

Abstract
Verticillium wilt, caused by the soil-borne fungus Verticillium dahliae, is a devastating disease of eggplant (Solanum spp.) and causes substantial losses worldwide. Although some genes or biological processes involved in the interaction between eggplant and V. dahliae have been identified in some studies, the underlying molecular mechanism is not yet clear. Here, we monitored the transcriptomic profiles of the roots of resistant S. sisymbriifolium plants challenged with V. dahliae. Based on the measurements of physiological indexes (T-SOD, POD and SSs), three time points were selected and subsequently divided into two stages (S_12 h vs. S_0 h and S_48 h vs. S_12 h). KEGG enrichment analysis of the DEGs revealed several genes putatively involved in regulating plant-V. dahliae interactions, including mitogen-activated protein kinase (MAPK) genes (MEKK1 and MAP2K1), WRKY genes (WRKY22 and WRKY33) and cytochrome P450 (CYP) genes (CYP73A/C4H, CYP98A/C3′H and CYP84A/F5H). In addition, a subset of genes that play an important role in activating V. dahliae defence responses, including Ve genes as well as genes encoding PR proteins and TFs, were screened and are discussed. These results will help to identify key resistance genes and will contribute to a further understanding of molecular mechanisms of the S. sisymbriifolium resistance response to V. dahliae.
Electronic supplementary material
The online version of this article (10.1007/s12298-019-00666-4) contains supplementary material, which is available to authorized users.
Keywords: Solanum sisymbriifolium, Transcriptome, Verticillium dahliae, Defence-response genes
Introduction
Eggplant (Solanum melongena L.) is the third solanaceous fruit crop of outstanding importance worldwide after potato (Solanum tuberosum) and tomato (Solanum lycopersicum) (Polignano et al. 2010). The global production of eggplants has increased considerably, especially in Asian and African countries. According to data of the Food and Agriculture Organization of the United Nations (http://faostat.fao.org), eggplant production has increased from 29 to 49 million tons in the last decade. Eggplant verticillium wilt (EVW), which is caused by Verticillium dahliae, is a devastating disease of eggplant worldwide and causes substantial yield losses (Collonnier et al. 2001). Verticillium dahliae is a destructive plant fungal pathogen that is widely distributed in agricultural soils, and more than 400 plant species, including many economically important crops, are hosts for this pathogen (Bhat and Subbarao 1999); however, no effective fungicides or chemicals are available to cure infected commercial plants. Cultivation methods such as solarization, soil amendments and rotation also have very limited effects on EVW (Yadeta et al. 2014). Thus, genetic resistance still constitutes the most ideal strategy for controlling plant pathogenic fungi with respect to economic benefits and environmental protection, but genetic resistance has received little attention due to a lack of genetic resources (Fradin and Thomma 2006).
Some wild relatives of eggplant, such as Solanum torvum, Solanum aculeatissimum, and Solanum sisymbriifolium, are resistant to EVW (Bletsos et al. 2003; Collonnier et al. 2001; Liu et al. 2015; Zhou et al. 2016). However, fertile F1s are rarely obtained, as the above wild Solanum species and S. melongena are not cross compatible (Borua 1990; Gramazio et al. 2016; Plazas et al. 2016; Tamura et al. 2002; Toppino et al. 2008). Recently, efforts have been made to uncover the mechanisms underlying the molecular interactions between V. dahliae and Arabidopsis (Pantelides et al. 2010), tomato (Diwan et al. 1999; Fradin et al. 2009; Kawchuk et al. 2001), potato (Yang et al. 2015) and cotton (Sun et al. 2013; Xu et al. 2011; Zhang et al. 2012, 2013) plants. In studies on the responses of eggplant to V. dahliae, several resistance-related genes have been isolated and studied (Chai et al. 2003; Fei et al. 2004), and RNA-Seq (RNA sequencing) technologies have been applied to analyze gene expression profiles in eggplant defence responses (Yang et al. 2013, 2014; Zhou et al. 2016). However, these data are insufficient to elucidate the defence mechanism of eggplant challenged with V. dahliae. As such, it is necessary and urgent to excavate useful resistance genes for eggplant improvement.
Solanum sisymbriifolium is a wild relative of S. melongena and is resistant to EVW (Bletsos et al. 2003; Zhou et al. 2016). However, compared with other wild relatives of eggplant, S. sisymbriifolium has been very rarely studied. For better utilization of this potential material, more genomic information is demanded. Therefore, the first transcriptome sequencing of S. sisymbriifolium with and without V. dahliae infection was performed using Illumina next-generation sequencing technology both to provide an overview of the S. sisymbriifolium root transcriptome and to identify the genes and pathways involved in the pathogen-plant interaction (PPI) system. The results generated in this study can provide important fundamental information for understanding the control of plant development and disease resistance in eggplant.
Materials and methods
Plant material and inoculation
The highly toxic pathogenic V. dahliae strain QZ-S was isolated from the roots of a diseased eggplant in the Kunming area of China (Wu et al. 2017). The isolate was cultured on potato dextrose agar (PDA) plates for 15–20 days, after which it was transferred to Czapek’s liquid medium in a shaker at 110 rpm under a temperature of 25 °C for 5–7 days. The suspension liquid was then filtered and adjusted to approximately 1 × 107 spores/mL with sterilized distilled water prior to inoculation.
Seeds of S. sisymbriifolium were obtained from the Kunming Institute of Botany, Chinese Academy of Science. After the seeds were sterilized, they were allowed to germinate in a growth chamber at 30 °C. The germinated seeds were subsequently transplanted to sterilized soil and cultivated in the greenhouse at 25/15 °C (day/night). When they had two pairs of leaves, the seedlings were inoculated by dipping their roots into the fungal spore suspension for 30 min. The roots of the V. dahliae-treated plants were then harvested after 6, 12, 24, 48 and 72 h. Control plants were not inoculated but were treated with distilled water in the same manner. At each time point, 18 plants were randomly divided into three groups, and the roots of the six plants in each group were pooled (n = 3 groups). All of the samples were rapidly frozen in liquid nitrogen and stored at − 80 °C.
Measurement of physiological indexes
For enzyme extracts and assays, the roots (0.1 g) were ground in liquid nitrogen, and were then suspended in a 0.9-mL solution containing 10 mM phosphate buffer (pH 7.4). The homogenate was then centrifuged at 2500 rpm at 4 °C for 10 min, after which the resulting supernatant was collected for determining the T-SOD and POD activities and the SS content. The T-SOD activity was measured using a total superoxide dismutase assay kit (hydroxylamine method, A001-1), the POD activity was measured using a POD assay kit (A084-3), and the SS content was assayed using a plant SS content test kit (A145). The commercial assay kits mentioned above were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China), and a DDS-306 electrical conductivity meter (Fangzhou Company, Chengdu, China) was used for measuring all of the indexes.
RNA isolation, library construction and sequencing
The total RNA of each sample was extracted using RNAout 1.0 (Tianenze, Beijing, China). RNA degradation and contamination were monitored on 1% agarose gels, and a NanoPhotometer® spectrophotometer (Implen, CA, USA) was used to check the RNA purity. A Qubit® RNA Assay Kit in conjunction with a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA) was used to measure the RNA concentrations, and RNA integrity was assessed using an RNA Nano 6000 Assay Kit together with an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). To construct a cDNA library for each time point, each group of three replicates was sampled at each time point. All cDNA library construction and Illumina sequencing were performed by the Beijing Novogene Bioinformatics Technology Co., Ltd.
Briefly, poly-T oligo-conjugated magnetic beads were used to purify the mRNA from the total RNA. Fragmentation was then carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5 ×). First-strand cDNA synthesis was primed by random hexamer and transcribed by M-MuLV Reverse Transcriptase (RNase H). Second-strand was subsequently synthesized using DNA Polymerase I and RNase H. Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. The remaining overhangs were converted to blunt ends via exonuclease/polymerase activities. After the 3′ ends of the DNA fragments were adenylated, NEBNext adaptors having a hairpin loop structure were ligated in preparation for hybridization. To select cDNA fragments that were preferentially 150–200 bp in length, an AMPure XP system (Beckman Coulter, Beverly, USA) was used to purify the library fragments. Then, 3 µl of USER Enzyme (NEB, USA) was used with size-selected adaptor-ligated cDNA at 37 °C for 15 min followed by 95 °C for 5 min prior to PCR. Phusion High-Fidelity DNA polymerase, universal PCR primers and index (X) primer were then used to perform PCR. The PCR products were purified (via the AMPure XP system) a final time, after which the Agilent Bioanalyzer 2100 system was used to assess the library quality.
Clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina) in accordance with the manufacturer’s instructions. After the clusters were generated, the library preparations were sequenced on an Illumina HiSeq 2000 platform, after which paired-end reads were generated.
De novo transcriptome assembly and gene functional annotation
In-house Perl scripts were used to first process the raw data (raw reads) in fastq format. During this step, clean data (clean reads) were obtained by removing reads containing adaptors, reads containing poly-N sequences and low-quality reads (Q ≤ 20) from the raw data. At the same time, Q20 and Q30 values, the GC content and the sequence duplication level of the clean data were calculated. All downstream analyses were based on high-quality clean data. Besides, the reads of V. dahlia were removed by comparing the clean data with the Nt database. The clean reads were subsequently assembled using Trinity software (version r20140413p1) as described for de novo transcriptome assembly in the absence of a reference genome (Kanehisa et al. 2008). To avoid redundant transcripts, in-house Perl scripts were applied to extract the longest transcripts as unigenes. The duplicate genes were removed by TGICL (TGI Clustering tools) software (Pertea et al. 2003) to attain the final set of unigenes.
Gene function was annotated based on the following databases: Nr, Nt, Pfam, KOG/COG, Swiss-Prot (a manually annotated and reviewed protein sequence database), KEGG orthologue (KO), and GO.
Differential expression analysis
The DESeq R package (v1.10.1) was used to analyse the differential expression of both groups. Using a model based on negative binomial distribution, DESeq provides statistical routines to determine differential expression among digital gene expression data. For the samples with biological replicates, the DEGs were filtered for a corrected P value threshold of 0.05 (the P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate (FDR)). Functional enrichment analyses including the use of GO and KEGG were performed to identify which GO terms or metabolic pathways were significantly enriched in DEGs. The GO enrichment analysis of the DEGs was implemented through the GOseq R package (v1.10.0); the analysis was based on the Wallenius non-central hypergeometric distribution (Young et al. 2010), and the GO terms with a corrected P value < 0.05 were considered significantly enriched. KOBAS (Mao et al. 2005) software (v2.0.12) was used to test the statistical enrichment of the DEGs among the KEGG pathways. Those pathways with an FDR value of ≤ 0.05 were defined as containing genes with significant differential expression.
