

Artificial intelligence (AI) has been described as the machine for the fourth industrial revolution. Without exception, AI is predicted to change the face of every industry. In the field of drug development, AI is employed to enhance efficiency. Pharmacometrics and systems pharmacology play a vital role in the drug‐development decision process. Thus, it is important to recognize and embrace the efficiencies that AI can bring to the pharmacometrics community.
The Dartmouth summer research project (1955) of AI proceeded on the basis that every aspect of learning or any other feature of intelligence can be described so precisely that a machine can be created to simulate it.1 Deep learning (DL) and machine learning (ML) algorithms constitute the essential building blocks of AI systems. DL represents learning using multiple processing layers, whereas ML techniques use learning algorithms that self‐learn and improve efficiency for a period of time.2 There have already been significant contributions of AI in medicine, and some notable examples include the diagnosis of tuberculosis,3 metastasis of breast cancer4 and retinal changes as a result of diabetes.5
AI/DL/ML methods have made their way into a handful of areas in drug development. Some of these inroads are described later and include pharmacometric modeling and drug repurposing. We believe that these methods, which have proven valuable in other fields, have the promise to make important contributions to a wide range of drug‐development disciplines. This commentary describes some of the applications of AI/DL/ML in this space and calls for the examination of these methods in other areas.
ML Methods for Pharmacometrics
Model selection in pharmacometrics is often described as a linear process, starting with “structural” features and followed by random effects and covariate effects, each tested one at a time. In the optimization field, this is known as a “greedy” or local search algorithm. Similar to other greedy algorithms (such as the quasi‐Newton used for parameter optimization), this algorithm is at risk for arriving at local minima. A global search method called Genetic Algorithm (GA) can be proposed as a better alternative. GA creates a user‐defined “search space” of candidate models representing all hypotheses to be tested (e.g., number of compartments, covariates, random effects). This set of possible hypotheses are then coded into a “genome” consisting of a string of 0s and 1s, with genes representing each hypothesis (Figure 1). GA then searches this space for the optimal combination of “features.” GA uses the mathematics of “survival of the fittest” with mutation and cross‐over for the optimal combination of “features” (e.g., compartment, random effect, covariate effects, initial parameter estimates) based on a user‐provided function that describes the quality of the model, typically based on log‐likelihood and a parsimony penalty. This user‐provided function is known as the “fitness function” in GA. A prototype software has been developed to implement GA using NONMEM for the parameter estimation.6 Initial experience suggests that GA consistently finds a better model (based on Akaike information criteria and a likelihood ratio test) than human pharmacometricians.7, 8 Such an approach with a global search algorithm offers an objective and robust method for identifying optimal pharmacometric models.
Figure 1.
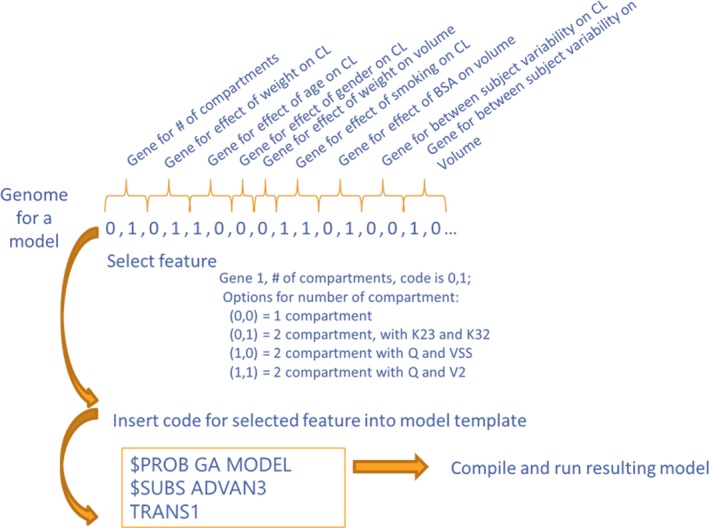
Coding of model options into a genome using Genetic Algorithm. CL‐ Clearance, BSA‐ Body surface area, K23 and K32‐ micro rate constants, Q‐ intercompartmental clearance, VSS‐ steady‐state volume of distribution, V2‐volume of distribution
DL for Drug Repurposing Efforts
Hit identification is the first and a crucial step in identifying a drug against a biological target of interest. Excelra has developed a multilayered AI‐driven platform aimed at identifying novel chemical hits (Figure S1). This platform screens drug candidates by passing them through a sequential process of filtration using ML and DL techniques integrated with chemo‐informatics. The filtered candidates are then passed through the target‐based data points to define potential drug candidates. Below are several key components required to build such AI/ML models:
An appropriate data set (including active and inactive compounds)
An optimized set of target specific descriptors
DL and ML algorithms
Perfect match of algorithms and parameters
