

Abstract
Background
Kindler Syndrome (KS) is a rare genodermatosis characterized by skin fragility, skin atrophy, premature aging and poikiloderma. It is caused by mutations in the FERMT1 gene, which encodes kindlin-1, a protein involved in integrin signalling and the formation of focal adhesions. Several reports have shown the presence of non-melanoma skin cancers in KS patients but a systematic study evaluating the risk of these tumors at different ages and their potential outcome has not yet been published. We have here addressed this condition in a retrospective study of 91 adult KS patients, characterizing frequency, metastatic potential and body distribution of squamous cell carcinoma (SCC) in these patients. SCC developed in 13 of the 91 patients.
Results
The youngest case arose in a 29-year-old patient; however, the cumulative risk of SCC increased to 66.7% in patients over 60 years of age. The highly aggressive nature of SCCs in KS was confirmed showing that 53.8% of the patients bearing SCCs develop metastatic disease. Our data also showed there are no specific mutations that correlate directly with the development of SCC; however, the mutational distribution along the gene appears to be different in patients bearing SCC from SCC-free patients. The body distribution of the tumor appearance was also unique and different from other bullous diseases, being concentrated in the hands and around the oral cavity, which are areas of high inflammation in this disease.
Conclusions
This study characterizes SCCs in the largest series of KS patients reported so far, showing the high frequency and aggressiveness of these tumors. It also describes their particular body distribution and their relationship with mutations in the FERMT-1 gene. These data reinforce the need for close monitoring of premalignant or malignant lesions in KS patients.
Keywords: Kindler syndrome, SCC, Skin cancer, Bullous disease, Prevalence
Background
Kindler Syndrome (KS) is a rare autosomal recessive genodermatosis, considered a subtype of epidermolysis bullosa (EB), characterized by skin fragility with photosensitivity and acral blisters formation in young patients [1]. As they age, they develop progressive skin atrophy, premature aging, poikiloderma, palmoplantar hyperkeratosis, and pseudosyndactyly [2]. Mucosal manifestations are also common and include haemorrhagic mucositis and gingivitis, periodontal disease, premature loss of teeth, and labial leukokeratosis [3]. Interestingly, unlike other skin bullous diseases, KS is characterized by sensitivity to UV and dysregulation of oxidative stress [4], [5], [6].
KS is caused by mutations in the FERMT1, a gene that codes for kindlin-1, a protein associated to integrin and focal adhesions [7], [8].
Reports in the literature have documented the presence of SCCs in KS patients suggesting a predisposition of these patients to this neoplasia [9], [10]. However, a systematic study of cancer frequency and aggressiveness in a series of KS patients has not been carried out. Therefore, the relative risk of SCC at different ages cannot be predicted from the current published data on KS. Furthermore, although SCCs in KS have been described in individual case reports, published studies do no provide common behaviours, body location and the existence or lack of mutations in FERMT1 that can be directly associated with cancer in KS.
We have previously reported the largest series of KS patients from different countries and with the associated FERMT 1 (KIND1) genotype in patients of different ethnicities. The development of cutaneous cancer was noted in some patients; however, tumor characteristics and follow-up data were not provided [2].
In this paper we present a series of 91 cases, 69 cases previously published [2], [11] and 22 new cases unpublished, with the primary goal of establishing the incidence of SCC in KS patients, at different ages. We have also used this series, in some cases supplemented with data from the literature, to investigate other characteristics of SCCs in KS, such as tumoral prognosis and outcome, body distribution of the tumors and the existence or lack of KS mutations associated to SCC appearance. To our knowledge, this represents the first study published that characterizes as a whole, SCC in Kindler syndrome.
Results
Patients and development of SCC
Information about our patients including gender, age, genetic data and the presence of muco-cutaneous SCC is provided (Table 1).
Table 1.
Characterization of the SCCs developed in the KS adult patients of this study. A total series of 91 KS patients is shown, 69 of them previously published and 22 new unpublished cased. SCC: Squamous Cell Carcinoma; WD: Well differentiated; AS: Altered splicing; RT: Reduced transcription. UD: Unpublished data; **: FERMT1 mutation confirmed (manuscript in preparation)
| Pat # | Gender & Age | Mutation DNA nucleotide change (cDNA) |
Mutation (gDNA) in Chromosome 20 |
Mutation (gDNA) in gene FERMT1 |
Protein | Epithelial Cancer/ Histopathology | Cancer onset (Age) | Outcome | Refs |
|---|---|---|---|---|---|---|---|---|---|
| 1. | M, 15 | c.1718 + 2 T > C | g.6084038A > G | 35516 | AS | – | [2] | ||
| 2. | F, 16 | c.190G > T | g.6116006C > A | 3548 | p.E64X | – | UD | ||
| 3. | M, 17 | c.614G > A | g.6110430C > T | 9124 | p.W205X | – | [7] | ||
| 4. | M, 17 | c.328C > T | g.6115868G > A | 3686 | p.R110X | – | [12] | ||
| 5. | M, 18 | c.1051G > T | g.6096940C > A | 22614 | p.E351X | – | [2] | ||
| 6. | F, 18 | c.676dupC | g.6110368dupG | 9186 | p.Q226PfsX17 | – | [2] | ||
| 7. | M, 18 | c.910G > T | g.6097571C > A | 21983 | p.E304X | – | [2] and [13] | ||
| 8. | M, 20 |
g.70250_74168del/ c.-20A > G g.6116239_ 6120157 |
g.6122775 T > C | − 3221 | p.P381Rfs: PTC/RT | – | [14] | ||
| 9. | F, 20 | c.957 + 1G > A | g.6097523C > T | 22031 | AS: In intron 7 at donor splice site | – | [15] | ||
| 10. | M, 22 | c.1729delA | g.6079567delT | 39987 | p.S577AfsX14 | – | [2] | ||
| 11. | F, 23 | c.994_995delCA | g.6096996-6096997delTG | 22558 | p.Q332fsX9 | – | [15] | ||
| 12. | M, 23 | ** | – | – | – | – | UD | ||
| 13. | M, 24 | c.1161delA | g.6089068delT | 30486 | p.A388LfsX14 | – | [2] | ||
| 14. | M, 24 | c.1161delA | g.6089068delT | 30486 | p.A388LfsX14 | – | [2] | ||
| 15. | F, 24 | c.676dupC | g.6110368dupG | 9186 | p.Q226PfsX17 | – | [2] and [16] | ||
| 16. | M, 24 | c.1139 + 2 T > C / c.889A > G | g.6094937A > G/ g.6097592 T > C | 21962 | AS/ p.R297G | – | [15] | ||
| 17. | F, 24 | c.910G > T | g.6097571C > A | 21983 | p.E304X | – | [15] | ||
| 18. | M, 25 |
g.-711-1241del g.6045219_6047230del |
g.6088988-6110333del | 30566 | RT | – | [14] and [17] | ||
| 19. | M, 25 | c.170C > A | g.6116026G > T | 3528 | p.S57X | – | [2] and [16] | ||
| 20. | F, 25 |
g.70250_74168del g.6116239_ 6120157del |
g.6084280del | 35274 |
AS Large deletion p.P381Rfs PTC |
– | [2] | ||
| 21. | M, 25 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 22. | M, 26 | c.1718 + 2 T > C | g.6084038A > G | 35516 | AS | – | [15] and [18] | ||