RT-qPCR analysis of DEGs
Eighteen co-expressed DEGs were chosen for validation using RT-qPCR. The total RNA was isolated from the roots of both V. dahliae-treated and control plants. First-strand cDNA was synthesized using a Transcript® All-in-one First-Strand cDNA Synthesis SuperMix for qPCR kit (Trans, Beijing). The primers used for RT-qPCR were designed with Primer Premier 5.0 software (Supplementary Table S1). All primer pairs were custom ordered from Tsingke, Kunming. The 18S RNA gene of eggplant served as an internal control. RT-qPCR was performed on an ABI 7500 Real-time PCR system using a SYBR Green-based PCR assay. All RT-qPCRs for each gene were performed for three biological replicates, and each biological replicate included three technical replicates. A reaction containing 10 μl of 2 × TransStart® Tip Green qPCR SuperMix, 0.4 μl of passive reference dye (50 ×), 1 μl of diluted cDNA, and 0.4 μl (10 μM) of each primer in a final volume of 20 μl was performed. The amplification programme consisted of one cycle of 94 °C for 30 s, followed by 40 cycles of 94 °C for 5 s and 60 °C for 30 s. The relative gene expression levels were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001).
Results
Changes in physiological indexes of S. sisymbriifolium infected with V. dahliae
To investigate the physiological responses of S. sisymbriifolium challenged with V. dahliae and to choose the key time points for the transcriptome analysis, the total superoxide dismutase (T-SOD) and peroxidase (POD) activities as well as the soluble sugar (SS) content were measured at 0 (mock control), 6, 12, 24, 48 and 72 h post inoculation (hpi).
After the plants were infected with V. dahliae, the T-SOD, POD and SS enzymes within the S. sisymbriifolium roots showed greater activity than did those within the control roots (0 hpi). The maximum T-SOD (Fig. 1a) was observed at 12 hpi, and a second, smaller increase was detected at 48 hpi. The POD enzyme activity increased markedly after the plants were inoculated with the pathogen (Fig. 1b) and exhibited two similar peaks at 12 and 48 hpi. The SS content also increased sharply after the plants were inoculated; it peaked at 12 hpi, after which it decreased (Fig. 1c).
Fig. 1.
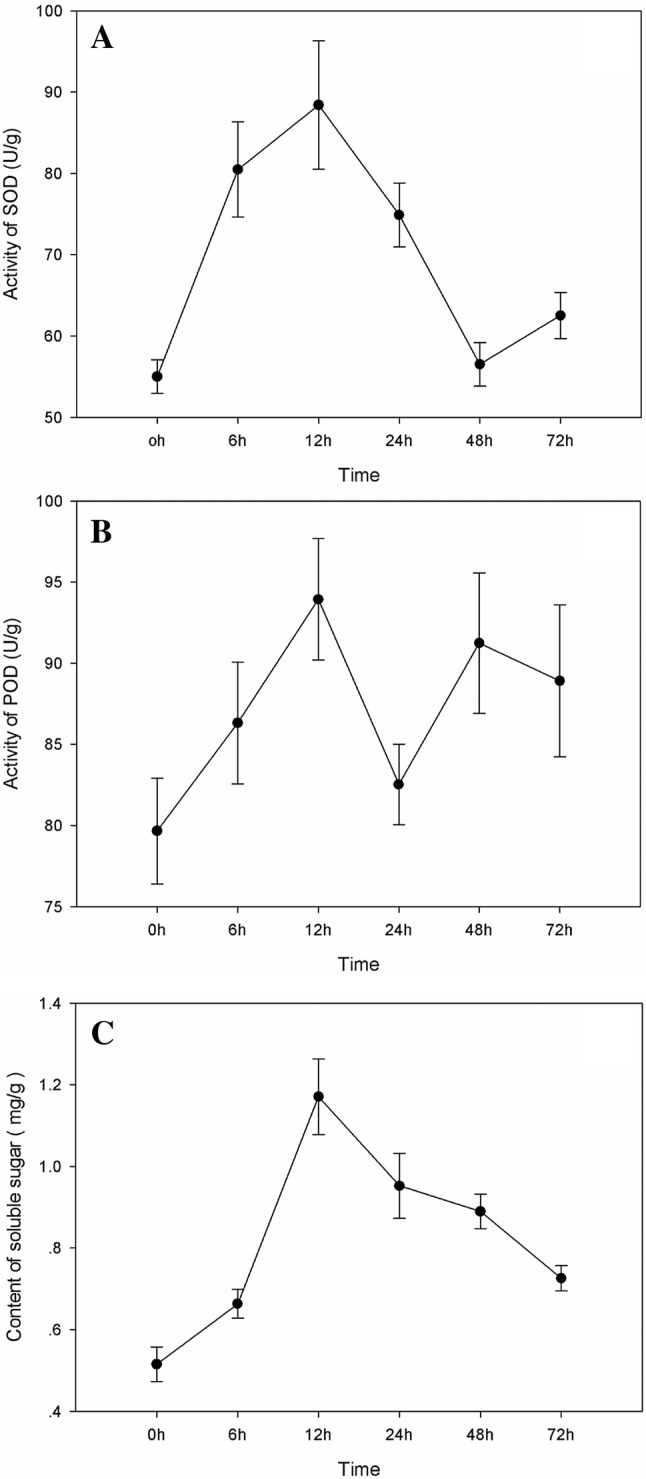
Determination of physiological indexes. The activities of T-SOD (a) and POD (b) as well as the SS content (c) in the roots of plants challenged with V. dahliae were measured at 0, 6, 12, 24, 48 and 72 hpi
Based on the above results, we can conclude that the physiological activity of S. sisymbriifolium changed markedly under stress from V. dahliae, especially at 12 and 48 hpi. To identify the resistance-related genes, samples at three time points (0, 12 and 48 hpi) of those examined above were selected for RNA-Seq.
Illumina sequencing and de novo assembly
Nine transcriptome libraries by using an Illumina HiSeq™ 2000 platform for S. sisymbrifolium, three time points (0, 12 and 48 hpi) and their biological replicates were constructed and sequenced. A total of 480,282,756 raw reads were generated from the nine libraries. After removing the reads containing ploy-N, reads containing adaptor sequence fragments and low quality reads by stringent quality checks and data cleaning, 454,990,956 clean reads with a mean Q20 of 96.87% were obtained from all of the libraries (Supplementary Table S2). The de novo assembly of these clean reads produced 430,269 transcripts and 221,695 unigenes by using Trinity softwore (r20140413p1) (Grabherr et al. 2011). And the N50 of total unigenes was 1772 bp. The sequence size distribution is presented in Figure S1. RNA-seq read data have been deposited in the NCBI Sequence Read Archive (SRA) database under accession No. PRJNA495012 (https://www.ncbi.nlm.nih.gov/sra/PRJNA495012). The sequencing and assembly results suggest that the unigene data were highly reliable for further analysis.
Functional annotation and classification
The all-unigenes were tested via BLAST analysis against seven databases (the NCBI non-redundant protein sequences (Nr) and non-redundant nucleotide sequences (Nt), Swiss-Prot, Protein family (Pfam), Clusters of Orthologous Groups of proteins (KOG/COG), Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases) with an e-value threshold of 10−5. The results indicated that the unigenes (221,695) with homologous sequences were distributed among the above databases as follows: 141,213 (63.69%), 170,754 (77.02%), 107,976 (48.70%), 50,281 (22.68%), 111,391 (50.24%), 59,105 (26.66%) and 110,326 (49.76%) unigenes were matched to the Nr, Nt, Pfam, KOG, Swiss-Prot, KEGG and GO databases, respectively. Among these unigenes, 192,578 (86.86%) with homologous sequences were present in at least one database. The Nr database annotation results indicated that the majority of the data could be compared with and mapped to S. tuberosum (35.1%) or S. lycopersicum (17%).
We used the GO database as an international standardized classification system to classify the possible functions of the unigenes based on the Nr annotation. A total of 110,326 unigenes were classified via sequence homology into three major functional categories (biological process (BP), cellular component (CC) and molecular function (MF)) and 57 subcategories (Supplementary Fig. S2). Within the BP category, the top three categories were ‘cellular process’ (63,727), ‘metabolic process’ (61,791) and ‘single-organism process’ (48,950). Within the CC category, the most frequent categories were ‘cell’ (36,204), ‘cell part’ (36,191) and ‘organelle’ (25,126). Genes in the MF category primarily sorted into ‘binding’ (59,763), ‘catalytic activity’ (51,536) and ‘transporter activity’ (8245) categories.
KOG is a database for protein orthology classification. The alignment of the all-unigenes to the KOG database resulted in the identification of 50,281 unigenes assigned to 26 function clusters (Supplementary Fig. S3). Among these categories, the largest group was ‘posttranslational modification, protein turnover, and chaperones’ (6928 unigenes, 13.78%), then the following groups were ‘translation, ribosomal structure and biogenesis’ (6221 unigenes, 12.37%), ‘general function prediction only’ (5980 unigenes, 11.89%), ‘energy production and conversion’ (3889 unigenes, 7.73%) and ‘signal transduction mechanisms’ (3595 unigenes, 7.15%).
The KEGG database was used for the functional classification and pathway assignment of the all-unigenes. A total of 59,105 unigenes were assigned to 130 KEGG pathways. The first 19 pathways are shown in Supplementary Fig. S4. The main pathways were ‘translation’ (7034, 11.90%), ‘carbohydrate metabolism’ (6070, 10.27%), ‘overview’ (5101, 8.63%), ‘folding, sorting and degradation’ (4484, 7.59%) and ‘energy metabolism’ (3445, 5.83%). The KEGG pathway analysis also revealed that the ‘plant-pathogen interaction’ (1479, 2.50%), ‘plant hormone signal transduction’ (1080, 1.83%) and ‘phenylpropanoid biosynthesis’ (835, 1.41%) clusters were the largest subcategories that were involved in the ‘environmental adaptation’, ‘signal transduction’ and ‘biosynthesis of secondary metabolites’ pathways, respectively.
Differential expression analysis
A comparison of the three time points identified 6218 (2958 up-regulated, 3260 down-regulated), 7059 (4137 up-regulated, 2922 down-regulated) and 10,402 (5831 up-regulated, 4571 down-regulated) differentially expressed genes (DEGs) in the S_12 h versus S_0 h, S_48 h versus S_0 h and S_48 h versus S_12 h pairs, respectively (Fig. 2).
Fig. 2.
