

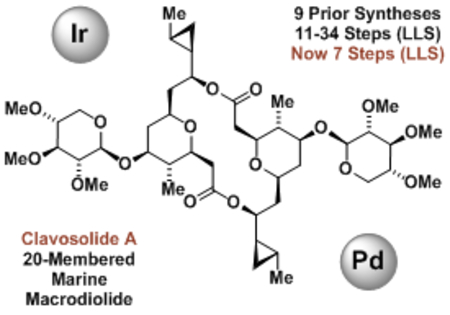
Graphical Abstract
Less is more. The 20-membered marine macrodiolide clavosolide A is prepared in 7 steps (LLS) in the absence of protecting groups or chiral auxiliaries via enantioselective alcohol-mediated carbonyl addition. In 9 prior total syntheses, 11-34 steps (LLS) were required.
Keywords: Enantioselective, C-C Bond Formation, Iridium, Hydrogen Transfer, Homogenous Catalysis
Polyketide natural products and their semisynthetic congeners are used more frequently in human medicine than any other class of secondary metabolites.[1,2] With one exception (eribulin®),[3] manufacturing routes to commercial polyketide drugs invariably rely on fermentation. These data suggest available methods for de novo polyketide construction do not adequately meet the challenges evoked by the synthesis of such stereochemically complex compounds. Carbonyl additions, in particular asymmetric aldol reactions[4] and allylmetallations,[5] figure prominently among methods for polyketide total synthesis, yet often require multi-step preparations of preformed enol(ate) or allylmetal reagents that involve numerous sacrificial reagents. To address these limitations, our laboratory has developed a suite of metal-catalyzed carbonyl reductive couplings and related hydrogen auto-transfer processes that convert lower alcohols to higher alcohols.[6] The redox-economy of these processes and the ability to directly deploy feedstock pronucleophiles in a stereo- and site-selective manner has been shown to streamline polyketide construction, enabling total syntheses in significantly fewer manipulations than previously required.[7] The bidirectional asymmetric allylation of 1,3-diols[8] represents one especially valuable method of this type, as it directly assembles triketide substructures, and has no counterpart in classical carbonyl addition chemistry as the corresponding 1,3-dialdehydes are unstable. For example, a 7-step total synthesis of cyanolide A was completed via bidirectional asymmetric allylation of neopentyl glycol[9g] - the shortest among 8 other total syntheses ranging between 12-19 steps (Figure 1).[9] Clavosolide A is a structurally related but more complex macrodiolide, for which 9 total syntheses exist that range between 11-34 steps (LLS) (Figure 1).[10,11] Here, we report a 7-step (LLS) total synthesis of clavosolide A in the absence of protecting groups[12] or chiral auxiliaries via bidirectional asymmetric allylation of 2-methyl-1,3-propane diol.
Figure 1.
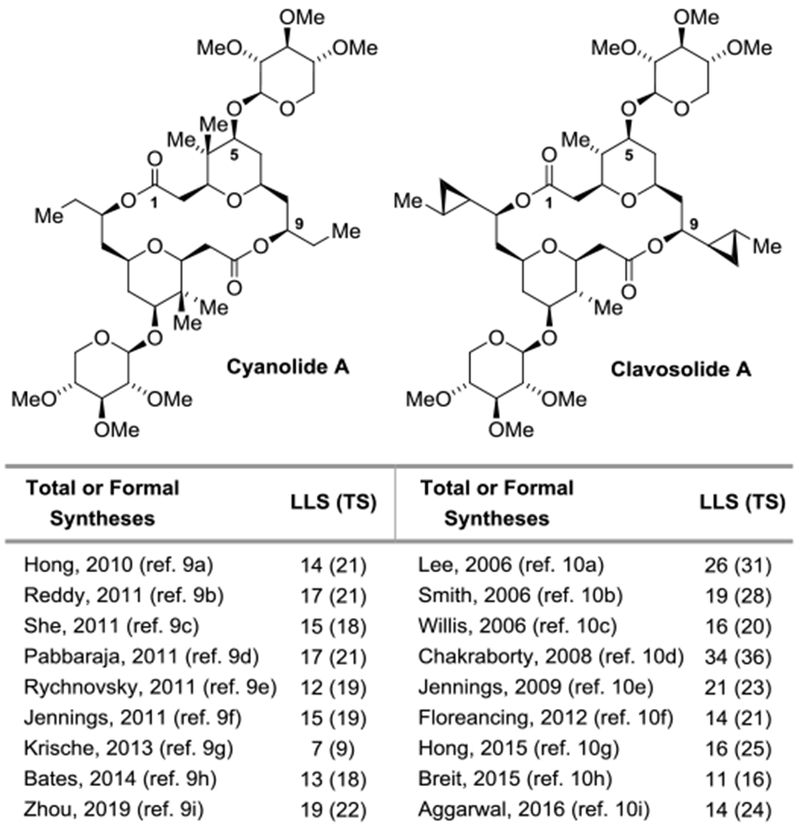
Summary of prior total and formal syntheses of cyanolide A and clavosolide A. For graphical summaries of total syntheses, see Supporting Information. Longest Linear Sequence (LLS), Total Steps (TS).