| 23. | M, 26 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 24. | F, 26 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 25. | F, 27 |
g.70250_74168del g.6116239_ 6120157del |
g.6084280del | 35274 |
Large deletion p.P381Rfs PTC |
– | [2] | ||
| 26. | M, 27 | c.1161delA | g.6089068delT | 30486 | p.A388LfsX14 | – | [2] | ||
| 27. | M, 27 | c.910G > T | g.6097571C > A | 21983 | p.E304X | – | [15] | ||
| 28. | F, 28 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 29. | F, 29 | c.1719-1G > A | g.6079578C > T | 39976 | AS | – | [2] | ||
| 30. | F, 29 |
c.1139 + 740G > A (IVS9 + 740G > A) |
g.6094199C > T | 25355 | pseudoexon | – | [19] | ||
| 31. | F, 29 | c.889A > G/ c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 32. | M, 29 | c.1718 + 2 T > C | g.6084038A > G | 35516 | AS | – | [15] and [18] | ||
| 33. | F, 29 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 34. | M, 29 | c.1383C > A | g.6085276G > T | 34278 | p.Y461X | – | [15] | ||
| 35. | F, 29 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 36. | F, 30 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 37. | F, 31 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | 2 SCC: WD& early invasive (dorsal hand, between 4th and 5th fingers) | 29 | Alive | UD |
| 38. | M, 31 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 39. | M, 31 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 40. | M, 31 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 41. | F, 31 | ** | – | – | – | – | UD | ||
| 42. | F, 32 | c.1714_1715insA | g.6084043-6084044insT | 35511 | p.V572DfsX4 | – | [2] | ||
| 43. | M, 32 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 44. | F, 32 | c.1383C > A | g.6085276G > T | 34278 | p.Y461X | – | [15] | ||
| 45. | F, 32 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 46. | M, 32 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 47. | F, 33 | c.889A > G/ c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 48. | M, 33 | c.328C > T | g.6115868G > A | 3686 | p.R110X | – | [12] | ||
| 49. | M, 34 | c.958-1G > A | g.6097034C > T | 22520 | AS | – | [2] | ||
| 50. | M, 34 | c.456dupA | g.6112553dupT | 7001 | p.D153RfsX4 | SCC: WD, early infiltrating, Oral mucosa, extending to tongue, lymph node metastases | 33 | Deceased at 35 years | [2] |
| 51. | M, 34 | c.1718 + 1G > A | g.6084039C > T | 35515 | AS | – | [2] | ||
| 52. | M, 34 | c.1383C > A | g.6085276G > T | 34278 | p.Y461X | – | [15] | ||
| 53. | M, 34 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 54. | F, 36 | c.1209C > G | g.6089020G > C | 30534 | p.Y403X | – | Breast cancer | 39 years | [2] and [20] |
| 55. | M, 36 | c.1176 T > G | g.6089053A > C | 30501 | p.Y392X | – | [15] | ||
| 56. | M, 36 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 57. | M, 36 | ** | – | – | – | – | UD | ||
| 58. | M, 37 | c.1365–1371 + 3del | g.6087774-6087783del | 31780 |
AS exon11 |
– | [2] and [21] | ||
| 59. | F, 37 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 | – | [15] | ||
| 60. | M, 37 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 61. | M, 39 | g.6109607-6112272del | g.6109607-6112272del | 9947 | Deletion of exon4 | – | [15] | ||
| 62. | F, 40 | c.550-551insA | g.6110493-6110494insT | 9061 | p.S184LfsX1 |
2 Metastasizing SCC to lymph nodes Fingers and hand |
38 | Deceased at 40 years | [15] |
| 63. | M, 40 | c.889A > G / c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 64. | M, 40 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 65. | F, 40 | c.676C > T | g.6110368G > A | 9186 | p.Q226X | – | UD | ||
| 66. | M, 42 | c.373delT | g.6115823delA | 3731 | p.C125AfsX4 | – | [2] | ||
| 67. | M, 42 | c.1179G > A | g.6089050C > T | 30504 | p.W393X | – | UD | ||
| 68. | F, 43 |
g.6000982_6009222del (IVS13_15del) |
g.6076879_6084040del | 42675 | PTC (exon14) | – | [2] | ||
| 69. | F, 45 |
g.70250_74168del g.6116239_ 6120157del |
g.6084280del | 35274 |
p.P381Rfs Large deletion |
– | [2] | ||
| 70. | M, 47 |
g.6000982_6009222del (IVS13_15del) |
g.6076879_6084040del | 42675 | PTC (exon14) | – | [2] | ||
| 71. | M, 47 | c.889A > G / c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 72. | M, 47 | ** | – | – | – | – | UD | ||
| 73. | M, 48 | c.958-1G > A | g.6097034C > T | 22520 | AS |
2 Infiltrating SCC: WD Upper Lip and dorsum of the hand |
43 | Alive | [2] [22] |
| 74. | M, 49 | c.676dupC | g.6110368dupG | 9186 | p.Q226PfsX16 | – | UD | ||
| 75. | M, 50 | c.910G > T | g.6097571C > A | 21983 | p.E304X |
Metastasizing and poorly differentiated SCC on the foot. Diffuse metastases (skin & lung) |
48 years | Deceased at 50 years | [2] |
| 76. | M, 50 | c.958-1G > A | g.6097034C > T | 22520 | AS | – | [2] | ||
| 77. | M, 51 | c.889A > G / c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 78. | M, 52 | c.328C > T | g.6115868G > A | 3686 | p.R110X | Metastasizing SCC to lymph nodes (Lip) | 48 | Deceased at 52 years | [15] |
| 79. | F, 60 |
g.6000982_6009222del (IVS13_15del) |
g.6076879_6084040del | 42675 |
Large deletion and PTC in exon14 PTC (exon14) |
2 SCC: WD & early invasive Lower lip and dorsum of the hand |
45, 53 | Deceased at 60 years | [2], [11] and [23] |
| 80. | F, 56 | c.152-2delAGinsCT | g.6116046delinsAG | 3508 | AS | – | [2] | ||
| 81. | M, 56 | c.299-301del | g.6115895-6115897del | 3659 | p.R100del | – | [2] and [24] | ||
| 82. | F, 58 | c.96-97delAG | g.6119458-6119459delCT | 96 | p.R32SfsX63 | Poorly differentiated SCC. Left arm | 52 | Amputation of left arm | [2] |
| 83. | M, 65 | c.1209C > G | g.6089020G > C | 30534 | p.Y403X | 1 SCC: WD Invasive, left hand | 57 | Alive | [2] |
| 84. | M, 59 | c.889A > G / c.1139 + 2 T > C | g.6097592 T > C/ g.6094937A > G | 21962 | AS/ p.R297G | – | [15] | ||
| 85. | M, 60 | c.958-1G > A | g.6097034C > T | 22520 |
AS Intron7 |
1–3 Infiltrating SCC, all WD; 4, 5 in situ SCC; 1 upper lip; 2 third right finger; 3 right hand dorsum; 4 lower lip; 5 fourth interdigital space of the left hand | 43 (44, 45, 45, 56) | Alive | [2] and [22] |
| 86. | M, 61 |
c.1139 + 740G > A (IVS9 + 740G > A) |
g.6094199C > T | 25355 | Pseudoexon | [19] | |||
| 87. | F, 63 | c.1761 T > A | g.6079535A > T | 40019 | p.Y587X | [25] | |||
| 88. | M, 65 | g.70250_74168del | g.6084280del | 35274 |
p. P381R fsX36 PTC (large deletion: exons 10 and 11) |
Infiltrating SCCs, all WD 1 lower gingival mucosa and regional lymph nodes; 2 scalp; 3 lower lip |
52 (53, 55) | Alive | [2] |
| 89. | M, 71 | c.862C > T | g.6097619G > A | 21935 | p.R288X | UD | |||
| 90. | M, 71 | c.328C > T | g.6115868G > A | 3686 | p.R110X |
Multiple in situ SCC: Hyperkeratotic SCC in situ. Face and hands. |
Start at 50 years | Alive | [2] |
| 91. | F, 79 | c.328C > T | g.6115868G > A | 3686 | p.R110X |
Invasive SCC, BCC, in situ SCC. Face and hands. |
Start at 66 years | Alive | [2] |
In our series of 91 patients, 13 developed non-melanoma skin tumors. Most of these patients presented multiple tumors, which resulted in a total of 25 tumors in the skin and 2 in the oral mucosa. With the exception of one BCC, all of the neoplastic lesions were SCC of variable histological grades but mostly well differentiated SCC.
