

Abstract
Background
Colorectal cancer (CRC) has been shown to acquire RAS and EGFR ectodomain mutations as mechanisms of resistance to epidermal growth factor receptor (EGFR) inhibition (anti-EGFR). After anti-EGFR withdrawal, RAS and EGFR mutant clones lack a growth advantage relative to other clones and decay; however, the kinetics of decay remain unclear. We sought to determine the kinetics of acquired RAS/EGFR mutations after discontinuation of anti-EGFR therapy.
Patients and methods
We present the post-progression circulating tumor DNA (ctDNA) profiles of 135 patients with RAS/BRAF wild-type metastatic CRC treated with anti-EGFR who acquired RAS and/or EGFR mutations during therapy. Our validation cohort consisted of an external dataset of 73 patients with a ctDNA profile suggestive of prior anti-EGFR exposure and serial sampling. A separate retrospective cohort of 80 patients was used to evaluate overall response rate and progression free survival during re-challenge therapies.
Results
Our analysis showed that RAS and EGFR relative mutant allele frequency decays exponentially (r2=0.93 for RAS; r2=0.94 for EGFR) with a cumulative half-life of 4.4 months. We validated our findings using an external dataset of 73 patients with a ctDNA profile suggestive of prior anti-EGFR exposure and serial sampling, confirming exponential decay with an estimated half-life of 4.3 months. A separate retrospective cohort of 80 patients showed that patients had a higher overall response rate during re-challenge therapies after increasing time intervals, as predicted by our model.
Conclusion
These results provide scientific support for anti-EGFR re-challenge and guide the optimal timing of re-challenge initiation.
Keywords: colorectal cancer, anti-EGFR therapy, circulating tumor DNA, clonal decay
Key Message
RAS and EGFR clones decay exponentially with a cumulative half-life of 4.4 months. A retrospective cohort of 80 patients showed that patients had a higher overall response rate and progression-free survival during re-challenge therapies after increasing time intervals. These results provide scientific support for anti-EGFR re-challenge and guide the optimal timing of re-challenge initiation.
Introduction
Patients with KRAS/NRAS (RAS) wild-type metastatic colorectal cancer (mCRC) have improved survival when treated with anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibodies. However, these agents do not benefit patients with oncogenic RAS mutations [1–4]. Though less common, alterations in BRAF/HER2 and MAP2K1 (MEK) are additional biomarkers of primary resistance to anti-EGFR [5–7]. Among patients who initially respond to anti-EGFR, acquired abnormalities eventually develop and result in secondary resistance. Growing utilization of plasma circulating tumor DNA (ctDNA) testing [8] has allowed for the non-invasive detection of heterogeneous molecular alterations, which underlie the evolution of resistance to targeted therapies in mCRC [6, 9]. Such analyses have uncovered the role of acquired RAS mutations in resistance to anti-EGFR but have also implicated subclonal mutations in the EGFR ectodomain (ECD) in the development of acquired resistance to anti-EGFR [6, 9–13].
Our group has previously shown that in the absence of continued selective pressure from EGFR inhibition, the prevalence of KRAS and EGFR mutant clones declines [14]. Accordingly, we hypothesize that as the clone declines, sensitivity to anti-EGFR is restored. These data are consistent with prior reports of clinical benefit with anti-EGFR re-challenge [15, 16]. However, the dynamics of mutant clones after progression on anti-EGFR have not been previously validated and is of paramount importance in guiding the timing of re-challenge therapies. Here, we show that RAS and EGFR mutant alleles decay exponentially after discontinuation of anti-EGFR in an institutional cohort and validate these findings in a large external dataset. Further, we show that the efficacy of re-challenge strategies may be influenced by these clonal dynamics and effectively guided by the monitoring of serial ctDNA analysis.
Methods
Patients
We first analyzed a retrospective cohort of 135 patients at MD Anderson Cancer Center (MDACC) with RAS/EGFR/BRAFWT mCRC who were treated with and progressed on anti-EGFR, and from whom plasma samples had been collected for sequencing of ctDNA on a platform optimized for very low allele frequencies (Guardant360®, Guardant Health, Inc., Redwood City, CA). An external cohort of 4465 patients with mCRC was analyzed to validate our institutional findings. Of these, 496 patients with mCRC had serial ctDNA sequencing (Guardant360®), defined as ≥2 tests with the same targeted next-generation sequencing (NGS) assay between 2 June 2014 and 26 December 2017. Seventy-three of 496 patients had serial plasma testing and alterations highly consistent with prior anti-EGFR and were used for this analysis. As treatment histories were not known for this cohort, we evaluated patients with molecular abnormalities that would be most likely consistent with prior anti-EGFR exposure. We included patients with at least one of the following: a subclonal RAS mutation [defined as relative mutant allele frequency (rMAF) of <50%], any EGFR mutation, or multiple concurrent RAS mutations.
