

Abstract
Motivation
Neuronal analyses such as transcriptomics, epigenetics and genome-wide association studies must be assessed in the context of the human brain to generate biologically meaningful inferences. It is often difficult to access primary human brain tissue; therefore, approximations are made using alternative sources such as peripheral tissues or in vitro-derived neurons. Gene sets from these studies are then assessed for their association with the post-mortem human brain. However, most analyses of post-mortem datasets are achieved by building new computational tools each time in-house, which can cause discrepancies from study to study. The field is in need of a user-friendly tool to examine spatiotemporal expression with respect to the postmortem brain. Such a tool will be of use in the molecular interrogation of neurological and psychiatric disorders, with direct advantages for the disease-modeling and human genetics communities.
Results
We have developed brainImageR, an R package that calculates both the spatial and temporal association of a dataset with post-mortem human brain. BrainImageR identifies anatomical regions enriched for candidate gene set expression. It further predicts the developmental time point of the sample, a task that has become increasingly important in the field of in vitro neuronal modeling. These functionalities of brainImageR enable a quick and efficient characterization of a given dataset across normal human brain development.
Availability and implementation
BrainImageR is released under the Creative Commons CC BY-SA 4.0 license and can be accessed directly at brainimager.salk.edu or the R code can be downloaded through github at https://github.com/saralinker/brainImageR.
1 Introduction
Many neurological studies do not use primary tissue for their analysis. Genome-wide association studies are taken from the whole genome, which is largely independent of the tissue of origin, and transcriptomics and epigenetics studies are increasingly being assayed using in vitro-derived neurons. Therefore, it is important to have a clear and reproducible way to assess the relationship of this information to the primary tissue of the human brain. A common method is to compare genetic datasets to the post-mortem human brain using the ABA reference data (Hawrylycz et al., 2012; Miller et al., 2014). Some elegant packages and on-line tools are available to analyze this information (Bahl et al., 2017; Grote et al., 2016; Stein et al., 2014), but these tools either lack the ability to predict developmental time, or do not provide informative visualizations of the dataset with respect to the human brain. Therefore, researchers have turned to repeatedly building new computational tools in-house to perform spatiotemporal enrichment analyses (Lathe and Haas, 2017; Paşca et al., 2015;Pollen et al., 2014). This repeated effort indicates that the field requires an analysis suite that is easy to implement and is flexible across data types to identify the spatiotemporal relationship to the human brain. BrainImageR provides flexible and intuitive tools as both a web resource and within the R environment.
2 Software description
2.1 Spatial gene set enrichment
The Allen Brain Atlas (ABA) assays gene expression across multiple regions of the brain and provides this data as a microarray resource and an RNA-seq resource (Hawrylycz et al., 2012; Miller et al., 2014). The microarray dataset is optimal for spatial analysis as the microdissected tissues provide high resolution of the structures underlying each larger brain region (developing = 327 regions; adult = 193 regions). The RNA-seq resource is optimal for temporal analysis as it samples only 26 brain regions but spans developmental time from 8 weeks post-conception to 40 years of age. We therefore used the microarray datasets for the spatial gene set enrichment (SGSE) analysis (adult = 6 samples: H0351.2001, H0351.2002, H0351.1009, H0351.1012, H0351.1015 and H0351.106, 15 pcw = 1 sample: H376_IIIA_02) (Fig. 1A and the RNA-seq dataset for temporal predictions. The ABA microarray reference set notes the presence or absence of a microarray probe signal for each gene. Many genes are assessed by multiple probes; a gene is defined as expressed within a given tissue if ≥80% of the respective probes detect expression, denoted as 1 in the presence–absence file. For the adult dataset, genes were required to be expressed in three or more individuals (≥80% of probes = 1).
Fig. 1.
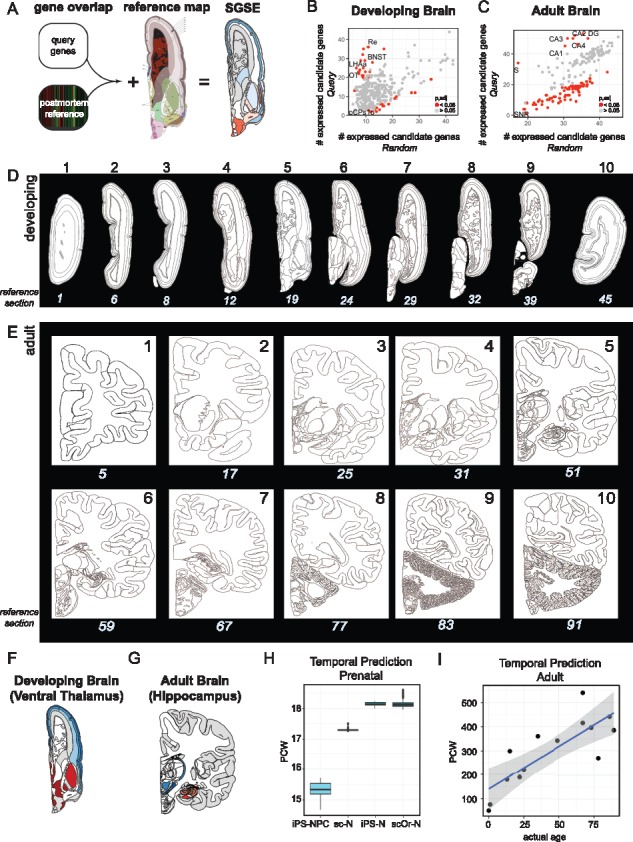
SGSE with brainImageR. (A) BrainImageR identifies gene set enrichment with respect to postmortem expression data from the ABA. (B–C) Enrichment per region, x-axis: background gene set, y-axis: query gene set for the ventral thalamus of the developing brain (B) and the hippocampus of the adult brain (C). Red dots indicate enrichment scores with a bootstrapped Padj < 0.05. Reference images for the developing (D) and adult (E) brain. The top number is used as input into brainImageR to plot the enrichment data. The bottom number is the section for the respective image in the ABA and can be used to look up additional information about each section. (F and G) SGSE for values in (B) and (C), respectively. (H) Predicted age in pcw from iPSC-neural progenitors (iPS-NPC), single-cell RNA-seq from post-mortem 16–18 pcw neurons (sc-N), 2D iPSC-neurons (iPS-N) and single-cell RNA-seq from 3D organoid neurons (scOr-N). (I) Predicted age in pcw from human post-mortem prefrontal cortex
The brainImageR function, SpatialEnrichment, calculates the number of query genes that are expressed above baseline within each tissue of the developing or the adult brain and compares this value to either the entire background gene set or a user-defined background list. Significance can then either be calculated with a Fisher’s exact test or a bootstrapping procedure. The bootstrapping procedure calculates significance by estimating the number of times gene overlap occurs at random when compared with the provided gene list and calculates a one-tailed P-value from this enrichment information that is adjusted for multiple-testing correction using the p.adjust function. As a proof-of-concept, we curated a list of genes that were over-represented in either the ventral thalamus of the developing brain (Fig. 1B) or the hippocampus of the adult brain (Fig. 1C). BrainImageR returned the enrichment for every microdissected region along with the bootstrapped significance estimate, which was indeed significant (Padj < 0.05) within the relevant thalamus and hippocampal regions.
