

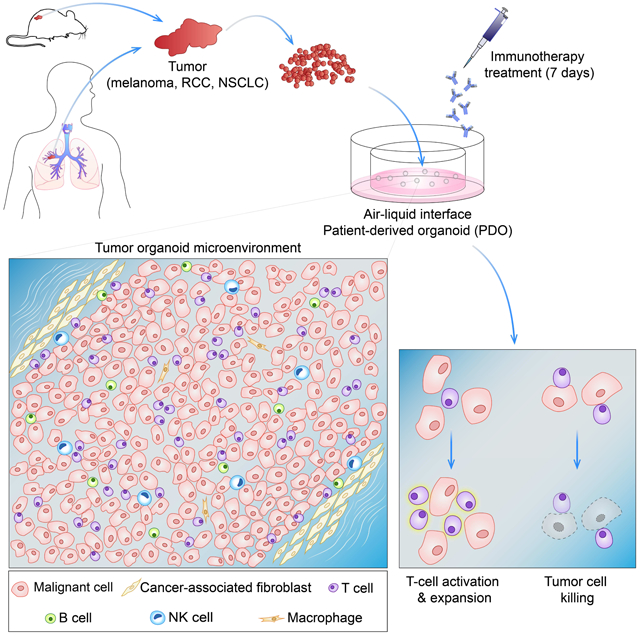
SUMMARY
In vitro cancer cultures, including 3-dimensional organoids, typically contain exclusively neoplastic epithelium but require artificial reconstitution to recapitulate the tumor microenvironment (TME). The co-culture of primary tumor epithelia with endogenous, syngeneic tumor-infiltrating lymphocytes (TILs) as a cohesive unit has been particularly elusive. Here, an air-liquid interface (ALI) method propagated Patient-Derived Organoids (PDOs) from >100 human biopsies or mouse tumors in syngeneic immunocompetent hosts as tumor epithelia with native embedded immune cells (T, B, NK, macrophages). Robust droplet-based, single cell simultaneous determination of gene expression and immune repertoire indicated that PDO TILs accurately preserved the original tumor T cell receptor (TCR) spectrum. Crucially, human and murine PDOs successfully modeled immune checkpoint blockade (ICB) with anti-PD-1- and/or anti-PD-L1 expanding and activating tumor antigen-specific TILs and eliciting tumor cytotoxicity. Organoid-based propagation of primary tumor epithelium en bloc with endogenous immune stroma should enable immunooncology investigations within the TME and facilitate personalized immunotherapy testing.
Graphical Abstract
In Brief
The tumor immune microenvironment is modeled using a patient-derived organoid approach that preserves the original tumor T cell receptor spectrum and successfully models immune checkpoint blockade
INTRODUCTION
The vast heterogeneity within cell types of the tumor microenvironment (TME) crucially impact treatment responses (Junttila and de Sauvage, 2013; Klemm and Joyce, 2015; Palucka and Coussens, 2016). The recent promise of therapies manipulating tumor-infiltrating immune cells has created a particular exigency for human cancer models that recapitulate this TME diversity. There is, however, a dearth of models, 2D or 3D, that represent the in vivo interaction of tumor and immune cells in the TME. Immune cells from blood or patient tumors have been reconstituted with heterologous established cancer cell lines in traditional monolayer, spheroid (Feder-Mengus et al., 2008) (Hirt et al., 2014) or primary organoid cultures (Dijkstra et al., 2018). However, such in vitro models of tumor immunity do not robustly retain the complex full diversity and physical architecture of the TME and particularly do not allow the co-culture of primary tumor epithelium with their native infiltrating immune populations en bloc without reconstitution. Short-term preservation of murine macrophages (Chen et al., 2018) and several human immune cell types (Finnberg et al., 2017) have not evidenced presence or functionality of T cells. Alternatively, custom microfluidic devices with human tumor suspension-derived microspheroids containing immune cells exhibit response to immunotherapeutics, but without tumor-immune specificity (Deng et al., 2018; Jenkins et al., 2018). Despite shortcomings, these studies increasingly support the use of 3-dimensional organoid models for holistic study of the immune TME.
We previously reported an air-liquid interface (ALI) murine organoid model containing tightly integrated epithelial and stromal compartments that recapitulates multi-hit tumorigenesis within normal stomach, pancreas and colon organoids (Li et al., 2014; Ootani et al., 2009). Here we extend this method to culture clinical tumor samples as Patient-Derived Organoids (PDOs) (Neal and Kuo, 2016; Vlachogiannis et al., 2018), in distinction to in vivo patient-derived xenograft tumors (PDX). These PDOs propagate primary diverse human and mouse tumors by a single method that preserves the complex histological TME architecture with tumor parenchyma and stroma, including functional, tumor-specific TILs. The ALI PDO method is thus distinct from organoid models lacking stroma (Boj et al., 2015; Dijkstra et al., 2018; Fujii et al., 2016; Sato et al., 2011; van de Wetering et al., 2015; Vlachogiannis et al., 2018). Importantly, this PDO system allows in vitro modeling of TME-intrinsic immune cell responses as opposed to those driven by peripheral immune populations, which remains a key obstacle to understanding mechanisms of checkpoint blockade (Wei et al., 2017).
RESULTS
Patient-Derived Organoids (PDO) from diverse tumor histologies preserve integrated stroma
We previously generated organoids from mouse wild-type tissues including intestine, stomach and pancreas by plating mechanically dissociated tissue fragments in Type I collagen matrix air-liquid interface (ALI) culture (Li et al., 2014; Ootani et al., 2009). Here, we adapted this method to establish PDOs from surgically-resected primary and metastatic tumors. We successfully established ALI PDO cultures from 100 individual patient tumors representing 14 distinct tissue sites and 28 unique disease subtypes (Figure 1A, Mendeley Figures 1–5; Table S1, STAR Methods). Organoid growth could be obtained with fetal calf serum alone, but was improved by rich growth factor supplementation with R-spondin1 as with wild-type ALI intestinal organoids (Ootani et al., 2009) or by WNT3A, EGF, NOGGIN and RSPO1 (WENR) for tumor epithelial organoids grown in submerged Matrigel (Boj et al., 2015; Fujii et al., 2016; Sato et al., 2011; van de Wetering et al., 2015). Thus, we utilized WENR base medium to expand and serially passage mechanically processed tumor fragments as ALI organoids. These PDOs included common tumor sites such as colon, pancreas, and lung, and rarer histologies such as bile duct ampullary adenocarcinoma, brain schwannoma and salivary gland pleomorphic adenoma (Figures 1B–D, Mendeley Figures 1–5; Table S1) for which cell lines are scarce (Barretina et al., 2012). PDOs typically recapitulated the parental tumor histology (Figure 1C, Mendeley Figures 1–5); establishment tended to be more robust with rapidly growing high-grade tumors. Growth under hypoxic conditions was not advantageous.
Figure 1. Air-liquid interface culture of human patient-derived tumor organoids (PDOs).

(A) Air-liquid interface PDO cultures from diverse tumor types and histologies.
(B) PDO primary culture and secondary passage. Stereomicroscopy, human colon adenocarcinoma PDO.
(C) PDOs from primary colorectal adenocarcinoma or lung adenocarcinoma recapitulate original tumor histology.
(D) Phase contrast (top), H&E (middle), and marker staining (bottom) of diverse day 30 PDOs.
(E) Cryorecovery and serial passage of representative PDOs.
(F) PDOs can be xenografted and re-derived as ALI organoids. Pancreatic ductal adenocarcinoma (PDAC) original histology (upper left) is recapitulated by PDO (upper right). PDOs grafted s.c. in NOG mice (lower right) generated tumors with PDAC histology which was preserved in organoids derived from the xenograft (lower left).
(G) Successful PDO culture irrespective of disease stage. Bars represent TNM staging for tumors used for PDOs. Crosshatched bars indicate unavailable staging information. Each column represents a distinct tumor.
(H) Significantly altered genes in 100 PDOs from end-to-end targeted exome and hot-spot exome PCR sequencing, with single nucleotide variation (SNV) and copy number alterations (CNA).
Abbreviations: P = passage number. d=culture day.
Each PDO was established via primary plating in ALI and then passaged once to confirm viability, with an overall 73% success rate at one-month culture across tumor histologies (STAR Methods). The variable presence of necrotic tumor in freshly plated tumor samples was substantially removed by serial passage. We assayed 15 of the most rapidly growing PDOs for cryopreservation and 12/15 (80%) could be cryorecovered and serially re-propagated every few weeks (Figure 1E, Mendeley Figure 6A). Continued growth (>4 passages, >100 days) did not always maintain complex tissue architecture resulting in some PDOs eventually exhibiting a simple, cystic morphology (Figure 1E). Alternatively, PDOs could be xenografted into immunocompromised mice and re-derived as organoids (Figure 1F; Mendeley Figure 6B), similar to submerged Matrigel organoids (Boj et al., 2015; van de Wetering et al., 2015). While this method utilized surgical samples, we have also established PDOs from smaller core needle biopsies.
Genetic characterization of PDOs
PDOs were generated from diverse tumor grades and metastatic status (Figure 1G). Targeted exome sequencing and copy-number variation analysis revealed expected PDO alterations such as APC loss in colorectal adenocarcinoma, KRAS codon 12 mutations in pancreatic ductal adenocarcinoma, TP53 loss in non-small cell lung cancers (NSCLC), VHL alterations in clear cell renal carcinoma (ccRCC) and BRAFV600E in thyroid carcinoma (ref. (Bailey et al., 2016; Cancer Genome Atlas, 2012) and www.cbioportal.org) (Figure 1H; Table S2, STAR Methods). In a subset of tumors with available clinical next generation sequencing, PDOs shared identical mutations such as EGFR L858R in lung PDOs (LT5 and LT18) and KRAS codon 12 mutations in pancreas PDOs (PT8) (Table S2C).
Preservation of fibroblast stroma within air-liquid interface PDOs
We previously observed that wild-type ALI organoids contain myofibroblasts closely associated with epithelium. This is potentially attributable to non-enzymatic processing, which generates larger contiguous sheets of cells allowing co-culture of epithelium and stroma without reconstitution (Li et al., 2014; Ootani et al., 2009), or alongside improved oxygenation in ALI (DiMarco et al., 2014).
Importantly, PDOs preserved tumor architecture and stroma expressing SMA and Vimentin (Figure 2A). In ~70% of cultures, stromal myofibroblasts progressively decreased (kidney, colon tumors) (Figure 2B–C, Mendeley Figure 7). In the remainder, particularly lung NSCLC, fibroblast proliferation was strongly stimulated by passage and could overgrow but could be separated PDOs by 70 micron cell filters. Fibroblasts could be present after cryorecovery (Figure 2D, Mendeley Figure 7) but were typically slowly lost over successive passage except for occasional overgrowth.
Figure 2. Human and mouse PDO cultures preserve integrated stromal cancer-associated fibroblasts.

(A) SMA+ and VIM+ cancer-associated fibroblasts (CAFs) in human PDOs. Top, SMA+ CAFs in human PDAC, colorectal adenocarcinoma and thyroid carcinoma PDOs. Tumor parenchyma (CK19, E-cadherin, thyroglobulin (TG)) and SMA immunofluorescence (IF) is shown. Bottom, VIM+ CAFs from human lung adenocarcinoma, pancreatic ductal adenocarcinoma and ampullary adenocarcinoma PDOs. IF for tumor parenchyma (TTF-1, CK19 and CK7) and VIM, culture d30.
(B) Time course of CAF preservation in representative human clear cell RCC PDO in fresh tumor (d0) and culture days 7 and 30. PanCK (magenta) and SMA (yellow) IF. DAPI (cyan). Scale bar = 20 μm. (C) Area quantitation of (B). N=4 areas, error bars +/− SEM, all values P<0.05 versus each other.
(D) Cryopreservation of PDOs preserves architecture and epithelial/stromal compartments. Lung adenocarcinoma PDO at day 30 and passage 2 day 21 after cryorecovery. H&E and IF for epithelium (E-cadherin) and stroma (SMA), scale bar = 50 μm.
(E) ALI cultures from the indicated s.c. mouse tumors borne in syngeneic immunocompetent hosts. Column 1: stereomicroscopy after passage 1 (P1) d10 (scale bar = 400 μm). Column 2: phase contrast of P0 d7 (scale bar = 200 μm). Column 3: H&E (scale bar = 100 μm). Columns 4 and 5: tumor lineage marker IF staining (E-cadherin, S100, CD20) or stroma (SMA, VIM) (4th and 5th column; scale bar = 50 μm). Columns 3–5 are culture d7.
