacts to conserved asparagines. Close-up view of contacts from asparagine side chains (magenta) to the threonine-rich region of HCF-1rep(Q) (cyan sticks; upper panel) reveals key interactions that anchor substrate in the TPR lumen; the same contacts are observed for a TAB1 peptide in the structure of an OGT4.5-TAB1 fusion protein (orange sticks; lower panel). (d) Cartoon representation of the structure of the TPR region of OGT implicated in substrate recognition. Asparagine residues in the TPR lumen are highlighted in magenta. e) Chemical mechanism of HCF-1 cleavage. Glycosylation on glutamate is followed by on-enzyme decomposition to an internal pyroglutamate, which undergoes spontaneous hydrolysis after release from OGT. The C-terminal cleavage product contains an N-terminal pyroglutamate.
The first structure of the catalytic region of human OGT was reported in 2011 (PDB 3PE4; Figure 2a) [25]. This structure, a complex with UDP and a peptide substrate (CKII), contained 4.5 TPRs. The TPRs in this structure partially overlap with the previously crystallized TPR region and a full-length model of OGT can be generated by superimposing these structures (Figure 2b, middle panel). All OGT structures obtained subsequently have used the 4.5 TPR construct as it crystallizes readily, and multiple structures of OGT complexed with substrates, substrate analogs, and products have been reported since 2011 (Figure 1d) [26–28]. Many of these are below 2.5 00C5 and include bound waters. To obtain ternary complexes that reflect how the substrates bind (i.e., pseudo-Michaelis complexes), it was necessary to use a hydrolysis-resistant analog of UDPGlcNAc in which the ring ether oxygen is replaced with sulfur (UDP-5SGlcNAc) and/or to substitute the reactive serine or threonine in the peptide substrate with alanine or aminoalanine [26]. Ternary glycopeptide product complexes were obtained simply by setting up crystals with UDP-GlcNAc or UDP-5SGlcNAc and a peptide substrate. Sulfur substitution does not alter the conformation of the substrates or products. Overall, the OGT substrate and product complexes have provided useful information about substrate preferences and reaction mechanisms.
The following picture has emerged for substrate recognition. OGT first binds UDP-GlcNAc and then the peptide substrate binds over it, making contact with the nucleotide-sugar for almost its entire length. The peptide backbone is anchored by polar contacts to OGT side chains, and there is also a polar contact between the α-phosphate of UDP-GlcNAc and the amide of the residue that becomes glycosylated, but there are almost no contacts from OGT to substrate side chains. This seemingly strange state of affairs is consistent with what is known about the lack of a strong consensus sequence for glycosylation. Some sequence preferences, such as prolines and β-branched residues in positions flanking the glycosite, have been identified from comprehensive glycosite mapping, but are insufficient to determine whether a site is glycosylated; these preferences can largely be explained by the requirement for the peptide to adopt an extended conformation over several residues in the active site [25,28,29••]
OGT complexes obtained with peptide substrates that bind in the TPR lumen of the 4.5 construct have provided important insight into how OGT substrates are selected. The first such complexes contained peptides derived from the HCF-1 cleavage region (PDBs 4N3A, 4N3B, 4N3C) (Figure 2b left panel) [30]. OGT cleaves HCF-1 within one of several, centrally-located, 26-amino acid repeats that contain a cleavage motif that binds in the active site and a threoninerich region that binds in the TPR domain [11]. The threonine-rich region is anchored in the TPR lumen by several asparagines that make bidentate hydrogen bonds to the peptide backbone (Figure 2c). Cleavage was abrogated when these asparagines were mutated to alanine [30]. These structures suggested that some glycosylation substrates may use a similar binding mode. Indeed, the structure of an OGT construct fused to a peptide derived from the glycosylation substrate TAB1 shows that the TAB1 peptide binds identically to the HCF-1 threonine-rich region in the TPR lumen (Figure 2c) [31]. Moreover, a global functional analysis has shown that a substantial fraction of OGT glycosylation substrates recognize the asparagine ladder [32••]. The evidence thus indicates that many OGT substrates are selected through extensive interactions both in the active site and to the contiguous TPR lumen. An unknown fraction of OGT substrates are thought to be directed to the active site through interactions with adaptor proteins that interaction with the TPR domain [20–22], and these adaptor proteins may also exploit the asparagine ladder for recognition. In this regard, it should be noted that the asparagine ladder lines the full length of the TPR lumen (Figure 2d) [23]. Other binding modes to OGT likely exist for both substrates and adapter proteins, and further structural and biochemical studies will be required to define these interactions.