Unbiased validation set to authenticate model performance
An optimized set of normalized chemical features (descriptors) represents the crucial component for building ML and DL models. Five different statistical algorithms were used to reduce noise and obtain a set of ~100 descriptors with no/fewer outliers. This information was subsequently used to construct ML (using ~30 algorithms) and DL models with their hyper‐parameterization, entailing an equal ratio of active (highly potent) and inactive compounds obtained from proprietary and public databases. Of the 150 models created for each target, 5 different reduction methods and more than 30 algorithms were employed to generate a performance score that was used to identify the “best model.” In addition, the multilayer ML and DL methods were integrated with traditional chemo‐informatics and molecular‐docking approaches to yield the best possible results. Finally, at the end of the pipeline, human intelligence is coupled with AI to achieve the best outcome using this multipronged approach. All models were validated using well‐curated proprietary (GOSTAR) and public (ChEMBL and PubChem) libraries. For example, the pipeline for Bruton's tyrosine kinase was built using this approach. A total of 52 randomly selected compounds were passed through the pipeline to assess its efficiency in differentiating between actives and inactives. These compounds were subsequently tested in vitro using a specific Bruton's tyrosine kinase activity assay. The pipeline was able to predict actives and inactives with an accuracy of 79%, with 80% sensitivity and 78% specificity (Table S1). Similarly, the models were built for two other targets with an accuracy ranging between 68%–87% (data not shown).
Accelerating Therapeutics for Opportunities in Medicine (ATOM)—A Multidisciplinary Effort for AI/ML/DL–Based Accelerated Drug Development
The convergence of recent advances in data science and computing is primed for applications in the pharmaceutical industry, and a multidisciplinary approach to integrating these fields will enable the rapid acceleration of drug development through applications of AI. Although pharmaceutical companies and clinical centers are transforming their data infrastructure to enable more value to be derived from the information collected, there is also an incredible opportunity to deliver more value to patients by sharing data, models, and cross‐industry expertise in AI.
ATOM is a public–private consortium formed to transform preclinical drug discovery into a rapid, parallel, patient‐centric model through an in silico–first, precompetitive platform. The founding members of the consortium are the Department of Energy's Lawrence Livermore National Laboratory, GlaxoSmithKline, the National Cancer Institute's Fredrick National Laboratory for Cancer Research, and the University of California, San Francisco. The consortium is open to other partners and is actively seeking new members.
ATOM aims to develop, test, and validate a multidisciplinary approach to drug discovery in which modern science, technology and engineering, supercomputing simulations, data science, and AI are highly integrated into a single drug‐discovery platform that can ultimately be shared with the drug‐development community at large. The ATOM platform integrates an ensemble of algorithms, built on shared public and private data, to simultaneously evaluate candidate drug molecules for efficacy, safety, pharmacokinetics, and developability. The current data set includes information on 2 million compounds from GlaxoSmithKline's drug discovery and development programs and screening collections. To date, ATOM has built thousands of models on that data, with an initial focus on pharmacokinetics and safety parameters that feed into human physiologically based pharmacokinetics and systems toxicology models. The ATOM active learning workflow, highlighted in Figure 2, will drive the acquisition of additional experimental and computational data where needed to improve prediction performance. Ultimately, this integrated approach is expected to lower attrition, reduce unproductive preclinical experimentation, and improve clinical translation that will result in better patient outcomes.
Figure 2.
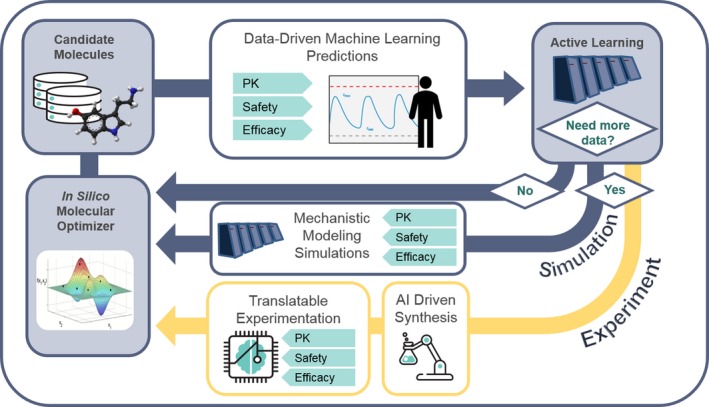
Multidisciplinary approach to the convergence of data science, drug discovery efforts, and translational modeling at the ATOM‐Accelerating Therapeutics for Opportunities in Medicine Consortium. PK, pharmacokinetics.