To evaluate the clinical significance of the clonal half-life determined by our exponential decay model, we separately identified 80 unique patients at our institution with RAS/BRAFWT mCRC who progressed on prior anti-EGFR-based therapy (cetuximab or panitumumab) and were subsequently re-challenged with ≥2 doses of anti-EGFR at MDACC. Previous anti-EGFR before retreatment was based upon retrospective review of the medical records. All patients had progressed on prior anti-EGFR and not taken off due to toxicity.
As we did not have treatment information available for the external cohort that was used to validate our half-life predictions, we ensured that these patients had molecular features characteristic of prior anti-EGFR exposure. Using the same Guardant360® assay, we analyzed a separate institutional cohort of 374 patients (who had no serial sampling) with mCRC and carried out a logistic regression to determine whether the presence of (i) multiple concurrent RAS mutations, (ii) any EGFR mutation, or (iii) subclonal RAS mutations (<50% rMAF) was predictive of prior anti-EGFR. In this cohort, 295 patients had previously received anti-EGFR and 79 had not. We further validated the specificity and positive predictive value (PPV) of this exposure signature using an additional cohort of 93 separate patients with and without anti-EGFR exposure (supplementary Table S1, available at Annals of Oncology online). Previous anti-EGFR-based therapy was based upon retrospective review of the medical records. As this was a retrospective study, informed consent was waived by the MDACC Institutional Review Board.
Clonal decay analysis
To compare clonal prevalence of resistance mechanisms over time, we compared changes in the rMAF of mutations over time. The rMAF approximates the percentage of tumor cells shedding the mutation of interest into the circulation and was defined as the MAF of a mutation (in this article any mutation in RAS/BRAF/EGFR and MAP2K1) divided by the maximum MAF for any gene detected in that sample. In patients with multiple detected RAS or EGFR mutations, we assume each mechanism of resistance is only in a single clone. Analysis was limited to the dominant clones, defined as those with the highest rMAF in the initial plasma sample. Our analysis was then expanded to include all known mechanisms of resistance, including all RAS and EGFR ECD mutations occurring per patient, plus any BRAF/MAP2K1 mutations (supplementary Table S2, available at Annals of Oncology online). Amplifications were excluded in this analysis.
A one-phase decay analysis was used to determine the non-linear fit of the exponential decay of RAS and EGFR using GraphPad Prism, version 7.03 (GraphPad Software, Inc., La Jolla, CA). The relationship between RAS and EGFR exponential decay in the external cohort was assessed by a Mann–Whitney-non-parametric test. Comparisons were considered significant if P ≤ 0.05.
In our external validation cohort, in which all patients had serial plasma sampling after anti-EGFR, we first calculated the slope between two time points (lambda) of plasma collection for each patient, by subtracting the rMAF at time A (earlier date of ctDNA collection) from the rMAF at time B (later date), then dividing this by the time between samples in months. To calculate the half-life, we then divided the ln of 2 by the calculated value of lambda as above. Thus, this half-life calculation is accurate despite the lack of clinical information regarding exact timing of anti-EGFR.
The modeling of rMAF versus time also allows estimates of rMAF at the time of progression, defined as the y-intercept of the model. The use of this rMAF allows estimates of the relative proportion of heterozygous mutant-to-wild-type clones in the tumor, such that an rMAF of 15% for KRASG12V would imply that 15% of the tumor cells demonstrate this resistance mechanism.
Data collection
Clinical information including age, race, the date of initial diagnosis and staging, KRAS/NRAS/BRAF/MAP2K1, and PIK3CA mutational status of the tumor specimen, microsatellite status, prior treatment history, and tumor sidedness were collected at the initiation of anti-EGFR retreatment. Responses upon re-challenge were determined for each patient and categorized per RECISTv1.1 [17]. Clinical benefit on anti-EGFR retreatment was defined as complete response, partial response, or stable disease. Between-group comparisons were carried out using the chi-square test. Patients without progression were censored on 5 November 2018. Progression-free survival (PFS) was graphed with the Kaplan–Meier method and compared with the log-rank test for trend. Between-group comparisons were carried out using a Cox-regression model with those patients who started an anti-EGFR re-challenge <1 half-life from their prior anti-EGFR as the reference group.