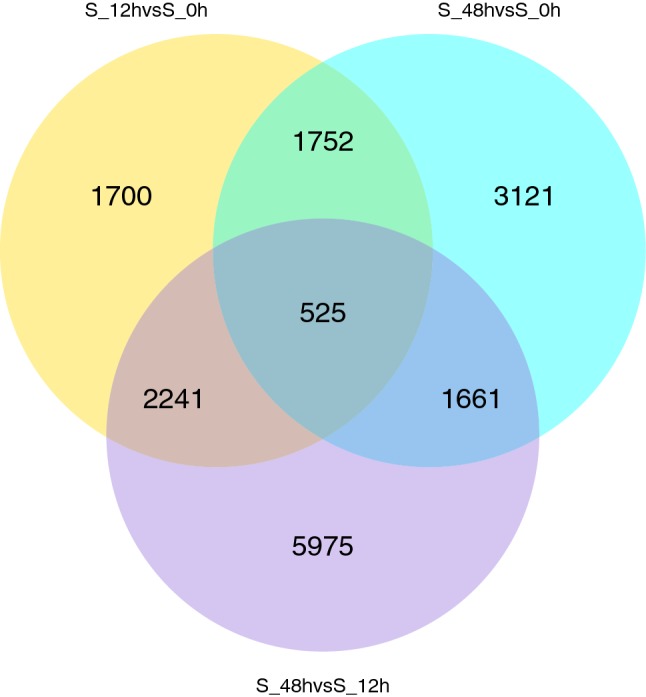
Venn diagram showing DEGs from the comparison of three time points. Yellow, blue and purple represent the DEGs of S_12 h versus S_0 h, S_48 h versus S_0 h and the S_48 h versus S_12 h, respectively. The overlapping values correspond to the number of DEGs in two/three comparisons (color figure online)
To analyse the gene expression trends between the different time points, we divided the three time points into stage 1 (S_12 h vs. S_0 h) and stage 2 (S_48 h vs. S_12 h). A comparative analysis between these two stages revealed 13,854 DEGs, including 2766 unigenes, that were significantly regulated during both periods, with 3452 and 7636 unigenes differentially regulated in stage 1 and stage 2, respectively.
The DEGs were hierarchically clustered according to the fragments per kilobase of transcript per million mapped reads (FPKM) values of the samples; this clustering allowed us to observe the overall gene expression pattern. The results (Fig. 3) indicated that the expression patterns of the DEGs varied greatly at the different time points and that the gene expression trends at 0 hpi and 48 hpi were more similar than those at 12 hpi. At the same time, after the plants were inoculated with V. dahliae, the majority of the DEGs were expressed at only one or two time points, and a small subset of DEGs were co-expressed at all three time points. The majority of the DEGs exhibited similar expression patterns at 0 hpi and 48 hpi, but these patterns were opposite from those at 12 hpi.
Fig. 3.
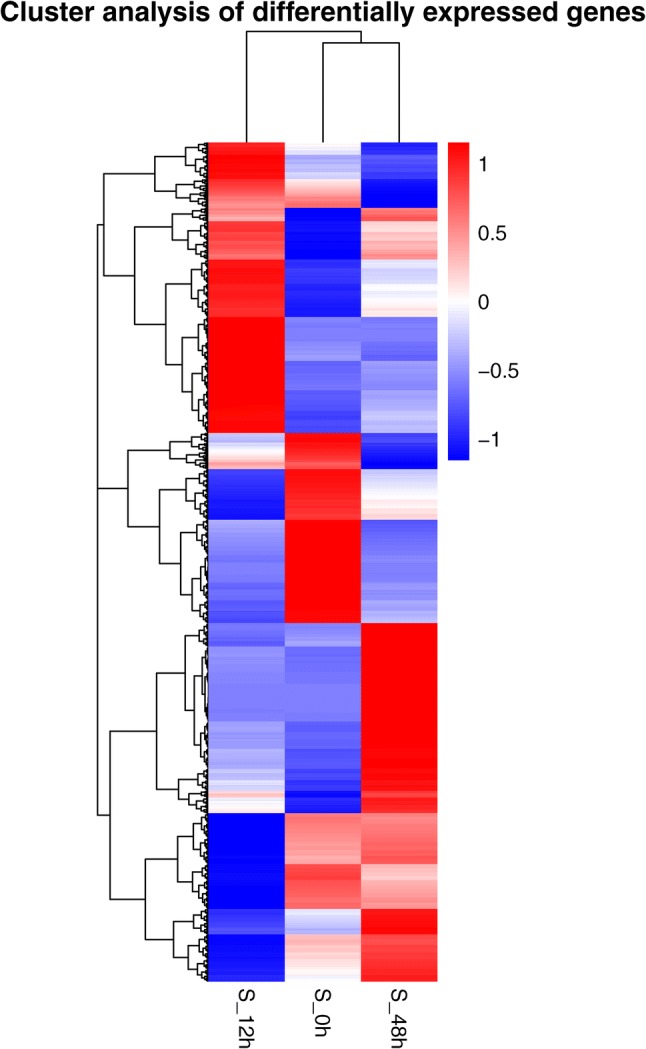
Expression profiles of DEGs at both stages. The graph indicates the similarity in the tendencies for changes in gene expression. The shorter the line is, the more similar the genes. Red colour indicates a high expression level, while blue colour indicates a low expression level (color figure online)
GO assignments were used to classify the functions of the predicted S. sisymbriifolium genes. The enriched GO terms (P < 0.05) are listed in Supplementary Fig. S5. During stage 1, in the BP category, ‘oxidation–reduction process’, ‘phosphorylation’ and ‘single-organism carbohydrate metabolic process’ were significantly overrepresented. In the CC category, ‘transcription factor complex’, ‘extracellular matrix’ and ‘cytosolic part’ were the top three significantly enriched terms. In the MF category, many genes were involved in ‘cation binding’, ‘metal ion binding’ and ‘oxidoreductase activity’. During stage 2, ‘single-organism process’, ‘single-organism metabolic process’ and ‘oxidation–reduction process’ were the most enriched in the BP category, and ‘chloroplast part’, ‘plastid part’ and ‘thylakoid membrane’ were the top three enriched terms in the CC category. For the MF category, ‘catalytic activity’, ‘ion binding’ and ‘cation binding’ were the most enriched terms.
We performed a KEGG analysis of the DEGs to explore their involvement in biological pathways. The pathways that were significantly enriched in both stages are listed in Supplementary Table S3; the top 20 are shown in Fig. 4. The KEGG pathway enrichment analysis indicated that the major pathways for both stages involved the metabolism and biosynthesis of secondary metabolites. For stage 1 (Fig. 4a), ‘oxidation–reduction process’, ‘phosphorylation’ and ‘single-organism carbohydrate metabolic process’ were the three most enriched terms. For stage 2 (Fig. 4b), ‘single-organism process’, ‘single-organism metabolic process’ and ‘oxidation–reduction process’ were the most enriched. These results are consistent with those of the GO enrichment analysis. In particular, some pathways that play important roles in plant resistance to fungal pathogens, such as ‘plant hormone signal transduction’, ‘plant-pathogen interaction’ and ‘phenylpropanoid biosynthesis’, contained many genes and were significantly enriched in both stages. In contrast, several pathways were enriched in only one stage. For example, ‘arginine and proline metabolism’, ‘cutin, suberin and wax biosynthesis’, ‘flavonoid biosynthesis’ and ‘stilbenoid, diarylheptanoid and gingerol biosynthesis’ were enriched specifically in stage 1, whereas ‘diterpenoid biosynthesis’, ‘photosynthesis’, ‘regulation of autophagy’ and ‘zeatin biosynthesis’ were enriched only in stage 2.
Fig. 4.

KEGG pathway enrichment analysis of stage 1 (A) and stage 2 (B). The top 20 enriched KEGG pathways are listed. The X-axis indicates the enrichment factor of the KEGG term, and the Y-axis indicates the name of the KEGG term. The size of the points indicates the number of genes within the pathway term. The colour of each point indicates the degree of enrichment (color figure online)
To provide basic information for further study of the interaction between S. sisymbriifolium and V. dahliae, several sets of genes involved in important pathways, such as the ‘plant hormone signal transduction’, ‘plant-pathogen interaction’ and ‘phenylpropanoid biosynthetic’ pathways, were selected. Additionally, a subset of the genes that played important roles in activating V. dahliae defence responses, including Ve genes as well as those encoding pathogenesis-related (PR) proteins and transcription factors (TFs), were screened in this study.
Selection of the major genes related to resistance to V. dahliae
DEGs involved in the plant hormone signal transduction pathway
Many regulated genes that participate in the plant hormone signal transduction (PHST) pathway were identified based on their KEGG pathway assignment (Table 1, Supplementary Fig. S6). The results show that most of the PHST-related genes are involved in abscisic acid (ABA), auxin (AUX), ethylene (ET), cytokinin (CTK) and salicylic acid (SA) biosynthesis during both time periods; however, stage 2 involved more genes than did stage 1.
Table 1.