When the patients were stratified by age, and the cumulative incidence calculated, our data showed that the earliest SCC appeared in a 29 year old woman and the cumulative risk of SCC increased with age to reach 66.7% in patients over 60 years of age.
The SCC development cumulative risk in KS was compared to that reported for other types of EB patients and spontaneous SCC (Fig. 1). The KS profile is different from recessive dystrophic EB (RDEB) in which SCCs develop earlier and reach a higher cumulative risk and are also different from the generalized non-Herlitz types of Junctional EB (JEB) patients profile [28], [29], which showed a lower incidence and later onset. Fig. 1 shows the data from Fine et al., although this data has been somewhat controversial [29], [30]. Furthermore, in a series of one of our authors, the frequency appears much lower with only 1 SCC in 70 patients with Collagen XVII mutation and none in patients with Laminin322 or Integrin α6β4 mutations (Has, C. et al. unpublished results). In contrast, the incidence of sporadic SCC of sun exposed areas in the general population was much lower (0.001–0.005%), even in older patients (Fig. 1) [31], [26].
Fig. 1.
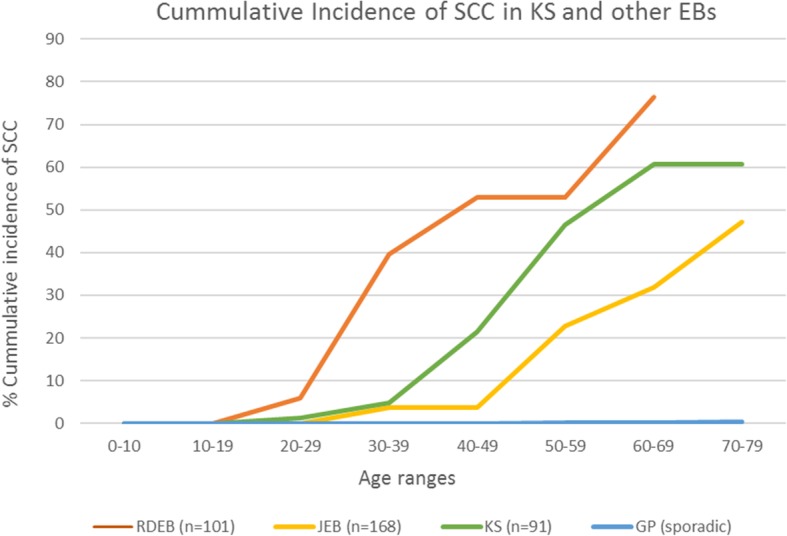
Calculated cumulative risk of the first squamous cell carcinoma in Kindler syndrome patients. For comparative purposes, we have also plotted the calculated cumulative risk described for RDEB, JEB and the general population which were taken from the literature [26], [27]
The outcome of SCCs (Table 1) in these patients has been variable leading to premature demise in five patients and arm amputation in another. Overall, 53.8% (7/13 cases) of the patients with SCC present metastasis .
Regarding mortality, 5/13 KS patients (38.5%), that developed SCC died as a direct consequence of the tumor, in a period that varied between 2 and 7 years, with an average of 40.8 months. This percentage(50%) is consistent with the previously reported data for KS-SCC [11], but much higher than the 5–10% of mortality described for sporadic tumors [31], [26]. Their detailed demographic, clinical and histopathological features are presented in Table 2.
Table 2.