(F) Passage and cryopreservation of B16-SIY mouse ALI tumor cultures. SMA IF is depicted (green) (scale bars: light microscopy = 200 μm; IF=50 μm).
(G) Area quantitation of (F), B16-SIY organoids vs. fresh tumor with SMA IF. n=5, average +/−SEM, * = P < 0.05 (organoids at indicated time vs. tumor).
Abbreviations: FT= fresh tumor, P=passage number, d=culture day.
See also Mendeley Figure 7.
ALI organoids were also robustly generated from mouse tumors implanted subcutaneously (s.c.) into syngeneic immunocompetent hosts: mouse B16 melanoma transduced with SIY peptide (Sivan et al., 2015) or mouse MC38 colon adenocarcinoma cells in syngeneic C57Bl/6 mice; or A20 B-cell lymphoma cells in syngeneic Balb/c mice. For all three mouse tumor systems, ALI organoids were easily generated, serially passaged, and exhibited integrated SMA- and Vimentin-positive stromal elements that declined over a 6-week period (Figure 2E–G). In contrast to human PDOs, fibroblast overgrowth did not occur in mouse ALI tumor cultures.
PDOs preserve diverse integrated immune elements
Given the myofibroblast stroma in ALI PDOs, we investigated an analogous presence of immune stroma. Human PDOs contained CD3-positive T-cells (tumor-infiltrating lymphocytes, TILs) integrally embedded in close proximity to tumor epithelium (Figures 3A–3C). PDOs also contained variable extents of CD14+ or CD68+ macrophages (Figure 3D). PDO FACS analysis revealed CD8+ (Tc) and CD4+ (Th) T cells, B-cells, natural killer (NK) and natural killer T (NKT) cells as well as infiltrating CD3+ T-cells expressing the immune checkpoint surface receptor programmed cell death protein-1 (PD-1) (Figure 3E). PDO TILs continually decreased over 1 month culture in WENR base medium (Figure 3F–G). Inclusion of interleukin-2 (IL-2) preserved intraorganoid PDO CD3+ TIL clusters and CD4+ and CD8+ subsets to approximately day 7 levels (Figure 3H–I) although TILs could not be supported beyond 60 days even with IL-2. Variation of glutamine concentrations did not alter PDO T cell subsets.
Figure 3. Immune components within human PDOs.

(A-C) IF staining of d14 PDOs from lung adenocarcinoma (A), clear cell renal cell carcinoma (ccRCC)
(B), and melanoma (C) identifies CD3+ TILs (yellow) closely associated with tumor epithelium (magenta, PanCK, S100), DAPI (cyan), scale bar = 25 μm.
(D) PDOs contain TAMs. PDO anti-CD14, E-Cad or CD68 IF staining in human clear cell renal cell carcinoma (ccRCC) (top) or human lung adenocarcinoma (middle, bottom). Scale bar = 50 μm.
(E) Diverse immune components upon FACS analysis of d7 lung adenocarcinoma and ccRCC PDOs.
(F) CD3+ TIL content in representative human ccRCC PDO in fresh tumor (d0) and culture days 7 and 30. PanCK (green) and CD3+ TIL (red) IF and DAPI (blue) co-stain. % area ratio of CD3+ cells indicated in red in the lower right corner. Scale bar = 20 μm.
(G) FACS quantitation of CD3, CD4 and CD8 TIL number/106 organoid cells from representative ccRCC PDO, +/− IL-2 for fresh tumor (FT) and culture d7 and d30.
(H) FACS analysis of IL-2-expanded organoid TILs. LC-1 – LC-4: independent lung NSCLC PDOs grown for 7d +/− IL-2. −, no IL-2; +, 600 IU/mL IL-2; ++, 6000 IU/mL IL-2. LC-5 and KT-1–4 (ccRCC) PDO-infiltrating T-cells persist at d21–28 without IL-2 but are significantly expanded with IL-2 (6000 IU/mL). ccRCC: d28 ccRCC PDO analyzed as in LC-1–5. Blue=CD8, Yellow=CD4.
(I) CD3+ TIL IF staining in representative d30 ccRCC PDO +/− IL-2 (100 IU/ml). CD3 (yellow), PanCK (magenta), DAPI (cyan). Scale bar = 20 μm.
ALI organoids from B16, MC38 and A20 s.c. tumors in syngeneic mouse hosts also preserved integrated immune populations without reconstitution. Organoids from all three mouse tumors contained CD3+ TILs and CD11b+ tumor-associated macrophages (TAMs) at day 7 (Figure 4A). IL-2 was needed to preserve TILs at longer time points and was routinely included. PDOs contained TILs and TAMs through serial passage at 42 days but progressively declined and again did not persist beyond 60 days (Figures 4B–D).
Figure 4. Immune components within mouse ALI tumor organoids.

(A) Mouse ALI organoids from syngeneic s.c. mouse MC38, B16-SIY and A20-OVA tumors retain integrated TILs and TAMs at culture d7. Rows 1–3: IF for CD3 (red, left column), CD8 (green, middle column) or merge (yellow, right column). Row 4: CD11b IF (red). DAPI (blue). Scale bar = 50 μm. d=culture day.
(B) Integrated TILs persist in mouse B16-SIY ALI organoids after serial passage and extended time points. CD3 (red) and CD8 (green) IF, DAPI (blue). Scale bar = 50 μm.
(C,D) Area quantitation of B16-SIY organoids vs. fresh tumor. NN=2 sections from 2 biological replicates (N=4 total), error bars +/− SEM, *= P < 0.05 (CD8:P1 d28 vs. CD8:tumor) or (CD11b:P1 d28 vs. CD11b:tumor).
Droplet-based simultaneous cell gene expression and immune repertoire profiling from single cells
We sought to confirm that PDOs faithfully represented the immune diversity and T cell receptor (TCR) repertoire of the original tumor biopsies. We thus developed the Chromium Single Cell Immune Profiling Solution, a robust droplet-based assay simultaneous determination of simultaneously determining gene expression profiles, T cell receptor and B cell immunoglobulin repertoires from single cells in the same input sample (Figure 5A, S1, STAR Methods). Briefly, single cell suspensions were loaded onto the Chromium controller, and full-length cDNA generated in Gelbeads in Emulsion (GEMs). The purified cDNA was amplified before being divided into 3 aliquots for 5’ Gene Expression (GEX) library preparation, and T and B cell enrichment for V(D)J library preparation (Table S3A). This utilizes the same microfluidics as our previously described Chromium Single Cell Gene Expression Solution (3’ GEX), with similarly low doublet rate (<1% for every 1,000 cells) and high cell capture efficiency (up to ~65%), which is important for analysis of limiting organoid material (Yan et al., 2017; Zheng et al., 2017).
Figure 5. Droplet-based tandem single cell 5’ V(D)J and 5’ RNA-seq of immune cells.

(A) Chromium single cell Immune Profiling Solution. Single cell 5’ GEX and enrichment libraries can be generated from the same sample.
(B) t-SNE plot of 5’ scRNA-seq human healthy donor PBMCs.
(C, D) (Left) t-SNE plot of (B) with PBMCs having rearranged TCR (C) or Ig (D) clonotypes by single cell 5’ V(D)J-seq and T cell enrichment assay (magenta). (Right) Top 10 paired TCR (C) or Ig (D) clonotypes from T or B cells, respectively.
(E) t-SNE plots of 5’ RNA-seq of human ccRCC CD45+ FACS-sorted cells from fresh tumor (FT, left) or day 7 PDO (right).
(F) t-SNE plots of single cell 5’ V(D)J-seq of human ccRCC CD45+ cells from FT (left) vs. day 7 PDO (right). Cells with detected TCR clonotypes by 5’ V(D)J T cell enrichment assay from (E) are colored in magenta and correspond to T cell identity by 5’ scRNA-seq in (E).
(G) Observed frequency of top 10 TCR clonotypes in FT vs. PDO from (E) (ranked by order in FT).
(H) Scatter plot of cell counts between matching FT and PDO clonotypes from (F) (log scale). Circles with larger sizes indicate multiple overlapping data points, i.e., distinct clonotypes having same frequencies in fresh tumor and organoid. Concordance (R2 = 0.719) between FT and PDO is significant (p < 0.01, permutation test).
(I) Paired TCRαβ chain sequences and exact cell counts of the number of individual TILs expressing each unique TCR clonotype in F and G. The top 3 clonotypes are denoted by dark red, green and blue dots.
(J) t-SNE plots denoting the top 2 clonotypes in FT and PDO localize to exhausted T cells identified by 5’ RNA-seq in (E).
See also Figures S1–S5, Mendeley Figures 8–9, Table S3 and STAR Methods.
As proof of concept, we applied the Chromium Immune Profiling Solution on ~3,800 human heathy donor peripheral blood mononuclear cells (PBMCs) and obtained GEX, T and B cell enrichment from the same sample (Table S3B). At least 12 sub-populations of T (Naïve CD4+, CD4+ Effector and Memory, Naïve CD8+, CD8+ Effector, and CD8+ Memory), B (Naïve B and B Memory), monocytes and dendritic cells were detected by 5’ GEX, of which 1,744 (~45%) were T cells and 556 (~14%) B cells, based on well-characterized markers (Figure 5B, S1B–C, STAR Methods). Greater than 85% of T and B cells from the 5’ GEX sample were also detected by VDJ enrichment assays, linking cell type identification and immune repertoires from the same cells. Among 1,744 T cells from the 5’ GEX assay, 86% (1,495/1,744) had at least a productive, full-length TCR α or β chain, with 73% (1,096/1,495) showing both α and β chains. Most T cells contained unique TCRs, with the most abundant TCRs shared by 0.4% (7/1,495) CD8 effector cells (Figure 5C). Among 556 B cells from the 5’ GEX assay, 99% (551/556) had at least a productive, full-length immunoglobulin (Ig) heavy or light (λ or κ) chain, with 92% (509/551) showing paired Ig heavy and light chains (Figure 5D). Similar to T cells, most B cells contained unique Ig receptors, with the most abundant Ig shared by 0.4% (2/551) memory B cells. This lack of clonal enrichment is expected from healthy donor T and B cells.
To assess sensitivity and accuracy, we profiled pan T and CD19+ B cells mixed with Jurkat and GM12878 cells which have well-characterized TCRs and Ig chains (Croce et al., 1985) (Table S3C). Paired Jurkat TCR chains were detected at expected frequencies of 0.1% (10/8,263 cells) and 1% (9/781 cells), with paired clonotype accuracy of at least 89% (8/9) and 91% (10/11), respectively (Table S3D). B and GM12878 mixes behaved similarly, accurately detecting clonotype frequencies as low as 0.1% (~10 cells) (Table S3D). This high sensitivity and accuracy enables immune repertoire assessment when clonal amplification is limited in tumor or organoid samples (Table S3B).
TILs within PDOs recapitulate the TCR repertoire of the original tumor
We next employed the Chromium Immune Profiling Solution to compare 5’ GEX and TCR repertoires of original tumors versus corresponding organoids. 5’ GEX of the FACS-sorted CD45+ fraction of a human clear cell renal cell carcinoma (ccRCC-1) fresh tumor (9,914 CD45+ cells) versus day 7 PDO (10,377 CD45+ cells) revealed common shared diverse immune populations including Tc, Th, B and NK cells. A CD8A/CD8B+ cell cluster expressing mRNAs such as LAG3, TIGIT, HAVCR2/TIM3 and PDCD1/PD1 marked exhausted T cells (Tex); Treg cells expressing FOXP3 and IL2RA were also present (Figures 5E, S2, Tables S3B and S3E). Macrophages were extensively of the M2 phenotype (CD206+ CD163+ INOS-) (Figures 5E, S2). The inferred cell types in the CD45+ fraction were consistent across ccRCC samples from 3 additional patients: one with a fresh tumor/organoid pair ccRCC (ccRCC-2) and ccRCC organoids from two further individuals (ccRCC-A, ccRCC-B). (Figure S3, Mendeley Figure 8, Tables S3B and S3E). The immune diversity of T, B and NK cells was generally reproduced across distinct PDOs and/or corresponding fresh tumor (FT), except for variable macrophage content (Figures 5E, S2–S3, Mendeley Figure 8), consistent with other reports (Azizi et al., 2018).