Clavosolide A, a C2-symmetric 20-membered glycosidic macrodiolide, was isolated in trace quantities from the marine sponge Myriastra clavosa, which is found in the Philippines.[13] The structure initially proposed for clavosolide A was misassigned and later revised by total syntheses.[10a,14] While the biological properties of clavosolide A have yet to be determined, the structurally related natural product cyanolide A[9,15] displays potent molluscicidal activity (LC50 = 1.2 μM) against the water snail Biomphalaria glabrata, a vector of the human parasitic disease schistosomiasis. Additionally, in 2016, the closely related macrodiolide xylopyranoside was discovered, cocosolide (not shown), which exhibits immunosuppressive activity.[16] The structure of cocosolide was verified by total synthesis through a 17-step route (LLS).[16]
Retrosynthetically, clavosolide A was envisioned to arise through dimerization of the hydroxy acid 11, which is prepared from pyran 3 by glycosylation using the permethylated D-xylose trichloroacetimidate 4 and subsequent introduction of the cyclopropyl moiety using iodide 8 (Scheme 1). Pyran 3 is potentially accessible in only two steps via bidirectional asymmetric allylation of 2-methyl-1,3-propane diol[8a] 2 followed by Fenton-Semmelhack alkoxypalladation-carbonylation of the resulting C2-symmetric diol 2.[17,18,19] The feasibility of assembling pyran 3 in this manner was rendered uncertain due to the presence of the methyl-bearing chirotopic nonstereogenic center at C4. However, in related halocyclizations of olefinic alcohols, highly stereoselective desymmetrization is achieved owing to the preference of the latent methyl-bearing stereocenter to reside equatorially in the nascent pyran.[8b,20]
Scheme 1.
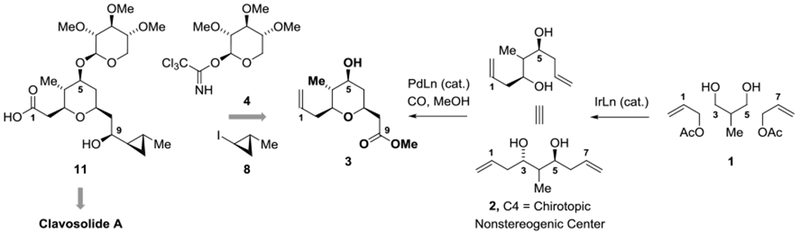
Retrosynthetic analysis of clavosolide A via bidirectional allylation of 2-methyl-1,3-propane diol.