In addition to providing substantial insight into substrate recognition, structural studies have revealed important details about mechanisms of glycosylation and HCF-1 cleavage [25,27,30]. Activation of the nucleotide-sugar is accomplished through interactions of the β-phosphate with the N-terminus of an α-helix as well as side chain contacts, including a contact with K842, an essential catalytic residue (Figure 2a middle panel). The previously mentioned contact from the α-phosphate to the amide of the glycosylated residue likely plays a crucial role in catalysis by positioning the side chain in the immediate vicinity of the anomeric carbon and providing additional activation of the leaving group [25–27]. The structures have also provided strong evidence for an electrophile migration mechanism in which the sugar rotates up and away from the leaving group and towards the nucleophile during the reaction. Surprisingly, the structures of substrate complexes did not show a proximal “catalytic base” that could serve to deprotonate the nucleophile, and two hypotheses have been proposed for how the proton is removed during the reaction. In one, an α-phosphate oxygen serves as the base [26]. In the other, the proton is translocated through two ordered water molecules to D554, a residue ~7 Å from the site of reaction (Figure 2a middle and right panel) [27]. While the issue of the catalytic base is unresolved, D554 is catalytically important for glycosylation, but not for HCF-1 cleavage, a difference consistent with its function as a base required for Ser/Thr glycosylation.
The structures of OGT substrate complexes showed that peptide backbones of glycosylation and cleavage substrates are superimposable (Figure 2b right panel). Combined with biochemical evidence consistent with a reaction mechanism involving a glutamyl ester, the structural evidence suggested that HCF-1 cleavage initiates when the invariant glutamate in the cleavage motif (Gln in Figure 2b) attacks the anomeric carbon [25,30]. No base is required in this reaction as glutamate is deprotonated at the reaction pH. Other studies have led to the mechanism for cleavage shown in Figure 2e [33••]. It has been found that aspartate-containing substrates can also be glycosylated by OGT; this results in isomerization to isoaspartate via a succinimide intermediate [34••]. Whether aspartate to isoaspartate isomerization is physiologically relevant for OGT, either as a side reaction requiring repair or as a switch mechanism for activity is not yet known. OGT can also occasionally transfer GlcNAc to cysteines in cellular proteins [35••]. OGT has thus revealed that a range of post-translational modifications (O-glycosylation, S-glycosylation, peptide backbone cleavage, and peptide backbone isomerization) are possible for a glycosyltransferase only on the identity of the glycosylated residue.
In order to decipher OGT’s cellular functions in mammals, OGT mutants lacking particular functions must be evaluated in cells. Based on structural studies, progress has already been made in identifying mutants that can affect only O-GlcNAcylation or HCF-1 cleavage; mutants lacking both functions are also available to evaluate the sufficiency of a scaffolding function for OGT. Recent work has also led to a better understanding of how the TPR region of OGT is involved in substrate recognition, which will make it possible to assess how changes to this region impact cell survival.