Furthermore, PDOs faithfully recapitulated the TCR repertoire of the original tumor biopsies. In matched fresh tumor/PDO pairs, parallel single cell 5’ V(D)J sequencing of TCR a and b chains from FACS sorted TILs (single CD45+ cells having TCR rearrangements) (Figure 5F) revealed that TCR clonotype composition was strongly preserved between PDO and FT, regardless of clone definition by TCR α, β or αβ chains. Indeed, (a) the most expanded clone in a given FT was consistently the most expanded clone in the corresponding PDO (Figures 5G–I, S4), and (b) the cell count of the observed clones was significantly correlated between FT and PDO (p < 0.01, permutation test) (Figures 5H, S4). Notably, the highest represented TCR clonotypes in both FT and PDOs were identically concentrated in exhausted T cells (Figures 5I–J, S5). We also independently confirmed strong TCR repertoire concordance in FT versus day 7 PDO by previously described SMART-seq2 TCR α and β CDR3 single cell sequencing (Han et al., 2014; Picelli et al., 2014) in two individual human NSCLC clinical samples (NSCLC-1, NSCLC-2, p < 0.01, permutation test) (Mendeley Figure 9, Table S3E). Overall, TCR analysis by the Chromium Immune Profiling Solution revealed ~300 FT/PDO overlapping TCR clonotypes, or ~10x more than by Smart-Seq2 (Han et al., 2014) (Figures 5G–5I, S4, Mendeley Figure 9), attributable to the former method capturing more than 10x the cell number in FT/PDO with corresponding deeper TCR profiling and higher confidence in the observed concordance.
In vitro PD-1/PD-L1 immune checkpoint blockade expands and activates antigen-specific TILs within murine tumor organoids
We next examined TIL functionality within murine organoids from MC38, B16-SIY and A20-OVA s.c. tumors in syngeneic immunocompetent hosts (Figures 2E, 4). The corresponding MC38, B16-SIY and A20-OVA tumor organoids were treated with function-blocking monoclonal anti-murine PD-1 or PD-L1 versus control IgG for 7 days (Figure 6 and STAR Methods). Both anti-PD-1 and anti-PD-L1 strongly increased CD8+ TILs per total organoid CD3+ TILs (Figure 6A) or total organoid cells (including tumor epithelium) (Figure 6B), paralleling CD8+ TIL expansion by anti-PD-1 within MC38 and B16 tumors in vivo and in patient peripheral blood (Huang et al., 2017; Kamphorst et al., 2017; Wei et al., 2017). Induction of mRNA for T-cell activation marker interferon-gamma (Ifng) and the cytolytic markers Perforin-1 (Prf1) and Granzyme B (Gzmb) are each associated with clinical responses to anti-PD-1/PD-L1 checkpoint blockade (Herbst et al., 2014). Accordingly, anti-PD-1 and anti-PD-L1 each activated CD8+ TILs in MC38, B16-SIY and A20-OVA organoids, with prominent stimulation of Ifng, Prf1 and Gzmb mRNA (Figure 6C) even after cryopreservation and recovery (Mendeley Figure 10). Both anti-PD-L1 strongly promoted tumor epithelial cell killing in B16-SIY organoids, with 2–3-fold increased Annexin-V(+)/7-AAD(−) early apoptotic cells and 7–14 fold induction of Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells versus control IgG (Figures 6D–E, S6A).
Figure 6. A functional PD-1/PD-L1 immune checkpoint in organoids derived from s.c. mouse tumors in syngeneic immunocompetent hosts.

(A) FACS analysis of CD3+CD8+ T cells in B16-SIY, A20-OVA and MC38 mouse tumor-derived organoids after 7 days in vitro αPD-1/αPD-L1 treatment vs. IgG, from a single representative experiment from n=3 biological replicates for each tumor line.
(B) FACS quantification of CD8+ TILs/106 organoid cells from (A) after 7 days organoid αPD-1 and αPD-L1 treatment.
(C) Quantitative RT-PCR (qRT-PCR) analysis of FACS-sorted CD3+ TILs from (A) after 7 days of αPD-1 and αPD-L1 treatment. Ifng, Gzmb and Prf1 mRNA expression normalized to control IgG. N=3. Error bars, +/− SEM * Ifng, Ϯ Gzmb or Δ Prf1 control vs. αPD-1/αPD-L1 = P<0.05.
(D) αPD-1 and αPD-L1 induce tumor organoid epithelial cell cytotoxicity. B16-SIY ALI organoids from syngeneic C57BL/6 s.c tumors were cultured for 7d +/− αPD-1, αPD-L1 or IgG. Tumor epithelial cell death was analyzed by FACS using anti-melanoma antibody pre-gating to denote Annexin-V(+)/7-AAD(−) early apoptotic (orange) and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells (pink). Representative of n=3 independent experiments.
(E) FACS histogram plot of 7-AAD staining within late apoptotic/necrotic Annexin-V(+) cells from (D).
(F) SIY tetramer FACS staining of antigen-specific T cells per total CD8+ T cells in freshly dissociated parental B16-SIY tumor (upper left) vs, B16-SIY organoids (panels 2–4) after 7d αPD-1, αPD-L1 or IgG treatment. Negative control SIIN tetramer was devoid of signal. Representative of n=3 independent experiments.
(G) Quantification of SIY tetramer-reactive CD8+ TILs per total organoid cells from (F).
(H) qRT-PCR of FACS-sorted SIY tetramer-reactive CD8+ TILs from (F). N=3 technical replicates. Error bars, +/− SEM, * Ifng, Ϯ Gzmb or Δ Prf1 control vs.: αPD-1/αPD-L1 = vs. IgG control, P<0.05.
(I) αPD-1 and αPD-L1 induce SIY-specific TILs in passage 2 B16-SIY organoids, culture d42. FACS of CD8+ SIY tetramer-reactive TILs. Representative of n=3 independent experiments.
(J) qRT-PCR analysis of CD8+ SIY tetramer-reactive TILs from (I). N=3 technical replicates. Error bars, +/− SEM, *= P<0.05 vs. IgG.
See also Figure S6, Mendeley Figure 10 and STAR Methods.
The stable transduction of the B16-SIY melanoma line with the immunogenic SIYRYYGL peptide (SIY) allows FACS enumeration of organoid CD8+ TILs bearing SIY-reactive TCR via SIY peptide-loaded H-2Kb tetramer staining (Sivan et al., 2015). In B16-SIY organoids from s.c. tumor-bearing C57Bl/6 mice, the proportion of TILs with SIY-reactive TCRs was well maintained between B16-SIY fresh tumor and organoid (Figure 6F). Notably, anti-PD-1 or PD-L1 expanded SIY tetramer-reactive CD8+ TILs in B16-SIY organoids (Figures 6F, 6G) with lack of staining with negative control SIINFEKL (SIIN) peptide tetramers. Anti-PD-1 and PD-L1 activated SIY-recognizing CD8+ organoid TILs with Ifng, Prf1 and Gzmb mRNA induction (Figure 6H). SIY-reactive TILs persisted after serial passage at culture day 42 and were expanded and activated by anti-PD-1 and anti-PD-L1 (Figures 6I, 6J). Anti-PD-1 and PD-L1 responses were highly reproducible amongst biological replicates, indicating that ALI organoid TILs are both functional and robustly recapitulate the PD-1/PD-L1 immune checkpoint.
Human organoid TILs functionally recapitulate the PD-1-dependent immune checkpoint
The PDO preservation of primary tumor epithelium en bloc with native endogenous TILs affords an opportunity for in vitro human immunotherapy modeling. We thus investigated the functional human PDO response to anti-PD-1 within a clinically actionable 7-day time frame. PDOs were established from 20 additional distinct surgically resected tumors representing the immunotherapy-responsive neoplasms NSCLC (n=9), ccRCC (n=8), and melanoma (n=3) and treated with either the therapeutic PD-1 blocking antibody nivolumab (Topalian et al., 2012) or isotype human IgG4 control (Figure 7 and STAR Methods). Nivolumab extinguished PD-1 FACS signal on CD3+ T cells, indicating nivolumab saturation of TIL cell-surface PD-1 via antibody competition against the distinct anti-PD-1 monoclonal used in FACS. Significantly, in 6 of 20 PDOs nivolumab elicted high-grade induction (>5-fold) of IFNG, PRF1, and/or GZMB within organoid FACS-sorted CD3+ TILs, denoting functional in vitro recapitulation of checkpoint inhibition (Figure 7A). PDO TIL activation responses to nivolumab (6/20) spanned NSCLC (33%, 3/9), RCC (25%, 2/8) and melanoma (33%, 1/3) PDOs, concordant with anti-PD-1/PD-L1 response rates in clinical NSCLC trials (Borghaei et al., 2015; Brahmer et al., 2015; Garon et al., 2015; Herbst et al., 2016), RCC (Motzer et al., 2015) and melanoma (Ribas et al., 2015; Robert et al., 2015; Weber et al., 2015). In parallel, nivolumab also expanded PDO CD8+ TILs by >35% in 5/6 activation responders versus 2/14 non-responders, with variable CD4+ TIL induction (Figure S7A–C).
Figure 7. In vitro recapitulation of the PD-1-dependent immune checkpoint in PDOs derived from human surgical tumor resections.

(A) IFNG, GZMB, and PRF1 qRT-PCR of FACS-sorted CD3+ TILs from NSCLC, RCC and melanoma PDOs, after 7 days nivolumab or control IgG4 treatment, nivolumab-treated normalized to IgG4. 28–8 PD-L1 IHC % is depicted (n/a: original tumor not available for PD-L1 IHC), N=3 technical replicate determination. Error bars, +/− SEM.
(B) Clone 28–8 PD-L1 IHC of fresh tumor from PDO responders from (A). Original histology from a sixth responding PDO was not available for analysis.
(C) FACS T-cell profiling of PDOs versus qRT-PCR +/− nivolumab with % PD-1 T cells, % T cells per total viable organoid cells, and CD4:CD8 ratio.
(D) Anti-PD-1/nivolumab induction of PDO tumor epithelial cell death. 7-AAD FACS histogram of Annexin V(+) tumor epithelial cells. Human ccRCC or bladder urothelial carcinoma PDOs received nivolumab or IgG4 with anti-CD3 + anti-CD28 for 7 days.
(E) FACS analysis of CD3+, CD8+ and CD4+ PDO TILs per 106 organoid cells from (D).
(F) qRT-PCR of PRF1, GZMB and IFNG from FACS-sorted CD3+ PDO TILs from (D) as fold-change mRNA for nivolumab vs. IgG4 (all significant at P<0.001). N=3 technical replicates, error bars, +/− SEM.
See also Figures S6B–D, S7 and STAR Methods.
Tumor IHC using an anti-PD-L1 antibody (clone 28–8) is a companion diagnostic for nivolumab treatment19 and analogous PD-L1 IHC testing is performed for the PD-1 antibody pembrolizumab. These can be confounded by antibody specificity, tumor PD-L2 expression, PD-L1 expression on multiple TME cell types (Topalian et al., 2016) and significant false-positive and false-negatives (Carbognin et al., 2015; Garon et al., 2015). Thus, we compared the PDO response to nivolumab against the CLIA-certified PD-L1 clone 28–8 IHC assay. Notably, PDO TIL activation markers did not correlate with the 28–8 PD-L1 IHC assay except for GZMB (r=.51, p=.03) (Figures 7B, S7D–G). Further, the IFNG/PRF1/GZMB TIL activation response was not correlated with PDO CD4/CD8 ratio or T-cell frequency but was correlated with PD-1 expression frequency on PDO TILs (Figures 7C, S7D–G). We further examined anti-PD-1-dependent tumor cell killing in an independent cohort of 10 additional PDOs encompassing surgically resected ccRCC (5), NSCLC (3), bladder urothelial carcinoma (1) and melanoma (1) alongside anti-CD3 and anti-CD28 antibody treatment to expand organoid TILs. Although baseline tumor necrosis was present in these freshly plated cultures, 2/10 PDOs (ccRCC, bladder urothelial carcinoma) exhibited nivolumab-dependent tumor cytotoxicity in parallel with TIL expansion and activation (Figures 7D–F, S6B–D). Overall, these studies strongly indicate that human PDO TILs functionally recapitulate the PD-1-dependent immune checkpoint.
