

Abstract
Background
The recognition of distinct molecular subgroups within cholangiocarcinoma (CC), along with the increasing availability of targeted therapies, suggests that further characterization of the prevalence and prognosis of frequently occurring subgroups may assist with the development of more effective treatment approaches for the management of CC. A systematic review was performed to investigate the prevalence of isocitrate dehydrogenase 1 (IDH1) mutations (mIDH1) in patients with CC, the possible clinical and prognostic significance of mIDH1, and the presence of co-mutations in tumors with mIDH1.
Methods
This review was conducted using the Cochrane dual-reviewer methodology and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses protocol (PRISMA-P) guidelines. Searches were performed in Embase, MEDLINE, the Cochrane Central Trials Register and Database of Systematic Reviews, and other Cochrane Library assets using terms for CC and mIDH1 with no language or date restrictions for articles published up to December 31, 2017. Searches were also performed of abstracts presented at the following conferences in 2016 and 2017: American Society of Clinical Oncology (ASCO), ASCO-Gastrointestinal Cancers Symposium (ASCO-GI), the European Society for Medical Oncology (ESMO), and ESMO-Asia. Screening was performed separately by two reviewers and cross-checked. Any discrepancies between reviewers were resolved by a senior researcher. Data from all selected references were recorded in a data extraction grid.
Results
A total of 46 publications met the inclusion criteria and were included in the systematic review. Of these publications, 45 reported the frequency of mIDH1 among a total sample of 5,393 patients with CC. mIDH1 was enriched in intrahepatic CC (ICC), with 552 (13.1%; 95% CI, 12.1–14.2) of the 4,214 patients with ICC having the mutation compared with 9 (0.8%; 95% CI, 0.4–1.5%) of the 1,123 patients with extrahepatic CC (ECC). The percentage of females with mIDH1 CC (66.2%; 95% CI, 57.7–73.7%) was higher than in the overall CC population (44.4%). The frequency of mIDH1 in patients with ICC reported in individual studies ranged from 4.5–55.6%, and a significantly higher frequency was reported in non-Asian centers compared with Asian centers (weighted mean, 16.5% vs. 8.8%; P<0.001). The prevalence of mIDH1 in patients with ICC at USA centers was 18.0% (95% CI, 16.4–19.8%). Eleven publications reported the prevalence of co-mutations in patients with mIDH1 ICC, with the most frequent being AT-rich interactive domain-containing protein 1A (ARID1A) (22.0%), BRCA1-associated protein 1 (BAP1) (15.5%), and PBRM1 (13.3%). Eight publications investigated the possible prognostic significance of mIDH1. None of the studies reported a statistically significant association between mIDH1 and overall survival (OS), progression-free survival (PFS), or time to progression.
Conclusions
This systematic review substantiates the prevalence of mIDH1 in CC and further characterizes clinical, pathologic, and genetic covariates within this sub-population. Co-mutation data may inform future studies of mechanisms of response and resistance to mIDH1-targeted therapies.
Keywords: Cholangiocarcinoma (CC), isocitrate dehydrogenase 1 (IDH1), prevalence, clinical prognosis, systematic review
Introduction
Cholangiocarcinomas (CC) are a heterogeneous group of biliary epithelial tumors arising from the intrahepatic, perihilar, and distal biliary tree. Each anatomical subtype has a distinct clinical presentation, though prognosis is poor for all three subtypes (1). Although an uncommon disease [overall incidence <1 per 100,000 in the USA and Europe (2)], the global incidence of CC, particularly intrahepatic CC (ICC), has increased in recent years (1-3). The main risk factors for CC include chronic biliary inflammation from hepatitis B and hepatitis C virus infections, primary sclerosing cholangitis, inflammatory bowel disease, liver fluke infestation, obesity and diabetes (4,5). Other etiologic factors associated with CC include cirrhosis, alcohol, smoking, hepatolithiasis, fatty liver disease, and cholelithiasis (6).