The synthesis of pyran 3 begins with the bidirectional allylation of 2-methyl-1,3-propane diol (Scheme 2). The reported procedure involves generation of the cyclometallated catalyst in situ and utilizes (S)-Cl,MeO-BIPHEP as ligand.[8a] To improve scalability, an effort was made to optimize the reaction using the inexpensive ligand (S)-BINAP. Ultimately, it was found that using the preformed π-allyl-C,O-benzoate complex modified by (S)-BINAP, (S)-Ir-BINAP (10 mol%), and 4-cyano-3-nitrobenzoic acid (20 mol%) as an additive, the desired C2-symmetric diol 2 could be obtained in 53% yield on 10 mmol scale, which is roughly equivalent to the yield previously obtained using (S)-Cl,MeO-BIPHEP. Notably, an earlier synthesis of the corresponding desmethyl diol required a 7-step sequence involving three protecting group manipulations, two separate alcohol oxidations and two separate carbonyl asymmetric allylborations was required.[21] Initial attempts at Fenton-Semmelhack alkoxypalladation/carbonylation[17,18,19] of the C2-symmetric diol 2 using Pd(OAc)2/CuCl2 in acetonitrile-methanol solvent provided the desired pyran 3, but with poor levels of diastereocontrol at the C4 methyl-bearing chirotopic nonstereogenic center. Improved stereocontrol (6:1 dr) was achieved upon slow addition of diol 2 (over 10 min) and use of benzonitrile as cosolvent. Lower diastereoselectivity was observed using other Pd catalysts (PdCl2, PdI2, Pd(dppf)Cl2, Pd(PPh3)2Cl2).
Scheme 2.
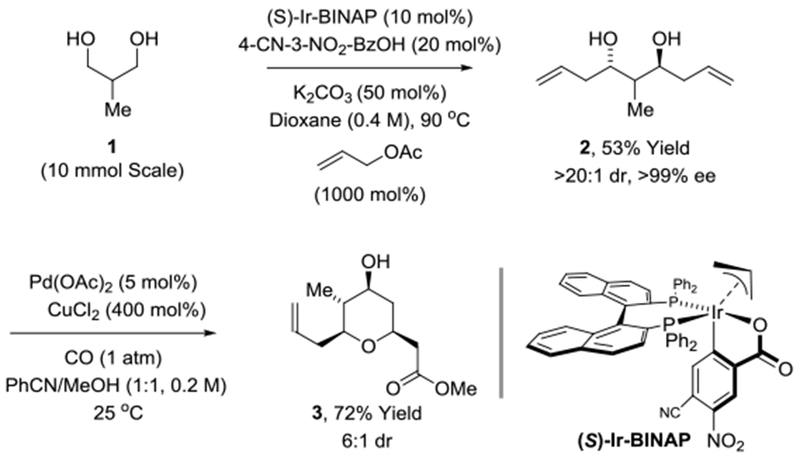
Enantioselective synthesis of pyran 3.
aYields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
In prior syntheses of clavosolide A,[10] direct formation of the trimethyl xylopyranoside moiety suffered from poor levels of β-selectivity. Specifically, in Schmidt-type glycosylations of trichloroacetimidates promoted by BF3•OEt2 or TMSOTf, a 1:1 ratio of α- vs β-anomers was observed.[10a,b,d,f] Aggarwal’s synthesis exploited neighbouring group participation in a tribenzoyl-protected xylopyranosyl donor to guide β-anomer formation,[10i] however, this approach incurred two additional chemical manipulations to transform the tribenzoate into permethylated xylopyranoside. A recent report from the Fairbanks group showed that chiral phosphoric acids catalyze β-selective Schmidt-type glycosylations of trichloroacetimidates.[22] Inspired by this work, glycosylation of pyran 3 catalyzed by (R)- and (S)-PA-I and PA-II were attempted in dichloromethane and toluene solvent using the trimethyl xylopyranoside trichloroacetimidate 4 (Scheme 3).[10a] Reactions catalyzed by the parent BINOL phosphoric acids (R)- and (S)-PA-I were inefficient and did not significantly influence β-selectivity. However, in accord with Fairbanks’ report, glycosylations catalyzed by (R)-PA-II displayed good levels of β-diastereoselectivity, with reactions conducted in toluene displaying slightly higher yields. The structure of β-xylopyranoside 5 was corroborated by single crystal X-ray diffraction analysis.
Scheme 3.