DISCUSSION
We describe an organoid methodology facilitating in vitro TME study by preserving primary tumor epithelium en bloc with endogenous immune and non-immune stromal elements. Such PDOs allow human in vitro immunotherapy modeling via unified culture of tumor epithelium together with their native syngeneic, autologous tumor-reactive TILs, as opposed to epithelial-only organoid models (Boj et al., 2015; Fujii et al., 2016; Sato et al., 2011; van de Wetering et al., 2015), or co-culture reconstitution of peripheral blood or TILs with cancer cell lines (Feder-Mengus et al., 2008; Hirt et al., 2014) or organoids (Dijkstra et al., 2018). Here, the in vivo association between native TILs and tumor cells is preserved in vitro, with MHC tetramer detection of tumor antigen-specific T cells. Importantly, both human and mouse tumor organoid TILs functionally exhibit activation, expansion and cytotoxicity responses to PD-1/PD-L1 checkpoint blockade with rapid 7−day assessment. Our method notably preserves diverse endogenous immune cell types besides T cells, including macrophages, B and NK cells, versus reconstitution of clonally expanded or TCR-engineered TIL populations added to tumor cells (Feder-Mengus et al., 2008; Hirt et al., 2014) (Dijkstra et al., 2018; Feldman et al., 2015; Forget et al., 2017; Tran et al., 2016).
Mouse and human ALI tumor organoid cultures differ in several respects. Cell line-based mouse organoids exhibit rapid and reproducible doubling time and serial passage. In contrast, human PDOs exhibit highly variable growth correlating with high-versus low-grade tumor histology and initial condition of the tumor biopsy (tumor viability, pre- or post-treatment, sample acquisition delay). Necrotic tissue is thus differentially present in the primary plating, from which viable organoids proliferate. TIL activation, expansion and cytotoxicity responses are stereotyped in immunogenic mouse tumor organoids but are differ broadly in human PDOs from well-documented intrinsic intrapatient differences in tumor and immune composition and resistance to checkpoint inhibition. The immune and fibroblast stroma in both human and mouse organoids progressively decline over a 1–2 month period. Although TIL loss can be slowed by IL-2 or anti-CD3/anti-CD28, future optimization is needed to preserve TILs beyond the current 60 days limit.
Tumor immunity arises from concerted action of actively communicating peripheral and intratumoral components (Pardoll, 2012; Spitzer et al., 2017; Wei et al., 2017). However, current in vivo systems can not resolve the intratumoral immune response into distinct contributions from immune cells in the periphery versus those intrinsically resident in the TME. The present PDOs represent a minimal system allowing identification of such local events within the immune TME. It has been uncertain whether anti-PD-1 antibodies expand intratumoral exhausted-like CD8+ T cells via primary action on peripheral versus tumor-infiltrating populations since PD-1 blockade increases exhausted-like TILs (Wei et al., 2017) but also proliferation of peripheral blood PD-1+ CD8+ T cells (Huang et al., 2017; Kamphorst et al., 2017). Accordingly, anti-PD-1 and anti-PD-L1 activation of TILs within human and mouse PDOs identify PD-1 axis blockade within the TME as sufficient to elicit both TIL expansion and activation. We also find expanded ccRCC TIL TCR clonotypes concentrated within exhausted T cells, which may facilitate tumor progression.
The present PDOs, although encapsulating immune effectors within the TME, by no means exclude essential peripheral immune system contributions that are clearly essential for optimal anti-tumor immunity (Huang et al., 2017; Kamphorst et al., 2017; Spitzer et al., 2017). Certainly, combining ALI PDOs with immune components from lymph node or blood (Dijkstra et al., 2018) may generate even more holistic models that model bidirectional communication between tumor and periphery. Equivalent organoids could enable study of epithelial-immune interactions in inflammatory, infectious or autoimmune conditions.
The accurate modeling of intratumoral aspects of cancer immunotherapy is strongly supported by faithful recapitulation of TCR repertoires between fresh tumors and corresponding PDOs. Here, we introduce a robust single cell-based methodology allowing tandem gene expression and immune profiling from 10,000s of individual cells. Similar to our prior 3’ single cell RNA-seq workflow (Zheng et al., 2017), this Chromium Immune Profiling Solution exhibits high single cell capture efficiency without FACS sorting, greatly streamlining the single cell sequencing of paired TCR α and β chains, or Ig heavy and light chains. The low multiplet rate (<1% for every 1,000 cells) allows simultaneous profiling of 1000s of cells; in our PDO samples with larger number of cells, we can further computationally filter doublets by our robust analysis workflow, which simultaneously considers UMI counts, gene counts as well as immune markers from single cell transcriptomes.
Simultaneous determination of single cell transcriptome and TCR or Ig rearrangements links immune repertoire to various states of T and B cells, while strongly enhancing their discrimination from other immune cell types. As applied here, the single cell transcriptome assay demonstrates that PDOs from multiple donors preserve of all main immune lineages PDOs and is complementary to CyTOF-based profiling of the immune TME (Chevrier et al., 2017; Lavin et al., 2017). Similarly, the deep V(D)J enrichment analysis strongly infers that dominant TCR clonotypes from initial tumor biopsies are both extremely well represented in daughter organoids and identically concentrated within exhausted T cells. This high sensitivity and cell throughput confidently detects even rare TCR clonotypes that correspond between fresh tumor and PDO as low as ~0.1% and up to 10,000 organoid TILs having TCR V(D)J rearrangements. Indeed, our computational workflow strongly reproduces similar immune landscapes across samples with clonotype patterns that remarkably coincide with individual transcriptomes. Recent paired single cell TCR/transcriptome measurements have also found breast cancer clonotype phenotypes (Azizi et al., 2018) but required cells from individuals to be pooled and computationally clustered to identify phenotypic patterns; in contrast, we leverage abundant cell counts and confidently detected clonotypes, so each sample independently manifests clonotype phenotypes based on cell type markers. This enables unbiased analysis of concordance between matched fresh tumor and PDO at the resolution of distinct clonotypes. A prior single cell FACS method (Han et al., 2014) confirms that organoids accurately recapitulate the dominant TCR repertoire of the original tumor, albeit at lower cellular throughput (250–600 TILs) and power.
Extending the substantial benefits of immunotherapy is an unmet need, as only a minority of unstratified patients respond to PD-1/PD-L1 targeting (Gandini et al., 2016). PDOs could be applied to study additional immunotherapies/immune checkpoints including targeting of B, NK or macrophages, cell-based immunotherapies such as chimeric antigen receptor T cells, iPSC-based organoids with stromal elements (Spence et al., 2011) or mediation of anti-PD-1 effects by TAMs (Gordon et al., 2017) and peripheral blood monocytes (Krieg et al., 2018). Additional studies will be required to extend long-term PDO preservation of TILs and other immune cell types and define functional sequelae of their long-term co-culture with tumor epithelium. PDOs could also facilitate predictive assessment of individualized patient responses to clinically approved immune therapies or combinations. Our rapid functional in vitro organoid assessment of anti-PD-1 activity within a clinically-relevant 7-day time frame could complement descriptive biomarkers such as PD-L1 IHC, neoantigen burden or peripheral blood monocyte content (Anagnostou et al., 2017; Carbognin et al., 2015; Garon et al., 2015; Krieg et al., 2018; Rizvi et al., 2015). As we used organoids derived from “curative-intent” surgical resection samples without parallel patient treatment, future prospective studies will be required to establish definitive correlations between organoid and patient immunotherapy responses, by analogy to microfluidic culture approaches (Deng et al., 2018; Jenkins et al., 2018). PDOs or analogous methods may also allow cryopreservable biobanks of the human immune TME for research and drug discovery, with potential relevance to adoptive cell transfer TIL-based immunotherapies (Feldman et al., 2015; Forget et al., 2017; Tran et al., 2016). Ultimately, PDOs incorporating immune and other stromal components may help actualize the promise of precision cancer therapies.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Calvin Kuo (cjkuo@stanford.edu).
Experimental Model and Subject Details
Human specimens
Primary and metastatic tumor tissues were obtained through the Stanford Tissue Bank from patients undergoing surgical resection at Stanford University Medical Center (SUMC). All experiments utilizing human material were approved by the SUMC Institutional Review Board and performed under protocols #28908, #26213 and #17425. Written informed consent for research was obtained from donors prior to tissue acquisition. Samples were obtained from adult male or female patients who were treatment-naïve and accrued continuously to saturate a diversity of tumor histologies and to observe in vitro responses to checkpoint inhibitors. Specific collection of age and gender information was not performed for the samples of Figures 1 and 7A–C, which were anonymized, but was collected for Figure 5 (Table S3E) and 7D–F (Table S4). Analysis of influence of gender identity upon experiments was not performed. Samples were confirmed to be tumor by pathological assessment at SUMC and further validated by targeted exome sequencing (see below). Additional sample information is available in Table S1A, S1B. Samples were not allocated to distinct experimental groups because PDOs typically were subdivided and received both a control and a test treatment (i.e. checkpoint inhibition).
Mouse models
Female C57BL/6 Mice or BALB/c Mice were used for subcutaneous tumor implantation (Mice were obtained from Taconic Biosciences) in accordance with NIH and Stanford Administrative Panel on Laboratory animal Care (APLAC). Mice were housed in pairs and used for experimentation at 4–8 weeks of age. Animals were maintained on a 12-hour light/dark cycle, in a temperature- and humidity-controlled room with food and water.
Method Details
Preparing ALI organoid plates
Inserts containing a permeable, membranous bottom (PICM03050, Millicell-CM, Millipore) were inserted into tissue culture dishes as described (Li et al., 2014; Ootani et al., 2009). Collagen gel matrices were prepared for transwell inserts by mixing collagen matrix (Cellmatrix type I-A or Rat Collagen I), 10 X concentrated sterile culture medium (Ham’s F-12), and sterile reconstitution buffer (2.2 g NaHCO3 in 100 ml of 0.05 N NaOH and 200 mM HEPES) on ice at a ratio of 8:1:1 until use. After mixing collagen matrix and concentrated culture medium, reconstitution buffer was added and mixed again, avoiding bubbles. This reconstituted collagen solution was kept on ice (4°C) to prevent gel formation until added to the insert. 1 ml of reconstituted collagen solution was added to the insert under sterile conditions, serving as a bottom layer gel without tissue. The bottom layer was left to solidify for 30 min either in the hood or in a 37°C incubator.
Human PDO culture
Tumor tissues were minced finely on ice, washed twice in ADMEM/F12 (Invitrogen) containing 1X Normocin (InvivoGen), resuspended in 1 ml of Type I collagen gel (Trevigen), and layered on top of pre-solidified 1 ml collagen gel within a 30 mm, 0.4 mm inner transwell to form the double dish air-liquid culture system as described above. The transwell containing tumor tissue and collagen was placed into an outer 60 mm cell culture dish containing 1.0 ml of medium (ADMEM/F12 supplemented with 50% Wnt3a, RSPO1, Noggin-conditioned media (L-WRN, ATCC) was supplemented with HEPES (1 mM, Invitrogen), Glutamax (1X, Invitrogen), Nicotinamide (10 mM, Sigma), N-Acetylcysteine (1 mM, Sigma), B-27 without vitamin A (1X, Invitrogen), A83–01 (0.5 M, Tocris), Pen-Strep Glutamine (1X, Invitrogen), Gastrin (10 nM, Sigma), SB-202190 (10 M, Sigma), and EGF (50 ng/mL, Invitrogen) followed by replacing the lid of the outer dish. Organoids were passaged every 14–30 days by dissociation with 200 units ml−1 collagenase IV (Worthington) at 37 °C for 30 min, followed by three 5 minute washes with 100% FBS and replating at a 1:4 split into new air-liquid interface collagen gels. Additionally, in some cases, media was supplemented with recombinant human IL-2 (Peprotech) at 100, 600 or 6000 IU/ml as indicated.