All the KS patients that have developed SCC, ordered by the age at which the first tumor appeared. Yellow patients have been published previously to this study. SCC: Squamous Cell Carcinoma; WD: Well differentiated; MD: Moderately differentiated; AS: Altered splicing; RT: Reduced transcription; PTC: Premature termination codon; FS: Frameshift; UD: Unpublished data
| Gender, Age | Mutation | Protein | Age of cancer | Localisation of SCC | Histopath. of SCC | Outcome | Refs |
|---|---|---|---|---|---|---|---|
| M, 16 |
ISV9-6 T > A c.1140-6 T > A |
FS and PTC | 16 | Right lower leg | WD aggressive SCC |
Present for 10 years as a hyperkeratotic plaque, then progress in 2 months. Deceased. |
[32] |
| F, 23 | UD | UD | 23 | Right hand | Ulcerative and invasive MD SCC | Methastasic and Nodes affected | [33] |
| M, 40 | c.1761 T > A and c.1089del/c.1089 + 1del | p.Y587X and Exon 8 skipped. (p.L363 fs) | 27 | Palm of the right hand | SCC recurrent 4 times | Recurrent SCC | [34] |
| F, 31 | c.676 C > T | p.Q226X | 29 | Fingers and dorsal hand | WD early agressive | Still alive | UD |
| M, 34 | c. 456dupA | p.D153R fsX4 | 33 | Oral mucosa, extending to tongue, lymph node metastases | WD, early infiltrating SCC with metastases | Deceased at 35 years | [2] |
| F, 42 | c.750G > A | p.W250X | 34 | Hard palate | WD SCC: unresectable treated with radiation | 2 years follow up no recurrence. Now deceased. | [35] |
| F, 40 | c. 550-551insA | p. S184 L fsX1 | 38 | Fingers and hand | SCC metastasic to limph nodes | Deceased at 40 years | [15] |
| M, 49 | c.910G > T | p. E304X | 42, [20, 24] | Right shoulder, upper lip, dorsum of the right hand | SCC | UD | [36] |
| M, 60 | c.676insC | p.W250X | 43, 45 | Lip and later in penis | SCC | UD | [37] |
| M, 48 | c.958-1G > A |
AS Intron 7 |
43 | Upper Lip and dorsum of the hand | 2 infiltrating WD SCC | Still alive | [2] and [22] |
| M, 60 | c.958-1G > A |
AS Intron 7 |
43 (44, 45, 45, 56) |
1 upper lip; 2 third right finger; 3 right hand dorsum; 4 lower lip 5 fourth interdigital space of the left hand |
1–3 Infiltrating SCC All WD; 4, 5 in situ SCC. |
Still alive | [2] |
| F, 60 | g.6000982_6009222del |
IVS13_15del Large deletion and PTC in exon14 |
45, 53 |
1 SCC of the left hand 2 SCC of the lower lip |
1 WD 2 WD early invasive |
Deceased at the age of 60 years | [23] and [11] |
| M, 49 | c.910G > T | p. E304X | 48 | Foot |
Poorly differentiated SCC Metastasizing Diffuse metastases (skin & lung) |
Deceased at the age of 50 years | [2] |
| M, 52 | c.328C > T | p. R110X | 48 | lip | SCC metastasic to limph nodes | Deceased at the age of 52 years | [15] |
| M, 71 | c.328C > T | p. R110X | 50 | Face and hands |
Hyperkeratotic SCC in situ Multiple in situ SCCs |
Still alive | [38] |
| F, 58 | c.96_97delAG | p. R32S fsX63 | 52 | Left arm | Poorly differentiated SCC | Amputation of left arm | [2] |
| F, 52 | c.95_96delAG | p.R32fsX63 | 52 | Lower lip | SCC | No follow up | [39] |
| M, 65 | g.70250_74168del | p. P381R fsX36 | 52 (53, 55) |
1 lower gingival mucosa and regional lymph nodes; 2 scalp; 3 lower lip |
Infiltrating SCC, all WD | Still alive | [2] |
| F, 55 | UD | UD | 55 | buccal mucosa extended to both lips | SCC | Treated with radiation therapy with partial response | [40] |
| M, 57 | c.328 C > T | p. R110X | 57 | Left hand | moderate-to-poorly differentiated deeply invasive SCC | Recurrent, requiring axillary node clearance and amputation. | [10] and [41] |
| M, 65 | c.1209C > G | p. Y403X | 57 | Left Hand | Invasive SCC WD | Still alive | [2] |
| F, 78 | c.328C > T | p. R110X | 66 | Face and hands | Invasive SCC, BCC, In situ SCC | Still alive | [38] |
The information in Table 2, includes grading of KS-SCC in 26/37 tumors (17/22 KS patients with histological data available). SCC considered as well-differentiated forms were the majority of the cases (22/26: 84.6%), moderately to poorly differentiated SCCs, were only presented in 4/26 (15.38%).
In order to study gender influence on the development of the KS-SCC, Table 3 was expanded to include previously published studies as well as the patients in this study. Our analysis did not reveal a significant difference in the development of SCC in male and female KS patients (Table 3).
Table 3.
Gender and age distribution of SCCs appearance in all the KS patients that has been reported
| Patients with carcinoma (including bibliography) | |||
|---|---|---|---|
| Age range | Male | Female | Total |
| 16–19 | 1 | 0 | 1 |
| 20–29 | 1 | 2 | 3 |
| 30–39 | 1 | 2 | 3 |
| 40–49 | 6 | 1 | 7 |
| 50–59 | 4 | 3 | 7 |
| 60–69 | 0 | 1 | 1 |
| 70–79 | 0 | 0 | 0 |
| Total | 13 | 9 | 22 |
Even though a difference in the total number of tumors in men and women cannot be observed, there seems to be a tendency in women to develop tumors at earlier ages (4/9: 44% before the 40’s), whereas in men, there was an abrupt increase after 40 years of age (10/13: 77%).
Location of tumors
The location of the tumors in the body is shown (Fig. 2). All the KS-SCC described in the literature were also included in the figure [11], thus, a total of 37 SCC was used for this analysis. Furthermore, when tumors of this study and tumors from the literature were segregated, results were nearly identical (data not shown).
Fig. 2.
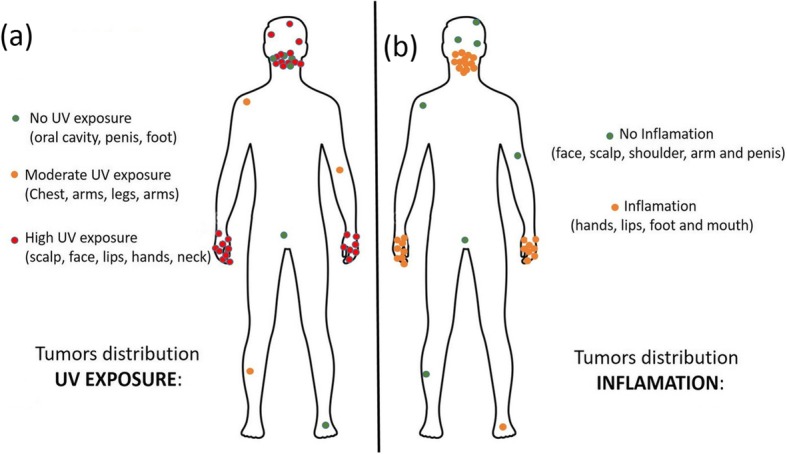
Representation of the body distribution of KS-SCCs. a The key color, classifies every tumor in different areas according to its´ UV exposure: Non-exposed (green), Moderately-exposed (orange) and Highly-exposed (red). b The key color, classifies every tumor in different areas according to whether the zone has been described as chronically inflamed in KS: no inflammation (green) and chronic inflammation (orange)
It can be observed that the majority of the tumors (28/37) developed in sun exposed areas, with the exception of 6 SCC of the oral mucosa, foot and penis, with 3 of them in moderately sun exposed areas (Fig. 2a). Surprisingly, most tumors were restricted to just two areas of the body; the face, particularly around the mouth (13/37 tumors), and hands (16/37 tumors); that together represented 78% of the KS-SCCs. Other sun-exposed areas were minimally affected, which resulted in a unique distribution, that overlaps with areas of chronic inflammation in KS patients [42], [43]. This pattern is different from sporadic [26] JEB [29] and RDEB associated SCCs [44].
The body distribution of SCC was represented with a key color. These classified every tumor if the zone was considered as a sun exposed area (Fig. 2a) and if the zone had been described as chronically inflamed in KS (Fig. 2b). It can be observed that, in KS-SCC there is a perfect coincidence between areas of chronic inflammation and tumor development.