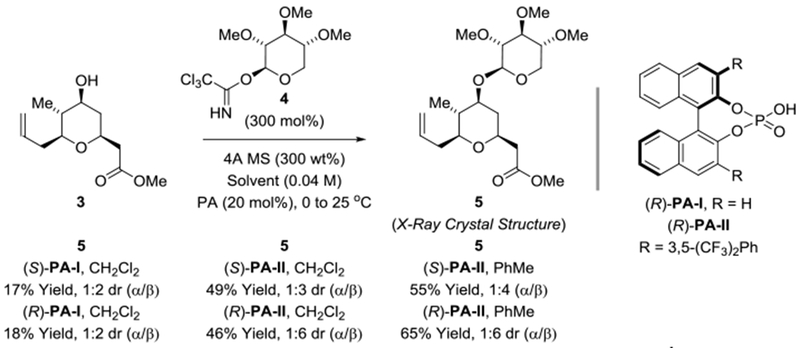
Chiral phosphoric acid-catalyzed Schmidt glycosylation of pyran 3 to form trimethyl xylopyranoside 5.a
aYields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. See Supporting Information for further experimental details.
With xylopyranoside 5 in hand, a carbonyl addition strategy for stereoselective introduction of the cyclopropane moiety spanning C10-C13 was undertaken, which necessitated preparation of cyclopropyl iodide 8 (Scheme 4). To this end, allyl alcohol 6 was subjected to Charette’s enantioselective iodocyclopropanation mediated by the indicated commercially available boronate ester.[23] The resulting cyclopropylcarbinyl alcohol 7 was converted to the tosylate and exposed to LiBH4 in ether solvent to furnish cyclopropyl iodide 8. Cyclopropyl iodides participate in lithium-halogen exchange and may be captured by diverse electrophiles with complete retention of configuration.[23b,24] Conversion of 8 to the corresponding cyclopropyl lithium followed by treatment with aldehyde 9 (prepared via DIBAL reduction of xylopyranoside 5) delivered the desired carbonyl addition product 10, but as an equimolar mixture of C9 epimers across a range of solvents. Based on chelation control or the Cram-Reetz model, additions to β-alkoxy aldehydes are anticipated to favor the undesired 1,3-anti-diastereomer.[25] Hence, the observed lack of diastereoselectivity was actually encouraging as it suggested the feasibility of utilizing a chirally modified cyclopropyl lithium reagent to bias formation of the desired 1,3-syn-diastereomer. Indeed, after screening a range of chiral diamines, including (1S,2S)-N1,N1,N2,N2-tetramethylcyclohexane-1,2-diamine[26] and (1S,2S)-N1,N1,N2,N2-tetramethyl-1,2-diphenylethane-1,2-diamine,[27] it was found that aldehyde addition in the presence of (−)-sparteine[28] enabled formation of cyclopropylcarbinyl alcohol 10 in a 4:1 diastereomeric ratio. The major and minor diastereomers could be separated by column chromatography on basic alumina. Exposure of 10 to KMnO4/NaIO4 affected chemoselective alkene oxidative cleavage to form carboxylic acid 11 without oxidation of the C9 alcohol.[29] Finally, mixed anhydride-mediated esterification-macrolactonization delivered clavosolide A in 7 steps (LLS).[30]
Scheme 4.
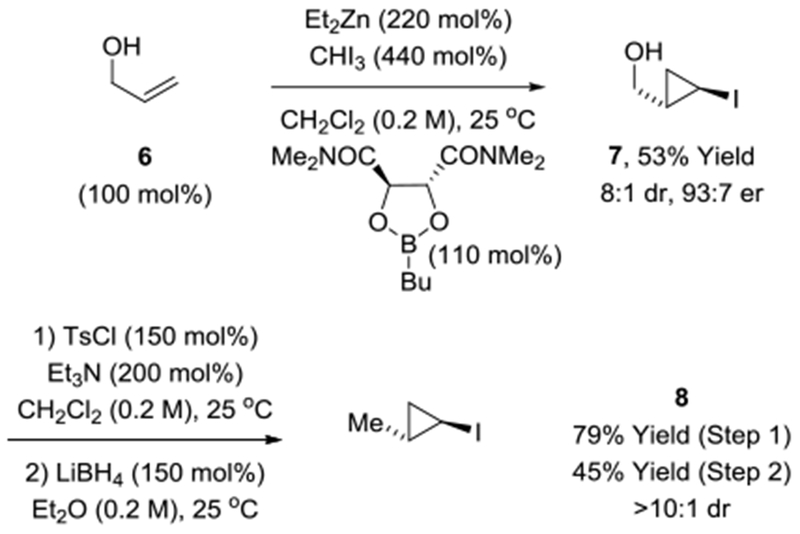
Enantioselective synthesis of iodide 8.
