

Abstract
Aims
Knockout (KO) of the mitochondrial Ca2+ uniporter (MCU) in mice abrogates mitochondrial Ca2+ uptake and permeability transition pore (PTP) opening. However, hearts from global MCU-KO mice are not protected from ischaemic injury. We aimed to investigate whether adaptive alterations occur in cell death signalling pathways in the hearts of global MCU-KO mice.
Methods and results
First, we examined whether cell death may occur via an upregulation in necroptosis in MCU-KO mice. However, our results show that neither RIP1 inhibition nor RIP3 knockout afford protection against ischaemia-reperfusion injury in MCU-KO as in wildtype (WT) hearts, indicating that the lack of protection cannot be explained by upregulation of necroptosis. Instead, we have identified alterations in cyclophilin D (CypD) signalling in MCU-KO hearts. In the presence of a calcium ionophore, MCU-KO mitochondria take up calcium and do undergo PTP opening. Furthermore, PTP opening in MCU-KO mitochondria has a lower calcium retention capacity (CRC), suggesting that the calcium sensitivity of PTP is higher. Phosphoproteomics identified an increase in phosphorylation of CypD-S42 in MCU-KO. We investigated the interaction of CypD with the putative PTP component ATP synthase and identified an approximately 50% increase in this interaction in MCU-KO cardiac mitochondria. Mutation of the novel CypD phosphorylation site S42 to a phosphomimic reduced CRC, increased CypD-ATP synthase interaction by approximately 50%, and increased cell death in comparison to a phospho-resistant mutant.
Conclusion
Taken together these data suggest that MCU-KO mitochondria exhibit an increase in phosphorylation of CypD-S42 which decreases PTP calcium sensitivity thus allowing activation of PTP in the absence of an MCU-mediated increase in matrix calcium.
Keywords: Mitochondria , Calcium , Cyclophilin D , Phosphorylation , Necroptosis
1. Introduction
Calcium enters mitochondria via the mitochondrial calcium uniporter (MCU)1–3 and can activate dehydrogenases and several complexes of the electron transport chain to enhance mitochondrial adenosine triphosphate (ATP) production.4 In addition, high levels of mitochondrial calcium can cause cell death via activation of the mitochondrial permeability transition pore (PTP).5 Several groups have generated mice lacking the MCU, including mice with a global germline loss of MCU,6 a cardiac-specific dominant-negative MCU expressed at birth,7 and a cardiac-specific tamoxifen-inducible loss of MCU in adult.8,9 Isolated mitochondria from all these mouse models exhibit a loss of rapid mitochondrial calcium uptake and a loss of calcium-activated PTP opening. During cardiac ischaemia and reperfusion (I/R) an increase in cytosolic calcium is thought to lead to calcium entry into the mitochondria via the MCU, triggering activation of the PTP and resultant necrotic cell death.5,10–12 Consistent with this hypothesis, cardiac-specific tamoxifen-inducible loss of MCU in adults is associated with a reduction in cardiac I/R injury.8,9 However, despite the loss of rapid mitochondrial calcium uptake and calcium-activated PTP opening, adult mice lacking MCU or expressing a myocardial-specific dominant-negative MCU from birth are not protected from cardiac I/R injury.6,7 Overall, results to date suggest that acute deletion of MCU in adult mice protects from cardiac ischaemic injury, but this cardioprotection is lost when MCU is deleted before birth or with expression of a cardiac-specific dominant-negative MCU prior to birth. We tested the hypothesis that the lack of protection from cardiac I/R injury in mice lacking MCU prior to birth is likely due to some adaptation or compensatory change in mitochondrial cell death pathways. A better understanding of adaptations in cell death pathways could be important for understanding how to protect the heart from I/R-mediated death.
Cardiac I/R injury occurs primarily via necrosis, which is thought to occur via two routes: calcium-sensitive PTP opening and receptor interacting protein (RIP) kinase-activated necroptosis.13–15 The mechanism through which calcium stimulates PTP opening remains unclear, but cyclophilin D (CypD) plays a key role in mediating PTP opening. CypD is thought to bind to the PTP and enhance its opening, as loss of CypD decreases PTP calcium sensitivity and protects from cardiac I/R injury.16 Further, post-translational modifications of CypD, such as phosphorylation,17 acetylation,18 and S-nitrosylation,19 have been suggested to alter the regulation of PTP opening. Cell death via necroptosis has been defined as a form of regulated necrosis involving RIP kinases, and knock out of RIP kinases decreases I/R injury.14,20 Data in kidney suggest that both PTP opening and the RIP kinase pathway play a role in I/R-mediated death and that these pathways are additive.14 Since mitochondria isolated from the germline mitochondrial Ca2+ uniporter (MCU) knockout (KO) mice lack rapid calcium uptake and PTP opening, we considered that cell death in these hearts is occurring via an upregulation of necroptosis. We also examined a second hypothesis that cell death is still occurring via a modified PTP. This study highlights key alterations that occur in response to loss of MCU-mediated mitochondrial calcium uptake in the heart.
2. Methods
See ‘Supplementary material online, Methods section’ for further information.
2.1 Animals
All animals were treated and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health), and protocols were approved by the NHLBI Animal Care and Use Committee. Global MCU-KO mice, as described previously,6 were maintained on a CD1 background. Genotyping was performed as described in Ref.21 RIP3-KO mice were obtained from Dr Vishva Dixit at Genentech.22 For experiments, mice were anaesthetized with an intraperitoneal injection of sodium pentobarbital (200 mg/kg; Sigma). Heparin (3000 U/kg; Sigma) was co-administered to prevent blood clotting during the isolation procedure. Induction of anaesthesia was confirmed within 5 min of injection by the absence of physical responses, including pedal withdrawal and corneal reflexes.