O-GlcNAcase
Human OGA is a multidomain protein with an N-terminal domain similar to glycoside hydrolase family 84 (GH84) enzymes, a stalk domain, a C-terminal pseudo histone acetyltransferase (HAT) domain, and several low-complexity regions (Figure 1f) [36]. A splice variant that lacks the HAT domain is less abundant and less active than OGA [37]. Although human OGA was first cloned and biochemically characterized in 2001 [36], it did not yield to structural studies until 2017. In the meantime, studies of bacterial homologs possessing a GH84 domain provided valuable insights into the hydrolytic mechanism and allowed the rational design of potent and specific OGA inhibitors, including GlcNAcstatin and thiamet-G [38–44].
In 2017, three independent research groups reported structures of human OGA, in apo form and in complex with small molecule inhibitors (Figure 1g) [45–47••]. One group also reported a series of structures of OGA complexed with different glycopeptide substrates [47••,48••], and these have begun to reveal general principles for substrate recognition. Key to obtaining the structures was the identification of stable OGA constructs for protein crystallization. Different approaches were used to generate stable OGA constructs (Figure 1f), but all maintained activity comparable to full-length OGA [45–47••]. The structures showed that the catalytic domain has the classic (β/α)8-barrel conformation characteristic of GH84 hydrolases, and also revealed an unusual dimeric structure not observed for previously studied bacterial OGA homologs (Figures 3a and 3b). The stalk domain forms a helical bundle comprising four main α-helices, three from the same OGA monomer and the fourth (labeled “arm” in Figure 3a) contributed by the sister monomer via a flexible loop. The dimer is stabilized by burial of a large surface area with formation of an extensive network of hydrogen bonds, salt bridges, and hydrophobic interactions. Residues on the dimerization interface are evolutionarily conserved in eukaryotes, and biophysical studies have confirmed that mammalian OGA exists as a stable dimer in solution [46••,47••].
Figure 3. Human OGA structures provide insights into substrate recognition and inhibitor design.

(a) OGA forms an “arm-in-arm” homodimer (PDB: 5TKE). The catalytic domain and stalk domain of one monomer are shown as ribbons in orange and marine color, respectively; the sister monomer is shown as ribbon in white color. An α-helix from one monomer (arm) forms tight binding with the sister monomer. (b) Ribbon representation of the overlay of the structures obtained from four different constructs. PDB 5TKE (green), 5UHK (cyan), and 5M7R (yellow) show nearly identical dimerization while 5UHP (magenta) shows a more relaxed conformation. (c) OGA binds inhibitors in the sugar binding pocket through strong hydrogen bonding and hydrophobic interactions. The complexes of OGA with thiamet-G (Left panel), PUGNAc and PUGNAc-imidazole (Middle panel), and VV347 (Right panel) are presented with inhibitors shown as sticks in indicated colors. The catalytic domain of one monomer and the stalk domain of the sister monomer are represented as surface in white and marine color, respectively; interacting residues from the catalytic domain and stalk domain are shown as grey and marine sticks, respectively. Hydrogen bonds and hydrophobic interactions are shown as dashed lines. (d) OGA binds glycopeptide substrates in a bi-directional yet conserved conformation. Left panel: α-crystallin B chain glycopeptide (green spheres) is bound in the substrate-binding cleft of OGA. Middle panel: Five different peptides (ribbon in indicated colors) are bound in the cleft in bidirectional but conserved conformation despite their distinct sequences (p53: QLWVDSTPPPG;α-crystallin B chain: FPTSTSLSPFYLR; TAB1: VPYSSAQS; ELK1: FWSTLSPI; Lamin B1: KLSPSPSSRVTVS. Residues with clear electron density are highlighted in bold and the O-GlcNAcylation site for each peptide is underlined). The N- or C-terminus of each peptide is highlighted in bold in corresponding color. Right panel: Glycopeptides are bound as Vshape in the substrate-binding cleft of OGA and form side-chain specific interactions with cleft surface residues. The interacting residues from the catalytic domain and stalk domain of OGA are highlighted as pink and green, respectively.