DEGs involved in the PHST pathway
| Terms | Stage 1 (S_12 h vs. S_0 h) | Stage 2 (S_48 h vs. S_12 h) | ||
|---|---|---|---|---|
| Hormone category | Gene name | Gene definition | Unigenes annotated | Unigenes annotated |
| Auxin | 22 (9 up-, 13 down-regulated) | 24 (16 up-, 8 down-regulated) | ||
| AUX1 | auxin influx carrier (AUX1 LAX family) | 1 (up-regulated) | 2 (down-regulated) | |
| ARF | auxin response factor | 2 (down-regulated) | 4 (up-regulated) | |
| IAA | auxin-responsive protein IAA | 6 (5 up-, 1 down-regulated) | 5 (1 up-, 4 down-regulated) | |
| GH3 | auxin responsive GH3 gene family | 5 (1 up-, 4 down-regulated) | 6 (up-regulated) | |
| SAUR | SAUR family protein | 8 (2 up-, 6 down-regulated) | 6 (5 up-, 1 down-regulated) | |
| TIR1 | transport inhibitor response 1 | 0 | 1 (down-regulated) | |
| Cytokinin | 11 (5 up-, 6 down-regulated) | 11 (8 up-, 3 down-regulated) | ||
| A-ARR | two-component response regulator ARR-A family | 5 (up-regulated) | 2 (down-regulated) | |
| CRE1 | arabidopsis histidine kinase 2/3/4 (cytokinin receptor)[EC:2.7.13.3] | 4 (down-regulated) | 5 (4 up-, 1 down-regulated) | |
| B-ARR | two-component response regulator ARR-B family | 2 (down-regulated) | 2 (up-regulated) | |
| AHP | histidine-containing phosphotransfer peotein | 0 | 2 (up-regulated) | |
| Gibberellin | 1 (down-regulated) | 2 (up-regulated) | ||
| PIT4 | phytochrome-interacting factor 4 | 1 (down-regulated) | 0 | |
| GID1 | gibberellin receptor GID1[EC:3.-.-.-] | 0 | 2 (up-regulated) | |
| Abscisic acid | 33 (9 up-, 24 down-regulated) | 32 (21 up-, 11 down-regulated) | ||
| PYL | abscisic acid receptor PYR/PYL family | 7 (up-regulated) | 10 (down-regulated) | |
| PP2C | protein phosphatase 2C [EC:3.1.3.16] | 16 (down-regulated) | 9 (8 up-, 1 down-regulated) | |
| ABF | ABA responsive element binding factor | 3 (down-regulated) | 5 (up-regulated) | |
| SnRK2 | serine/threonine-protein kinase SRK2 [EC:2.7.11.1] | 7 (2 up-, 5 down-regulated) | 8 (up-regulated) | |
| Ethylene | 18 (18 up-regulated) | 18 (6 up-, 12 down-regulated) | ||
| ETR | ethylene receptor [EC:2.7.13.-] | 6 (up-regulated) | 6 (1 up-, 5 down-regulated) | |
| CTR1 | serine/threonine-protein kinase CTR1 [EC:2.7.11.1] | 3 (up-regulated) | 2 (1 up-, 1 down-regulated) | |
| EBF1_2 | EIN3-binding F-box protein | 7 (up-regulated) | 5 (down-regulated) | |
| ERF1 | ethylene-responsive transcription factor 1 | 2 (up-regulated) | 1 (down-regulated) | |
| EIN2 | ethylene-insensitive protein 2 | 0 | 1 (up-regulated) | |
| EIN3 | ethylene-insensitive protein 3 | 0 | 3 (up-regulated) | |
| Brassinosteroid | 2 (2 down-regulated) | 6 (5 up-, 1 down-regulated) | ||
| BKI1 | BRI1 kinase inhibitor 1 | 2 (down-regulated) | 3 (up-regulated) | |
| BSK | BR-signalling kinase [EC:2.7.11.1] | 0 | 2 (up-regulated) | |
| BIN2 | protein brassinosteroid insensitive 2 [EC:2.7.11.1] | 0 | 1 (down-regulated) | |
| Jasmonic acid | 3 (down-regulated) | 3 (up-regulated) | ||
| JAR1 | jasmonic acid-amino synthetase | 1 (down-regulated) | 3 (up-regulated) | |
| MYC2 | transcription factor MYC2 | 2 (down-regulated) | 0 | |
| Salicylic acid | 7 (2 up-, 5 down-regulated) | 21 (17 up-, 4 down-regulated) | ||
| NPR1 | regulatory protein NPR1 | 3 (1 up-, 2 down-regulated) | 5 (4 up-, 1 down-regulated) | |
| TGA | transcription factor TGA | 4 (1 up-, 3 down-regulated) | 13 (10 up-, 3 down-regulated) | |
| PR1 | pathogenesis-related protein 1 | 0 | 3 (up-regulated) | |
| Total | 97 (43 up-, 54 down-regulated) | 117 (78 up-, 39 down-regulated) | ||
Additionally, most of the enzymes or TFs emerged in both time courses but with contrasting expression trends. For example, the expression of AUX1, A-ARR, PYL, EBF1_2 and ERF1 was up-regulated in stage 1 but down-regulated in stage 2, and the expression of ARF, B-ARR, BKI1 and JAR1 was down-regulated in stage 1 but up-regulated in stage 2. Some enzymes or TFs were detected only in one time course. All of the genes related to the PIT4 and MYC2 TFs were down-regulated and were detected specifically in stage 1. For the genes that emerged only in stage 2, the expression of AHP, GID1, EIN2, EIN3, BSK and PR1 increased, but the expression of TIR1 and BIN2 decreased.
DEGs involved in the PPI pathway
In the PPI pathway, 70 and 83 DEGs were identified in stage 1 and stage 2, respectively, and the majority of those genes were down-regulated in both stages (Table 2, Supplementary Fig. S7). In stage 1, the expression of RBOH, NOA1, Pti4, Pti1, RPM1, EIX1_2, and CERY1 increased, but that of CDPK, WRKY33, FLS2, MEKK1, NHO1, and RPS2 decreased. In stage 2, the MEKK1, MAP2K1, NHO1, and PR1 genes were up-regulated, but the NOA1, WRKY33, WRKY22, Pti4, Pti5, Pti1, RIN4, PBS1, and EIX1_2 genes were down-regulated.
Table 2.
DEGs involved in the PPI pathway
| Terms | Stage 1 (S_12 h vs. S_0 h) | Stage 2 (S_48 h vs. S_12 h) | |
|---|---|---|---|
| Gene name | Gene description | Unigenes annotated | Unigenes annotated |
| Rboh | respiratory burst oxidase [EC:1.6.3.- 1.11.1.-] | 7 (up-regulated) | 5 (4 up-, 1 down-regulated) |
| NHO1 | glycerol kinase [EC:2.7.1.30] | 2 (down-regulated) | 2 (up-regulated) |
| CaM | calmodulin | 20 (5 up-, 15 down-regulated) | 21 (6 up-, 15 down-regulated) |
| MAP2K1 | mitogen-activated protein kinase kinase 1 [EC:2.7.12.2] | 0 | 2 (up-regulated) |
| CNGC | cyclic nucleotide gated channel, plant | 12 (8 up-, 4 down-regulated) | 5 (2 up-, 3 down-regulated) |
| HSP90 | heat shock protein 90 kDa beta | 2 (1 up-, 1 down-regulated) | 4 (1 up-, 3 down-regulated) |
| SGT1 | suppressor of G2 allele of SKP1 | 0 | 2 (1 up-, 1 down-regulated) |
| CDPK | calcium-dependent protein kinase | 8 (down-regulated) | 8 (6 up-, 2 down-regulated) |
| MEKK1 | mitogen-activated protein kinase kinase kinase 1 [EC:2.7.11.25] | 1 (down-regulated) | 1 (up-regulated) |
| FLS2 | LRR receptor-like serine/threonine-protein kinase FLS2 [EC:2.7.11.1] | 2 (down-regulated) | 2 (1 up-, 1 down-regulated) |
| WRKY33 | WRKY transcription factor 33 | 5 (down-regulated) | 1 (down-regulated) |
| WRKY22 | WRKY transcription factor 22 | 0 | 1 (down-regulated) |
| NOA1 | NOA1,nitric-oxide synthase, plant [EC:1.14.13.39] | 1 (up-regulated) | 1 (down-regulated) |
| CERK1 | chitin elicitor receptor kinase 1 | 2 (up-regulated) | 10 (1 up-, 9 down-regulated) |
| PBS1 | disease resistance protein RPS2 | 0 | 1 (down-regulated) |
| Pti4 | pathogenesis-related genes transcriptional activator PTI4 | 1 (up-regulated) | 1 (down-regulated) |
| Pti5 | pathogenesis-related genes transcriptional activator PTI5 | 0 | 2 (down-regulated) |
| Pti1 | pto-interacting protein 1 [EC:2.7.11.1] | 1 (up-regulated) | 3 (down-regulated) |
| PR1 | pathogenesis-related protein 1 | 0 | 3 (up-regulated) |
| RIN4 | RPM1-interacting protein 4 | 0 | 2 (down-regulated) |
| RPM1 | disease resistance protein RPM1 | 3 (up-regulated) | 2 (1 up-, 1 down-regulated) |
| RPS2 | disease resistance protein RPS2 | 2 (down-regulated) | 2 (1 up-, 1 down-regulated) |
| EIX1_2 | EIX receptor 1/2 | 1 (up-regulated) | 1 (down-regulated) |
| Total | 70 (30 up-, 40 down-regulated) | 83 (32 up-, 50 down-regulated) | |
Furthermore, in the PPI pathway, more genes participated in stage 2 than in stage 1, such as MAP2K1, WRKY22, Pti5, PR1, PBS1, RIN4 and SGT1. The majority of the enzymes and proteins encoded by the unigenes were identified in both stages, but their gene expression patterns were very different. For example, the expression of EIX1_2, NOA1, Pti1 and Pti4 increased in stage 1 but decreased in stage 2, and, the expression of NHO1 and MEKK1 increased in stage 1 but decreased in stage 2.
DEGs involved in phenylpropanoid biosynthetic (PB) pathway-related genes
Several sets of genes assigned to the PB pathway were also detected in this study (Table 3, Supplementary Fig. S8). In stage 1, several genes encoding enzyme transcripts, including COMT, C3′H, C4H, and CCoAOMT, were up-regulated; however, the genes encoding F5H and CALDH were down-regulated. In stage 2, the expression of BGLU, PAL, CCoAOMT, 4CL, C4H, HCT, and F5H increased, but the expression of C3′H and CALDH decreased.
Table 3.
DEGs involved in the PB pathway
| Terms | Stage 1 (S_12 h vs. S_0 h) | Stage 2 (S_48 h vs. S_12 h) | |
|---|---|---|---|
| Gene name | Gene description | Unigenes annotated | Unigenes annotated |
| CYP73A/C4H | trans-cinnamate 4-monooxygenase [EC:1.14.13.11] | 2 (up-regulated) | 2 (up-regulated) |
| COMT | caffeic acid 3-O-methyltransferase [EC:2.1.1.68] | 3 (up-regulated) | 5 (3 up-, 2 down-regulated) |
| CYP98A/C3′H | coumaroylquinate(coumaroylshikimate) 3′-monooxygenase [EC:1.14.13.36] | 3 (up-regulated) | 2 (down-regulated) |
| CCoAOMT | caffeoyl-CoA O-methyltransferase [EC:2.1.1.104] | 1 (up-regulated) | 2 (up-regulated) |
| CYP84A/F5H | ferulate-5-hydroxylase [EC:1.14.-.-] | 1 (down-regulated) | 1 (up-regulated) |
| CALDH | coniferyl-aldehyde dehydrogenase [EC:1.2.1.68] | 1 (down-regulated) | 0 |
| 4CL | 4-coumarate–CoA ligase [EC:6.2.1.12] | 4 (1 up-, 3 down-regulated) | 2 (up-regulated) |
| HCT | shikimate O-hydroxycinnamoyltransferase [EC:2.3.1.133] | 4 (1 up-, 3 down-regulated) | 1 (up-regulated) |
| CAD | cinnamyl-alcohol dehydrogenase [EC:1.1.1.195] | 7 (3 up-, 4 down-regulated) | 13 (8 up-, 5 down-regulated) |
| BGLU | beta-glucosidase [EC:3.2.1.21] | 3 (2 up-, 1 down-regulated) | 6 (up-regulated) |
| POD | peroxidase [EC:1.11.1.7] | 27 (18 up-, 9 down-regulated) | 29 (23 up-, 6 down-regulated) |
| PAL | phenylalanine ammonia-lyase [EC:4.3.1.24] | 0 | 2 (up-regulated) |
| UGT72E | coniferyl-alcohol glucosyltransferase [EC:2.4.1.111] | 0 | 1 (down-regulated) |
| CCR | cinnamoyl-CoA reductase [EC:1.2.1.44] | 0 | 4 (1 up-, 3 down-regulated) |
| Total | 56 (34 up-, 22 down-regulated) | 70 (51 up-, 19 down-regulated) | |
More genes in the PB pathway were identified in stage 2 than in stage 1, and most of those genes were up-regulated. Regarding the enzymes that were present in both stages, the expression of C4H and CCoAOMT increased continuously during the two stages, but the expression of F5H and C3′H showed the opposite pattern. In addition, one CALDH gene was down-regulated and differentially regulated only in stage 1, whereas two up-regulated PAL genes were detected only in stage 2.