2.2 Langendorff perfused hearts
Hearts were excised quickly from mice, cannulated via the aorta and connected to a Langendorff perfusion apparatus, through which the heart was perfused retrogradely with Krebs buffer at a constant pressure of 100 cm H2O. For all experiments, hearts were first equilibrated for 20 min, and then subjected to ischaemia for 25 min followed by reperfusion for 2 h. In some cases, hearts were exposed to 30 µM necrostatin-1 prior to ischaemia. Left ventricular developed pressure (LVDP), rate pressure product (RPP), and infarct size were measured for each heart.
2.3 Human induced pluripotent stem cell differentiation
Human induced pluripotent stem cells (iPSC) were acquired as previously described.23 The MCU was knocked-out using CRISPR. Confirmation of MCU-KO clones was done by sequencing and western blot against MCU (Abcam, Cambridge, MA, USA, ab121499, 1:1000). Wildtype and MCU-KO iPSC were differentiated into cardiomyocytes using a 10-day differentiation protocol.24 An adenoviral vector (Serotype 5) expressing the wildtype (RIP3 WT) or a kinase-dead form of RIP3 (RIP3-D160N) was generated. On Day 15 after the end of differentiation, cardiomyocytes were infected with the above adenoviruses [multiplicity of infection (MOI) 20–100]. After an incubation period of 48 h, lactate dehydrogenase (LDH) activity in culture medium was spectrophotometrically assessed using enzymatic procedure SPPYRU01 from Sigma. Cell monolayers were lysed using radioimmunopeptide assay (RIPA) buffer supplemented with protease and phosphatase inhibitors (Roche Complete Mini). Western blotting against RIP3 (Abcam ab56164, 1:1000) and β-actin (Santa Cruz sc-47778, 1:1000) was performed.
2.4 CRC and swelling assays
Calcium-induced mitochondrial swelling was measured in isolated cardiac mitochondria as a decrease in absorbance at 540 nm using a microplate reader (FLUOstar Omega, BMG Labtech, Waltham, MA, USA). Calcium uptake and retention capacity were examined by using the fluorescent calcium indicator Calcium Green-5N (1 µM, Molecular Probes), which measures extramitochondrial calcium. Calcium concentrations between 2.5 and 10 µM were repeatedly added. In some experiments, the calcium ionophore ETH-129 (10 µM) was added to individual wells of mitochondria. ETH-129 transports calcium in an electrogenic manner, as opposed to commonly used ionophores A23187 or ionomycin, which are electroneutral.25 In some experiments, mitochondria were first depleted of their endogenous calcium.
2.5 Mass spectrometry with tandem mass tags
Protein levels were compared in 5 WT and 5 MCU-KO hearts using quantitative tandem mass tags (TMT10plex #90110, Pierce). Identified peptides were then quantified based on the intensities of TMT reporter ions using Proteome Discoverer 2.2 (Thermo Fisher Scientific, Walham, MA, USA). Abundance of phosphorylated proteins was examined using a similar method; however, samples consisted of isolated cardiac mitochondria.
2.6 Immunoprecipitations
For immunoprecipitation (IP) of phosphorylated proteins, mitochondria (300 µg) were incubated with anti-phosphoserine/threonine/tyrosine antibody (#MA1-38450, SPM101, Thermo Fisher Scientific, Waltham, MA, USA) overnight at 4°C with shaking, and then with magnetic Protein G beads (#LSKMAGG10, Millipore). Phospho IP samples were analysed by western blotting with anti-CypD (#ab110324, Abcam, Cambridge, MA, USA). In some experiments, anti-mouse IgG (#sc2025, Santa Cruz Biotechnology, Dallas, TX, USA) was used as a control. The amount of CypD was normalized to the amount of IgG in each lane, and total CypD was confirmed to be unchanged between groups in each experiment.
For pull-down of F1F0 ATP synthase, isolated mitochondria (500 µg) were resuspended in buffer and mitochondrial membranes were solubilized with 1% digitonin. Samples were incubated with ATP synthase immunocapture beads already coupled to agarose beads (#109715, Abcam) overnight at 4°C with shaking. ATP synthase IP samples were analysed by western blotting with anti-CypD (#ab110324, Abcam, Cambridge, MA, USA) and anti-ATP5α (#ab14748, Abcam, Cambridge, MA, USA). In some experiments, anti-mouse IgG was used as a control. The amount of CypD was normalized to the amount of ATP5α in each lane, and total CypD was confirmed to be unchanged between groups in each experiment.
2.7 Mouse embryonic fibroblasts
CypD KO mouse embryonic fibroblasts (MEFs) were obtained from the lab of John Elrod (Temple University, PA, USA), and immortalized by transfection with pBABE-neo large TcDNA (Addgene 1780). CypD WT and S42A and S42D mutants were purchased from GenScript and sub-cloned into the lentiviral vector PLVX-puro (Clontech, Palo Alto, CA, USA). Lentiviruses were produced, purified by ultracentrifugation and tittered in Lenti-X293T cells (Clontech). CypD KO MEFs were infected with lentivirus (MOI of 1–5) in the presence of polybrene (8 µg/mL) for 24 h. Infected MEFs were selected with puromycin (3 µg/mL) and maintained in media with 1 µg/mL puromycin.