Organoid culture from subcutaneous mouse tumors
B16-SIY, MC38, A20-OVA cells were cultured in DMEM with 10% FCS (Invitrogen) and 1X Pen-Strep Glutamine (Invitrogen, Cat No. 10379). 106 cells were implanted s.c. to 4–6 week old female C57Bl6 (B16-SIY, Thomas Gajewski (Sivan et al., 2015); MC38, Ron Levy, Stanford) or BALB/c (A20-OVA, Mark Davis, Stanford) mice. Tumors of approximately 500~1000 mm3 were dissected, minced finely, washed twice in F12 (Invitrogen) containing 1X Normocin (InvivoGen), resuspended in 1 ml of type I collagen gel (Trevigen), and layered on top of 1 ml of pre-reconstructed collagen within a 30 mm, 0.4 mm inner transwell. The inner transwell was placed into an outer 60 mm tissue culture dish containing 1.0 ml of growth medium (F12 (Invitrogen), 20% FBS, 1X Normocin supplemented with murine IL-2 (Peprotech) at 500 IU/m)l and the lid of the outer dish was replaced as previously described (Li et al., 2014; Ootani et al., 2009). Organoids were passaged every 7–14 days by dissociation with 300 units ml−1 collagenase IV (Worthington) at 37 °C for 30 min, followed by three 5 minute washes with 100% FBS and propagation at a 1:4 ratio into new air-liquid interface collagen gels. Growth media was replaced once every week.
Targeted exome sequencing of organoid cultures
Organoid cultures were sequenced using a commercial targeted resequencing assay for 131 cancer genes and software (TOMA SIGNOME Tumor Profiling System, Foster City, CA). Alternatively, for the data obtained with the TOMA SIGNOME panel, SNV and indels where identified using the TOMA SIGNOME Analysis pipeline (http://tomabio.com/products/toma-signome-tumor-profiling-system/) to identify somatic mutations from germline variants. For this study, raw variants produced by the pipeline were filtered by quality to include both germline and somatic variants for downstream analyzes.
Alternatively, organoid cultures were sequenced with the Broad Institute Rapid Cancer Detection Panel in which DNA samples were sequenced using TruSeq Custom Amplicon (TSCA; Illumina Inc.) technology. To build the targeted sequencing panel, Rapid Cancer Detection panel, we designed 3566 amplicon primers (Table S3). The Broad Institute Genomic Platform performed library construction and sequencing was performed using a Nextseq sequencer.
Sequencing of original patient tumors was carried out by Stanford Hospital using the Stanford Actionable Mutation Panel (STAMP). This assay detects potentially clinically actionable mutations, as well as additional genes that are frequently mutated in cancers. STAMP is a targeted next generation sequencing method optimized for ultra-deep sequencing of formalin fixed tumor biopsy tissue specimens. The workflow includes acoustic shearing of isolated genomic DNA, followed by efficient preparation of sequencing libraries and a target enrichment approach to capture genomic regions of interest for sequencing. The enrichment is done using custom designed libraries of capture oligonucleotides that target a specific set of genomic regions. This panel targets 130 genes, either in part or fully, with the genes selected on the basis of their known impact as actionable targets of existing and emerging anti-cancer therapies, their prognostic features, and/or their mutation recurrence frequency across patients with known cancer types. These genomic features are interrogated to achieve a minimum analytic detection-limit of at least 5%. Pooled libraries are sequenced on the Illumina MiSeq system. This test covers 130 genes, either in part or fully, at a minimum analytic detection limit of 5%.
Organoid sample quality checking
We utilized 3 quality control (QC) points, including mouse species contamination, sequencing coverage, and fingerprinting to ensure the purity of human materials, sequencing result quality, and patient unique identity of individual samples before further mutation analyses. To exclude mouse contamination in human PDOs, we analyzed 10 SNPs that uniquely identify the mouse strain used. If the genotype for more than 8 of the SNPs matched with greater than 75 % to that of the mouse, the sample was labeled as contaminated. For coverage QC, we extracted coverage info using the DepthofCov algorithm (https://software.broadinstitute.org/gatk/documentation/tooldocs/current/org_broadinstitute_gatk_tools_walkers_coverage_DepthOfCoverage.php) and excluded any sample with mean target coverage less than 50x. For fingerprinting QC, we analyzed the samples for fingerprinting matches against all previous sequenced samples by comparing the genotype at each of the 82 Fluidigm SNPs with a cutoff percent matching of 70%.
Mutation analyses
SNVs and Indels were extracted using MuTect and the HG19/GRC37 genome build (Cibulskis et al., 2013, full instructions available at https://software.broadinstitute.org/cancer/cga/mutect_run), and Indelocator (http://archive.broadinstitute.org/cancer/cga/indelocator), respectively. All computing processes were through Broad Firehose, the Broad Institute’s analysis platform <http://www.broadinstitute.org/cancer/cga/Firehose>. The SNVs and Indels mutation data were then combined into a single MAF file. In order to remove common sequencing artifacts or residual germline variation, each mutation in the combined MAF file was subjected to a “Panel of Normals” filtering using a panel of over 4000 BAM files from normal samples. We next filtered out mutations with low read count (< 20 reads), or ones classified as RNA, UTR, IGR, Intron, Silent, or Flank as well as any possible germline event from the ExAC and dbSNP databases, and mutations with low tumor fraction (< 3%). Finally, we rescued known cancer mutations found in COSMIC and TCGA databases. The log2 (T/N) copy number variation was generated from the coverage per interval and normalized it using a panel of normal (30 samples) with >= 50X coverage. We determined whether a sample has a clear copy number alteration (CNA) based on either a) the percentage of altered (insertion/deletion) intervals is significantly greater than the percentage altered in normal samples or b) there is any chromosome with a significantly longer continuous region of altered intervals compared to normal. Therefore, we characterized that the sample is verified tumor if it passes all three quality control steps and either has clear SNVs/Indels or has clear CNA.
Histological analysis and immunofluorescence
Organoids were fixed with 4% paraformaldehyde overnight, paraffin embedded and sectioned (4–5 m). Sections were deparaffinized and stained with H&E for histological analysis. Human immunofluorescence analysis was carried out using the following primary antibodies: anti-CK19 (DSHB, TROMA-III, 1:25), anti-SMA (Abcam, ab5694, 1:1000), anti-vimentin (Millipore, AB1620, 1:1000), anti-E-cadherin (BD, 610181, 1:1000), anti-p63 (Abcam, ab63881, 1:50), anti-CAIX (Novus, NB100–417, 1:1000), anti-S100 (Abcam, ab52642, 1:200), anti-CK7 (Abcam, ab9021, 1:100), anti-pan-cytokeratin (Abcam, ab7753, 1:200), anti-chromogranin A (Abcam, ab15160, 1:200). Mouse immunofluorescence analysis utilized anti-Mouse CD8a (4SM16, BD, 1:100), anti-mouse CD3e (145–2C11, BD, 1:100), anti-mouse CD4 (4SM95, BD, 1:100), anti-CD11b (ab1333357, Abcam, 1:2000), anti-Vimentin (Ab5733, EMD, 1:2000), anti-CD20(Abcam, 1:100). All secondary antibodies were used at 1:500. All images were captured on a Zeiss Axio-Imager Z1 with ApoTome attachment.
Immunohistochemistry
CLIA-certified PD-L1 immunohistochemistry analyses were performed by NeoGenomics Laboratories using the FDA-approved Dako 28–8 pharmDx companion diagnostic antibody. CD68 immunohistochemistry was performed using anti-CD68 (DAKO, M0814, clone KP1) using Ventana CC1 solution (pH 8.5) for heat-induced epitope retrieval and Optiview detection kit, using the Ventana Ultra Instrument.
Activation and analysis of mouse organoid TILs by anti-PD-1 or anti-PD-L1
Mouse organoids were grown using F12(Invitrogen), 20% FBS, 1X Normocin, and murine IL-2 (Peprotech, 500 UI/ml) in a double dish ALI system (Li et al., 2014; Ootani et al., 2009). Growth media was supplemented with anti-mouse-CD274 (α-PD-L1, B7–H1, Biolegend, 10 μg/ml) or anti-mouse-CD279 (α-PD-1, 29F.1.A12, Biolegend, 10 mg/ml) at day 0 of primary plating or passaging. After 7 days of antibody treatment, single cells were prepared by dissociation with 300 units ml−1 collagenase IV (Worthington) at 37 °C for 30 min, followed by t hree 5-min washes with 100% FBS. FACS staining cocktails for murine cells contained 10 ml anti-CD45 (30-F11, BD), 3 μl anti-CD3e (500A2, BD), anti-CD4 (RM4–5, BD) and anti-CD8a (53–6.7, BD), Anti-mouse-CD274 (B7–H1, Biolegend, 10 mg/ml) and anti-mouse-CD279 (29F.1.A12, Biolegend, 10 mg/ml). SIYRYYGL/SIINFEKL-loaded MHC peptides were synthesized by Elim Biopharmaceuticals. MHC tetramers were prepared by mixing enzymatically biotinylated MHC molecules with APC-streptavidin (Life Technologies, S32362)/PE-streptavidin (Life Technologies, S21388). Primers for qRT-PCR analysis of murine TILs were as follows:
Ifng F: 5’- GACAGAAGTTCTGGGCTTCT-3’
Prf1 F: 5’- ATGACTACTGTGCCTGCAGCAT-3’
Gzmb F: 5’- AGATCATCGGGGGACATGAAGT-3’
QRT-PCR was performed on a StepOnePlus instrument (Applied Biosystems) using anneal/extension temperature at 60°C for 35 cycles. Paired Student’s T-Test was used to calculate P values.
Assay for anti-PD-1-dependent human TIL activation within PDOs
Organoid cultures were established as above and supplemented with organoid medium containing 10 g/mL nivolumab (Bristol-Myers Squibb) or 10 g/mL control human IgG4 (Abcam). Organoids were grown for 7 days, dissociated in 200 units ml−1 collagenase IV (Worthington) at 37°C for 30 min, washed twice in ADMEM/F12, and digested in Liberase-TL (Roche; 50 g/mL final concentration) at 37°C for 15 min. Samples were washed twice in ADMEM /F12, triturated with a P1000 pipet to dissociate further, and passed over a 40 M filter. Single cells were then pelleted, resuspended in 100 FACS Buffer (PBS plus 2 mM EDTA and 0.1% BSA) and stained for FACS. FACS staining cocktail contained 50 L Brilliant Stain Buffer (BD), 10 μL anti-CD45 (2D1, BD), 10 L anti-CD279 (PD-1 clone EH12.1, BD), 3 μL anti-CD-3 (UCHT1, BD), 3 μL anti-CD4 (RPA-T4, BD), and 10 L 7-AAD (BD). An additional subset of samples also contained 3 μL anti-CD19 (SJ25C1, BD) and 3 μL anti-CD56 (B159, BD). T-cells were sorted on a BD Aria II flow cytometer into RNA extraction buffer, and RNA was extracted using the Arcturus PicoPure kit (Applied Biosystems). Extracted RNA was converted to cDNA using the iScript cDNA synthesis kit (Bio-Rad), and cDNA was subjected to 10–12 rounds of preamplification using SsoAdvanced PreAmp Supermix (Bio-Rad). cDNA was used for quantitative real-time PCR on a StepOnePlus instrument (Applied Biosystems) using TaqMan probe/primer sets for TBP, IFNG, GZMB and PRF1 (Applied Biosystems).
Tumor cytotoxicity analysis
For murine cytotoxicity analysis, B16-SIY organoids were treated with IgG/α-PD-L1/α-PD-1 as indicated. Organoids were dissociated to single cells and pelleted as described above. Cells were incubated with anti-Melanoma antibody (Abcam, 1:80) for 30 minutes on ice. For human tumor cell cytotoxicity assay, PDOs were cultured for 1 week in the presence of anti-CD3 (clone HIT3a, 2 mg/ml) (cat no: 300332, BioLegend) and anti-CD28 (clone CD28.2, 2 μg/ml) (cat no: 302923, BioLegend) with either 10 μg/ml anti-PD-1 (nivolumab) or IgG4 control. For both mouse and human analysis, cells were washed twice with cold Cell Staining Buffer, and resuspended in Annexin V Binding Buffer (Biolegend, FITC Annexin V Apoptosis Detection Kit) at a concentration of 0.25−1.0 × 107 cells/ml. 100 μl of cell suspension were transferred in a 5 ml test tube and incubated with 5 μl of Annexin V-FITC and 5 μl of 7-AAD Viability Staining Solution for 15 min at room temperature (25°C) in the dark and analyzed by FACS for Annexin V and 7-AAD, yielding Annexin-V(+)/7-AAD(−) early apoptotic and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells. The Annexin-V, 7-AAD double positive tumor cells were pre-gated based on forward and side scatter properties to enrich for tumor epithelium, eliminate hematopoietic and debris populations and further pre-gated for single cells.