Ve genes and PR protein-encoding genes
Ve genes (Fradin et al. 2009; Kawchuk et al. 2001) and PR protein-encoding genes (Zhang et al. 2013, 2016a, b; Zhou et al. 2016) play important roles in defence reactions against verticillium wilt.
In this study, the Nr annotation described two non-DEGs (Cluster-33922.112117 and Cluster-33922.87359) as Ve genes (Table 4), and NCBI queries revealed that those genes shared high identity with the S. torvum genes StVe1 (93.0%, DQ020574.1) and StVe2 (95.6%, KT946796.1), respectively.
Table 4.
Expression profiles of several known unigenes in response to infection by V. dahlia
| Gene name | Unigene ID | Gene length | Description | FPKM | ||
|---|---|---|---|---|---|---|
| 0 hpi | 12 hpi | 48 hpi | ||||
| Ve2 | Cluster-33922.87359 | 3756 | verticillium wilt disease resistance protein precursor [Solanum torvum] | 29.09 | 10.12 | 13.85 |
| Ve1 | Cluster-33922.112117 | 3615 | verticillium wilt disease resistance protein [Solanum torvum] | 1.28 | 0.63 | 1.04 |
| PR10 | Cluster-33922.86915 | 1578 | pathogenesis-related protein 10 [Solanum virginianum] | 0.87 | 11.07 | 36.84 |
| PR10 | Cluster-33922.88265 | 1767 | pathogenesis-related protein 10 [Solanum virginianum] | 2.77 | 17.23 | 21.99 |
| PR5 | Cluster-33922.53642 | 1413 | PREDICTED: pathogenesis-related protein 5 [Solanum lycopersicum] | 4.45 | 1.47 | 7.36 |
| PR5 | Cluster-33922.53641 | 1423 | PREDICTED: pathogenesis-related protein 5 [Solanum lycopersicum] | 6.55 | 2.79 | 14.21 |
| PR10 | Cluster-33922.86911 | 896 | pathogenesis-related protein 10 [Solanum virginianum] | 382.66 | 641.74 | 1909.39 |
| PR | Cluster-33922.69006 | 1717 | PREDICTED: pathogen-related protein-like [Solanum tuberosum] | 0.13 | 0.46 | 0.00 |
| PR1 | Cluster-33922.28369 | 716 | pathogenesis related protein 1 | 2.64 | 1.40 | 48.66 |
| PR1 | Cluster-20063.0 | 874 | pathogenesis-related protein 1 | 0.06 | 0.00 | 2.18 |
| PR1 | Cluster-33922.147658 | 1145 | pathogenesis-related protein 1 | 0.02 | 0.19 | 2.37 |
Seven unigenes that were described as or predicted to be PR proteins of Solanum species on the basis of their Nr description and that were expressed differentially after the plants were inoculated with V. dahliae were screened (Table 4). Among those unigenes, three were annotated as PR10 and were up-regulated in both stages, and two were annotated as PR5 and were down-regulated in stage 1 but up-regulated in stage 2. In addition, three PR1 genes that are involved in both the PHST (Table 1) and PPI pathways (Table 2) exhibited different expression trends.
TFs
TFs have been known to play significant roles in plant responses to biotic stresses as important regulatory proteins. In the present study, querying the unique transcripts of the DEGs resulted in the identification of 499 and 663 TFs in stage 1 and stage 2, respectively, and their classification into 57 and 63 families. The top 20 groups of TFs are listed in Fig. 5. Analysis of the TFs revealed that the groups and numbers of TFs were similar between the two stages, and the MYB, AP2-EREBP, Orphan, WRKY, bHLH and bZIP groups were the six largest groups in both stages.
Fig. 5.

Classification of TFs in stage 1 (A) and stage 2 (B). The top 20 groups of TFs are listed. Different colours represent different TF types. Because the different stage has different TF groups, the same TF type may have different colour in graph A and B (color figure online)
Validation of DEGs by real-time quantitative PCR (RT-qPCR)
To confirm the assembled sequences and the expression profiles of RNA-Seq, RT-qPCR was performed on a subset of 18 select DEGs (Fig. 6, Supplementary Table S4). These genes were differentially expressed in both stages and consisted of nine PHST genes, seven PPI genes and two phenylpropanoid biosynthesis genes (Supplementary Table S5). For all of the detected genes, the trends observed from the RT-qPCR expression were in agreement with those from the RNA-Seq data.
Fig. 6.

RT-qPCR validation of stage 1 (A) and stage 2 (B). Eighteen co-expressed DEGs were selected from the PHST, PPIand PB pathways. The Y-axis indicates the normalized expression patterns based on RNA-Seq and RT-qPCR, which are transformed by either the log2 FC and the mean relative quantities (RQ), respectively. Error bars indicate SD. The calculation of RQ were controlled by 0 hpi in stage 1 (S_12 h vs. S_0 h) and by 12 hpi in stage 2 (S_48 h vs. S_12 h)
Discussion
Physiological and biochemical studies on EVW have been conducted in several plants (Chen et al. 2015; Derksen et al. 2013; Faino et al. 2012), and some verticillium wilt resistance genes have been cloned (Chai et al. 2003; Diwan et al. 1999; Fradin et al. 2009). Nevertheless, the molecular mechanism of wild eggplant resistance to V. dahliae is still unclear; the disease resistance mechanism is complex, and there are several resistance genes involved. Moreover, previous studies on the eggplant defence response have not provided enough information to determine the complex transcriptomic dynamics. In recent years, the accessibility of high-throughput sequencing technologies has provided a good opportunity to thoroughly analyse the large-scale expression profile of the defence mechanism, despite uncharacterized genome sequences (Grabherr et al. 2011). In this study, we chose S. sisymbriifolium, which has proven to be a valuable resistance resource against verticillium wilt, as the subject of a transcriptome analysis, both to investigate its response process under stress from V. dahliae and to explore new resistance genes and resistance mechanisms for overcoming EVW. Considering the large differences between the cultivated and wild species of eggplant, the genomic sequence of eggplant was not used in this study despite being published.
Selection of time points
Signal perception, transduction and exchange in plant defense responses usually occur during the early periods of pathogen infection (Jones and Dangl 2006; Kunkel and Brooks 2002), especially within the first 12 hpi, which often corresponds to fungal spore germination and fungal penetration of plant epidermal cells (Fradin and Thomma 2006). Additionally, 48 hpi with V. dahliae has also been used as a key time point for RNA-Seq analysis in previous studies (Gayoso et al. 2010; Sun et al. 2013; Tan et al. 2015).
In the present study, measurements of physiological activity of the S. sisymbriifolium roots following infection with V. dahliae (Fig. 1) revealed that the maximum T-SOD and POD activities and SS content occurred at 12 hpi and that a second increase in both T-SOD and POD activities occurred at 48 hpi. GO enrichment analysis of the DEGs revealed that all six SOD-related unigenes were up-regulated in stage 1 but down-regulated in stage 2. Regarding POD-related genes, there were 25 up-regulated and 14 down-regulated unigenes in stage 1 but 48 up-regulated and 7 down-regulated unigenes in stage 2. The same trend was observed for unigenes related to glucose (54 up-regulated and 9 down-regulated unigenes in stage 1; 28 up-regulated and 56 down-regulated unigenes in stage 2) and fructose (35 up-regulated and 3 down-regulated unigenes in stage 1; 22 up-regulated and 36 down-regulated unigenes in stage 2), which are the main classes of SSs. This result coincided with the physiological measurement results of SOD, POD and the SSs. Those results indicated that 12 hpi and 48 hpi were the key inflection points during the interaction between S. sisymbriifolium and V. dahliae, reflecting the reliability and coherence of the physiological measurements and transcriptomic data.
Next, three time points (0, 12, and 48 hpi) were chosen and divided into two stages (S_12 h vs. S_0 h; S_48 h vs. S_12 h) that coincided with the crucial stages of fungal infection and the analysis of the basal gene expression patterns between the different time periods, according to the measurements of the physiological indexes. This setup enabled us to avoid any confounding factors, focus on the key time points, and accurately identify the critical genes from among the enormous amounts of RNA-Seq information.
Identification of DEGs during the S. sisymbriifolium response to V. dahliae
Comparison of S_12 h versus S_0 h (stage 1) revealed 6218 DEGs, including 2958 up-regulated unigenes whose mean log2 fold change (FC) was 4.79 and 3260 down-regulated unigenes whose mean log2 FC was − 4.24. For S_48 h versus S_12 h (stage 2), 5831 up-regulated unigenes whose mean log2 FC was 2.88 and 4571 down-regulated unigenes whose mean log2 FC was − 2.42 were revealed in this study. The results indicated that stage 2 has more DEGs than does in stage 1, especially up-regulated DEGs, but the change in amplitude of gene expression was larger in stage 1 than in stage 2. In addition, the hierarchical clustering of the DEGs showed that the expression patterns of the DEGs at 0 hpi and 48 hpi were more similar than those at 12 hpi (Fig. 3). These results indicate a major change at 12 hpi and the crucial role of that time point following inoculation of S. sisymbriifolium with V. dahliae; these results were consistent with those of the physiological experiments (Fig. 1). The results also indicated that negative regulation of the genes occurred promptly in the early stage but that positive regulation was pronounced in the later stage of the S. sisymbriifolium response to V. dahliae. Here, we predicted the important roles of the down-regulated genes during plant resistance to V. dahliae, and our predictions were consistent with those of Guo et al. (2017) in sunflower.
DEGs involved in the main pathways
In this study, both the GO and KEGG enrichment analyses of the DEGs in both stages indicated that many genes are involved in the three most important pathways (the PHST, PPI and PB pathways), which indicates that these processes play an important role in the response of S. sisymbriifolium to V. dahliae. These pathways have previously been studied with respect to the interaction between V. dahliae and cotton (Sun et al. 2013; Zhang et al. 2017), tomato (Tan et al. 2015) and sunflower (Guo et al. 2017), but this is the first time a comprehensive and detailed investigation has been conducted in eggplant.