The three structures of OGA-thiamet-G complexes are superimposable and reveal active site residues that engage in tight interactions with the inhibitor which is a transition state mimic (Figure 3c left panel) [45–47••]. An ordered water molecule positioned near the carbon corresponding to the anomeric position of the sugar reveals the trajectory of nucleophilic attack for glycoside hydrolysis. The structures provide direct structural evidence to support the proposed substrate-assisted mechanism for OGA [49] and unveil a set of ancillary residues contributing to the hydrolytic mechanism. A hydrophobic pocket that affords favorable contacts to the N-ethyl substituent of thiamet-G provides a structure-based rationale for selectivity against other β-hexosaminidases. PUGNAc, a potent but more promiscuous inhibitor, cannot leverage this deep pocket to achieve specific targeting of OGA (Figure 3c middle panel) [45••]. VV347, an especially potent OGA inhibitor, makes favorably contacts with OGA surface residues (Figure 3c right panel) [46••]. Taken together, the structures of OGA complexed with these compounds provide valuable knowledge to guide future efforts in inhibitor design, both for O-GlcNAc functional investigation, and potential therapeutic applications such as in Alzheimer’s disease [10].
The OGA-glycopeptide structures provide critical insight into how OGA recognizes diverse O-GlcNAcylated proteins. Structures of a catalytically impaired OGA mutant (D175N) complexed to five distinct glycopeptide substrates have been reported (Figure 1g) and show that all of these glycopeptides are bound in the substrate-binding cleft created by the dimerization of OGA (Figure 3d) [47••,48••]. Regardless of the sequence flanking the glycosylated residue or whether that residue is Ser or Thr, the GlcNAc moieties overlap perfectly and engage in significant interactions with active site residues. This sugar binding mode explains the absolute selectivity of OGA for hydrolyzing β-O-GlcNAc substrates over either α-O-GlcNAc or β-O-GalNAc substrates. More strikingly, although all of the peptides adopt a similar “V”-shaped binding conformation near the glycosylation site, the peptide backbones can be oriented in opposite directions (Figure 3d middle panel) [48••]. Some of the binding conformations are further stabilized by intramolecular peptide interactions, and some peptide side-chain specific interactions are observed with the OGA cleft, such as the W(−3) subsite of the p53 glycopeptide with F223 and W679 (Figure 3d right panel). Mutation of these contact residues substantially impaired the binding of the p53 glycopeptide for OGA. Notably, different cleft surface residues of OGA participate in interactions with distinct glycopeptides [48••]. These five glycopeptide structures thus illuminate a general mechanism for how human OGA recognizes and processes diverse protein substrates. The conserved strong interactions between the GlcNAc moiety and OGA active site residues are critical for recognizing and anchoring glycopeptides in the substrate-binding cleft, but contacts beyond the immediate catalytic pocket can enhance the binding affinity for individual substrates and may affect hydrolysis rates. In support of this, recent proteomic analysis revealed that protein OGlcNAcylation turnover rates varied dramatically in cells, depending on both the protein and the residue modified [50]. These results, in agreement with the structural discoveries, suggest that OGA favorably removes O-GlcNAc from certain substrates during dynamic O-GlcNAc regulation.
Conclusion
The past few years have witnessed substantial progress in understanding the structural mechanisms of substrate recognition and the reactions catalyzed by the O-GlcNAc-cycling enzymes, but more work remains. In particular, future research will be needed to establish how OGT and OGA interact with protein substrates and to advance understanding of substrate specificity. Structural work that reveals new binding modes and guides the design of cellular experiments to deconvolute OGT and OGA’s functions will continue to play a crucial role in linking in vitro biochemistry to cellular function. In addition, regulatory mechanisms of OGT and OGA that limit futile O-GlcNAc cycling remain to be uncovered.
Acknowledgements
This work was supported by NIH grants R01 GM094263, R01 GM121718, and R01 GM126300.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest statement
Nothing declared.