2.8 Statistical analyses
Sigmaplot (v13.0, Systat Software Inc., San Jose, CA, USA) was used for all statistical analyses. To estimate the sample size required for each experiment, statistical powering was performed using GPower 3.1 and SigmaPlot 11.0. All data are expressed as mean ± standard error measurement (SEM), and differences were tested with either the Student’s unpaired t-test or two-way analysis of variance (ANOVA) for datasets with more than two groups. Post hoc comparisons were performed using the Holm–Sidak method. Statistical significance was defined as P < 0.05.
3. Results
3.1 Cell death via necroptosis is not up-regulated in MCU-KO hearts
We first tested the hypothesis that loss of MCU-mediated calcium-activated PTP leads to upregulation of the RIP/necroptosis cell death pathway.14,15 Previous studies in kidney have suggested that inhibition of the RIP kinase pathway partially reduces cell death and that inhibition of PTP also partially reduces cell death, whereas inhibition of both pathways was additive.14 We, therefore, considered the hypothesis that deletion of MCU before birth leads to upregulation of necroptosis. If the hypothesis that necroptosis is up-regulated in MCU-KO mice is correct, we would expect inhibition of the necroptosis pathway to show enhanced protection in the MCU-KO hearts. To test the contribution of RIP signalling in cell death in WT and MCU-KO mice, hearts were perfused with the RIP1 inhibitor necrostatin-1 or dimethyl sulfoxide (DMSO) as a vehicle control, and post-ischaemic recovery of LVDP and infarct size were assessed following 25 min of ischaemia and 2 h of reperfusion (Figure 1A). Interestingly, post-ischaemic recovery of LVDP and RPP were improved by necrostatin-1 in WT hearts but remained unchanged in MCU-KO in comparison to control (Figure 1A and B). Necrostatin-1 decreased infarct size in hearts from WT mice, but not in hearts lacking MCU (Figure 1C). These results suggest that RIP1 inhibition protects against I/R injury in WT hearts but offers no protection in hearts from MCU-KO mice.
Figure 1.

Necroptosis is not up-regulated in MCU-KO hearts. (A) The top panel shows the ischaemia-reperfusion protocol, including treatment with 30 µM necrostatin-1 for 5 min prior to ischaemia. RIP1 inhibition with necrostatin-1 increased LVDP in WT, but not in hearts from MCU-KO mice. (B) RPP was increased in WT with necrostatin-1 treatment, but had no effect in MCU-KO. (C) Infarct size was decreased by necrostatin-1 in WT hearts, but remained unchanged in MCU-KO. Data are represented as mean ± SEM. *P < 0.01 (two-way ANOVA), n = 4–5 mice per group for necrostatin-1 experiments. (D) The top panel depicts the ischaemia-reperfusion protocol followed in these experiments. KO of RIP3 increased LVDP at the end of reperfusion, but double KO of RIP3 and MCU had no effect in comparison to WT. (E) RPP followed the same trend, in that RIP3-KO increased functional recovery in WT hearts, but had no effect in RIP3 and MCU double KO hearts. (F) Infarct size was reduced in RIP3-KO hearts, but this protection was not present in hearts from double KO mice. Data are represented as mean ± SEM. *P < 0.01 (two-way ANOVA), n = 5–7 mice per group for RIP3-KO experiments.
To further investigate the hypothesis that the RIP pathway might be contributing to cell death in MCU-KO mice, we examined whether KO of RIP3, the downstream binding partner of RIP1, would be cardioprotective in MCU-KO hearts. Hearts from RIP3-KO mice had improved post-ischaemic LVDP (Figure 1D) and rate-pressure product (Figure 1E), as well as decreased infarct size (Figure 1F) in comparison to WT hearts. However, crossing the RIP3-KO mice with the MCU-KO mice provided no improvement in functional recovery (Figure 1D and E) nor in infarct size (Figure 1E) in comparison to WT or MCU-KO hearts. These results suggest that while RIP3-KO is protective against I/R injury in WT hearts, loss of the RIP pathway does not reduce I/R injury in hearts from MCU-KO mice. Therefore, upregulation of RIP-mediated necroptosis is not responsible for the lack of cardioprotection in MCU-KO hearts following I/R injury. Further, it appears that RIP-mediated cell death either requires MCU or an adaptation in the MCU-KO mice interferes with the necroptosis pathway.
As RIP3 overexpression has been shown to lead to increased cell death,26 we examined a potential role of up-regulated RIP3-mediated death in MCU-KO cells. iPSC-derived cardiomyocytes lacking MCU were generated (Figure 2A). To examine the effect of increasing RIP3 expression, cardiomyocytes were infected with either RIP3 adenovirus or green fluorscent protein (GFP) control adenovirus. Western blotting was performed to confirm successful incorporation of the vector as overexpression of RIP3; Figure 2B shows that cells infected with RIP3 virus express RIP3 and that overexpression levels are similar with a MOI of either 20 or 100. Conversely, cardiomyocytes infected with GFP do not express RIP3. To examine whether RIP3 overexpression altered cardiomyocyte death, LDH release was assayed after infection with RIP3 or GFP adenovirus. Cell death was increased in WT cardiomyocytes infected with RIP3 in comparison to GFP control (Figure 2C). However, this detrimental effect of RIP3 expression did not occur in MCU-KO cardiomyocytes. These results suggest that overexpression of RIP3 causes cardiomyocyte death in WT but not in MCU-KO cardiomyocytes.
Figure 2.