DEGs involved in the PHST pathway
As signaling molecules, plant hormones are considered to play an important role in modulating signaling networks involved in plant responses to various biotic stresses (Bari 2009). For example, SA, which suppresses JA-mediated defences against necrotrophic signals, is activated in cotton upon V. dahliae infection (Gao et al. 2013; Pieterse et al. 2009, 2012). ET has complex functions in the response to verticillium wilt; for example, ET triggers induced systemic resistance (ISR) in plants (Pieterse et al. 1996; Wang et al. 2011), and the release of ET induces cotton susceptibility to verticillium wilt and can cause leaves to wilt (Beyer 1979; Veronese et al. 2003). AUX is involved in general transcriptional induction during the response of cultivated eggplant to V. dahliae infection (Yang et al. 2013). Several recent papers also have reported that ABA plays important roles in plant defence responses (Adie et al. 2007; de Wit 2007; Mohr and Cahill 2003).
In this study (Table 1), the largest group of ABA-related genes and the pathway ‘CRE1 → AHP → B-ARR → A-ARR → Stomatal closure’ were detected in both stages, suggesting an important role of ABA during the response of S. sisymbriifolium to V. dahliae. In contrast, two other important pathways, ‘SA → NPR1 → TGA → PR-1 → Disease resistance’ and ‘AUX1 → TIR1 → AUX/IAA → ARF → Cell enlargement/Plant growth’, of PHST were found in stage 2. These pathways were also reported in the transcriptome profiling of the defence responses of both S. aculeatissimum (Zhou et al. 2016) and cotton (Zhang et al. 2013) against V. dahliae, and the presence of these pathways might indicate that SA and AUX play important functions in resistance to V. dahliae.
Regarding the SA-mediated signal transduction pathway, NPR1 plays a key role in the induction of defence genes by activating the transcription factor TGA (Zhou et al. 2000; Johnson et al. 2003). In the present study, the NPR1 and TGA genes also showed a similar expression trend of down-regulation in stage 1 followed by up-regulation in stage 2, a pattern that was especially pronounced for the TGA genes. In addition, all of the ET genes were up-regulated in stage 1, but most of them were down-regulated in stage 2. In addition, only one JAR1 gene and two genes for MYC2 transcription factors in the JA-mediated signal transduction pathway were identified, all of which were down-regulated in stage 1 and up-regulated in stage 2. These results indicated that, after the plants were infected with the hemibiotrophic pathogen V. dahliae, SA-mediated defence was induced in S. sisymbriifolium, but the JA/ET signalling pathways were suppressed. SA-dependent responses are involved in responses to biotrophic or hemibiotrophic pathogens, whereas the JA/ET pathways are activated during resistance to necrotrophic pathogens (Glazebrook 2005; Pieterse et al. 2009).
In general, the majority of the PHST genes were involved in the biosynthesis of ABA, AUX, ET, CTK and SA, but their expression patterns varied. This variation indicated important roles for these hormones and their differential regulation during the interaction between S. sisymbriifolium and V. dahliae. Nevertheless, the mechanisms underlying the protective effects of these hormones remain largely unknown; the role of these hormones appears to be complex, and additional studies are still needed.
DEGs involved in the PPI pathway
The PPI pathway is one of the most important pathways during the plant defence response to pathogens, including V. dahliae. During the PPI process, PAMPs (or MAMPs) are recognized by plants through pattern recognition receptors (PRRs), resulting in PAMP-triggered immunity (PTI) that can halt further colonization as the first layer of the response (Brutus and He 2010). A second layer, effector-triggered immunity (ETI), specifically recognizes the effector; this recognition occurs via NB-LRR proteins (Nürnberger et al. 2004; Zhang and Zhou 2010). ETI is an accelerated response, while PTI is amplified, resulting in disease resistance and usually a hypersensitive response (HR) at the infection site (Dodds and Rathjen 2010; Jones and Dangl 2006).
In the present study, the majority of genes previously reported to be involved in the PPI pathway during plant and V. dahliae interactions (Ekengren et al. 2003; Guo et al. 2017; Zhang et al. 2017) were obtained in our library (Table 2). These genes include the PRR proteins EIX receptor 1/2 (EIX1_2), chitin elicitor receptor kinase 1 (CERK1), and LRR receptor-like serine/threonine-protein kinase (FLS2)(Eulgem and Somssich,2007), and plant cyclic nucleotide-gated channels (CNGCs), as well as subfamilies of Ca2+ sensors (CaM, CML and CDPK) (Boudsocq et al. 2010). RIN4 and serine/threonine-protein PBS1, which contribute to V. dahliae resistance in sunflower (Guo et al. 2017) and cotton (Zhang et al. 2016a, b), were also detected in this study. In addition, the HR during the interaction between S. sisymbriifolium and V. dahliae was activated via Pti1, SGT1 and HSP90B and was mediated by RBOH and NOS (Supplementary Fig. S5), which is consistent with studies on sunflower (Guo et al. 2017) and cotton (Zhang et al. 2013, 2017). Moreover, the two disease-resistance proteins RPM1 and RPS2 were only detected in stage 2, during which they were down-regulated. Three pathogenesis-related genes, the transcriptional activators Pti1, Pti4 and Pti5, were also identified and showed the same expression trend during the interaction between S. sisymbriifolium and V. dahliae.
In addition, MAPK cascades are associated with resistance to V. dahliae (Asai et al. 2002; Tena et al. 2001), and mitogen-activated protein kinase kinase kinase 1 (MEKK1) in both stages and mitogen-activated protein kinase kinase 1 (MAP2K1) in stage 2 were identified in the present study.
Furthermore, WRKY22 and WRKY33, which are linked to MAPK cascades, activated the expression of defence-related genes in cotton (Zhang et al. 2016a, b; 2017) and they were also shown to be involved in the PPI pathway in the present study. Among those TFs, WRKY22 expression was specifically detected and increased in stage 2. In contrast, three WRKY33-related genes were down-regulated in both stages, indicating negative regulation of those genes during interactions between S. sisymbriifolium and V. dahliae.
DEGs involved in the PB pathway
Phenylpropanoids function as pre-formed and inducible antimicrobial compounds during plant–microbe interactions (Naoumkina et al. 2010). The phenylpropanoid pathway plays a critical role in the plant defence response to V. dahliae (Gayoso et al. 2010; Sun et al. 2013; Xu et al. 2011; Zhang et al. 2013). Important enzymes of the PB pathway, such as PAL, POD, CAD, 4CL, COMT, HCT and CCoAOMT, have been characterized in plants following infection with V. dahliae (Gayoso et al. 2010; Pomar et al. 2004; Sun et al. 2013; Xu et al. 2011).
In the present study (Table 3), all of the abovementioned genes were detected, and the majority of them were up-regulated after the plants were inoculated with V. dahliae. It also has been reported that hundreds of cytochrome P450 (CYP) genes (Sun et al. 2014) and CCoAOMT enzymes (Schilmiller et al. 2009) are involved in the PB pathway. The present study identified CYP73A (C4H), CYP98A (C3′H) and CYP84A (F5H), which are members of the CYP gene family, as differentially regulated, and the genes encoding CYP73A and CCoAOMT were up-regulated in both stages, indicating their important functions during interactions between S. sisymbriifolium and V. dahliae.
As reported previously, HCT and 4CL can affect lignification in cotton (Sun et al. 2013) and alfalfa (Shadle et al. 2007), and lignin plays a central role in the cotton defence response (Xu et al. 2011). In this study, all of the HCT and 4CL genes demonstrated increased expression in stage 2, so the same function may exist in eggplant. PAL, which functions as a catalyst for phenylpropanoid metabolism, is the first enzyme in the PB pathway. Several studies have reported increased PAL expression and activity in response to V. dahliae (Gayoso et al. 2010; Xu et al. 2011). In the present study, two PAL genes were detected, and their expression increased in stage 2, indicating their positive regulation during the S. sisymbriifolium response to V. dahliae. Peroxidases are also involved in the polymerization of monolignols into lignin and in the reinforcement of plant cell wall after pathogen infection or wounding (Marjamaa et al. 2009). In the present study, PODs were the largest group in the PB pathway, and the expression of POD-related genes changed markedly, especially in stage 1. These results coincided with those of the physiological index measurements, indicating their important role in the response of eggplant to V. dahliae.
Ve genes and PR protein-encoding genes
The only locus responsible for effective resistance to race 1 of V. dahliae, Ve 1, has been isolated and cloned from tomato (S. lycopersicum) (Fradin et al. 2009; Kawchuk et al. 2001). The Ve locus comprises two genes, Ve1 and Ve2, both of which encode cell surface receptors that are members of the extracellular leucine-rich repeat class of receptor-like proteins (eLRR-RLPs) (Kawchuk et al. 2001). In the present study (Table 4), two unigenes (Cluster-33922.87359 and Cluster-33922.112117) shared high identity with the S. torvum genes StVe1 and StVe2. This result indicated that these unigenes were Ve homologues. Although both Ve1 and Ve2 provide resistance when expressed in close relatives of potato (Gayoso et al. 2010; Kawchuk et al. 2001), only Ve1 provides resistance in tomato (Fradin et al. 2009). Whether a single Ve gene (and which one) or both dominate the resistance of eggplant to V. dahliae remains unclear.
PR proteins also play important roles in the defence reaction against verticillium wilt (Wang et al. 2011; Zhang et al. 2016a, b). Several PR proteins, such as PR1, PR2, PR5 and PR10, have a wide range of roles, specifically in biotic and abiotic stresses (Agarwal and Agarwal 2014; van Ooijen et al. 2007; van der Hoorn and Kamoun 2008). In the present study (Table 4), PR1-, PR5- and PR10-related genes were detected and expressed in different patterns, which is consistent with the results of a study on S. aculeatissimum (Zhou et al. 2016). These findings probably indicate that these PR proteins are critical during interactions between eggplant and V. dahliae.
TFs
TFs act as master switches. By slight changes in their expression, TFs control the expression of a set of genes to regulate different aspects of plant development or responses to biotic and abiotic stresses (Singh et al. 2002). It is reported that both of basal disease resistance and gene-for-gene resistance are mediated by several classes of TFs, such as AP2-EREBPE (Gutterson and Reuber 2004) and bZIP (Scholz et al. 2018). Many WRKY family TFs have also been reported to play important roles in mediating plant responses to stress factors (Yang et al. 2012; Zhang et al. 2017). Moreover, bHLH family TFs often interact with MYB family proteins to form complexes that then regulate the downstream expression of target genes (Zhang et al. 2013).