Results
Modeling of RAS and EGFR clonal dynamics in patients with mCRC at a single institution
We analyzed 565 blood specimens from patients at The University of Texas MDACC with mCRC who underwent testing with a targeted NGS ctDNA assay (Guardant360®, Guardant Health, Inc.) between 23 June 2014 and 7 September 2017. Of these, 135 samples represented assays from patients who had been previously treated with and progressed on anti-EGFR.
To explore the hypothesis that RAS and EGFR clones decay over time from last anti-EGFR, we carried out mathematical modeling to describe the clonal dynamics. We found an inverse relationship between the rMAF of RAS and EGFR and time since last treatment with anti-EGFR (Figure 1A–C). There was no decay in truncal APC and TP53 mutations from time since last treatment. Our analysis showed that the decline in the RAS and EGFR rMAF is best described by an exponential decay model (r2=0.93 for RAS and 0.94 for EGFR) (Figure 1A–C).
Figure 1.

(A) Sum of exponential decay of median RAS and median EGFR rMAF (t1/2=4.4 ± 2.90 months, r2=0.94). (B) Exponential decay of the median RAS allele over time after discontinuation of anti-epidermal growth factor receptor (EGFR) therapy (t1/2=3.4 months; r2=0.93). (C) Exponential decay of the median EGFR allele over time after discontinuation of anti-EGFR therapy (t1/2=6.9 months; r2=0.94).
Pooled together, these clones exponentially decayed with a cumulative half-life of 4.4 months ± SEM of 2.90 months. The half-lives for the individual RAS and EGFR alleles were identified as 3.4 and 6.9 months, respectively; however, these were not significantly different (Figure 1A–C). At the time of progression on anti-EGFR, the modeled median RAS rMAF was 10.5%, and the modeled median EGFR rMAF was 10.6% (Figure 1B and C).
Modeling of exponential decay of RAS and EGFR clones using an external validation cohort
To validate our institutional findings, we analyzed 4465 patients with mCRC at multiple institutions who underwent ctDNA sequencing. Of these, 496 patients had serial ctDNA testing, defined as two or more tests with the same targeted NGS assay between 2 June 2014 and 26 December 2017. Seventy-three of 496 patients had serial plasma testing and alterations highly consistent with prior anti-EGFR and were used for this analysis. As treatment histories were not known for this cohort, we evaluated patients with molecular abnormalities that would be most likely consistent with prior anti-EGFR exposure. We included patients with at least one of the following: subclonal RAS mutation (rMAF of < 50%), any EGFR mutation, or multiple concurrent RAS mutations. In an institutional cohort of 374 mCRC patients with and without anti-EGFR exposure, the presence of any one of these variables was highly predictive of prior anti-EGFR exposure [PPV 98.3%, 95% confidence interval (CI) 93.4%–99.6%; specificity 98.7% CI 95.3%–99.8%.). All patients included in the validation cohort had at least one of these variables and 89% of patients had two or more of these characteristics. We further validated the specificity and PPV of this exposure signature in a separate institutional cohort of 93 mCRC patients with and without anti-EGFR exposure (supplementary Table S1, available at Annals of Oncology online).
When fit to an exponential model, we found that the RAS and EGFR clones decayed with a cumulative half-life of 4.3 months (Figure 2A), which was similar to the 4.4 months in our institutional cohort (Figure 3). When individual clones were analyzed, RAS clones decayed with a half-life of 3.7 months, and EGFR clones decayed with a half-life of 4.7 months (Figure 2B and C). There was no significant difference between the decay rates of these two populations (P = 0.11).
Figure 2.

(A) Estimated exponential decay of the average of the RAS plus EGFR allele on an external cohort of patients (t1/2=4.3 months). (B) Estimated exponential decay of the RAS allele on an external cohort of patients (t1/2=3.7 months). (C) Estimated exponential decay of the EGFR allele on an external cohort of patients (t1/2=4.7 months). P = 0.11 for a difference between RAS and EGFR.
Figure 3.