ML‐Based Methods in Regulatory Review
Applications of AI/DL/ML in the biomedical field are not limited to academia or industry. There are examples in which the US Food and Drug Administration has accelerated the approval of AI‐based devices and algorithms for diagnostic purposes, e.g., detecting diabetes‐related retinopathy and wrist fractures.9 The potential for the application in regulatory reviews is also being explored.10 ML could serve as a powerful tool for pharmacometrics analysis with its capacity to leverage high‐dimensional data and describe nonlinear relationships. This was illustrated by a simulation case study employing ML‐based techniques in the exposure–response analysis.
The case study was based on a simulation system in which both drug clearance and treatment outcome were described by highly nonlinear functions. In addition, drug clearance was designed to be independently associated with treatment response via confounders. The objective of this simulation was to assess whether ML‐based techniques were able to estimate the causal relationships between drug exposure and treatment outcome, without bias, when data from only one dose level were available.
Two analysis strategies involving ML were evaluated. Strategy A was based on a marginal structural model with inverse probability weighting, in which ML was employed to improve model robustness. In strategy A, pseudo‐subjects were grouped into five quintiles based on individual exposure; the propensity scores for each subject were estimated. Here, the propensity score represents the probability of the subject being assigned to their observed exposure quintile given a set of covariates. An unbiased propensity score is required to correctly generate the inverse probability weighting and assess the causal effects of exposure in marginal structural models. The simulation showed that, in a nonlinear system, ML proved more robust than traditional multinomial logistic regression in estimating the propensity score, thus correctly recovering the exposure–response relationship. Strategy B estimated the exposure–response relationship by employing an artificial neural network as a universal function approximator to recover the data‐generating mechanism without the requirement of accurately hand‐crafting the whole simulation system. A fully connected “feed forward” network was trained based on data simulated from one dose level. The results demonstrated that the trained network was able to correctly predict the treatment effects across a certain range of adjacent dose levels. In contrast, the traditional regression provided biased predictions even when all confounders were included in the model.
Conclusion
Given the Bayesian framework of pharmacometrics, it seems to be a natural evolution to embrace AI/DL/ML as additional tools and opportunities for collaboration. It is critical to actively expand our understanding of AI/ML/DL to appreciate their impact at various stages of drug development. Failure to acknowledge or shying away from AI/DL/ML may not be an option. On the contrary, pharmacometricians are poised to be a natural fit to the AI/DL/ML space and advance this field to the next level.
Funding
No funding was received for this work.
Conflict of Interest
M.S. is an employee of Nuventra, N. Gattu is an employee of Excelra, and N. Goyal and S.C.‐T. are employees of GlaxoSmithKline. All other authors declared no competing interests for this work.
Supporting information
Supplement: Figure S1 and Table S1.
Acknowledgment
The authors thank Scott R. Penzak (Auburn University) for providing comments to revise the manuscript.
References
- 1. McCarthy, J. , Minsky, M.L. , Rochester, N. & Shannon, C.E. A proposal for the Dartmouth summer research project on artificial intelligence <http://www-formal.stanford.edu/jmc/history/dartmouth/dartmouth.html> (1955). Accessed March 13, 2019.
- 2. Ghahramani, Z. Probabilistic machine learning and artificial intelligence. Nature 521, 452–459 (2015). [DOI] [PubMed] [Google Scholar]
- 3. Rajpurkar, P. et al Deep learning for chest radiograph diagnosis: a retrospective comparison of the CheXNeXt algorithm to practicing radiologists. PLoS Med. 15, e1002686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ehteshami, B. et al Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 318, 2199–2210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sayres, R. et al Using a deep learning algorithm and integrated gradients explanation to assist grading for diabetic retinopathy. Ophthalmology 126, 552–564 (2019). [DOI] [PubMed] [Google Scholar]
- 6. Bies, R. et al A genetic algorithm‐based, hybrid machine learning approach to model selection. J. Pharmacokinet. Pharmacodyn. 133, 195–221 (2006). [DOI] [PubMed] [Google Scholar]
- 7. Sherer, E.A. et al Application of a single‐objective, hybrid genetic algorithm approach to pharmacokinetic model building. J. Pharmacokinet Pharmacodyn. 39, 393–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ismail, M. et al. An open source software solution for automating pharmacokinetic/pharmacodynamic model selection. Annual Meeting of the Population Approach Group in Europe, Montreux, Switzerland, May 29, 2018.
- 9. Topol, E.J. High‐performance medicine: the convergence of human and artificial intelligence. Nat. Med. 25, 44–56 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Gottlieb, M. FDA's comprehensive effort to advance new innovations: initiatives to modernize for innovation <https://www.fda.gov/NewsEvents/Newsroom/FDAVoices/ucm619119.htm> (2018). Accessed March 13, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement: Figure S1 and Table S1.