Mutations in SCC bearing KS patients
In order to discern if the predisposition to develop SCC was related with a pattern in the mutational spectra, gDNA mutations were represented (Fig. 3). In our series of KS patients, no hot spot, at genomic level, associated to SCC development was identified. Our data showed that SCC developed in patients with 10 different FERMT1 mutations. In most cases, there was one patient with SCC, for each mutation, with the exception of two mutations present in multiple patients. Three patients with the mutations c.328C > T (p.R110X; g.6115868G > A) developed a total of 5 tumors and 2 patients with the mutation c.958-1G > A (altered splicing in intron 7; g.6097034C > T) developed 7 tumors. Thus, these two mutations were characterized not only for the higher frequency of patients with SCC, but also for a higher multiplicity of tumors. Another interesting mutation was c.96-97delAG (p.R32SfsX63 g.6119458-6119459delCT) presented in a single SCC bearing KS patient, while all other mutations in patients with SCC were also presented in patients free of neoplasia. We have represented the mutations for all our 91 case series (only 87 with mutational information available) for those who are free of neoplasias and those that developed SCC (Fig. 3). When we compared the spectra of mutations of patient bearing tumors with the tumor free patients we found an interesting pattern.
Fig. 3.

Distribution of the mutations in our patient series, along the gDNA of FERMT1. In the X axis, it is represented by the length of the FERMT1 gene, considering the + 1 position as the ATG. For that reason, the gene is from − 3990 to + 44709 positions. The Y-axis, shows the number of patients presenting with each mutation, in orange (free of SCC) and in blue (developed SCC). Four patients with no mutational information available were excluded from this figure
From our data (Fig. 3 and Table 1), we can see that 10 out of 13 KS patients that developed SCCs, presented mutations that resulted in stop codons being located closer to the 5’end of FERMT1, which resulted in short transcripts that were likely to be degraded. The other three mutations were also frame shifts or alternative splicing which are likely to generate an unstable protein. In contrast, the spectra of mutations reported in the literature, in patients with and without tumors, showed a more even distribution all along the gene and with a higher representation at the sequence encoding carboxy terminus. In addition, according to the literature, non-sense mutations represent 37.5% of the total KS mutations [2] which is consistent with our study (36.6%). In patients with SCC, this proportion increases up to 60% (6 out of 10 reported mutations). Taking together all of this data, suggests than even though specific mutations do not have a predictive value in the development of SCC, there is a different profile of mutations in patients bearing tumors and tumor free patients.
Discussion
In a case series of 91 KS patients, we have determined that KS patients have a high risk for developing SCC. The data from our case series indicated that SCCs appeared relatively early compared with sporadic SCC, but occurred about 10 to 20 years later than in RDEB, which is the type of EB with the highest risk of cancer [28]. On the other hand, SCC in JEB showed a higher latency and a lower incidence than KS.
The youngest patient that developed SCC in our series was a 29 year old female. It is worth noting that consistent with our study, there are only three younger KS patients of 16, 23 and 27 described with SCC in the literature [32], [33], [34]. It can be inferred that development of SCC in young patients is rare and in the case of paediatric patients is extremely rare or non-existent.
The cumulative incidence of SCC in KS increases after the early onset at 29 years old, to reach a maximum at 60 years old with a cumulative risk of 67%.
Of note, the body localization of cutaneous cancers in KS is also unique. Unlike RDEB and JEB, in which, the preponderance of lesions in the lower extremities is clear, KS tumors arose mainly in the mouth and hands. Interestingly, KS has been shown to have an oversensitivity to UV, but other areas of the body, highly exposed to the sun, were minimally affected [45]. This particular distribution was confirmed including 9 other KS patients that developed SCCs which were taken from the literature. Tumors in non-exposed areas were less common and were restricted to 6 patients; 4 in the oral mucosa, one, in the penis and another in the foot. RDEB and JEB SCCs are associated with chronically non-healing ulcerations, while in KS, the cutaneous condition improves with age. The adult skin in KS is characterized by atrophy, poikiloderma as well as inflammation of the mouth, hands and feet [42], [41]. These differential distributions support the notion that distinct pathogenic mechanisms may underlie cancer development in KS as compared to the other major EB types. Photosensitivity is a unique feature of KS among the EB disease spectrum, and loss of kindlin-1 is associated with an up-regulation of pro-inflammatory cytokines in keratinocytes, at least in part mediated by UV exposure with subsequently impaired DNA repair [4]. Furthermore, in KS we have observed mitochondrial damage with an increase in oxidative stress [5]. KS has also been characterized by strong inflammation in the mouth and hands [42], [43], [41]. Thus, SCC seems to develop by a synergy of UV exposure together with the stimulus of inflammation, in the mouth and hands, which results in the peculiar distribution of these tumors.
Interestingly, as in other EB related cancers, the SCC of the KS patients has the potential to be highly aggressive [28] leading to amputation [32], [33], [10] and early demise [32]. In agreement, in our series, five patients died as a consequence of the tumors, and another patient showed local metastasis which resulted in an amputation of an arm.
The mechanisms involved in the aggressive nature and early onset of SCC associated with ulcerative disease and chronic inflammation have not been completely elucidated. The putative role of Kindlin-1 has been widely discussed by Rognoni et al. (2014) which focused on the activation of TGF-β–mediated growth-inhibitory signals in the KS mouse model [46]. Moreover, TFG-beta has been described as a crucial factor in other bullous diseases modifying disease severity in RDEB, through the promotion/inhibition of a fibrotic matrix [47]. More recently, our group has underscored the role of the fibroblasts of KS, RDEB, and XPC, in the pathogenesis of these diseases, observing a higher activation of the TGF-β signalling pathway [48]. Taking together all these data and consistent with the body distribution described in this study, a potential mechanistic role of the TGF-β in the aggressive nature of KS-SCC becomes even more important, and should be further characterized.
In the last few years, a role of the stroma has been considered determinant and at least partially responsible for the early onset and aggressiveness of these tumors [48], [49], [50]. More recently; however, careful genetic studies by Cho et al. (2018) have shown that driver mutations seem to be shared between RDEB and sporadic skin tumors in sun exposed areas and other highly aggressive SCC, but unlike the sporadic tumor genes, RDEB showed a profile of endogenous mutations associated with APOBEC [51]. The alteration of these genes seems to be related with inflammation and is probably responsible of the differences between sporadic tumors and tumors associated with chronic ulcerative diseases and inflammation. Based on this data, we speculate that similar mechanism may occur in KS, in which tumors appear in areas that are both exposed to UV and present higher inflammation (i.e. mouth and hands).
In regard to the nature of FERMT1 mutations in patients that developed SCC, we showed that there are no clear hot spots or even predominant mutations in our cohort. Only two mutations are found in two and three patients, respectively. Although some mutations appear in this study more frequently associated with SCC (96, 3731, 22558 and 30534), there is not enough statistical significance to consider that they have a predictive value. In addition, none of the mutations seems to be carcinogenic ¨per se¨ since several mutations are found in patients bearing cancer and also in patients in which SCC have not been detected. However, the mutational distribution in FERMT1 and the type of mutation in the KS patients with SCC, was clearly different from that of the patients that did not develop SCC. While mutations in the overall population are distributed along the whole length of the gene, the mutations in the patients bearing SCC are mostly present in the N terminal part of the gene. Furthermore, most of them result in stop codons, which lead to short mRNA that are likely to be degraded. Taken together, these data suggest that most of the patients were null for kindlin-1, which is a paradoxical result, taking into account that there is a reasonable body of literature suggesting a pro-carcinogenic role of this protein. Overexpression of kindlin 1 has been reported in breast, lung, colon and esophageal cancers [52]. Furthermore, overexpression of this protein has also been associated with a poor prognosis in osteosarcoma [53] as well as breast and lung carcinomas and pancreatic cancer [54]. Based on these results, we conclude that kindlin1 may have a pro-carcinogenic or anticarcinogenic potential depending on the context as has been shown in other genes such as E2F, which can act as a tumor suppressor gene or an oncogene depending on the context [55].