References
Papers of particular interest, published within the period of the review, have been highlights as:
•• of outstanding interest
- 1.Torres CR, Hart GW: Topography and polypeptide distribution of terminal Nacetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for Olinked GlcNAc. Journal of Biological Chemistry 1984, 259:3308–3317. [PubMed] [Google Scholar]
- 2.Levine ZG, Walker S: The biochemistry of O-GlcNAc transferase: which functions make it essential in mammalian cells? Annual Review of Biochemistry 2016, 85:631–657. [DOI] [PubMed] [Google Scholar]
- 3.Janetzko J, Walker S: The making of a sweet modification: structure and function of OGlcNAc transferase. Journal of Biological Chemistry 2014, 289:34424–34432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X, Qian K: Protein O-GlcNAcylation: emerging mechanisms and functions. Nature Reviews Molecular Cell Biology 2017, 18:452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW: Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress: a survival response of mammalian cells. Journal of Biological Chemistry 2004, 279:30133–30142. [DOI] [PubMed] [Google Scholar]
- 6.Martinez MR, Dias TB, Natov PS, Zachara NE: Stress-induced O-GlcNAcylation: an adaptive process of injured cells. Biochemical Society Transactions 2017, 45:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaidyanathan K, Wells L: Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. Journal of Biological Chemistry 2014, 289:34466–34471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsh SA, Collins HE, Chatham JC: Protein O-GlcNAcylation and cardiovascular (patho)physiology. Journal of Biological Chemistry 2014, 289:34449–34456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slawson C, Hart GW: O-GlcNAc signalling: implications for cancer cell biology. Nature Review Cancer 2011, 11:678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuzwa SA, Vocadlo DJ: O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chemical Society Reviews 2014, 43:6839–6858. [DOI] [PubMed] [Google Scholar]
- 11.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W: O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 2011, 144:376–388. [DOI] [PubMed] [Google Scholar]
- 12.Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, Affar EB: Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proceedings of the National Academy of Sciences 2011, 108:2747–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S: Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proceedings of the National Academy of Sciences 2003, 100:845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lairson LL, Henrissat B, Davies GJ, Withers SG: Glycosyltransferases: structures, functions, and mechanisms. Annual Review of Biochemistry 2008, 77:521–555. [DOI] [PubMed] [Google Scholar]
- 15.Hanover JA, Yu S, Lubas WB, Shin S-H, Ragano-Caracciola M, Kochran J, Love DC: Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Archives of Biochemistry and Biophysics 2003, 409:287–297. [DOI] [PubMed] [Google Scholar]
- 16.Lubas WA, Frank DW, Krause M, Hanover JA: O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. Journal of Biological Chemistry 1997, 272:9316–9324. [DOI] [PubMed] [Google Scholar]
- 17.Kreppel LK, Hart GW: Regulation of a cytosolic and nuclear O-GlcNAc transferase: role of the tetratricopeptide repeats. Journal of Biological Chemistry 1999, 274:32015–32022. [DOI] [PubMed] [Google Scholar]
- 18.Allan RK, Ratajczak T: Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress & Chaperones 2011, 16:353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyer SPN, Hart GW: Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. Journal of Biological Chemistry 2003, 278:24608–24616. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Zhang F, Kudlow JE: Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 2002, 110:69–80. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, Wong J: Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-Linked β-N-acetylglucosamine transferase (OGT). Journal of Biological Chemistry 2014, 289:5986–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW: O-Linked β-Nacetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. Journal of Biological Chemistry 2008, 283:33935–33941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E: The superhelical TPRrepeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin [alpha]. Nature Structural & Molecular Biology 2004, 11:1001–1007. [DOI] [PubMed] [Google Scholar]
- ••24.Gundogdu M, Llabrés S, Gorelik A, Ferenbach AT, Zachariae U, van Aalten DMF: The OGlcNAc transferase intellectual disability mutation L254F distorts the TPR helix. Cell Chemical Biology 2018, 25:513–518.e514.This study presents the first structure of a mutation (L254F) within the tetratricopeptide repeat domain that is implicated in brain development. This structure gives us insight into how distortion of the TPRs could cause interruptions in the O-GlcNAc proteome.