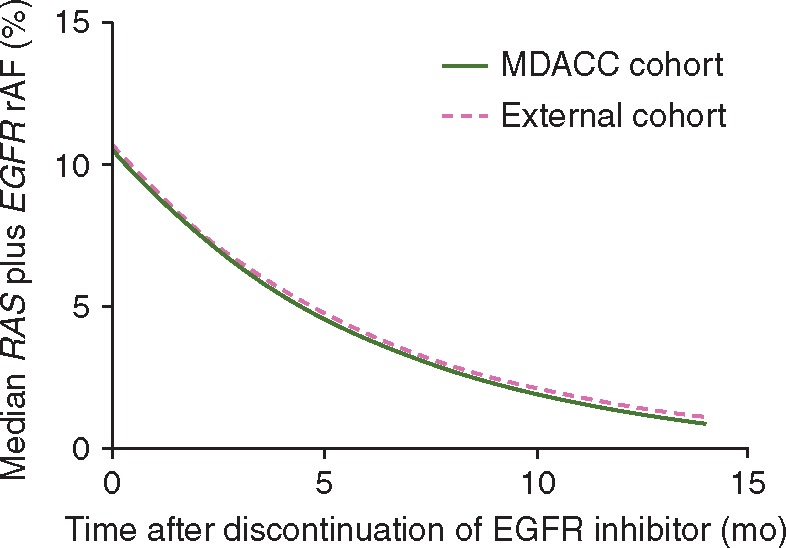
Comparison of the exponential decay of the median RAS plus EGFR rMAF in the MD Anderson Cancer Center (MDACC) cohort (t1/2=4.4 months) and validation cohort (t1/2=4.3 months).
Response to anti-EGFR re-challenge
We identified 80 separate patients with RAS/BRAFWT mCRC at MDACC who received anti-EGFR treatments in more than one line of therapy. Baseline characteristics and treatment response information are summarized in supplementary Table S4, available at Annals of Oncology online. The median number of therapies between the first and second anti-EGFR treatments was 1 [interquartile range (IQR): 1–2] and the median time between date of first progression on anti-EGFR and re-challenge with anti-EGFR was 5.1 months (IQR 2.7–10.6 months). Median PFS on first and second anti-EGFR was 5.2 and 3.1 months, respectively (P = 0.040). The objective response rate (ORR) to anti-EGFR re-challenge was 23%.
Utilizing the half-life of t = 4.4 months from our institutional cohort, we stratified treatment response and PFS by the time passed since prior anti-EGFR. Median PFS was longer with a greater interval between initial anti-EGFR treatment and re-challenge but not statistically different when stratifying patients by number of half-lives that elapsed between initial exposure and re-challenge (log-rank test for trend P = 0.44) (Figure 4A). ORR was highest in patients who waited >2 half-lives (ORR = 32%) from prior anti-EGFR compared with patients who waited <1 half-life (16%) or 1–2 half-lives (20%); however this was not significantly different between groups (Figure 4B).
Figure 4.

Impact of time from prior anti- epidermal growth factor receptor (EGFR) on (A) progression-free survival and (B) response rates of metastatic colorectal cancer patients undergoing re-challenge with a second regimen containing an anti-EGFR agent. CI, confidence interval.
Discussion
The growing utilization of ctDNA is a valuable resource in guiding treatment decisions in patients with mCRC. Here, we demonstrate that genomic profiling of known acquired resistance mutations to anti-EGFR antibodies by ctDNA can predict clonal decay and potentially guide re-challenge therapies. In our analysis of the NGS data from ctDNA of 135 patients with RAS/BRAFWT mCRC at our institution who were treated with and progressed on anti-EGFR antibodies, we showed that the decline in RAS/EGFR rMAF is best described by an exponential decay model. Thus, our translational data provide strong support for the feasibility and validity of genomic profiling of known acquired resistance mutations to anti-EGFR antibodies by ctDNA to predict clonal decay and provide strong support for re-challenge with anti-EGFR antibodies. In addition to providing a validated predictive model for the exponential decay of RAS and EGFR-resistant clones, our work provides further clarification of a still novel area of interest in the understanding of acquired resistant mutations to anti-EGFR. The role of EGFR ECD mutations in driving resistance to anti-EGFR has been documented through ctDNA by several groups [6, 9, 10, 12, 18–20]. Both our institutional and external ctDNA database analyses confirmed the recurrent alteration of these previously identified genomic alterations (supplementary Table S2 and Figure S3, available at Annals of Oncology online). However, now that we have found that the exponential decay of the EGFR ECD mutations is no different than that of the RAS clones, we must use this information to better predict optimal patients and adequate timing for re-challenge therapies.