iPSC-derived cardiomyocytes lacking MCU are protected against RIP3-induced necroptosis. (A) KO of MCU in iPSC clones was confirmed with Western blot, where no MCU protein is present in comparison to WT control. β-actin was used as a loading control. (B) Representative western blot analysis of RIP3 protein levels in iPS-cardiomyocytes infected with Ad-GFP (used as control) or Ad-RIP3. Similar expression of RIP3 was seen using an MOI of 20 or 100. (C) Cell viability as assessed by LDH activity after infection with Ad-GFP or Ad-RIP3; data obtained with an MOI of 20 or 100 were pooled since RIP3 expression was similar. Overexpression of RIP3 increased cell death in WT but not in MCU-KO cells. Data are represented as mean ± SEM. *P < 0.0001 (two-way ANOVA), n = 10 samples.
3.2 PTP opening can occur in the absence of MCU and has altered calcium sensitivity
As upregulation of RIP signalling does not appear to be responsible for the lack of cardioprotection in germline MCU-KO mice, we considered whether altered regulation of the PTP might occur in the MCU-KO hearts. To test this hypothesis, we first determined whether pore opening occurs in mitochondria lacking MCU if calcium is able to enter the matrix. To examine this, we used a calcium ionophore, ETH-129, to allow electrophoretic calcium uptake in MCU-KO mitochondria. Calcium uptake assays were performed using Calcium Green-5N to measure extramitochondrial calcium. Figure 3A shows that, in the presence of ETH-129, MCU-KO mitochondria are able to take up repeated additions of calcium. Mitochondrial swelling was then measured to examine whether pore opening occurs in MCU-KO mitochondria in response to a large bolus of calcium. As indicated by a decrease in mitochondrial absorbance, PTP opening occurred in MCU-KO when calcium uptake was facilitated by ETH-129 (Figure 3B). Pore opening is also depicted at the end of the calcium uptake experiment in Figure 3A, when matrix calcium is released. These results indicate that PTP opening occurs in MCU-KO hearts if calcium entry is enabled with ETH-129.
Figure 3.

PTP opening can occur in the absence of MCU. (A) The calcium ionophore ETH-129 permitted calcium entry into mitochondria from MCU-KO. (B) MCU-KO underwent mitochondrial swelling in the presence of ETH. (C) Following depletion of endogenous calcium, mitochondria from MCU-KO underwent pore opening at a lower concentration of calcium than WT. The inset shows that mean calcium retention capacity in MCU-KO is lower than WT. Data are represented as mean ± SEM. *P = 0.04 (Student’s t-test), n = 5 WT and 5 MCU-KO mice.
Next, we tested the hypothesis that less calcium is required to trigger PTP opening in MCU-KO hearts such that baseline matrix calcium may be sufficient to allow PTP opening during I/R, perhaps with the addition of an reactive oxygen species (ROS) trigger. Experiments were performed to measure the calcium sensitivity of PTP opening in mitochondria from WT and MCU-KO mice. To test this, isolated cardiac mitochondria were first depleted of endogenous calcium, to account for the lower basal matrix calcium that exists in the MCU-KO,6 and then exposed to the same extramitochondrial calcium concentration in the presence of the ionophore ETH-129. Calcium uptake assays revealed that the CRC was significantly lower in mitochondria from MCU-KO in comparison to WT (Figure 3C). Therefore, less calcium is required to trigger pore opening in the absence of MCU, suggesting that regulation of the PTP is altered.
3.3 CypD phosphorylation and interaction with ATP synthase is altered in MCU-KO
To gain insight into the mechanism responsible for the difference in calcium sensitivity of the PTP in MCU-KO mitochondria, we examined differences in the mitochondrial phosphoproteome of MCU-KO hearts using quantitative TMT. The phosphorylation state of many proteins was altered in the absence of MCU and the changes in mitochondrial protein phosphorylation following ablation of MCU are shown in Supplementary material online, Table S1. Previous studies have shown that increasing mitochondrial calcium decreases phosphorylation of pyruvate dehydrogenase (PDH) and increases phosphorylation of branched-chain alpha-keto acid dehydrogenase (BCKDH).27 Consistent with these results we find that the lower matrix calcium, which we previously reported in the MCU-KO hearts,6 is associated with a decrease in phosphorylation of BCKDH and an increase in phosphorylation of PDH.
From the mitochondrial phosphoproteome, a protein of interest for regulation of PTP opening that was identified was CypD (gene name peptidyl-prolyl cis-trans isomerase F; Ppif). CypD is a well-known regulator of PTP and this experiment suggested an increase in CypD phosphorylation in MCU-KO; Supplementary material online, Table S2 shows all the peptide spectral matches for phospho-CypD peptides identified in the discovery phosphoproteomic screen. This discovery screen provided insufficient quantitative values for phospho-CypD, and we therefore, performed targeted mass spectrometry by doing multiple reaction monitoring based on mass accuracy and retention time to confirm this phosphorylation site of CypD. The resulting peptides are shown in Table 1. The site of interest consists of three consecutive serines (S40, S41, and S42) located after the mitochondrial targeting sequence and before the cyclophilin isomerase domain. CypD (aka PPIF) is conserved in eukaryotes, and this novel phosphorylation sequence is conserved in most mammals (i.e. human, chimp, mouse, rat, cow); amino acid sequences were aligned using UCSC Genome Browser. In-depth analysis of the fragmentation pattern of the identified peptides (using Scaffold PTM software) predicts with 92% probability that S42 is phosphorylated (P = 0.047), and there is an increase in this phosphorylation in MCU-KO cardiac mitochondria (1.2-fold increase in MCU-KO vs. WT). A difference in CypD phosphorylation cannot be explained by altered CypD protein, as mass spectrometry and Western blotting indicate that CypD protein levels are similar between WT and MCU-KO (Figure 4A).