An advantage of BrainImageR over other methods is its ability to map this gene set enrichment information to the reference brain, thereby creating a visualization of SGSE. This visualization is achieved by referencing a conversion between the reference brain map and each tissue assayed by microarray in the ABA. CreateBrain will use this information to transform the regional enrichment values into a brain map. The user can specify which section along the rostral-caudal axis they would like to plot within either the developing (Fig. 1D) or adult (Fig. 1E) brain. The user plots this composite graph as a tiff image with PlotBrain. For example, genes enriched in the relevant regions are colored dark red (Fig. 1F and G), indicating enrichment in these areas.
2.2 Predicting developmental time given gene expression data
Developmental time point prediction is particularly useful for in vitro modeling of neurons using induced pluripotent stems cells (iPSCs) both in 2D culture and 3D organoids. Importantly, when modeling neurons in vitro, it is difficult to identify what age in development the neurons represent. BrainImageR solves this problem by predicting the relative developmental time point by comparing the sample transcriptome information with the developmental transcriptome from the ABA. The predict_time function is a wrapper for a random forest (Liaw and Wiener, 2002) regression model that scales the reference expression data, builds and tests a model to predict developmental time, and then applies this model to the query expression set. Applying predict_time to iPSC-derived neuronal progenitor cells (Li et al., 2017) identifies a consistent prenatal signature with an increase in age as the progenitors are matured either into neurons in the 2D culture setting (Schwartz et al., 2015) or into a 3D organoid setting assayed with single-cell RNA-sequencing (Camp et al., 2015) (Fig. 1H). Furthermore, single-cell RNA-sequencing from post-mortem fetal cortex (16–18 pcw) was also predicted to be an average of 17.3 pcw (Darmanis et al., 2015) (Fig. 1H), indicating the high accuracy of temporal prediction within the prenatal brain. The predict_time function can also be applied to adult post-mortem data, even though the temporal dynamics of the adult brain are limited compared with the developing brain. Although the accuracy is somewhat reduced in the adult predictions, the precision remains high and robust. For example, predicting developmental time from adult prefrontal cortex RNA-seq samples ranging from newborn to 89 years of age (Mertens et al., 2015) accurately identifies a linear association of predicted time with age (Fig. 1I), indicating the robust precision of temporal estimates.
BrainImageR provides a suite of tools for spatiotemporal analysis with respect to the human brain. The results are visualized in an easy-to-interpret anatomical map. With these tools, human genetics and neurological modeling studies will have a consistent framework within which to understand how a given dataset relates to gene expression within the human brain.
Acknowledgements
We would like to thank Mary Lynn Gage for assistance in editing. Image credit: Allen Institute.
Funding
This research was supported by a fellowship from the Lynn and Edward Streim (S.B.L.) Foundation.
Conflict of Interest: none declared.
References
- Bahl E., et al. (2017) cerebroViz: an R package for anatomical visualization of spatiotemporal brain data. Bioinformatics, 33, 762–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp J.G., et al. (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development . Proc. Natl. Acad. Sci. USA, 112, 15672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S., et al. (2015) A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA, 112, 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote S., et al. (2016) ABAEnrichment: an R package to test for gene set expression enrichment in the adult and developing human brain. Bioinformatics, 32, 3201–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz M.J., et al. (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature, 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathe R., Haas J.G. (2017) Distribution of cellular HSV-1 receptor expression in human brain. J. Neurovirol., 23, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., et al. (2017) Transcriptome analysis reveals determinant stages controlling human embryonic stem cell commitment to neuronal cells. J. Biol. Chem., 292, 19590–19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regression by randomForest. R News, 2, 18–22. [Google Scholar]
- Mertens J., et al. (2015) Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell, 17, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.A., et al. (2014) Transcriptional landscape of the prenatal human brain. Nature, 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca A.M., et al. (2015) Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods, 12, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollen A.A., et al. (2014) Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol., 32, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.P., et al. (2015) Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. U.S.A., 112, 12516–12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein J.L., et al. (2014) A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron, 83, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]