aYields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR analysis. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
To summarize, through bidirectional asymmetric diol allylation, a 7-step (LLS) total synthesis of the 20-membered marine macrodiolide clavosolide A has been achieved in the absence of protecting groups or chiral auxiliaries. In the longest linear sequence, 5 of 7 reactions represent skeletal construction events, 3 of which are stereoselective catalytic processes. The remarkably concise nature of this route is underscored by the fact that in 9 previously reported total syntheses 11-34 (LLS) were required. Future studies will focus on the development of related metal-catalyzed C-C bond formations that streamline polyketide construction by directly transforming lower alcohols to higher alcohols using abundant π-unsaturated pronucleophiles.[30]
Supplementary Material
Scheme 5.

Total synthesis of clavosolide A.
aYields are of material isolated by silica gel chromatography. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. See Supporting Information for further experimental details.
Footnotes
The Welch Foundation (F-0038), the NIH (RO1-GM069445) and the UT Austin Center for Green Chemistry and Catalysis are acknowledged for partial support of this research. Dr. Wonchul Lee and Mr. Jeremy Nicolai are acknowledged for skillful technical assistance.
References
- [1].For selected reviews on polyketide natural products, see:; a) O’Hagan D, The Polyketide Metabolites, Ellis Horwood Ltd., Chichester, 1991 [Google Scholar]; b) Rimando AM, Baerson SR, Polyketides: Biosynthesis, Biological Activity, and Genetic Engineering, ACS Symposium Series 955, 2007. [Google Scholar]
- [2].For selected reviews on the impact of natural products on drug development, see:; a) Newman DJ, Cragg GM, J. Nat. Prod. 2007, 70, 461–477 [DOI] [PubMed] [Google Scholar]; b) Cragg GM, Grothaus PG, Newman DJ, Chem. Rev. 2009, 109, 3012–3043. [DOI] [PubMed] [Google Scholar]
- [3].For a review on the synthesis of eribulin®, see:; Yu MJ, Zheng W, Seletsky BM, Nat. Prod. Rep 2013, 30, 1158–1164. [DOI] [PubMed] [Google Scholar]
- [4].For selected reviews on the asymmetric aldol reaction, see:; a) Knochel P, Molander GA, Comprehensive Organic Synthesis, Elsevier Ltd., Amsterdam, Vol. 2, 2014 [Google Scholar]; b) Arya P, Qin H, Tetrahedron 2000, 56, 917–947 [Google Scholar]; c) Romea P, Urpí F, Modern Methods in Stereoselective Aldol Reactions, (Ed.: Mahrwald R), Wiley-VCH, Weinheim, 2013, pp. 1–81 [Google Scholar]; d) Yamashita Y, Yasukawa T, Yoo W-J, Kitanosono T, Kobayashi S, Chem. Soc. Rev 2018, 47, 4388–4480. [DOI] [PubMed] [Google Scholar]
- [5].For selected reviews on asymmetric carbonyl allylation, see:; a) Ramachandran PV, Aldrichim. Acta 2002, 35, 23–35 [Google Scholar]; b) Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794 [DOI] [PubMed] [Google Scholar]; c) Yu C-M, Youn J, Jung H-K, Bull. Korean Chem. Soc 2006, 27, 463–472 [Google Scholar]; d) Marek I, Sklute G, Chem. Commun 2007, 1683–1691 [DOI] [PubMed] [Google Scholar]; e) Hall DG, Synlett. 