- 25.Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S: Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469:564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schüttelkopf AW, Navratilova I, Aristotelous T, Albarbarawi O, Robinson DA, et al. : O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis. Nature Chemical Biology 2012, 8:969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazarus MB, Jiang J, Gloster TM, Zandberg WF, Whitworth GE, Vocadlo DJ, Walker S: Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nature Chemical Biology 2012, 8:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, Schüttelkopf AW, Albarbarawi O, van Aalten DMF: The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nature Structural & Molecular Biology 2015, 22:744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••29.Woo CM, Lund PJ, Huang AC, Davis MM, Bertozzi CR, Pitteri S: Mapping and quantification of over 2,000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (IsoTaG). Molecular & Cellular Proteomics 2018.This study presents the most comprehensive glycosite mapping data of the O-GlcNAc proteome in human T cells. The data confirms that there are little residue preferences near the glycosylation site in OGT substrates.
- 30.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S: HCF-1 Is cleaved in the active site of O-GlcNAc transferase. Science 2013, 342:1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rafie K, Raimi O, Ferenbach AT, Borodkin VS, Kapuria V, van Aalten DMF: Recognition of a glycosylation substrate by the O-GlcNAc transferase TPR repeats. Open Biology 2017, 7:170078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••32.Levine ZG, Fan C, Melicher MS, Orman M, Benjamin T, Walker S: O-GlcNAc transferase recognizes protein substrates using an asparagine ladder in the tetratricopeptide repeat (TPR) superhelix. Journal of the American Chemical Society 2018, 140:3510–3513.This study developed a chemoenzymatic protein microarray system to interrogate the contributions of a conserved asparagine ladder within OGT’s TPR lumen to substrate selection. Analysis of >9000 mammalian proteins showed that OGT recognizes the majority of its substrates using this conserved asparagine ladder proximal to OGT’s catalytic active site.
- ••33.Janetzko J, Trauger SA, Lazarus MB, Walker S: How the glycosyltransferase OGT catalyzes amide bond cleavage. Nature Chemical Biology 2016, 12:899.This study shows the biochemical mechanism of HCF-1 cleavage by OGT. HCF-1 cleavage occurs via glycosylation at a key glutamate followed by on-enzyme formation of an internal pyroglutamate. This pyroglutamate undergoes spontaneous backbone hydrolysis resulting in cleavage.
- ••34.Janetzko J, Walker S: Aspartate glycosylation triggers isomerization to isoaspartate. Journal of the American Chemical Society 2017, 139:3332–3335.This study discovers OGT’s ability to glycosylate asparatate residues, which results in aspartate to-isoasparate isomerization through a succinimide intermediate. This study reveals a new post translational modification by OGT in the same active site as Ser/Thr glycosylation and HCF-1 cleavage.
- ••35.Maynard JC, Burlingame AL, Medzihradszky KF: Cysteine S-linked N-acetylglucosamine (S-GlcNAcylation), a new post-translational modification in mammals. Molecular & Cellular Proteomics 2016, 15: 3405–3411.This study demonstrates S-glycosylation of mammalian proteins in cells by OGT. Additionally, OGA is unable to hydrolyze the S-GlcNAc modification. This study demonstrates a new and more stable post-translational modification by OGT.