A limitation of the present study is the relatively low number of cases, particularly in older patients. However, since the total population of reported patients with KS is less than 300, the report of 91 patients can be considered as a representative sample for such a rare disease. Similarly, in ideal circumstances, the risk of a disease should be assessed in a more homogeneous population. Our series included people from different countries and ethnicities. However, given the low prevalence of this disease, it would be impossible to do a study of these characteristics in a homogeneous population. Furthermore, our study analyzed 26 carcinomas in 13 different patients being so far 19, the total number of KS-SCC previously reported in the literature [11].
Conclusions
Overall, this study predicts that more than half of patients with KS will develop SCC in their lifetime and among them, 53.8% of patients will develop metastatic disease with a high possibility of a lethal outcome. Thus, it is important to stress the need for close monitoring of these patients, aiming at early SCC detection (at pre-malignant stages and/or initial developmental stages) to avoid progression of the tumor, particularly in older patients when the symptoms of the disease are less severe and therefore, the monitoring may be less strict.
Methods
Patients and tumors
Patients for this retrospective study were recruited from different institutions in Europe and the United States. The core of our series are 34 patients previously described by Has et al. [2] to which, we included 57 new patients who were diagnosed with KS by clinical and molecular methods and who had records of tumor development as well as their characteristics [19]; [14]; [15]. All the patients included in this study were older than 16 years, the age of the youngest KS patient who developed a SCC [32]. In four patients from our series, FERMT1 mutations were not available and therefore, these patients were not included in the mutational distribution study (Fig. 3).
This series of 91 patients was used primarily to determine the incidence of SCC in KS, which has not been previously reported. In order to investigate other characteristics of KS-SCC, we have supplemented the information of our series with data from the literature.
For the SCC body distribution study, we have used 37 tumors, coming from 13 patients of our series and 9 additional KS patients from the literature [11]. To our knowledge, all the tumors and information available reported until now, are included here. Information regarding pedigree was not available for this study. To summarize, different groups of patients have been included in each analysis depending on the information available from them (mutations, follow up, histopathological characterization of the tumors,…). For cumulative risk calculation, 91 KS patients have been included (13 of which developed SCC) as was mentioned before and are shown in Table 1. For mutation distribution analysis (Fig. 3), only 87 KS patients were included from our 91 patients series, because 4 patients have no mutational information available (Patients #12, 41, 57 and 72 from Table 1). The SCC body distribution analysis shown in Fig. 2 was analyzed in 22 KS patients (from the bibliography and from our series) who developed a total of 37 tumors. The same group of patients was used to elaborate Table 2 that describes all the SCC bearing patients, but only 26/37 tumors had complete histopathological data available. For gender and age distribution analysis (Table 3), the same group of 22 SCC-KS patients was studied.
Distribution of mutations along the FERMT1 gene
The position of the mutations (Table 1 and Fig. 3) are expressed in both c.DNA and g.DNA. For cDNA numbering, + 1 corresponds to the A of the ATG translation initiation codon in the reference sequence. cDNA variations in nucleotides are in accordance with GenBank entry NM_017671.4, while g.DNA positions were calculated from those using the position converter available in the web-based software Mutalyzer (build hg38, www.mutalyzer.nl) [56]. If only one mutation is mentioned, then it is in a homozygous state. The mapping along protein domains was not performed because most of the mutations are stop codons and likely to degrade the mRNA.
Statistical analysis
Cumulative risk was obtained by calculating the number of new cases over the population at risk for each age segment. To compare the distribution of the mutations along the FERMT1 gene, parametric and non-parametric tests (Chi square, q-q test, ANOVA and Kruskal-Wallis test) were performed using R package v.3.1.2. Differences between groups were considered significant at P < 0.05.
Acknowledgements
Angeles Mencia for the genetic discussions. Ellen Valentine for the stylistic and grammar corrections. Blanca Duarte, Almudena Holguín and Nuria Illera for their technical support.
Authors’ contributions
SGA analyzed and interpreted the patients data regarding the body distribution and the mutations analysis, elaborated the statistics, the tables and was a major contributor in writing the manuscript. CJC analyzed and interpreted the prevalence and histopathology of the SCCs and was a major contributor in writing the manuscript. MJE provided critical discussion of the manuscript and provided useful information for the mutations analysis. DC, GZ, LY, HV, PI, GT, IY, LM and JU provided data from their patients and recollected clinical data for the manuscript. LR provided critical discussion of the research. CH provided data from her patients and was a major contributor in writing the manuscript.CH, CJC and SGA coordinated all the study. MDR provided critical discussion of the research and participated on the reviewing of the manuscript. All authors contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Funding
Science and Innovation Ministry of Spain (SAF2013–43475); Comunidad de Madrid (S2010/BMD-2420, B2017/BMD-3692); Instituto de Salud Carlos III (PI14/00931); DEBRA International and European Union (HEALTH-F2–2011-261392); CIBER-ER17PE01 and Italian Ministry of Health grant (Ricerca Corrente RC2017–1.2 to D.C).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Patient informed consents were obtained in agreement with the collaborative centers where the biopsies were available. Procedures were approved by their respective human subjects committee or institutional review board for each referral center in accordance with the institutional ethical standards on human experimentation and the project also adhered to the Helsinki Guidelines and further reviews (Fortaleza, 2013).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. doi: 10.1016/j.jaad.2014.01.903. [DOI] [PubMed] [Google Scholar]