2007, 1644–1655 [Google Scholar]; f) Hargaden GC, Guiry PJ, Adv. Synth. Catal 2007, 349, 2407–2424 [Google Scholar]; g) Lachance H, Hall DG, Org. React 2008, 73 [Google Scholar]; h) Han SB, Kim IS, Krische MJ, Chem. Commun 2009, 7278–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854 [DOI] [PubMed] [Google Scholar]; j) Moran J, Krische MJ, Asymmetric Synthesis – The Essentials II (Eds.: Christmann M, Bräse S), Wiley-VCH, Weinheim, 2012, pp. 187 [Google Scholar]; k) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2013, 113, 5595–5698 [DOI] [PubMed] [Google Scholar]; l) Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320 [Google Scholar]; m) Kumar P, Tripathi D, Sharma BM, Dwivedi N, Org. Biomol. Chem 2017, 15, 733–761 [DOI] [PubMed] [Google Scholar]; n) Spielmann K, Niel G, de Figueiredo RM, Campagne J-M, Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- [6].For selected reviews on hydrogen-mediated carbonyl reductive coupling and related hydrogen autotransfer reactions:; a) Bower JF, Krische MJ, Top. Organomet. Chem 2011, 34, 107–138 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hassan A, Krische MJ, Org. Proc. Res. Devel 2011, 15, 1236–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Moran J, Krische MJ, Pure Appl. Chem 2012, 84, 1729–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ketcham JM, Shin I, Montgomery TP, Krische MJ, Angew. Chem 2014, 126, 9294–9302; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 9142–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sam B, Breit B, Krische MJ, Angew. Chem 2015, 127, 3317–3325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2015, 53, 3267–3274 [Google Scholar]; f) Shin I, Krische MJ, Top. Curr. Chem 2016, 372, 85–101 [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Perez F, Oda S, Geary LM, Krische MJ, Top. Curr. Chem 2016, 374, 365–387 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354 (6310), aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For selected reviews on the application of hydrogen-mediated carbonyl addition in target-oriented synthesis, see:; a) Dechert-Schmitt A-MR, Schmitt DC, Gao X, Itoh T, Krische MJ, Nat. Prod. Rep 2014, 31, 504–513 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shin I, Montgomery TP, Krische MJ, Aldrichimica Acta. 2015, 48, 15. [PMC free article] [PubMed] [Google Scholar]; c) Feng J, Kasun ZA, Krische MJ, J. Am. Chem. Soc 2016, 138, 5467–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schwartz LA, Krische MJ, Isr. J. Chem 2018, 58, 45–51. [Google Scholar]
- [8].a) Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ, Angew. Chem 2009, 121, 5118–5121; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 5018–5021 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao X, Han H, Krische MJ, J. Am. Chem. Soc 2011, 133, 12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For total and formal syntheses of cyanolide A, see:; a) Kim H, Hong J, Org. Lett 2010, 12, 2880–2883 [DOI] [PubMed] [Google Scholar]; b) Hajare AK, Ravikumar V, Khaleel S, Bhuniya D, Reddy DS, J. Org. Chem 2011, 76, 963–966 [DOI] [PubMed] [Google Scholar]; c) Yang Z, Xie X, Jing P, Zhao G, Zheng J, Zhao C, She X, Org. Biomol. Chem 2011, 9, 984–986 [DOI] [PubMed] [Google Scholar]; d) Pabbaraja S, Satyanarayana K, Ganganna B, Yadav JS, J. Org. Chem 2011, 76, 1922–1925 [DOI] [PubMed] [Google Scholar]; e) Gesinski MR, Rychnovsky SD, J. Am. Chem. Soc 2011, 133, 9727–9729 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sharpe RJ, Jennings MP, J. Org. Chem 2011, 76, 8027–8032 [DOI] [PubMed] [Google Scholar]; g) Waldeck AR; Krische MJ Angew. Chem 2013, 125, 4566–4569; [Google Scholar]; Angew. Chem. Int. Ed 2013, 52, 4470–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Bates RW, Lek TG, Synthesis 2014, 46, 1731–1738 [Google Scholar]; i) Che W, Li Y-Z, Liu J-C, Zhu S-F, Xie J-H, Zhou Q-L, Org. Lett 2019, 21, 2369–2373. [DOI] [PubMed] [Google Scholar]