Chromium Single-cell 5’ VDJ and 5’ RNA-seq
Preparation of Single Cell Suspension
PBMCs, Pan T and CD19+ B cells used for the technical validation part of the immune profiling solution were obtained from AllCells (Catalog #: PB001, PB009–1F and PB010–0). Tumor and organoid samples were obtained as specified by “Human Specimen” and “PDO Culture” in earlier sections of the STAR Methods. Tumor or organoids were mechanically dissociated with Castro scissors on ice to generate ~1 mm × 1 mm pieces, resuspended in two pellet volumes RPMI with 10% FBS and partitioned for histology, snap frozen for DNA, organoid culture, and sorting (1:1:1:3 ratios). Minced tissues were subjected to sequential collagenase IV and Liberase TL digestion as per assay for anti-PD-1-dependent human TIL activation within PDOs except that DNAse I (Worthington; 100 Kunitz Units/mL final concentration) was added to the Liberase TL step and agitated gently at 37C for 30 minutes. After 30 minutes, pieces were triturated with a P1000 pipette ten times and passed through a 40 micron filter, centrifuged, subjected to TrypLE (Life Technologies) for 10 minutes at room temperature. The cell suspension was then centrifuged and resuspended in 10 pellet volumes of ACK lysing buffer (Lonza) for 5 minutes in the dark at room temp followed by centrifugation at 200g for 5 minutes. The cells were washed with FACS buffer (PBS, 2 mM EDTA, 0.1% FBS), incubated with Fc block (Biolegend) diluted 1:500 in 2 pellet volumes of FACS buffer, for 15 minutes on ice, followed by addition of labeling cocktail (FITC anti-CD45, 2 μg/mL (BD), 1:200 dilution Zombie Aqua live/dead stain (Biolegend)) in 2 pellet volumes of FACS buffer (CD45 antibody 1 μg/mL, 1:400 live/dead stain). The cells were incubated on ice for 30 minutes, washed thrice with FACS buffer, and sorted on an Aria Fusion (BD) with singlet discrimination, followed by live/dead gating, followed by CD45+ gating. For unfractionated cells, singlets and live/dead gating was used on both epithelial and mononuclear scatter populations.
Sequencing library construction using the 10x Chromium platform
Cellular suspensions were loaded on a Chromium Single Cell Controller instrument (10x Genomics, Pleasanton, CA, USA) to generate single-cell GEMs. Single-cell RNA-Seq libraries were prepared using the Chromium Single Cell 5’ Library & Gel Bead Kit (P/N 1000006, 10x Genomics). GEM-RT was performed in a C1000 Touch Thermal cycler with 96-Deep Well Reaction Module (Bio-Rad; P/N 1851197): 53 °C for 45 min, 85 °C for 5 min; held at 4 °C and stored at −20 °C. The GEMs were shipped to 10x Genomics on dry ice, then broken and the single-strand cDNA was cleaned up with DynaBeads MyOne Silane Beads (Thermo Fisher Scientific; P/N 37002D). Barcoded, full length cDNA was amplified using the C1000 Touch Thermal cycler with 96-Deep Well Reaction Module: 98 °C for 45 s; cycled 13 ×: 98 °C for 20 s, 67 °C for 30 s, and 72 °C for 1 min; 72 °C for 1 min; held at 4 °C. Amplified cDNA product was cleaned up with the SPRIselect Reagent Kit (0.6 × SPRI; Beckman Coulter; P/N B23318). Barcoded, full-length V(D)J segments were enriched from amplified cDNA with primers specific to TCR constant regions. The target enrichment 1 was performed in a C1000 Touch Thermal cycler with 96-Deep Well Reaction Module: 98 °C for 45 s; cycled 10 ×: 98 °C for 20 s, 67 °C for 30 s, and 72 °C for 1 min; 72 °C for 1 min; held at 4 °C. The target enrichment 1 was cleaned up with the SPRIselect Reagent Kit (0.8 × SPRI). The target enrichment 2 was performed in a C1000 Touch Thermal cycler with 96-Deep Well Reaction Module: 98 °C for 45 s; cycled 10 ×: 98 °C for 20 s, 67 °C for 30 s, and 72 °C for 1 min; 72 °C for 1 min; held at 4 °C. The target enrichment 2 was cleaned up twice with the SPRIselect Reagent Kit (0.5 × and 0.8 × SPRI). The enrichment primers can be found in Supplementary Table 4. 5’ gene expression and enriched libraries were constructed using the reagents in the Chromium™ Single Cell 3’/5’ Library Construction kit (P/N 1000020). For 5’ gene expression library construction, these steps were followed: (1) fragmentation, end repair and A-tailing; (2) post fragmentation, end repair and A-tailing cleanup with SPRIselect; (3) adaptor ligation; (4) post ligation cleanup with SPRIselect; (5) sample index PCR and cleanup. For the enriched library construction, these steps were followed: (1) fragmentation, end repair and A-tailing; (2) adaptor ligation; (3) post ligation cleanup with SPRIselect; (4) sample index PCR and cleanup. The barcode sequencing libraries were quantified by quantitative PCR (KAPA Biosystems Library Quantification Kit for Illumina platforms P/N KK4824). Sequencing libraries were loaded at concentrations on sequencers with the read configuration as specified in Table S3B.
Quantification and Statistical Analysis
Single Cell V(D)J Software Cell Ranger 2.1.0
The Cell Ranger (CR) Software Suite (version 2.1.0) was used for 5’ gene counting and V(D)J sequence assembly and paired clonotype calling. 5’ gene counting is similar to 3’ gene counting outlined previously, with some updates. (Zheng et al., 2017, https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/algorithms/overview). Briefly, reads were aligned to the genome using STAR. CR uses the transcript annotation GTF to bucket the reads into exotic, intronic and intergenic, and by whether the reads align confidently to the genome. For reads that align to a single exonic locus but also align to 1 or more non-exonic loci, the exonic locus is prioritized and the read is considered to be confidently mapped to the exonic locus with MAPQ 255. Cell Ranger further aligns exonic reads to annotated transcripts, looking for compatibility. A read that is compatible with the exons of an annotated transcript, and aligned to the same strand, is considered mapped to the transcriptome. If the read is compatible with a single gene annotation, it is considered uniquely (confidently) mapped to the transcriptome. Only reads that are confidently mapped to the transcriptome are used for UMI counting. VDJ assembly and paired clonotype calling have been added to CR. For V(D)J assembly, CR performs cell calling independently of V(D) J read filtering and assembly. In cell calling, UMIs are first filtered to contain a threshold of Read Pairs per UMI (for details, consult the software documentation page). Then a 2-component Gaussian mixture model was fit to the distribution of filtered UMIs per barcode. Cell barcodes are barcodes with posterior probability >0.5 of belonging to the higher-mean component. In V(D) J read filtering and assembly, CR first trims known adapter and primer sequences from the 5’ and 3’ ends of reads and then filters away reads that lacking at least one 15bp exact match against at least one of the reference segments (based on the T cell receptor, TRA and TRB, and B cell immunoglobulin, IGH, IGL and IGK gene annotations in Ensembl version 87). Next, CR performs de novo assembly, by building a De Bruijn graph of reads from each cell barcode independently. The assembler outputs the contig sequences associated with paths which are assigned at least one UMI with the mapping of the input read pairs that contributed to each contig. Each base in the assembled contigs is assigned a Phred-scaled Quality Value (QV), representing an estimate of the probability of an error at that base. The QV is computed with a hierarchical model that accounts for the errors in reverse transcription (RT), that will affect all reads with the same UMI, and sequencing errors which affect individual reads. Finally, each assembled contig in each cell is aligned against all of the germline segment reference sequences for annotation to V, D, J, C and 5’ UTR reference sequences. The presence of a CDR3 motif (Cys-to-FGXG/WGXG) is searched in in a frame defined by the start codon in the L+V region or all 6 frames in the absence of L+V region. A contig is considered productive if: 1) it fully spans the V and J segments; 2) it contains a recognizable start codon in the expected V location; 3) it contains a CDR3 region in-frame with the V start codon; 4) it contains no stop codons in the V-J spanning region. It is expected that each cell barcode typically contains two matching productive contigs, comprising either a TRA and a TRB, or a heavy chain (IGH) and a light chain (IGK or IGL). Additional productive contigs produced by the assembler are less likely to be legitimate. Each productive contig will be labeled as high or low confidence based on the number of UMIs supporting the contig. Cell barcodes are grouped together into clonotypes if they share the same set of productive CDR3 nucleotide sequences by exact match. Note that for B cells, somatic mutations within the CDR3 will break up clonotypes that are in fact clonally related. Cells with somatic mutations outside the CDR3 will be considered to share a clonotype.
Secondary analysis of the technical data
Seurat (Cibulskis et al., 2013) (version 2.1) was used to analyze the PBMC 5’ GEX data in Figure 4b. Genes with detected expression in at least 3 cells, and cells with at least 10 genes detected were used. Variable genes were identified with x.low.cutoff=0.05 and y.cutoff=0.1. The first 15 Principal Components were used for clustering (resolution=0.6) and for tSNE visualization. Clusters were identified based on genes that are enriched in a specific cluster (Yan et al., 2017). Classification of PBMCs was inferred from the annotation of cluster-specific genes; CD4 T (CD3E, IL7R) , CD8 T (CD3E, CD8A), CD4 Naïve (IL7R, CCR7), CD4 Memory (IL7R, S100A4), T Regulatory (CD3E, FOXP3), CD8 Naïve (CD8A, CCR7), T Exhausted (CD8 Effector (NKG7, CD8A, PRF1), CD8 Memory (NKG7, CD8A, S100A4, GZMK) and NK cells (NKG7 and CD3E negative).
To link 5’ RNA-seq data to 5’ VDJ data, barcodes between the PBMC 5’ GEX sample were overlapped with VDJ T and B enrichment libraries. There is a high fraction of barcode overlap between GEX and VDJ libraries. VDJ T enrichment assay detected 1,581 cells with productive, full length TCR a or b chains. 1,556 of 1,581 cells were also detected by 5’ GEX, representing a >98% overlap between GEX and VDJ T enrichment libraries. Of these 1,556 cells, 1,495 cells overlapped with the annotated T cell population. The remaining 61 cells are likely a combination of cell mis-classification, multiplets and chimeric molecules in GEX and VDJ libraries. Similarly, the barcode overlap between 5’ GEX and VDJ B enrichment libraries is >99%.
Loupe V(D)J Browser was used for the analysis of Pan T and CD19+ B cells mixed with Jurkat and GM12878 cells (whose clonotypes were termed Ground-Truth clonotypes). The observed number of cells with the correct Ground-Truth clonotype is the number of cells with at least 1 of the Ground-Truth chains. The number of cells with wrong CDR3 pairs is the number of cells with productive full-length pair and some, but not complete overlap with the provided ground truth. Multiplets (usually cells with two β chains or heavy chains) were not considered.
Secondary analysis of the tumor and organoid samples
Seurat (Cibulskis et al., 2013) (version 2.1) was used to perform basic quality control on the raw 5’ GEX matrices output from Cell Ranger. Prior to the analysis of Seurat, cells with less than 30 genes or more than 3000 genes were filtered out, and genes expressed in less than 3 cells were removed. Additionally, CD45− non-immune cells and CD3E+MS4A1+ (technical doublets with B and T cells) were removed. Samples were excluded if they had less 1000 cells or less than 5% CD3D+ CD3G+ CD3E+ CD247+ (putative T) cells to ensure sufficient number of cells for clonotype analysis. Then, gene dispersion analysis implemented in Seurat was used to select highly variable genes, preserving genes with logarithmic mean expression between 0.2 and 3.0 and with logarithmic dispersion greater than 0.5. The effects of total UMI counts per cell and mitochondrial percentage were regressed out from the gene expression.