- 2.Has C, Castiglia D, del Rio M, Diez MG, Piccinni E, Kiritsi D, et al. Kindler syndrome: extension of FERMT1 mutational spectrum and natural history. Hum Mutat. 2011;32(11):1204–1212. doi: 10.1002/humu.21576. [DOI] [PubMed] [Google Scholar]
- 3.Yildirim TT, Kaya FA, Taskesen M, Dundar S, Bozoglan A, Tekin GG, et al. Aggressive periodontitis associated with kindler syndrome in a large kindler syndrome pedigree. Turk J Pediatr. 2017;59(1):56–61. doi: 10.24953/turkjped.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Maier K, He Y, Wolfle U, Esser PR, Brummer T, Schempp C, et al. UV-B-induced cutaneous inflammation and prospects for antioxidant treatment in kindler syndrome. Hum Mol Genet. 2016;25(24):5339–5352. doi: 10.1093/hmg/ddw350. [DOI] [PubMed] [Google Scholar]
- 5.Zapatero-Solana E, Garcia-Gimenez JL, Guerrero-Aspizua S, Garcia M, Toll A, Baselga E, et al. Oxidative stress and mitochondrial dysfunction in kindler syndrome. Orphanet J Rare Dis. 2014;9:211. doi: 10.1186/s13023-014-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Luo S, Wu J, Zhang L, Wang WH, Degan S, et al. KIND1 loss sensitizes keratinocytes to UV-induced inflammatory response and DNA damage. J Invest Dermatol. 2017;137(2):475–483. doi: 10.1016/j.jid.2016.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS, Wilhelmsen KC, et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes kindler syndrome. Am J Hum Genet. 2003;73(1):174–187. doi: 10.1086/376609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jobard F, Bouadjar B, Caux F, Hadj-Rabia S, Has C, Matsuda F, et al. Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in kindler syndrome. Hum Mol Genet. 2003;12(8):925–935. doi: 10.1093/hmg/ddg097. [DOI] [PubMed] [Google Scholar]
- 9.Alper JC, Baden HP, Goldsmith LA. Kindler’s syndrome. Arch Dermatol. 1978;114(3):457. doi: 10.1001/archderm.1978.01640150085030. [DOI] [PubMed] [Google Scholar]
- 10.Emanuel PO, Rudikoff D, Phelps RG. Aggressive squamous cell carcinoma in kindler syndrome. Skinmed. 2006;5(6):305–307. doi: 10.1111/j.1540-9740.2006.05369.x. [DOI] [PubMed] [Google Scholar]
- 11.Saleva M, Has C, He Y, Vassileva S, Balabanova M, Miteva L. Natural history of kindler syndrome and propensity for skin cancer - case report and literature review. J Dtsch Dermatol Ges. 2018;16(3):338–341. doi: 10.1111/ddg.13435. [DOI] [PubMed] [Google Scholar]
- 12.Has C, Herz C, Zimina E, Qu HY, He Y, Zhang ZG, et al. Kindlin-1 is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am J Pathol. 2009;175(4):1442–1452. doi: 10.2353/ajpath.2009.090203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kern JS, Herz C, Haan E, Moore D, Nottelmann S, von Lilien T, et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin-1 isoforms. J Pathol. 2007;213(4):462–470. doi: 10.1002/path.2253. [DOI] [PubMed] [Google Scholar]
- 14.Has C, Chmel N, Levati L, Neri I, Sonnenwald T, Pigors M, et al. FERMT1 promoter mutations in patients with kindler syndrome. Clin Genet. 2015;88(3):248–254. doi: 10.1111/cge.12490. [DOI] [PubMed] [Google Scholar]
- 15.Youssefian L, Vahidnezhad H, Barzegar M, Li Q, Sotoudeh S, Yazdanfar A, et al. The kindler syndrome: a spectrum of FERMT1 mutations in Iranian families. J Invest Dermatol. 2015;135(5):1447–1450. doi: 10.1038/jid.2015.9. [DOI] [PubMed] [Google Scholar]
- 16.Mansur AT, Elcioglu NH, Aydingoz IE, Akkaya AD, Serdar ZA, Herz C, et al. Novel and recurrent KIND1 mutations in two patients with kindler syndrome and severe mucosal involvement. Acta Derm Venereol. 2007;87(6):563–565. doi: 10.2340/00015555-0314. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs-Telem D, Nousbeck J, Singer A, McGrath JA, Sarig O, Sprecher E. New intragenic and promoter region deletion mutations in FERMT1 underscore genetic homogeneity in kindler syndrome. Clin Exp Dermatol. 2014;39(3):361–367. doi: 10.1111/ced.12222. [DOI] [PubMed] [Google Scholar]
- 18.Fassihi H, Wessagowit V, Jones C, Dopping-Hepenstal P, Denyer J, Mellerio JE, et al. Neonatal diagnosis of kindler syndrome. J Dermatol Sci. 2005;39(3):183–185. doi: 10.1016/j.jdermsci.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Chmel N, Danescu S, Gruler A, Kiritsi D, Bruckner-Tuderman L, Kreuter A, et al. A deep-Intronic FERMT1 mutation causes kindler syndrome: an explanation for genetically unsolved cases. J Invest Dermatol. 2015;135(11):2876–2879. doi: 10.1038/jid.2015.227. [DOI] [PubMed] [Google Scholar]
- 20.Tonyali O, Tufan G, Benekli M, Coskun U, Buyukberber S. Synchronous bilateral breast cancer in a patient with kindler syndrome. Clin Breast Cancer. 2012;12(2):145–146. doi: 10.1016/j.clbc.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Mas-Vidal A, Minones-Suarez L, Toral JF, Mallo S, Perez-Oliva N. A novel mutation in the FERMT1 gene in a Spanish family with Kindler's syndrome. J Eur Acad Dermatol Venereol. 2010;24(8):978–979. doi: 10.1111/j.1468-3083.2009.03554.x. [DOI] [PubMed] [Google Scholar]
- 22.Ashton GH, McLean WH, South AP, Oyama N, Smith FJ, Al-Suwaid R, et al. Recurrent mutations in kindlin-1, a novel keratinocyte focal contact protein, in the autosomal recessive skin fragility and photosensitivity disorder, kindler syndrome. J Invest Dermatol. 2004;122(1):78–83. doi: 10.1046/j.0022-202X.2003.22136.x. [DOI] [PubMed] [Google Scholar]
- 23.Has C, Yordanova I, Balabanova M, Kazandjieva J, Herz C, Kohlhase J, et al. A novel large FERMT1 (KIND1) gene deletion in Kindler syndrome. J Dermatol Sci. 2008;52(3):209–212. doi: 10.1016/j.jdermsci.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Maier K, He Y, Esser PR, Thriene K, Sarca D, Kohlhase J, et al. Single amino acid deletion in Kindlin-1 results in partial protein degradation which can be rescued by chaperone treatment. J Invest Dermatol. 2016;136(5):920–929. doi: 10.1016/j.jid.2015.12.039. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi A, Kiniwa Y, Okuyama R, Kosho T, Suga T, Has C, et al. A case of kindler syndrome with severe esophageal stenosis. Int J Dermatol. 2015;54(4):e106–e108. doi: 10.1111/ijd.12715. [DOI] [PubMed] [Google Scholar]
- 26.Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018;78(2):237–247. doi: 10.1016/j.jaad.2017.08.059. [DOI] [PubMed] [Google Scholar]