There are several hypotheses to explain the regression of resistant clones after discontinuation of targeted therapy. Targeted therapies apply powerful selective pressure on the polyclonal and heterogeneous tumor microenvironment. This promotes the survival of cells with the highest fitness and ultimately leads to therapeutic failure, in line with Darwinian evolutionary theory [21]. However, as seen in preclinical work in BRCA2-mutated breast cancer, the fitness of this resistant population may be innately limited, allowing for the rapid growth of the wild-type cell population [22]. Furthermore, in BRAF-mutated melanoma, mechanistic studies with BRAF-inhibitor therapy alone suggested that hyperactivation of the MAPK pathway is not favorable to cell growth and leads to cell senescence, thus leading to resistant clones upon discontinuation of the targeted agent [23–25]. Similarly, in mCRC, Misale et al. demonstrated that, regardless of the mutation that confers resistance to EGFR-directed therapy, the outcome is always sustained activation of MEK and ERK [26]. These data have provided a rationale for the use of combined MEK inhibition and anti-EGFR to overcome resistance to EGFR antibodies and is currently being explored in clinical trials (NCT03087071).
Here, we provide a molecular explanation for the efficacy of anti-EGFR re-challenge therapies after a period off EGFR inhibition. The half-life of these RAS and EGFR clones may help guide the timing of re-challenge therapies and could be monitored by ctDNA. These data are consistent with previously published literature in patients with mCRC who were re-challenged with anti-EGFR antibodies, which found responses after a treatment interval of 4.6–6 months [15, 16]. Several randomized clinical trials are ongoing to further elucidate the role of subclonal RAS and EGFR mutations in acquired resistance to anti-EGFR antibodies. Some are monitoring the ctDNA of mCRC patients who progressed on anti-EGFR, and using a drop in the plasma RAS mutant population by 50% before re-challenging with anti-EGFR. Our data suggest that future re-challenge studies could utilize an EGFR treatment-free interval of at least two half-lives, or approximately 8 months. However, this represents a population average, and individual patient’s tumors may demonstrate different dynamics. Serial ctDNA monitoring may be a better guide than time if available. This will need to be further evaluated in prospective clinical trials.
A limitation of our study is the lack of clinical annotation for the external ctDNA cohort and the inability to directly confirm that these patients had received prior anti-EGFR. However, the ctDNA profile of these patients is unique and allow identification of these patients with high confidence. Further, EGFR ECD mutations have not been identified in CRC before EGFR inhibition, and, consequently, in the internal cohort where treatment histories were known, and treatment with anti-EGFR antibodies was universal, we found a very similar distribution of EGFR ECD mutations to that seen in the external cohort (supplementary Table S2 and Figure S3, available at Annals of Oncology online). Further, in approximately 50% of patients in our re-challenge analysis, anti-EGFR was combined with either an investigational agent or an investigational agent plus cytotoxic therapy, which may have confounded the results of our PFS analysis. Finally, no gene amplifications were included in this analysis and these too may contribute to EGFR resistance. However, the incidence of de-novo resistance to anti-EGFR due to KRAS amplifications, for example, is only 0.7% and has not shown statistical significance [27, 28].
In summary, our results support a molecular explanation for the efficacy of anti-EGFR re-challenge therapies. The half-life of RAS and EGFR clones may help guide the timing of re-challenge therapies and could be monitored by ctDNA. The absence of these clones in ctDNA collected at the time of re-challenge has already been shown to be a clinically reliable predictor of response [29]. Several randomized clinical trials are ongoing to further elucidate the role of subclonal RAS and EGFR mutations in acquired resistance to anti-EGFR antibodies and will certainly assist clinicians in timing of re-challenge therapies, as well as in the discovery of therapeutic efforts to reverse resistance to EGFR inhibitors.
Funding
CMP was funded by a Conquer Cancer Foundation Merit Award for this work (no grant number applies). SK was funded by National Institute of Health Grant (grant number R01 CA187238).
Disclosure
KCB, VMR, AAT and RBL are full-time employees and shareholders of Guardant Health. JHS is a consultant/advisory board member for Amgen, Bayer, Genentech/Roche, and OncoMed. RBC is a consultant/advisory board member for Amgen, Astex Pharmaceuticals, Avidity Biosciences, BMS, Fog Pharma, Genentech, LOXO, Merrimack, N-of-one, Roche, Roivant, Spectrum Pharmaceuticals, Symphogen, Taiho, and Warp Drive Bio and has received research funding from AstraZeneca and Sanofi. MJO receives research support from BMS and Amgen. All remaining authors have declared no conflicts of interest.