Table 1.
Multiple reaction monitoring of novel phosphorylated CypD peptide.
| Sequence: GANsssGNPLVYLDVGADGQPLGR | Abundance values (TMT tag, genotype) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Modifications | RT (min) | 126 KO | 127N WT | 127C KO | 128N WT | 128C KO | 129N WT | 129C KO | 130N WT | 130C KO | 131 WT |
| N-Term(TMT6plex); N3(Deamidated); S6(Phospho) | 101.3962 | 7.7 | 10.2 | 11.6 | 7.9 | 7 | 2 | 1.6 | 9.1 | 6.6 | 3.2 |
| N-Term(TMT6plex); N3(Deamidated); S4(Phospho) | 101.449 | 1.7 | 5.9 | 3.6 | 2.8 | 2.3 | 2.7 | 1.5 | 6.4 | 2.7 | 2.3 |
| N-Term(TMT6plex); S6(Phospho); N8(Deamidated) | 101.5204 | 10.6 | 9.2 | 11.9 | 12.7 | 10.2 | 7.9 | 11.2 | 13.3 | 11.6 | 7.1 |
| N-Term(TMT6plex); S6(Phospho); N8(Deamidated) | 101.5537 | 8.2 | 10.1 | 7.9 | 8.9 | 9 | 3.4 | 2.5 | 13.1 | 9.1 | 2.6 |
| N-Term(TMT6plex); S6(Phospho); N8(Deamidated) | 101.607 | 9.8 | 13.8 | 11.2 | 11.3 | 9.3 | 8.8 | 3.6 | 8.7 | 11.1 | 6.8 |
| N-Term(TMT6plex); S4(Phospho); N8(Deamidated) | 101.7375 | 6.7 | 2.4 | 3.2 | 8.4 | 2.8 | 3.2 | 6.3 | 9.2 | 2.6 | |
| N-Term(TMT6plex); N3(Deamidated); S5(Phospho) | 101.9492 | 2.6 | 1.7 | 1.3 | 1.4 | ||||||
Murine CypD peptide sequences identified by quantitative proteomics with TMT-labelling, indicating S40, S41 and S42 as possible phosphorylation sites in WT and MCU-KO cardiac mitochondria. (n = 5 WT, 5 MCU-KO); RT = retention time.
Figure 4.

Phosphoprotein immunoprecipitation showed an increase in CypD phosphorylation in MCU-KO vs. WT. (A) CypD protein levels were similar in mitochondria from WT and MCU-KO hearts (blot representative of n = 4). (B) Mitochondria immunoprecipitated for phospho-S/T/Y were probed for CypD, with IgG used as IP antibody control. (C) Mean data demonstrate that there was an increase in the amount of CypD pulled-down by phospho antibody in MCU-KO compared with WT. Data are represented as mean ± SEM. *P = 0.005 (Student’s t-test), n = 8 WT and 10 MCU-KO mice.
To confirm that CypD phosphorylation is altered at the protein level, IP experiments were performed on cardiac mitochondria using an antibody for phosphorylated proteins. Figure 4B shows a representative Western blot of IP samples that were probed for CypD, with IgG used as an IP control. Interestingly, MCU-KO mitochondria had a significant increase in the amount of CypD pulled down by the phospho antibody (Figure 4C). These results confirm the phosphoproteomics data, and show that the level of CypD phosphorylation is increased in MCU-KO mitochondria.
Phosphorylation of CypD has been reported to be associated with increased PTP opening,17 but the site of phosphorylation was not identified. As CypD has been shown to interact with the proposed PTP component F1F0-ATP synthase,28 experiments were performed to examine the interaction between CypD and ATP synthase. WT and MCU-KO cardiac mitochondria were incubated with an immunocapture antibody to pull-down ATP synthase. Figure 5A depicts IP samples probed for CypD and ATP5A as a pull-down control. Interestingly, results suggest that there was more CypD associated with ATP synthase in MCU-KO in comparison to WT (Figure 5B). Together, these results suggest that following global MCU deletion there is an increase in the association of CypD with ATP synthase.
Figure 5.

Pull-down of ATP synthase revealed an increase in association with CypD in MCU-KO mitochondria. (A) Representative blots of samples that were immunoprecipitated with an F1/F0-ATP synthase antibody, and probed for CypD and ATP synthase subunit α as a pull-down control. (B) There was a significant increase in the amount of CypD associated with ATP synthase in MCU-KO vs. WT. Data are represented as mean ± SEM. *P = 0.02 (Student’s t-test), n = 6 WT and 6 MCU-KO mice.