- 27.Souldi H., Bajja M.Y., Mahtar M. Kindler syndrome complicated by invasive squamous cell carcinoma of the palate. European Annals of Otorhinolaryngology, Head and Neck Diseases. 2018;135(1):59–61. doi: 10.1016/j.anorl.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the national EB registry experience, 1986-2006. J Am Acad Dermatol. 2009;60(2):203–211. doi: 10.1016/j.jaad.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 29.Yuen WY, Jonkman MF. Risk of squamous cell carcinoma in junctional epidermolysis bullosa, non-Herlitz type: report of 7 cases and a review of the literature. J Am Acad Dermatol. 2011;65(4):780–789. doi: 10.1016/j.jaad.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Fine Jo-David. Squamous cell carcinoma and junctional epidermolysis bullosa. Journal of the American Academy of Dermatology. 2012;66(5):856–857. doi: 10.1016/j.jaad.2012.01.045. [DOI] [PubMed] [Google Scholar]
- 31.Hollestein LM, de Vries E, Nijsten T. Trends of cutaneous squamous cell carcinoma in the Netherlands: increased incidence rates, but stable relative survival and mortality 1989-2008. Eur J Cancer. 2012;48(13):2046–2053. doi: 10.1016/j.ejca.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Arita K, Wessagowit V, Inamadar AC, Palit A, Fassihi H, Lai-Cheong JE, et al. Unusual molecular findings in kindler syndrome. Br J Dermatol. 2007;157(6):1252–1256. doi: 10.1111/j.1365-2133.2007.08159.x. [DOI] [PubMed] [Google Scholar]
- 33.Cardin-Langlois E, Hanna D, St-Amant M, Croteau F. Invasive squamous cell carcinoma of the hand in a patient with kindler syndrome: case report and literature review. Can J Plast Surg. 2010;18(3):e41–3. [PMC free article] [PubMed]
- 34.Natsuga K, Nishie W, Shinkuma S, Nakamura H, Matsushima Y, Tatsuta A, et al. Expression of exon-8-skipped kindlin-1 does not compensate for defects of kindler syndrome. J Dermatol Sci. 2011;61(1):38–44. doi: 10.1016/j.jdermsci.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Lotem M, Raben M, Zeltser R, Landau M, Sela M, Wygoda M, et al. Kindler syndrome complicated by squamous cell carcinoma of the hard palate: successful treatment with high-dose radiation therapy and granulocyte-macrophage colony-stimulating factor. Br J Dermatol. 2001;144(6):1284–1286. doi: 10.1046/j.1365-2133.2001.04262.x. [DOI] [PubMed] [Google Scholar]
- 36.Lanschuetzer CM, Muss WH, Emberger M, Pohla-Gubo G, Klausegger A, Bauer JW, et al. Characteristic immunohistochemical and ultrastructural findings indicate that Kindler's syndrome is an apoptotic skin disorder. J Cutan Pathol. 2003;30(9):553–560. doi: 10.1034/j.1600-0560.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 37.Techanukul T, Sethuraman G, Zlotogorski A, Horev L, Macarov M, Trainer A, et al. Novel and recurrent FERMT1 gene mutations in kindler syndrome. Acta Derm Venereol. 2011;91(3):267–270. doi: 10.2340/00015555-1063. [DOI] [PubMed] [Google Scholar]
- 38.Has C, Burger B, Volz A, Kohlhase J, Bruckner-Tuderman L, Itin P. Mild clinical phenotype of kindler syndrome associated with late diagnosis and skin cancer. Dermatology. 2010;221(4):309–312. doi: 10.1159/000320235. [DOI] [PubMed] [Google Scholar]
- 39.Has C, Wessagowit V, Pascucci M, Baer C, Didona B, Wilhelm C, et al. Molecular basis of kindler syndrome in Italy: novel and recurrent Alu/Alu recombination, splice site, nonsense, and frameshift mutations in the KIND1 gene. J Invest Dermatol. 2006;126(8):1776–1783. doi: 10.1038/sj.jid.5700339. [DOI] [PubMed] [Google Scholar]
- 40.Caldeira A, Trinca WC, Flores TP, Costa AB, Brito Cde S, Weigert KL, et al. A kindler syndrome-associated squamous cell carcinoma treated with radiotherapy. Rep Pract Oncol Radiother. 2016;21(6):532–536. doi: 10.1016/j.rpor.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai-Cheong JE, Tanaka A, Hawche G, Emanuel P, Maari C, Taskesen M, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160(2):233–242. doi: 10.1111/j.1365-2133.2008.08976.x. [DOI] [PubMed] [Google Scholar]
- 42.Wiebe CB, Petricca G, Hakkinen L, Jiang G, Wu C, Larjava HS. Kindler syndrome and periodontal disease: review of the literature and a 12-year follow-up case. J Periodontol. 2008;79(5):961–966. doi: 10.1902/jop.2008.070167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petricca G, Leppilampi M, Jiang G, Owen GR, Wiebe C, Tu Y, et al. Localization and potential function of kindlin-1 in periodontal tissues. Eur J Oral Sci. 2009;117(5):518–527. doi: 10.1111/j.1600-0722.2009.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallipeddi R. Epidermolysis bullosa and cancer. Clin Exp Dermatol. 2002;27(8):616–623. doi: 10.1046/j.1365-2230.2002.01130.x. [DOI] [PubMed] [Google Scholar]
- 45.Montaudie H, Chiaverini C, Sbidian E, Charlesworth A, Lacour JP. Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases. Orphanet J Rare Dis. 2016;11(1):117. doi: 10.1186/s13023-016-0489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rognoni E, Widmaier M, Jakobson M, Ruppert R, Ussar S, Katsougkri D, et al. Kindlin-1 controls Wnt and TGF-beta availability to regulate cutaneous stem cell proliferation. Nat Med. 2014;20(4):350–359. doi: 10.1038/nm.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Odorisio T, Di Salvio M, Orecchia A, Di Zenzo G, Piccinni E, Cianfarani F, et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum Mol Genet. 2014;23(15):3907–3922. doi: 10.1093/hmg/ddu102. [DOI] [PubMed] [Google Scholar]
- 48.Chacon-Solano E, Leon C, Diaz F, Garcia-Garcia F, Garcia M, Escamez MJ, et al. Fibroblasts activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br J Dermatol. 2019. 10.1111/bjd.17698. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 49.Mittapalli VR, Madl J, Loffek S, Kiritsi D, Kern JS, Romer W, et al. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res. 2016;76(4):940–951. doi: 10.1158/0008-5472.CAN-15-1348. [DOI] [PubMed] [Google Scholar]
- 50.Ng YZ, Pourreyron C, Salas-Alanis JC, Dayal JH, Cepeda-Valdes R, Yan W, et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012;72(14):3522–3534. doi: 10.1158/0008-5472.CAN-11-2996. [DOI] [PubMed] [Google Scholar]
- 51.Cho RJ, Alexandrov LB, den Breems NY, Atanasova VS, Farshchian M, Purdom E, et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci Transl Med. 2018;10(455). [DOI] [PubMed]
- 52.Zhan J, Zhang H. Kindlins: roles in development and cancer progression. Int J Biochem Cell Biol. 2018;98:93–103. doi: 10.1016/j.biocel.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 53.Ning K, Zhang H, Wang Z, Li K. Prognostic implications of Kindlin proteins in human osteosarcoma. Onco Targets Ther. 2017;10:657–665. doi: 10.2147/OTT.S125418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plow Edward F., Das Mitali, Bialkowska Katarzyna, Sossey-Alaoui Khalid. Of Kindlins and Cancer. Discoveries. 2016;4(2):e59. doi: 10.15190/d.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol. 1999;19(9):6408–6414. doi: 10.1128/MCB.19.9.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29(1):6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.