- [10].For total and formal syntheses of clavosolide A (revised structure):; a) Son JB, Kim SN, Kim NY, Lee DH, Org. Lett 2006, 8, 661–664 [DOI] [PubMed] [Google Scholar]; b) Smith AB III, Simov V, Org. Lett 2006, 8, 3315–3318 [DOI] [PubMed] [Google Scholar]; c) Barry CS, Elsworth JD, Seden PT, Bushby N, Harding JR, Alder RW, Willis CL, Org. Lett 2006, 8, 3319–3322 [DOI] [PubMed] [Google Scholar]; d) Chakraborty TK, Reddy VR, Gajula PK, Tetrahedron 2008, 64, 5162–5167 [Google Scholar]; e) Carrick JD, Jennings MP, Org. Lett 2009, 11, 769–772 [DOI] [PubMed] [Google Scholar]; f) Peh G, Floreancig PE, Org. Lett 2012, 14, 5614–5617 [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Baker JB, Kim H, Hong J, Tetrahedron Lett. 2015, 56, 3120–3122; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Haydl AM, Breit B, Angew. Chem 2015, 127, 15750–15754; [Google Scholar]; Angew. Chem. Int. Ed 2015, 54, 15530–15534; [DOI] [PubMed] [Google Scholar]; i) Millan A, Smith JR, Chen JL-Y, Aggarwal VK, Angew. Chem 2016, 128, 2544–2548; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2016, 55, 2498–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For a review on total syntheses of clavosolides A–D and cyanolide A, see:; For a review on total syntheses of clavosolides A–D and cyanolide A, see: Lee K, Lanier ML, Kwak J-H, Kim H, Hong J, Nat. Prod. Rep 2016, 33, 1393–1424. [DOI] [PubMed] [Google Scholar]
- [12].For selected reviews on site-selective catalysis and protecting group-free synthesis, see:; (a) Hoffmann RW, Synthesis 2006, 3531–3541; [Google Scholar]; (b) Young IS, Baran PS, Nature Chem. 2009, 1, 193–205; [DOI] [PubMed] [Google Scholar]; (c) Roulland E, Angew. Chem 2011, 123, 1260–1262; [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 1226–1227; [DOI] [PubMed] [Google Scholar]; (d) ‘Site-Selective Catalysis’ in Topics in Current Chemistry, Vol. 372 (Ed.: Kawabata T), Springer, Cham, Switzerland, 2016; [Google Scholar]; (e) Hartwig JF, Acc. Chem. Res. 2017, 50, 549–555; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Toste FD, Sigman MS, Miller SJ, Acc. Chem. Res. 2017, 50, 609–615; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dimakos V, Taylor MS, Chem. Rev. 2018, 118, 11457–11517. [DOI] [PubMed] [Google Scholar]
- [13].For isolation and structural assignment of clavosolides A–D, see:; a) Rao MR, Faulkner DJ, J. Nat. Prod 2002, 65, 386–388; [DOI] [PubMed] [Google Scholar]; b) Erickson KL, Gustafson KR, Pannell LK, Beutler JA, Boyd MR, J. Nat. Prod 2002, 65, 1303–1306. [DOI] [PubMed] [Google Scholar]
- [14].For total syntheses of the initially reported structure of clavosolide A, see:; Barry CS, Bushby N, Charmant JPH, Elsworth JD, Harding JR, Willis CL, Chem. Commun 2005, 5097–5099. [DOI] [PubMed] [Google Scholar]