3.4 Phosphorylation of residue S42 of CypD alters the PTP calcium sensitivity
Since Scaffold PTM software suggested that serine 42 was the most likely site of phosphorylation, we next tested whether mutation of S42 alters the regulation of PTP opening. S42 of CypD was mutated either to a phospho-resistant alanine (S42A), or a phosphomimic glutamine (S42D), and immortalized CypD KO MEFs were infected with lentivirus containing the mutant plasmids. Stable expression of CypD was confirmed by Western blot (see Figure 6A). The CypD-KO MEFs expressing the different CypD constructs were permeabilized with digitonin, and CRC was measured as an index of PTP opening. As shown in Figure 6B and C, CypD-KO MEFs transfected with CypD-S42A exhibited a CRC of approximately 80 nmol of calcium per mg of protein and there was no added benefit of cyclosporin A (CsA). In contrast, CypD-KO MEFs transfected with phosphomimic S42D had a significantly reduced CRC compared with S42A (CRC occurred at ∼50 nmol of calcium) and addition of CsA improved CRC to levels similar to those observed in CypD-S42A MEFs. To determine whether phosphorylation at CypD-S42 decreases CRC by altering the interaction between CypD and ATP synthase, IP experiments were performed to pull-down ATP synthase from mutant MEFs. Figure 6D shows that there was significantly more CypD interacting with ATP synthase in the phosphomimic S42D in comparison to the phospho-resistant S42A. To examine whether the CypD-S42 phosphorylation event affects cell death, assays were performed to measure cell death following 6 or 8 h of oxidative stress induced with H2O2. Cell death was significantly increased in the S42D mutant in comparison to S42A after 8 h exposure to 1 mM H2O2 (Figure 6E). Taken together, these data suggest that phosphorylation of CypD enhances PTP opening and cell death by increasing the binding of CypD to the putative PTP component ATP synthase and increasing the PTP calcium sensitivity, and that residue S42 is important in this regulation.
Figure 6.
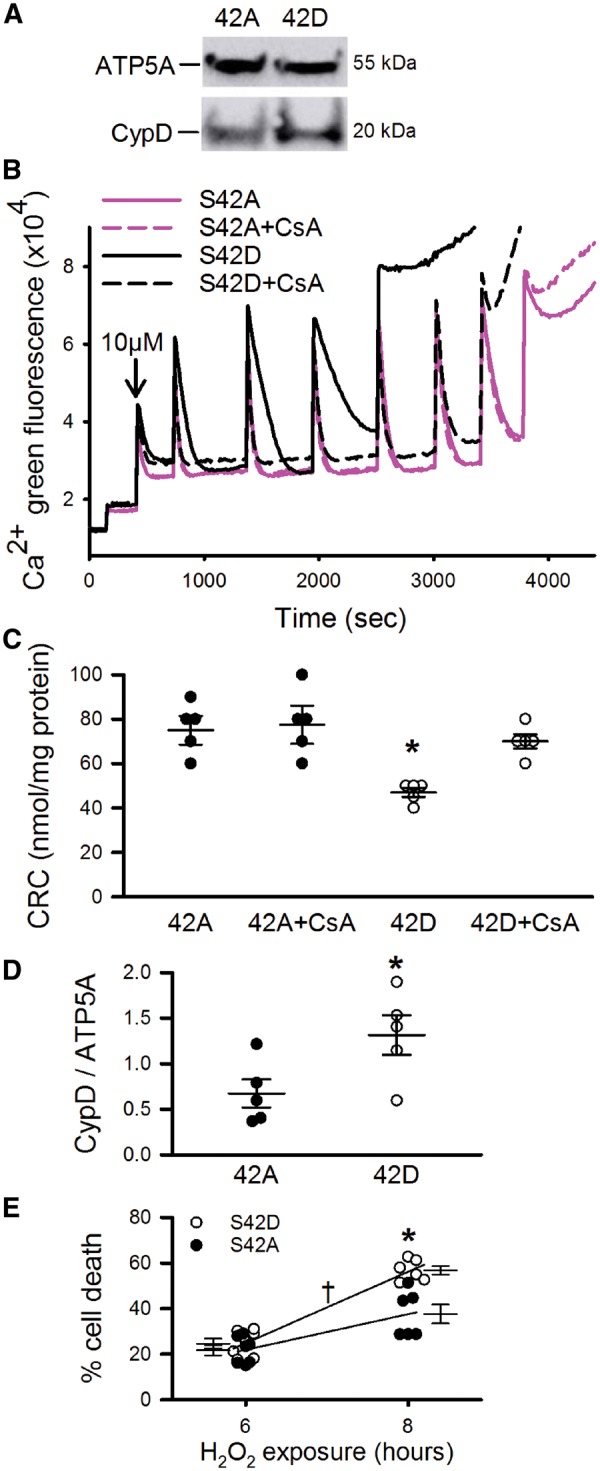
Phosphorylation of S42-CypD decreases CRC and increases CypD association with ATP synthase and cell death. (A) CypD levels were measured between different stable cell lines containing S42A or S42D mutations. ATP synthase subunit α was used as a loading control. (B) Representative calcium uptake assay in CypD-KO MEFs infected with lentivirus containing mutated CypD, either phospho-resistant S42A or phosphomimic S42D. (C) Mean CRC values, measured as nmol of calcium per mg of protein, indicated that phosphorylation of CypD at S42 decreased CRC. Data are represented as mean ± SEM. *P = 0.005 (two-way ANOVA), n = 5 independent experiments. (D) Immunoprecipitation of ATP synthase pulled down more CypD in the S42D mutant than S42A. Data are represented as mean ± SEM. *P = 0.02 (Student’s t-test), n = 5 independent experiments. (E) Oxidative stress induced with 1 mM H2O2 caused more cell death in the phosphomimic S42D mutant than in the phospho-resistant S42A mutant. P < 0.001 (Two-way ANOVA), *indicates within time point, and †indicates overall effect of cell line; n = 6–7 independent experiments.