For unbiased visualization of the 5’ GEX data, Principle Component Analysis (PCA), t-stochastic neighbor embedding (t-SNE) implemented in Seurat was used: the first 20 principle components were selected for all samples and no major differences were seen when using larger number of principle components. Given the 2D visualization by t-SNE, feature plots were generated by displaying cells that express particular genes, as well as cells with matching barcodes that had either TCRα or TCRβ sequenced. Next, each cell visualized in the t-SNE plots were annotated as one of M (macrophage) cell, Natural Killer (NK) cell, B cell, regulatory T (Treg) cell, exhausted T (Tex) cell, cytotoxic T (Tc) cell, or helper T (Th) cell based on its expression. Individual cells were classified by the enrichment of cell type markers: M (CD14, CD163, CD68, MRC1, MSR1, CSF1R), NK (CD3E, CD247, NCAM1, NKG7, TRDC, CD3D negative, CD3G negative), B (CD19, MS4A1), Treg (FOXP3, TIGIT, IL2RA, HAVCR2, NKG7 negative), Tex (PDCD1, LAG3, HAVCR2), Th (CD3D, CD3G,CD4, IL7R), and Tc (CD3D, CD3G, CD8A, CD8B). To classify a cell as a certain type, it was required that the total UMI counts for all the positive markers sum to at least 4 (with the exception of macrophages which requires 8 UMIs as more markers are used). Because canonical T cell markers may not be exclusive to T cells, the cells were classified sequentially in the order of M, NK, B, Treg, Tex, Tc, Th. As a result, the detected T cells must be negative for markers unique to M and B cells. Further, because NK cells can share many cell type markers with T cells, NK cells were first classified based on the absence of CD3D and CD3G, and then the remaining CD3D+ and CD3G+ cells were considered for candidate T cells. The sequential assignment has the benefit of reducing the doublets classified as T cells; the other cell types such as M, B and NK may have doublets, but a number of them were removed prior to running Seurat. Finally, only the classified T cells that had TCRα or TCRβ were considered for clonotype comparisons between matched fresh tumor and organoid samples. The visualization in the main figure only displays TCRαβ double-positive T cells, whereas the visualization in the supplementary figures distinguishes TCRα only, TCRβ only, and TCRαβ among all the CD45+ cells. The top three clonotypes shown in the t-SNE plots were based on the TCRαβ double-positive T cells for all the samples.
Smart-seq2-based single cell TCR sequencing
Single cell suspensions of tumor or organoid-resident T cells were isolated by sequential collagenase IV and Liberase-TL digestion as in the assay for anti-PD-1-dependent human TIL activation within PDOs (see above). Dissociated cells were subsequently stained with anti-CD3 (317310, BioLegend), anti-CD45 (304024, BioLegend), anti-CD4 (317436, BioLegend), and anti-CD8 (344711, BioLegend) antibodies for FACS. Dead cells were excluded from sorting with amine-reactive Zombie Aqua (423102, BioLegend). Single viable CD3+ T cells were directly sorted into 96-well plates. To obtain individual T cell clonal identities, sorted T cells were subjected to the Smart-seq2 protocol for the conversion of full transcriptome cDNA (Picelli et al., 2014). The variable CDR3 sequences of both TCR α and β chains were acquired through a modified version of the Han protocol (Han et al., 2014). Specifically, the RT step of the P1 reaction was omitted and instead only the 25-cycle pre-amplification was used. Subsequently, adapted, amplified CDR3 regions were sequenced on the Illumina MiSeq platform.
TCR clonotype comparison
To analyze the concordance between the observed clonotypes in a fresh tumor and that in the matched organoid, top ten clones were first compared between fresh tumor (FT) and organoid (OR) because the major clonotypes dominate majority of the cell counts. To further assess the entire clonotype landscape, pair-wise comparisons across all clonotypes were considered, and linear regression was applied to the pairs with the cell counts in FT as the predictor variable and the cell counts in OR as the response variable. (For visualization, the scatter plots and linear fit curve were transformed to be on log10(x+1)−log10(y+1) scale).
While the value of the R2 captures how well clonotypes in the organoid recapitulates those in the fresh tumor, the value explains the linear relation and is heavily influenced by (1) the many low frequency clonotypes and (2) the discrepancy between the total number of cells sequenced between FT and OR. To address this issue, we devised a permutation-based test to provide confidence about the concordance, with the null hypothesis being the FT and OR profiles are uncorrelated. Under this null hypothesis, permutations of the OR counts correspond to a random outcome, e.g., having random clonal captures and a few random clones being expanded. We constructed an empirical null distribution with 10,000 permutations and computed the permutation p-value based on the number of correlation values from the permutation that exceeded the observed correlation. We considered the observed correlation as significant if the p-value is less than 0.01. This procedure was repeated for the TCRα, TCRβ, and TCRαβ independently for each sample which had both FT and OR data available.
Quantification of cell fluorescence
Cell of interest measurements were performed by calculating area of all cells of interest (i.e. SMA+, TILs) divided by the fluorescence intensity of DAPI using ImageJ. N denotes the number of representative fields captured from respective samples.
DATA AND SOFTWARE AVAILABILITY
Deposited Data
Mendeley Figures 1–10 are available at https://data.mendeley.com/datasets/ykpbxfx2p9/2 Single cell sequencing has been deposited with accession number GSE111360. Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111360,
Supplementary Material
Figure S1. Technical validation of the Chromium immune Profiling Solution, related to Figure 5 and STAR Methods. (A) Assay and computational pipeline workflow.
(B) Feature plots of the PBMC sample, with population-specific markers.
(C) Normalized expression (centered) of the top variable genes (rows) from each of 12 clusters (columns) is shown in a heatmap. Numbers on the right indicate cluster numbers, with connecting lines indicating the hierarchical relationship between clusters. Gene symbols of markers from each cluster are shown on the bottom.
TABLE S2. Sequencing of PDO cultures, related to Figure 1 and STAR Methods. “TOMA” designates end-to-end targeted exome sequencing. “BROAD” designates PCR hotspot exome sequencing. “BOTH” designates the use of both “TOMA” and “BROAD” methods, (A) SNV mutational calls.
(B) Broad primer sequences. Regions covered by the primers used in the Broad PCR hotspot exome sequencing are denoted.
(C) PDOs broadly recapitulate genomic alteration landscape of tumors from which they are derived, related to Figure 1 and STAR Methods. Organoid cultures for which prior clinical sequencing data (of original tumor) was available are displayed. For each sample, genotypes are only displayed for loci assessed by both methods. All original tumor sequencing was performed using the Stanford Actionable Mutation Panel (STAMP, see methods), except sample CT17, which was only sequenced at the KRAS locus.
TABLE S3. Metrics for Chromium Immune Profiling Solution single cell sequencing, related to Figure 5, Figures S1–S6 and STAR Methods. (A) Primer sequences for the VDJ enrichment assays. Primer sequences for the TCR and Ig enrichment assays are denoted.
(B) Chromium single cell sequencing parameters. Sequencing library loading concentrations, read configurations and sequencing metrics are described.
(C) Ground-truth clonotype information of Jurkat and GM12878 cells.
(D) Sensitivity and accuracy of Chromium single cell immune sequencing assay. The assay performance allows assessment of immune repertoire even under conditions of limiting clonal amplification in tumor or organoid samples.
(E) Clinicopathologic information for tumor specimens used to generate organoids for single cell sequencing.
Figure S2. Summary of cell types profiled by Chromium Immune Profiling Solution in ccRCC-1 fresh tumor (A-D) versus day 7 organoid (E-H) CD45+ fraction, related to Figure 5. (A,E) Breakdown of major immune cell types.
(B,F) Unbiased visualization of single cells shown by t-SNE and colored by our cell type annotation.
(C,G) Cells detected with rearrangement of at least one of the TCRα or TCRβ chains.
(D,H) Gene feature plots of the cells supporting the assignment in (E) and (I).
Figure S3. Summary of cell types profiled by Chromium Immune Profiling Solution in ccRCC-2 fresh tumor (A-D) versus day 7 organoid (E-H) CD45+ fraction, related to Figure 5. (A,E) Breakdown of major immune cell types. (B,F) Unbiased visualization of single cells shown by t-SNE and colored by our cell type annotation.
(C,G) Cells detected with rearrangement of at least one of the TCRα or TCRβ chains.
(D,H) Gene feature plots of the cells supporting the assignment in (E) and (I).
Figure S4. Summary of single cell clonotype comparisons by Chromium single cell tandem 5’ V(D)J-seq between fresh tumor (FT) and organoid (OR) from ccRCC-1 (A-D) and ccRCC-2 (E-H), related to Figure 5. For the clonotypes defined by the TCRα, TCRβ and paired TCRαβ chains respectively, we observe the expanded clonotypes in FT (fresh tumor) to highly overlap with those in OR (organoid), and the top expanded clonotypes are consistent between FT and OR. Additionally, the expansion patterns are significantly correlated (p < 0.01, permutation test).
(A,E) TCRα clonotypes, FT vs OR.
(B,F) TCRβ clonotypes, FT vs OR.
(C,G) Paired TCR αβ clonotypes, FT vs OR.
(D,H) TCR CDR3s sequences and cell counts in FT and OR respectively of the most frequent clonotypes ranked in FT.
Figure S5. t-SNE visualization of cell type assignment and top 3 TCRαβ clonotypes in all six samples of human ccRCC CD45+ fraction, related to Figure 5. Cell type annotations were assigned according to the 5’ scRNA-seq data, as shown in Figures S10-S16. Clonotype assignments were derived from the 5’ scV(D)J-seq data simultaneously measured with the scRNA-seq data. The highlighted TCRαβ clonotypes are the 3 most frequent among the assigned T cells in respective samples and strongly co-localize with the Tex fraction (light green).
Figure S6. Anti-PD-1 or anti-PD-L1 treatment of ALI tumor organoids induces tumor cell killing, related to Figures 6 and 7. (A) Murine B16-SIY organoids prepared from syngeneic s.c. tumors were cultured for 7 or 14 days in the presence of IL-2 and either anti-mouse anti-PD-1, anti-PD-L1 or control IgG followed by FACS analysis with Annexin V-FITC and AAD. Pre-gating for tumor epithelium was performed by scatter properties. At both 7 and 14 days there is a decrease in viable Annexin-V(−)/7-AAD(−) cells and increased Annexin-V(+)/7-AAD(−) early apoptotic (orange) and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells (pink).
(B) Dual Annexin V-FITC/7-AAD FACS analysis of the human kidney ccRCC PDO from Figure 7D–E following treatment with nivolumab or control IgG4 for 7 days. Nivolumab decreased viable Annexin-V(−)/7-AAD(−) cells and increased Annexin-V(+)/7-AAD(−) early apoptotic (orange) and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells (pink).
(C) Anti-CD3 plus anti-CD28 augments nivolumab expansion of organoid TIL populations in the ccRCC PDO of Figure 7D–E.
(D) Expanded y-axis from (C) showing nivolumab expansion of organoid TIL populations without anti-CD3/CD28.
Figure S7. Comparison of TIL activation versus expansion in nivolumab-treated PDOs, related to Figure 7. (A) TIL activation by anti-PD-1/nivolumab in human PDOs, reprinted from Fig. 7A for reference.
(B) FACS enumeration of CD3+, CD4+ and CD8+ TILs per 106 total organoid cells in (A).
(C) Percent change of organoid TILs (+) nivolumab versus (−) nivolumab baseline in the samples of (A). A 35% threshold is indicated.
(D-G) Spearman correlations between Interferon-gamma (IFNG), Granzyme B (GZMB), and Perforin-1 (PRF1) mRNA expression levels after nivolumab treatment versus (D) PD-1+ T cell frequency, (E) total T cell frequency, (F) CD4+/CD8+ T-cell ratio and (G) pass/fail score for a clone 28–8 CLIA-certified PD-L1 test (>1% PD-L1 positivity in original tumor).
Highlights.