Supplementary Material
References
- 1. Amado RG, Wolf M, Peeters M. et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26(10): 1626–1634. [DOI] [PubMed] [Google Scholar]
- 2. Karapetis CS, Khambata-Ford S, Jonker DJ. et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359(17): 1757–1765. [DOI] [PubMed] [Google Scholar]
- 3. Douillard JY, Oliner KS, Siena S. et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369(11): 1023–1034. [DOI] [PubMed] [Google Scholar]
- 4. Van Cutsem E, Kohne CH, Hitre E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360(14): 1408–1417. [DOI] [PubMed] [Google Scholar]
- 5. Di Nicolantonio F, Martini M, Molinari F. et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008; 26(35): 5705–5712. [DOI] [PubMed] [Google Scholar]
- 6. Strickler JH, Loree JM, Ahronian LG. et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov 2018; 8(2): 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yonesaka K, Zejnullahu K, Okamoto I. et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011; 3(99): 99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thierry AR, Pastor B, Jiang ZQ. et al. Circulating DNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Clin Cancer Res 2017; 23(16): 4578–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pietrantonio F, Vernieri C, Siravegna G. et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res 2017; 23(10): 2414–2422. [DOI] [PubMed] [Google Scholar]
- 10. Arena S, Bellosillo B, Siravegna G. et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res 2015; 21(9): 2157–2166. [DOI] [PubMed] [Google Scholar]
- 11. Siravegna G, Mussolin B, Buscarino M. et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015; 21(7): 827. [DOI] [PubMed] [Google Scholar]
- 12. Van Emburgh BO, Arena S, Siravegna G. et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun 2016; 7: 13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siena S, Sartore-Bianchi A, Garcia-Carbonero R. et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann Oncol 2018; 29(1): 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morelli MP, Overman MJ, Dasari A. et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol 2015; 26(4): 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X, George GC, Tsimberidou AM. et al. Retreatment with anti-EGFR based therapies in metastatic colorectal cancer: impact of intervening time interval and prior anti-EGFR response. BMC Cancer 2015; 15: 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santini D, Vincenzi B, Addeo R. et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol 2012; 23(9): 2313–2318. [DOI] [PubMed] [Google Scholar]
- 17. Eisenhauer EA, Therasse P, Bogaerts J. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45(2): 228–247. [DOI] [PubMed] [Google Scholar]
- 18. Bertotti A, Papp E, Jones S. et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015; 526(7572): 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montagut C, Dalmases A, Bellosillo B. et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012; 18(2): 221–223. [DOI] [PubMed] [Google Scholar]
- 20. Vilar E, Tabernero J.. Cancer: pinprick diagnostics. Nature 2012; 486(7404): 482–483. [DOI] [PubMed] [Google Scholar]
- 21. Gillies RJ, Verduzco D, Gatenby RA.. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012; 12(7): 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rytelewski M, Maleki Vareki S, Mangala LS. et al. Reciprocal positive selection for weakness—preventing olaparib resistance by inhibiting BRCA2. Oncotarget 2016; 7(15): 20825–20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schreuer M, Jansen Y, Planken S. et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF(V600)-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol 2017; 18(4): 464–472. [DOI] [PubMed] [Google Scholar]
- 24. Das Thakur M, Salangsang F, Landman AS. et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013; 494(7436): 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petti C, Molla A, Vegetti C. et al. Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res 2006; 66(13): 6503–6511. [DOI] [PubMed] [Google Scholar]
- 26. Misale S, Arena S, Lamba S. et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med 2014; 6(224): 224ra226. [DOI] [PubMed] [Google Scholar]
- 27. Mekenkamp LJ, Tol J, Dijkstra JR. et al. Beyond KRAS mutation status: influence of KRAS copy number status and microRNAs on clinical outcome to cetuximab in metastatic colorectal cancer patients. BMC Cancer 2012; 12: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valtorta E, Misale S, Sartore-Bianchi A. et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer 2013; 133(5): 1259–1265. [DOI] [PubMed] [Google Scholar]
- 29. Rossini D, Cremolini C, Conca E. et al. Liquid biopsy to predict benefit from rechallenge with cetuximab (cet) + irinotecan (iri) in RAS/BRAF wild-type metastatic colorectal cancer patients (pts) with acquired resistance to first-line cet+iri: final results and translational analyses of the CRICKET study by GONO. J Clin Oncol 2018; 36(Suppl 15): 12007–12007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.