4. Discussion
Our results provide valuable insight into the mechanisms that regulate PTP and identify factors that may explain why the absence of rapid mitochondrial calcium uptake is not cardioprotective in hearts from mice lacking MCU prior to birth. Two studies reported that cardiac-specific tamoxifen-inducible loss of MCU in adults is protective against I/R injury.8,9 However, the cardiac-specific dominant-negative MCU-KO mouse was not protected.7 This dominant-negative MCU-KO model consists of myocardial-specific transgenic expression of a form of MCU that is unable to take up calcium. Therefore, these mice lack functional MCU in cardiomyocytes from birth. No protection was observed in dominant-negative MCU vs. WT, thus suggesting that the disparity in protection between MCU-KO models is dependent on the timing of MCU deletion and not the tissue-specificity. To understand the cell death pathways that may be underlying the lack of protection in germline MCU-KO, we investigated whether alterations were occurring in either necroptosis or PTP opening. The RIP3 cell death pathway does not appear to be a significant contributor to I/R injury in MCU-KO hearts, and instead the regulation of PTP opening may be altered. Specifically, we have identified an alteration in phosphorylation of CypD which sensitizes the PTP to calcium.
The present study shows that a lack of cardioprotection in germline MCU-KO mice is not due to upregulation of necroptosis, which is regulated necrosis involving RIP kinases, as inhibition or upregulation of necroptosis did not influence cell death in the absence of MCU. RIP3 is expressed in heart tissue, and overexpression in cardiomyocytes induces necroptosis, whereas loss of RIP3 is protective.29 Studies in kidney have suggested that necroptosis and PTP opening are independent mediators of cell death, as inhibition of ischaemic cell death by RIP3-KO is additive with protection afforded by CypD-KO.14 Consistent with previous studies, we find that inhibition of necroptosis by either RIP1 inhibition with necrostatin-1 or RIP3-KO was protective against I/R in WT, and upregulation of the RIP pathway increased cell death in WT cardiomyocytes. Interestingly, inhibition or upregulation of the RIP pathway had no effect on I/R injury or cell death in MCU-KO. The Xiao lab has recently shown that RIP3 mediates myocardial necroptosis by triggering PTP opening.30 Our data add to this and show that MCU is downstream of RIP3 in the same pathway. Further, RIP3 has recently been shown to act on PDH,31 which may be involved in the apparent absence of necroptosis in global MCU-KO mice where phosphorylation of PDH at S293 is greatly increased compared with WT.6 Overall, the results presented here indicate that upregulation of RIP-mediated cell death in MCU-KO mice is unlikely to be responsible for the lack of cardioprotection.
Our data suggest that loss of MCU may alter PTP opening such that less calcium is required to trigger PTP, which may be due to changes in CypD phosphorylation and pore regulation. We have shown that MCU-KO hearts are not protected from I/R injury, and PTP is present in the MCU-KO hearts. A key finding from the present study is that PTP opening can still occur in the absence of MCU. We use the calcium ionophore ETH-129, which has been used previously to allow calcium entry and PTP opening in yeast, as they do not possess MCU.32–34 Our results indicated that, in the presence of ETH-129, mitochondria lacking MCU can undergo calcium-triggered PTP opening. This observation indicates that MCU is not required for PTP formation or opening in the presence of ETH-129. Interestingly, the pore opened at a lower calcium concentration in MCU KO mitochondria than in WT, which would be consistent with PTP opening in MCU-KO hearts following I/R in response to lower matrix calcium concentration. It has been proposed that in I/R an increase in ROS is the primary trigger of PTP,35,36 but a trigger level of calcium is still likely to be required. Our results suggest that this trigger level of calcium may be lower in global MCU-KO hearts. It is possible that the basal matrix calcium concentration in MCU KO mitochondria is sufficient to trigger PTP in response to an increase in ROS, such as occurs following I/R.
Regarding possible mechanisms for the increase in calcium sensitivity in MCU-KO, our findings highlight CypD-S42 as a key regulator of mitochondrial CRC. Although phosphorylation of CypD has been suggested to increase PTP opening, this is the first study to report that phosphorylation of a specific residue of CypD alters PTP calcium sensitivity. We identified an increase in phosphorylation of CypD at S42 in MCU-KO mitochondria. Expression of CypD with a mutation of this site to a phosphomimic residue in CypD-KO MEFs decreased CRC by almost 50%, thus suggesting that increased phosphorylation of CypD-S42 in MCU-KO hearts is involved in increasing the PTP calcium sensitivity. We have also linked phosphorylation of CypD-S42 to an increase in cell death, which could be due to the sensitized PTP. Further, MCU-KO mitochondria have an increase in the association of CypD with the F1F0-ATP synthase, and this interaction appears to be largely regulated by CypD-S42 phosphorylation. Overall, our data suggest that increased phosphorylation of CypD-S42 in MCU-KO hearts increases the CypD-ATP synthase interaction and the calcium sensitivity of PTP. As such, it is possible that the PTP trigger calcium level is lower in MCU-KO hearts. These alterations in CypD signalling could be involved in explaining why germline MCU-KO mice are not protected from I/R injury.
As one of the biggest changes in the phosphoproteome of MCU KO hearts is an increase in PDH phosphorylation, it is tempting to speculate that the calcium-sensitive PDH phosphatase may be acting on CypD. Lower basal matrix calcium in MCU-KO vs. WT would cause less activation of PDH phosphatase6 and thus less dephosphorylation of its substrates. Alternatively, the increase in CypD-S42 phosphorylation in MCU-KO may be due to enhanced kinase activity. GSK-3β and Akt2 have both been previously proposed to phosphorylate CypD in cancer cells.17,37 These potential mechanisms underlying CypD-S42 phosphorylation are of interest for future studies.