- [15].For isolation and structural assignment of cyanolide A, see:; Pereira AR, McCue CF, Gerwick WH, J. Nat. Prod 2010, 73, 217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].For isolation, structural assignment and total synthesis of cocosolide, see:; Gunasekera SP, Li Y, Ratnayake R, Luo D, Lo J, Reibenspies JH, Xu Z, Clare-Salzler MJ, Ye T, Paul VJ, Luesch H, Chem. Eur. J 2016, 22, 8158–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Fenton DM, Steinwand PJ, J. Org. Chem 1972, 37, 2034–2035; [Google Scholar]; b) Semmelhack MF, Bodurow C, J. Am. Chem. Soc 1984, 106, 1496–1498. [Google Scholar]
- [18].For reviews on alkene alkoxycarbonylation in natural product total synthesis, see:; a) Bai Y, Davis DC, Dai M, J. Org. Chem 2017, 82, 2319–2328; [DOI] [PubMed] [Google Scholar]; b) Gehrtz PH, Hirschbeck V, Ciszek B, Fleischer I, Synthesis 2016, 48, 1573–1596; [Google Scholar]; c) Ma K; Martin BS, Yin X, Dai M, Nat. Prod. Rep 2019, 36, 174–219. [DOI] [PubMed] [Google Scholar]
- [19].For alkoxypalladation/carbonylation of an analogous C2-symmetric diol lacking a chirotopic nonstereogenic center, see:; Yang Z, Zhang B, Zhao G, Yang J, Xie X, She X, Org. Lett 2011, 13, 5916–5919. [DOI] [PubMed] [Google Scholar]
- [20].a) Gao X, Woo SK, Krische MJ, J. Am. Chem. Soc 2013, 135, 4223–4226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kasun ZA, Gao X, Lipinski RM, Krische MJ, J. Am. Chem. Soc 2015, 137, 8900–8903; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shin I, Hong S, Krische MJ, J. Am. Chem. Soc 2016, 138, 14246–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smith AB III, Minbiole KP, Verhoest PR, Schelhaas M, J. Am. Chem. Soc. 2001, 123, 10942–10953. [DOI] [PubMed] [Google Scholar]
- [22].a) Cox DJ, Smith MD, Fairbanks AJ, Org. Lett 2010, 12, 1452–1455; [DOI] [PubMed] [Google Scholar]; b) Kimura T, Sekine M, Takahashi D, Toshima K, Angew. Chem 2013, 125, 12353–12356; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2013, 52, 12131–12134. [DOI] [PubMed] [Google Scholar]
- [23].a) Beaulieu L-PB, Zimmer LE, Charette AB, Chem. Eur. J 2009, 15, 11829–11832; [DOI] [PubMed] [Google Scholar]; b) Beaulieu L-PB, Zimmer LE, Gagnon A, Charette AB, Chem. Eur. J 2012, 18, 14784–14791. [DOI] [PubMed] [Google Scholar]
- [24].a) Applequist DE, Peterson AH, J. Am. Chem. Soc 1961, 83, 862–865; [Google Scholar]; b) Corey EJ, Ulrich P, Tetrahedron Lett. 1975, 43, 3685–3688. [Google Scholar]
- [25].Open-chain and chelation models in β-alkoxy aldehyde addition both lead to 1,3-anti-diastereomers and cannot be distinguished:; Evans DA, Allison BD, Yang MG, Masse CE, J. Am. Chem. Soc 2001, 123, 10840–10852. [DOI] [PubMed] [Google Scholar]
- [26].Langer AW Jr., Polyamine-Chelated Alkali Metal Compounds, American Chemical Society, Washington, DC, 1974. [Google Scholar]
- [27].Tomioka K, Shindo M, Koga K, J. Am. Chem. Soc. 1989, 111, 8266–8268. [Google Scholar]
- [28].Nozaki H, Aratani T, Toraya T, Tetrahedron Lett. 1968, 9, 4097–4098. [Google Scholar]
- [29].Lemieux RU, Von Rudloff E, Can. J. Chem. 1955, 33, 1701–1709. [Google Scholar]
- [30].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi Bull M. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- [31].For a review on the use of feedstock pronucleophiles in metal-catalyzed carbonyl reductive coupling, see:; Doerksen RS, Meyer CC, Krische MJ, Angew. Chem 2019, 131, DOI: 10.1002/ange.201905532; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2019, 58, DOI: 10.1002/anie.201905532. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.