In the present study, in both stages (Fig. 6a, b), the six largest groups of TFs were the MYB, AP2-EREBP, Orphan, WRKY, bHLH and bZIP groups, indicating the significance of their roles during the response of S. sisymbriifolium to V. dahliae. The results of the transcriptome analysis revealed that the majority of TFs were suppressed after the plants were inoculated with the pathogen. This finding hinted that the negative regulation of some TFs to modulate the plant immune signal network might be required to set up the eggplant resistance to V. dahliae, as has been previously reported in cotton (Xu et al. 2011). Further study of the regulatory pattern of TFs during the response of S. sisymbriifolium to V. dahliae is needed.
Conclusions
This study provided profiles of the transcripts associated with the interaction between the resistant S. sisymbriifolium genotype and the highly toxic pathogen V. dahliae. The results revealed putative genes involved in the PHST pathway, the PPI pathway, and PB pathway and several crucial genes, including Ve genes, PR genes and WRKY genes, that regulate crucial points in the defence-related pathway and that may contribute to V. dahliae resistance in eggplant.
These findings could help us get a deeper understanding of the molecular interactions between eggplant and V. dahlia, lay foundations for solanaceae resistance breeding, and provide new insight into the interactions between plants and pathogens. Nevertheless, although some defence-related genes from S. sisymbriifolium were perceived, it is probably that extra biosynthetic pathways and more complicated signalling mechanisms in the roots are included in this defence system. Additional studies should be implemented to confirm the functions of the selected candidate DEGs involved in the present study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (31460512) and the Key Research and Development Plan of Yunnan Province (2018BB020).
Author contributions
L.Y.W. and G.H.D. performed the experiments, analysed the data, and drafted the manuscript. R.B. planted and managed the materials in the field. Z.B.L. inoculated the seedlings and helped with sampling. F.Y.L. and Y.J.G. conceived the study, participated in its design and coordination, and helped draft the manuscript. All authors read and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests, including financial and non-financial interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Liyan Wu and Guanghui Du have contributed equally to this work.
Contributor Information
Yaju Gong, Phone: 008613518738006, Email: gongyaju@sina.com.
Feihu Liu, Phone: 008613678709753, Email: dmzpynu@126.com.
References
- Adie BAT, Pérez-Pérez J, Pérez-Pérez MM, Godoy M, Sánchez-Serrano JJ, Schmelz EA, Solano R. ABA is an essential signal for plant resistance to pathogens affecting JA biosynthesis and the activation of defenses in Arabidopsis. Plant Cell. 2007;19:1665–1681. doi: 10.1105/tpc.106.048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P, Agarwal PK. Pathogenesis related-10 proteins are small, structurally similar but with diverse role in stress signaling. Mol Biol Rep. 2014;41:599–611. doi: 10.1007/s11033-013-2897-4. [DOI] [PubMed] [Google Scholar]
- Asai T, Tena G, Plotnikova J, Willmann MR, Chiu WL, Gomez-Gomez L, Boller T, Ausubel FM, Sheen J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature. 2002;415:977. doi: 10.1038/415977a. [DOI] [PubMed] [Google Scholar]
- Bari R. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69:473–488. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- Beyer EM. [14C]Ethylene metabolism during leaf abscission in cotton. Plant Physiol. 1979;64:971–974. doi: 10.1104/pp.64.6.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RG, Subbarao KV. Host range specificity in Verticillium dahliae. Phytopathology. 1999;89:1218–1225. doi: 10.1094/PHYTO.1999.89.12.1218. [DOI] [PubMed] [Google Scholar]
- Bletsos F, Thanassoulopoulos C, Roupakias D. Effect of grafting on growth, yield, and Verticillium wilt of eggplant. HortScience. 2003;38:183–186. doi: 10.21273/HORTSCI.38.2.183. [DOI] [Google Scholar]
- Borua PK. Failure in an interspecific cross between Solanum khasianum Clarke and Solanum mammosum L. Euphytica. 1990;46:1–6. doi: 10.1007/BF00057613. [DOI] [Google Scholar]
- Boudsocq M, Willmann MR, McCormack M, Lee H, Shan L, He P, Bush J, Cheng SH, Sheen J. Differential innate immune signalling via Ca2+ sensor protein kinases. Nature. 2010;464:418. doi: 10.1038/nature08794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutus A, He SY. Broad-spectrum defense against plant pathogens. Nat Biotechnol. 2010;28:330–331. doi: 10.1038/nbt0410-330. [DOI] [PubMed] [Google Scholar]
- Chai Y, Zhao L, Liao Z, Sun X, Zuo K, Zhang L, Wang S, Tang K. Molecular cloning of a potential Verticillium dahliae resistance gene SlVe1 with multi-site polyadenylation from Solanum licopersicoides. DNA Seq. 2003;14:375–384. doi: 10.1080/10425170310001605509. [DOI] [PubMed] [Google Scholar]
- Chen JY, Huang JQ, Li NY, Ma XF, Wang JL, Liu C, Liu YF, Liang Y, Bao YM, Dai XF. Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response to Verticillium wilt. BMC Plant Biol. 2015;15:148. doi: 10.1186/s12870-015-0508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collonnier C, Fock I, Kashyap V, Rotino GL, Daunay MC, Lian Y, Mariska IK, Rajam MV, Servaes A, Ducreux G, Sihachakr D. Applications of biotechnology in eggplant. Plant Cell Tissue Organ Cult. 2001;65:91–107. doi: 10.1023/A:1010674425536. [DOI] [Google Scholar]
- de Wit PJ. How plants recognize pathogens and defend themselves. Cell Mol Life Sci. 2007;64:2726–2732. doi: 10.1007/s00018-007-7284-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derksen H, Badawi M, Henriquez MA, Yao Z, El-Bebany AF, Daayf F. Differential expression of potato defence genes associated with the salicylic acid defence signalling pathway in response to weakly and highly aggressive isolates of Verticillium dahliae. J Phytopathol. 2013;161:142–153. doi: 10.1111/jph.12038. [DOI] [Google Scholar]
- Diwan N, Fluhr R, Eshed Y, Zamir D, Tanksley SD. Mapping of Ve in tomato: a gene conferring resistance to the broad-spectrum pathogen, Verticillium dahliae race 1. Theor Appl Genet. 1999;98:315–319. doi: 10.1007/s001220051075. [DOI] [Google Scholar]
- Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant–pathogen interactions. Nat Rev Genet. 2010;11:539. doi: 10.1038/nrg2812. [DOI] [PubMed] [Google Scholar]
- Ekengren SK, Liu Y, Schiff M, Dinesh-Kumar SP, Martin GB. Two MAPK cascades, NPR1, and TGA transcription factors play a role in Pto-mediated disease resistance in tomato. Plant J. 2003;36:905–917. doi: 10.1046/j.1365-313X.2003.01944.x. [DOI] [PubMed] [Google Scholar]
- Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10:366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Faino L, de Jonge R, Thomma BP. The transcriptome of Verticillium dahliae-infected Nicotiana benthamiana determined by deep RNA sequencing. Plant Signal Behav. 2012;7:1065–1069. doi: 10.4161/psb.21014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Chai Y, Wang J, Lin J, Sun X, Sun C, Zuo K, Tang K. cDNA cloning and characterization of the Ve homologue gene StVe from Solanum torvum Swartz. DNA Seq. 2004;15:88–95. doi: 10.1080/1042517042000199942. [DOI] [PubMed] [Google Scholar]
- Fradin EF, Thomma BPHJ. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol. 2006;7:71–86. doi: 10.1111/j.1364-3703.2006.00323.x. [DOI] [PubMed] [Google Scholar]
- Fradin EF, Zhang Z, Juarez Ayala JC, Castroverde CDM, Nazar RN, Robb J, Liu CM, Thomma BPHJ. Genetic dissection of Verticillium Wilt resistance mediated by tomato Ve1. Plant Physiol. 2009;150:320–332. doi: 10.1104/pp.109.136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Long L, Zhu LF, Xu L, Gao WH, Sun LQ, Liu LL, Zhang XL. Proteomic and virus-induced gene silencing (VIGS) analyses reveal that gossypol, brassinosteroids, and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae. Mol Cell Proteom. 2013;12:3690–3703. doi: 10.1074/mcp.M113.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso C, Pomar F, Novo-Uzal E, Merino F, de Ilarduya OM. The Ve-mediated resistance response of the tomato to Verticillium dahliae involves H2O2, peroxidase and lignins and drives PAL gene expression. BMC Plant Biol. 2010;10:232. doi: 10.1186/1471-2229-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005;43:205–227. doi: 10.1146/annurev.phyto.43.040204.135923. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramazio P, Blanca J, Ziarsolo P, Herraiz FJ, Plazas M, Prohens J, Vilanova S. Transcriptome analysis and molecular marker discovery in Solanum incanum and S. aethiopicum, two close relatives of the common eggplant (Solanum melongena) with interest for breeding. BMC Genom. 2016;17:300. doi: 10.1186/s12864-016-2631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zuo Y, Zhang Y, Wu C, Su W, Jin W, Yu H, An Y, Li Q. Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMC Genom. 2017;18:42. doi: 10.1186/s12864-016-3386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutterson N, Reuber TL. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr Opin Plant Biol. 2004;7:465–471. doi: 10.1016/j.pbi.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Johnson C, Boden E, Arias J. Salicylic acid and NPR1 induce the recruitment of trans-activating TGA factors to a defense gene promoter in Arabidopsis. Plant Cell. 2003;15:1846–1858. doi: 10.1105/tpc.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucl Acids Res. 2008;36:480–484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawchuk LM, Hachey J, Lynch DR, Kulcsar F, van Rooijen G, Waterer DR, Robertson A, Kokko E, Byers R, Howard RJ, Fischer R, Prufer D. Tomato Ve disease resistance genes encode cell surface-like receptors. Proc Natl Acad Sci. 2001;98:6511–6515. doi: 10.1073/pnas.091114198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel BN, Brooks DM. Cross talk between signaling pathways in pathogen defense. Curr Opin Plant Biol. 2002;5:325–331. doi: 10.1016/S1369-5266(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Zheng Z, Zhou X, Feng C, Zhuang Y. Improving the resistance of eggplant (Solanum melongena) to Verticillium wilt using wild species Solanum linnaeanum. Euphytica. 2015;201:463–469. doi: 10.1007/s10681-014-1234-x. [DOI] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- Marjamaa K, Kukkola EM, Fagerstedt KV. The role of xylem class III peroxidases in lignification. J Exp Bot. 2009;60:367–376. doi: 10.1093/jxb/ern278. [DOI] [PubMed] [Google Scholar]