In summary, our results provide valuable insight into the compensatory changes that occur when MCU is knocked-out from birth and provide understanding as to why the absence of rapid mitochondrial calcium uptake is not cardioprotective in germline MCU-KO hearts. We propose that increased phosphorylation of CypD sensitizes or primes the pore to opening and reduces the calcium sensitivity, such that it can now be activated by ROS at a lower calcium concentration. We have identified a novel phosphorylation site on CypD (S42) that regulates PTP sensitivity to calcium, which could enhance mitochondria-triggered cell death in I/R. These results identify how a specific phosphorylation event on CypD can have significant effects on PTP opening and cell death. If similar modifications are present in cardiac disease models, this could sensitize cardiomyocytes to ischaemic stress. Overall, our findings suggest that increased phosphorylation of CypD at S42 in global MCU-KO hearts may contribute to why these mice are not cardioprotected.
Supplementary Material
Acknowledgements
The authors would like to thank Mélanie Paillard (Lyon, France) for the phospho IP protocol, John Elrod (Temple University, PA, USA) for CypD-KO mouse embryonic fibroblasts, and the NHLBI iPSC core facility for producing the MCU-KO iPS-cells.
Conflict of interest: none declared.
Funding
This work was supported by intramural funding from the National Heart Lung and Blood Institute at the National Institutes of Health [grant number ZIA-HL002066 to E.M.]; and Fondation Leducq [grant number 16CVD04 to E.M. and P.B.].
Footnotes
Time for primary review: 36 days
This manuscript was handled by Consulting Editor, Godfrey Smith.
References
- 1. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK.. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011;476:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R.. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011;476:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwong JQ. The mitochondrial calcium uniporter in the heart- energetics and beyond. J Physiol (Lond) 2017;595:3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glancy B, Balaban RS.. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012;51:2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hunter DR, Haworth RA.. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 1979;195:453–459. [DOI] [PubMed] [Google Scholar]
- 6. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T.. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 2013;15:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME.. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA 2015;112:9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW.. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 2015;12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD.. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 2015;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duchen MR, McGuinness O, Brown LA, Crompton M.. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res 1993;27:1790–1794. [DOI] [PubMed] [Google Scholar]
- 11. Griffiths EJ, Halestrap AP.. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol 1993;25:1461–1469. [DOI] [PubMed] [Google Scholar]
- 12. Murphy E, Steenbergen C.. Preconditioning: the mitochondrial connection. Annu Rev Physiol 2007;69:51–67. [DOI] [PubMed] [Google Scholar]
- 13. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P.. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135–147. [DOI] [PubMed] [Google Scholar]
- 14. Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S.. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 2013;110:12024–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Linkermann A, Konstantinidis K, Kitsis RN.. Catch me if you can: targeting the mitochondrial permeability transition pore in myocardial infarction. Cell Death Differ 2016;23:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD.. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- 17. Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P.. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci USA 2010;107:726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA.. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010;2:914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E.. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 2011;286:40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD.. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS One 2015;10:e0130520.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E, Finkel T.. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol 2015;85:178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newton K, Sun X, Dixit VM.. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 2004;24:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beers J, Linask KL, Chen JA, Siniscalchi LI, Lin Y, Zheng W, Rao M, Chen G.. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci Rep 2015;5:11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin Y, Linask KL, Mallon B, Johnson K, Klein M, Beers J, Xie W, Du Y, Liu C, Lai Y, Zou J, Haigney M, Yang H, Rao M, Chen G.. Heparin promotes cardiac differentiation of human pluripotent stem cells in chemically defined albumin-free medium, enabling consistent manufacture of cardiomyocytes. Stem Cells Transl Med 2017;6:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang E, Erdahl WL, Hamidinia SA, Chapman CJ, Taylor RW, Pfeiffer DR.. Transport properties of the calcium ionophore ETH-129. Biophys J 2001;81:3275–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Q, Li G, Lan X, Zheng M, Chen KH, Cao CM, Xiao RP.. Receptor interacting protein 3 suppresses vascular smooth muscle cell growth by inhibition of the phosphoinositide 3-kinase-Akt axis. J Biol Chem 2010;285:9535–9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boja ES, Phillips D, French SA, Harris RA, Balaban RS.. Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation. J Proteome Res 2009;8:4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G.. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 2009;284:33982–33988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe H-J, Linkermann A, Wolf MJ, Rose-John S, Lüllmann-Rauch R, Adam D, Flögel U, Heikenwalder M, Luedde T, Frey N.. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 2014;103:206–216. [DOI] [PubMed] [Google Scholar]
- 30. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP.. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med 2016;22:175–182. [DOI] [PubMed] [Google Scholar]
- 31. Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, Chen X, Liang Y, Wu J, Zhao S, Zhou D, Han J.. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol 2018;20:186–197. [DOI] [PubMed] [Google Scholar]
- 32. Yamada A, Yamamoto T, Yoshimura Y, Gouda S, Kawashima S, Yamazaki N, Yamashita K, Kataoka M, Nagata T, Terada H, Pfeiffer DR, Shinohara Y.. Ca2+-induced permeability transition can be observed even in yeast mitochondria under optimized experimental conditions. Biochim Biophys Acta 2009;1787:1486–1491. [DOI] [PubMed] [Google Scholar]
- 33. Carraro M, Giorgio V, Šileikytė J, Sartori G, Forte M, Lippe G, Zoratti M, Szabò I, Bernardi P.. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 2014;289:15980–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung DW, Bradshaw PC, Pfeiffer DR.. Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J Biol Chem 1997;272:21104–21112. [DOI] [PubMed] [Google Scholar]
- 35. He L, Lemasters JJ.. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002;512:1–7. [DOI] [PubMed] [Google Scholar]
- 36. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ.. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, Seo JH, Kossenkov AV, Michalek RD, Schultz DC, Bosari S, Languino LR, Altieri DC.. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 2015;107:dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.