Air-liquid interface (ALI) patient-derived tumor organoids (PDO) retain immune cells
5’ V(D)J and RNA-seq from the same single cells allows robust immune characterization
T cell receptor repertoire is highly conserved between tumor and PDO
ALI PDOs functionally recapitulate the PD-1/PD-L1-dependent immune checkpoint
ACKNOWLEDGEMENTS
We are grateful to the Kuo lab, Ron Levy, Ed Engleman, Suki Padda, Cecile Chartier, Courtney Easley-Neal, Paul Ryvkin, Josephine Lee and Deanna Church for discussions. We thank Jerald Sapida, Luz Montesclaros and Osman Sharifi and Dhananjay Wagh for single cell library prep, Lisa Wagar for advice with FACS, Kathleen Cormier and Pauline Chu for histology, Danielle Leuenberger, Dharati Trivedi and Stanford Tissue Bank for sample collection and Qiang Gong for data submission. We thank Thomas Gajewski for B16-SIY cells. J.T.N. was supported by a Stanford Dean’s Fellowship, a Stanford Cancer Institute Fellowship, and American Cancer Society Postdoctoral Fellowship 124574-PF-13-296-01-TBG. J.Z. and C.S. were supported by the Stanford Discovery Innovation Fund and NSF DMS 1712800. L.M.S. was supported by a St. Baldrick’s Fellowship, A.R.S. by NHLBI T32 IT32HL098049 and F.Z. by the Ovarian Cancer Research Alliance. This work was also supported by the NCI Cancer Target Discovery and Development Network CA176299 (C.J.K.) and CA176058 (W.C.H.); NIDDK Intestinal Stem Cell Consortium U01DK085527 (C.J.K.); NAIAD Novel, Alternative Model Systems for Enteric Diseases U19AI116484 (C.J.K.); NCI Ras Synthetic Lethal Network U01CA199241 (C.J.K.) and U01DE025188 (J.B.S. and C.J.K.). We also thank the Parker Institute for Cancer Immunotherapy (M.M.D.), the John and Jill Friedenrich Foundation and the Emerson Collective (C.J.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
C.J.K. and J.T.N. are inventors on a patent describing the ALI PDO method. G.X.Y.Z and V.G. are employees and shareholders of 10X Genomics. C.J.K. is a founder, scientific advisory board member and shareholder of both TOMA Biosciences and Surrozen, Inc. C.J.K. is a consultant for BioTechne. D.M-V., F.M.DLV are employees and shareholders in TOMA Biosciences. WCH is a consultant for Thermo Fisher, AjuIB, MPM Capital, and Paraxel and receives research support from Deerfield. WCH is a founder, has equity and consults for KSQ Therapeutics.
REFERENCES
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. (2017). Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 7, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. (2015). Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 373, 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. (2015). Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med 373, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Calio A, Cuppone F, Sperduti I, Giannarelli D, Chilosi M, et al. (2015). Differential Activity of Nivolumab, Pembrolizumab and MPDL3280A according to the Tumor Expression of Programmed Death-Ligand-1 (PD-L1): Sensitivity Analysis of Trials in Melanoma, Lung and Genitourinary Cancers. PLoS One 10, e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lau BT, Andor N, Grimes SM, Handy C, Wood-Bouwens C, and Ji HP Single-cell transcriptome analysis identifies distinct cell types and intercellular niche signaling in a primary gastric organoid model. bioRxiv 190132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, et al. (2017). An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 169, 736–749.e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM, Isobe M, Palumbo A, Puck J, Ming J, Tweardy D, Erikson J, Davis M, and Rovera G (1985). Gene for alpha-chain of human T-cell receptor: location on chromosome 14 region involved in T-cell neoplasms. Science 227, 1044–1047. [DOI] [PubMed] [Google Scholar]
- Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, et al. (2018). CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 8, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. (2018). Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMarco RL, Su J, Yan KS, Dewi R, Kuo CJ, and Heilshorn SC (2014). Engineering of three-dimensional microenvironments to promote contractile behavior in primary intestinal organoids. Integr Biol (Camb) 6, 127–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder-Mengus C, Ghosh S, Reschner A, Martin I, and Spagnoli GC (2008). New dimensions in tumor immunology: what does 3D culture reveal? Trends in molecular medicine 14, 333–340. [DOI] [PubMed] [Google Scholar]
- Feldman SA, Assadipour Y, Kriley I, Goff SL, and Rosenberg SA (2015). Adoptive Cell Therapy--Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin Oncol 42, 626–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnberg NK, Gokare P, Lev A, Grivennikov SI, MacFarlane A.W.t., Campbell KS, Winters RM, Kaputa K, Farma JM, Abbas AE, et al. (2017). Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 8, 66747–66757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget MA, Tavera RJ, Haymaker C, Ramachandran R, Malu S, Zhang M, Wardell S, Fulbright OJ, Toth CL, Gonzalez AM, et al. (2017). A Novel Method to Generate and Expand Clinical-Grade, Genetically Modified, Tumor-Infiltrating Lymphocytes. Frontiers in immunology 8, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, Ohta Y, Toshimitsu K, Nakazato Y, Kawasaki K, et al. (2016). A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 18, 827–838. [DOI] [PubMed] [Google Scholar]
- Gandini S, Massi D, and Mandala M (2016). PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit Rev Oncol Hematol 100, 88–98. [DOI] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Glanville J, Hansmann L, and Davis MM (2014). Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol 32, 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, et al. (2016). Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt C, Papadimitropoulos A, Mele V, Muraro MG, Mengus C, Iezzi G, Terracciano L, Martin I, and Spagnoli GC (2014). “In vitro” 3D models of tumor-immune system interaction. Advanced drug delivery reviews 79–80, 145–154. [DOI] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2018). Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, and de Sauvage FJ (2013). Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. (2017). Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 114, 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F, and Joyce JA (2015). Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol 25, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, and Becher B (2018). High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med 24, 144–153. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169, 750–765.e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, Cantrell MA, Rack PG, Neal JT, Chan CW, et al. (2014). Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med 20, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. (2015). Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JT, and Kuo CJ (2016). Organoids as Models for Neoplastic Transformation. Annual review of pathology 11, 199–220. [DOI] [PubMed] [Google Scholar]
- Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, Sugihara H, Fujimoto K, Weissman IL, Capecchi MR, et al. (2009). Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med 15, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka AK, and Coussens LM (2016). The Basis of Oncoimmunology. Cell 164, 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, et al. (2015). Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol 16, 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. (2015). Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372, 320–330. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, et al. (2011). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, et al. (2011). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, and Anders RA (2014). Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20, 5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA, and Pardoll DM (2016). Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 16, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr., Lao CD, et al. (2015). Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16, 375–384. [DOI] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133.e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. (2017). Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature 545, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Technical validation of the Chromium immune Profiling Solution, related to Figure 5 and STAR Methods. (A) Assay and computational pipeline workflow.
(B) Feature plots of the PBMC sample, with population-specific markers.
(C) Normalized expression (centered) of the top variable genes (rows) from each of 12 clusters (columns) is shown in a heatmap. Numbers on the right indicate cluster numbers, with connecting lines indicating the hierarchical relationship between clusters. Gene symbols of markers from each cluster are shown on the bottom.
TABLE S2. Sequencing of PDO cultures, related to Figure 1 and STAR Methods. “TOMA” designates end-to-end targeted exome sequencing. “BROAD” designates PCR hotspot exome sequencing. “BOTH” designates the use of both “TOMA” and “BROAD” methods, (A) SNV mutational calls.
(B) Broad primer sequences. Regions covered by the primers used in the Broad PCR hotspot exome sequencing are denoted.
(C) PDOs broadly recapitulate genomic alteration landscape of tumors from which they are derived, related to Figure 1 and STAR Methods. Organoid cultures for which prior clinical sequencing data (of original tumor) was available are displayed. For each sample, genotypes are only displayed for loci assessed by both methods. All original tumor sequencing was performed using the Stanford Actionable Mutation Panel (STAMP, see methods), except sample CT17, which was only sequenced at the KRAS locus.
TABLE S3. Metrics for Chromium Immune Profiling Solution single cell sequencing, related to Figure 5, Figures S1–S6 and STAR Methods. (A) Primer sequences for the VDJ enrichment assays. Primer sequences for the TCR and Ig enrichment assays are denoted.
(B) Chromium single cell sequencing parameters. Sequencing library loading concentrations, read configurations and sequencing metrics are described.
(C) Ground-truth clonotype information of Jurkat and GM12878 cells.
(D) Sensitivity and accuracy of Chromium single cell immune sequencing assay. The assay performance allows assessment of immune repertoire even under conditions of limiting clonal amplification in tumor or organoid samples.
(E) Clinicopathologic information for tumor specimens used to generate organoids for single cell sequencing.
Figure S2. Summary of cell types profiled by Chromium Immune Profiling Solution in ccRCC-1 fresh tumor (A-D) versus day 7 organoid (E-H) CD45+ fraction, related to Figure 5. (A,E) Breakdown of major immune cell types.
(B,F) Unbiased visualization of single cells shown by t-SNE and colored by our cell type annotation.
(C,G) Cells detected with rearrangement of at least one of the TCRα or TCRβ chains.
(D,H) Gene feature plots of the cells supporting the assignment in (E) and (I).
Figure S3. Summary of cell types profiled by Chromium Immune Profiling Solution in ccRCC-2 fresh tumor (A-D) versus day 7 organoid (E-H) CD45+ fraction, related to Figure 5. (A,E) Breakdown of major immune cell types. (B,F) Unbiased visualization of single cells shown by t-SNE and colored by our cell type annotation.
(C,G) Cells detected with rearrangement of at least one of the TCRα or TCRβ chains.
(D,H) Gene feature plots of the cells supporting the assignment in (E) and (I).
Figure S4. Summary of single cell clonotype comparisons by Chromium single cell tandem 5’ V(D)J-seq between fresh tumor (FT) and organoid (OR) from ccRCC-1 (A-D) and ccRCC-2 (E-H), related to Figure 5. For the clonotypes defined by the TCRα, TCRβ and paired TCRαβ chains respectively, we observe the expanded clonotypes in FT (fresh tumor) to highly overlap with those in OR (organoid), and the top expanded clonotypes are consistent between FT and OR. Additionally, the expansion patterns are significantly correlated (p < 0.01, permutation test).
(A,E) TCRα clonotypes, FT vs OR.
(B,F) TCRβ clonotypes, FT vs OR.
(C,G) Paired TCR αβ clonotypes, FT vs OR.
(D,H) TCR CDR3s sequences and cell counts in FT and OR respectively of the most frequent clonotypes ranked in FT.
Figure S5. t-SNE visualization of cell type assignment and top 3 TCRαβ clonotypes in all six samples of human ccRCC CD45+ fraction, related to Figure 5. Cell type annotations were assigned according to the 5’ scRNA-seq data, as shown in Figures S10-S16. Clonotype assignments were derived from the 5’ scV(D)J-seq data simultaneously measured with the scRNA-seq data. The highlighted TCRαβ clonotypes are the 3 most frequent among the assigned T cells in respective samples and strongly co-localize with the Tex fraction (light green).
Figure S6. Anti-PD-1 or anti-PD-L1 treatment of ALI tumor organoids induces tumor cell killing, related to Figures 6 and 7. (A) Murine B16-SIY organoids prepared from syngeneic s.c. tumors were cultured for 7 or 14 days in the presence of IL-2 and either anti-mouse anti-PD-1, anti-PD-L1 or control IgG followed by FACS analysis with Annexin V-FITC and AAD. Pre-gating for tumor epithelium was performed by scatter properties. At both 7 and 14 days there is a decrease in viable Annexin-V(−)/7-AAD(−) cells and increased Annexin-V(+)/7-AAD(−) early apoptotic (orange) and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells (pink).
(B) Dual Annexin V-FITC/7-AAD FACS analysis of the human kidney ccRCC PDO from Figure 7D–E following treatment with nivolumab or control IgG4 for 7 days. Nivolumab decreased viable Annexin-V(−)/7-AAD(−) cells and increased Annexin-V(+)/7-AAD(−) early apoptotic (orange) and Annexin-V(+)/7-AAD(+) late apoptotic/necrotic cells (pink).
(C) Anti-CD3 plus anti-CD28 augments nivolumab expansion of organoid TIL populations in the ccRCC PDO of Figure 7D–E.
(D) Expanded y-axis from (C) showing nivolumab expansion of organoid TIL populations without anti-CD3/CD28.
Figure S7. Comparison of TIL activation versus expansion in nivolumab-treated PDOs, related to Figure 7. (A) TIL activation by anti-PD-1/nivolumab in human PDOs, reprinted from Fig. 7A for reference.
(B) FACS enumeration of CD3+, CD4+ and CD8+ TILs per 106 total organoid cells in (A).
(C) Percent change of organoid TILs (+) nivolumab versus (−) nivolumab baseline in the samples of (A). A 35% threshold is indicated.
(D-G) Spearman correlations between Interferon-gamma (IFNG), Granzyme B (GZMB), and Perforin-1 (PRF1) mRNA expression levels after nivolumab treatment versus (D) PD-1+ T cell frequency, (E) total T cell frequency, (F) CD4+/CD8+ T-cell ratio and (G) pass/fail score for a clone 28–8 CLIA-certified PD-L1 test (>1% PD-L1 positivity in original tumor).
Data Availability Statement
Deposited Data
Mendeley Figures 1–10 are available at https://data.mendeley.com/datasets/ykpbxfx2p9/2 Single cell sequencing has been deposited with accession number GSE111360. Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111360,