- 36.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW: Dynamic O-Glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-Nacetylglucosaminidase from human brain. Journal of Biological Chemistry 2001, 276:9838–9845. [DOI] [PubMed] [Google Scholar]
- 37.Kim EJ, Kang DO, Love DC, Hanover JA: Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydrate Research 2006, 341:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, et al. : A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nature Chemical Biology 2008, 4:483–490. [DOI] [PubMed] [Google Scholar]
- 39.Ostrowski A, Gundogdu M, Ferenbach AT, Lebedev AA, van Aalten DMF: Evidence for a functional O-linked N-acetylglucosamine (O-GlcNAc) system in the thermophilic bacterium Thermobaculum terrenum. Journal of Biological Chemistry 2015, 290:30291–30305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schimpl M, Borodkin Vladimir S, Gray Lindsey J, van Aalten Daan MF: Synergy of peptide and sugar in O-GlcNAcase substrate recognition. Chemistry & Biology 2012, 19:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao FV, Dorfmueller HC, Villa F, Allwood M, Eggleston IM, van Aalten DMF: Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. The EMBO Journal 2006, 25:1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dennis RJ, Taylor EJ, Macauley MS, Stubbs KA, Turkenburg JP, Hart SJ, Black GN, Vocadlo DJ, Davies GJ: Structure and mechanism of a bacterial β-glucosaminidase having OGlcNAcase activity. Nature Structural & Molecular Biology 2006, 13:365–371. [DOI] [PubMed] [Google Scholar]
- 43.Dorfmueller HC, Borodkin VS, Schimpl M, Shepherd SM, Shpiro NA, van Aalten DMF: GlcNAcstatin: a picomolar, selective O-GlcNAcase inhibitor that modulates intracellular O-GlcNAcylation levels. Journal of the American Chemical Society 2006, 128:16484–16485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schimpl M, Schüttelkopf Alexander W, Borodkin Vladimir S, van Aalten Daan M F: Human OGA binds substrates in a conserved peptide recognition groove. Biochemical Journal 2010, 432:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••45.Elsen NL, Patel SB, Ford RE, Hall DL, Hess F, Kandula H, Kornienko M, Reid J, Selnick H, Shipman JM, et al. : Insights into activity and inhibition from the crystal structure of human O-GlcNAcase. Nature Chemical Biology 2017, 13:613–615.This study presents the structures of human OGA in apo forms of two crystallization constructs: hOGA14–400/554–705 and hOGA57–400/544–705, as well as the structures of the second construct in complex with OGA inhibitor thiamet-G or PUGNAc.
- ••46.Roth C, Chan S, Offen WA, Hemsworth GR, Willems LI, King DT, Varghese V, Britton R, Vocadlo DJ, Davies GJ: Structural and functional insight into human O-GlcNAcase. Nature Chemical Biology 2017, 13:610–612.This study presents the structures of human OGA in apo form (construct named Split1) and in complex with each of the three small molecule inhibitors: thiamet-G, PUGNAc-imidazole, and VV347.
- ••47.Li B, Li H, Lu L, Jiang J: Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode. Nature Structural & Molecular Biology 2017, 24:362–369.This study presents the structures of human OGA in apo form (construct named OGAcryst) and in complex with thiamet-G, as well as the structure of a catalytically deficient mutant OGAcryst D175N in complex with a p53 glycopeptide substrate. The structure of OGAcryst-D175N-p53 complex shows for the first time that the glycopeptide is bound in a substrate-binding cleft created by the dimerization of human OGA.
- ••48.Li B, Li H, Hu C-W, Jiang J: Structural insights into the substrate binding adaptability and specificity of human O-GlcNAcase. Nature Communications 2017, 8:666.This study presents the structures of human OGAcryst-D175N apo form and its complex with each of four distinct glycopeptide substrates: α-crystallin B chain, TAB1, ELK1, and Lamin B1. These structures reveal general principles for OGA substrate recognition.
- 49.Çetinbaş N, Macauley MS, Stubbs KA, Drapala R, Vocadlo DJ: Identification of Asp174 and Asp175 as the key catalytic residues of human O-GlcNAcase by functional analysis of site-directed mutants. Biochemistry 2006, 45:3835–3844. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Yuan Z-F, Fan J, Karch KR, Ball LE, Denu JM, Garcia BA: A Novel quantitative mass spectrometry platform for determining protein O-GlcNAcylation dynamics. Molecular & Cellular Proteomics 2016, 15:2462–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]