- Mohr PG, Cahill DM. Abscisic acid influences the susceptibility of Arabidopsis thaliana to Pseudomonas syringae pv. tomato and Peronospora parasitica. Funct Plant Biol. 2003;30:461–469. doi: 10.1071/FP02231. [DOI] [PubMed] [Google Scholar]
- Naoumkina MA, Zhao Q, Gallego-Giraldo L, Dai X, Zhao PX, Dixon RA. Genome-wide analysis of phenylpropanoid defence pathways. Mol Plant Pathol. 2010;11:829–846. doi: 10.1111/j.1364-3703.2010.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nürnberger T, Brunner F, Kemmerling B, Piater L. Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev. 2004;198:249–266. doi: 10.1111/j.0105-2896.2004.0119.x. [DOI] [PubMed] [Google Scholar]
- Pantelides IS, Tjamos SE, Paplomatas EJ. Ethylene perception via ETR1 is required in Arabidopsis infection by Verticillium dahliae. Mol Plant Pathol. 2010;11:191–202. doi: 10.1111/j.1364-3703.2009.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;5:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- Pieterse CMJ, van Wees SCM, Hoffland E, van Pelt JA, van Loon LC. Systemic resistance in Arabidopsis induced by biocontrol bacteria is independent of salicylic acid accumulation and pathogenesis-related gene expression. Plant Cell. 1996;8:1225–1237. doi: 10.1105/tpc.8.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterse CMJ, Leon-Reyes A, van der Ent S, van Wees SCM. Networking by small-molecule hormones in plant immunity. Nat Chem Biol. 2009;5:308. doi: 10.1038/nchembio.164. [DOI] [PubMed] [Google Scholar]
- Pieterse CMJ, Leon-Reyes HA, van der Does D, Verhage A, Koornneef A, van Pelt JA, van Wees SCM. Networking by small-molecule hormones in plant immunity. IOBC-WPRS Bull. 2012;83:77–80. [Google Scholar]
- Plazas M, Vilanova S, Gramazio P, Rodriguez-Burruezo A, Fita A, Herraiz F, Ranil R, Fonseka R, Niran L, Fonseka H, Kouassi B, Kouassi A, Kouassi A, Prohens J. Interspecific hybridization between eggplant and wild relatives from different genepools. J Am Soc Hortic Sci. 2016;141:34–44. doi: 10.21273/JASHS.141.1.34. [DOI] [Google Scholar]
- Polignano G, Uggenti P, Bisignano V, Gatta CD. Genetic divergence analysis in eggplant (Solanum melongena L.) and allied species. Genet Resour Crop Evol. 2010;57:171–181. doi: 10.1007/s10722-009-9459-6. [DOI] [Google Scholar]
- Pomar F, Novo M, Bernal MA, Merino F, Barceló AR. Changes in stem lignins (monomer composition and crosslinking) and peroxidase are related with the maintenance of leaf photosynthetic integrity during Verticillium wilt in Capsicum annuum. New Phytol. 2004;163:111–123. doi: 10.1111/j.1469-8137.2004.01092.x. [DOI] [PubMed] [Google Scholar]
- Schilmiller AL, Stout J, Weng JK, Humphreys J, Ruegger MO, Chapple C. Mutations in the cinnamate 4-hydroxylase gene impact metabolism, growth and development in Arabidopsis. Plant J. 2009;60:771–782. doi: 10.1111/j.1365-313X.2009.03996.x. [DOI] [PubMed] [Google Scholar]
- Scholz SS, Schmidt-Heck W, Guthke R, Furch ACU, Reichelt M, Gershenzon J, Oelmüller R. Verticillium dahliae-Arabidopsis interaction causes changes in gene expression profiles and jasmonate levels on different time scales. Front Microbiol. 2018;9:217. doi: 10.3389/fmicb.2018.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadle G, Chen F, Reddy MSS, Jackson L, Nakashima J, Dixon RA. Down-regulation of hydroxycinnamoyl CoA: shikimate hydroxycinnamoyl transferase in transgenic alfalfa affects lignification, development and forage quality. Phytochemistry. 2007;68:1521–1529. doi: 10.1016/j.phytochem.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Singh KB, Foley RC, Oñate-Sánchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5:430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- Sun Q, Jiang H, Zhu X, Wang W, He X, Shi Y, Yuan Y, Du X, Cai Y. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 2013;14:852. doi: 10.1186/1471-2164-14-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zhu L, Xu L, Yuan D, Min L, Zhang X. Cotton cytochrome P450 CYP82D regulates systemic cell death by modulating the octadecanoid pathway. Nat Commun. 2014;5:5372. doi: 10.1038/ncomms6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Murata Y, Mukaihara T. A somatic hybrid between Solanum integrifolium and Solanum violaceum that is resistant to bacterial wilt caused by Ralstonia solanacearum. Plant Cell Rep. 2002;21:353–358. doi: 10.1007/s00299-002-0524-2. [DOI] [Google Scholar]
- Tan G, Liu K, Kang J, Xu K, Zhang Y, Hu L, Zhang J, Li C. Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-sequencing. Front Plant Sci. 2015;6:428. doi: 10.3389/fpls.2015.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tena G, Asai T, Chiu WL, Sheen J. Plant mitogen-activated protein kinase signaling cascades. Curr Opin Plant Biol. 2001;4:392–400. doi: 10.1016/S1369-5266(00)00191-6. [DOI] [PubMed] [Google Scholar]
- Toppino L, Valè G, Rotino GL. Inheritance of Fusarium wilt resistance introgressed from Solanum aethiopicum Gilo and Aculeatum groups into cultivated eggplant (S. melongena) and development of associated PCR-based markers. Mol Breed. 2008;22:237–250. doi: 10.1007/s11032-008-9170-x. [DOI] [Google Scholar]
- van der Hoorn RA, Kamoun S. From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell. 2008;20:2009–2017. doi: 10.1105/tpc.108.060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooijen G, van den Burg HA, Cornelissen BJC, Takken FLW. Structure and function of resistance proteins in solanaceous plants. Annu Rev Phytopathol. 2007;45:43–72. doi: 10.1146/annurev.phyto.45.062806.094430. [DOI] [PubMed] [Google Scholar]
- Veronese P, Narasimhan ML, Stevenson RA, Zhu JK, Weller SC, Subbarao KV, Bressan RA. Identification of a locus controlling Verticillium disease symptom response in Arabidopsis thaliana. Plant J. 2003;35:574–587. doi: 10.1046/j.1365-313X.2003.01830.x. [DOI] [PubMed] [Google Scholar]
- Wang FX, Ma YP, Yang CL, Zhao PM, Yao Y, Jian GL, Luo YM, Xia GX. Proteomic analysis of the sea-island cotton roots infected by wilt pathogen Verticillium dahliae. Proteomics. 2011;11:4296–4309. doi: 10.1002/pmic.201100062. [DOI] [PubMed] [Google Scholar]
- Wu LY, Guo ZX, Zeng L, Bao R, Li ZB, Gong YJ. Resistance identification of yunnan wild eggplant resources to verticillium wilt. J Plant Genet Res. 2017;6:1046–1054. [Google Scholar]
- Xu L, Zhu L, Tu L, Liu L, Yuan D, Jin L, Long L, Zhang X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J Exp Bot. 2011;62:5607–5621. doi: 10.1093/jxb/err245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadeta KA, Valkenburg DJ, Hanemian M, Marco Y, Thomma BPHJ. The brassicaceae-specific EWR1 gene provides resistance to vascular wilt pathogens. PLoS ONE. 2014;9:e88230. doi: 10.1371/journal.pone.0088230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CQ, Fang X, Wu XM, Mao YB, Wang LJ, Chen XY. Transcriptional regulation of plant secondary metabolism. J Integr Plant Biol. 2012;54:703–712. doi: 10.1111/j.1744-7909.2012.01161.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Jue D, Li W, Zhang R, Chen M, Yang Q. Identification of MiRNA from eggplant (Solanum melongena L.) by small RNA deep sequencing and their response to Verticillium dahliae infection. PLoS ONE. 2013;8:e72840. doi: 10.1371/journal.pone.0072840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cheng YF, Deng C, Ma Y, Wang ZW, Chen XH, Xue LB. Comparative transcriptome analysis of eggplant (Solanum melongena L.) and turkey berry (Solanum torvum Sw.): phylogenomics and disease resistance analysis. BMC Genom. 2014;15:412. doi: 10.1186/1471-2164-15-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mu X, Liu C, Cai J, Shi K, Zhu W, Yang Q. Overexpression of potato miR482e enhanced plant sensitivity to Verticillium dahliae infection. J Integr Plant Biol. 2015;57:1078–1088. doi: 10.1111/jipb.12348. [DOI] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou JM. Plant immunity triggered by microbial molecular signatures. Mol Plant. 2010;3:783–793. doi: 10.1093/mp/ssq035. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Jiang TF, Cui X, Qi FJ, Jian GL. Colonization in cotton plants by a green fluorescent protein labelled strain of Verticillium dahliae. Eur J Plant Pathol. 2012;135:867–876. doi: 10.1007/s10658-012-0131-1. [DOI] [Google Scholar]
- Zhang Y, Wang XF, Ding ZG, Ma Q, Zhang GR, Zhang SL, Li ZK, Wu LQ, Zhang GY, Ma ZY. Transcriptome profiling of Gossypium barbadense inoculated with Verticillium dahliae provides a resource for cotton improvement. BMC Genom. 2013;14:637. doi: 10.1186/1471-2164-14-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang W, Jian G, Qi F, Si N. The genes involved in the protective effects of phytohormones in response to Verticillium dahliae infection in Gossypium hirsutum. J Plant Biolo. 2016;59:194–202. doi: 10.1007/s12374-016-0568-4. [DOI] [Google Scholar]
- Zhang W, Zhang H, Qi F, Jian G. Generation of transcriptome profiling and gene functional analysis in Gossypium hirsutum upon Verticillium dahliae infection. Biochem Biophys Res Commun. 2016;473:879–888. doi: 10.1016/j.bbrc.2016.03.143. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang H, Liu K, Jian G, Qi F, Si N. Large-scale identification of Gossypium hirsutum genes associated with Verticillium dahliae by comparative transcriptomic and reverse genetics analysis. PLoS ONE. 2017;12:e0181609. doi: 10.1371/journal.pone.0181609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JM, Trifa Y, Silva H, Pontier D, Lam E, Shah J, Klessig DF. NPR1 differentially interacts with members of the TGA/OBF family of transcription factors that bind an element of the PR-1 gene required for induction by salicylic acid. Mol Plant Microbe Interact. 2000;13:191–202. doi: 10.1094/MPMI.2000.13.2.191. [DOI] [PubMed] [Google Scholar]
- Zhou X, Bao S, Liu J, Zhuang Y. De novo sequencing and analysis of the transcriptome of the wild eggplant species Solanum Aculeatissimum in response to Verticillium dahliae. Plant Mol Biol Report. 2016;34:1193–1203. doi: 10.1007/s11105-016-0998-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.