CC may be asymptomatic during the early stages of disease and is often diagnosed at advanced stages or as metastatic disease. For localized tumors without metastatic spread, surgical resection and—in rare cases—liver transplantation can be curative, though recurrence rates are high. Only about 10–15% of patients are eligible for curative surgery (7), and 5-year overall survival (OS) rates are reportedly low (30%) even with resection, reflecting the high rates of disease recurrence (4,8). Gemcitabine plus cisplatin is the current standard of care for patients with advanced stages of disease ineligible for surgery; however, the median survival remains less than 1 year (1), and there are no established treatments with survival benefit after failure of gemcitabine plus cisplatin. There is an urgent need for new treatment options and approaches for the management of CC.
Tumor molecular profiling has identified substantial genetic heterogeneity between and within anatomic subtypes of biliary cancers. For example, Javle et al. (9) compared the predominant mutations present in ICC, extrahepatic CC (ECC), and gallbladder CC. In ICC, tumor protein 53 (TP53) (27%), cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) (27%), KRAS proto-oncogene, GTPase (KRAS) (22%), AT-rich interactive domain-containing protein 1A (ARID1A) (18%), and isocitrate dehydrogenase 1 (IDH1) (16%) were the most frequently occurring mutations, while in ECC the following mutations occurred in more than 15% of tumors: KRAS (42%), TP53 (40%), CDKN2A/B (17%), SMAD family member 4 (SMAD4) (21%), and CDKN2A/B (19%). Jusakul et al. (10) also reported differences in the genetic profile and clinical characteristics between CC subtypes. These authors defined four subtypes based on different clusters of mutations: cluster 1 was defined as having higher frequencies of ARID1A and BRCA1/2 mutations; cluster 2 as having upregulated catenin beta 1 (CTNNB1), WNT5B, and AKT1 expression; cluster 3 as having upregulation of immune checkpoint genes [programmed cell death protein 1 (PD-1), programmed death-ligand 2 (PD-L2), and BTLA]; and cluster 4 as having BRCA1-associated protein 1 (BAP1) mutations, IDH1/2 mutations and FGFR alterations. Clusters 1 and 2 were largely found in ECC tumors while clusters 3 and 4 were more common in ICC tumors. Further analysis found that patients having tumors with cluster 1 or 2 subtypes had a worse OS than those with tumors of the cluster 3 or 4 subtype. Other authors have also reported differences in the mutation profiles between the two main CC anatomic subtypes, ICC and ECC (11-19).
For some molecular subgroups of CC, specific inhibitors offer potential for a targeted therapeutic approach. In particular, FGFR2 fusions, B-Raf proto-oncogene, serine/threonine kinase (BRAF), HER2, and microsatellite instability high (MSI-H) or mismatch repair deficiency are amenable to targeted therapies in other tumor types, suggesting these may be actionable in CC, as well. For example, the FGFR inhibitors ponatinib, dovitinib, and BGJ398 have been shown to inhibit cell proliferation and induce apoptosis in a mouse xenograft model derived from a CC tumor having an FGFR fusion protein (20). Moreover, Borad et al. have reported achievement of stable disease in a patient in response to the pan-FGFR inhibitor ponatinib (21). Responses to vemurafenib have been reported in two studies including patients with CC who had BRAF mutations (22,23), and responses to pembrolizumab have been reported in CC patients with MSI-H and mismatch repair deficient tumors (24,25).
Another emerging molecular target in a subset of CC is mutant IDH1. IDH1 is a NADP (+)-dependent metabolic enzyme that catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate. IDH1 mutations (mIDH1) result in a loss of function for normal oxidative decarboxylation of isocitrate, and a gain of function for the NADPH-dependent reduction of α-ketoglutarate to produce the oncometabolite D-2-hydroxyglutarate (2-HG) (26,27). Mutations in IDH1 have been identified in approximately 80% of lower-grade gliomas and secondary glioblastoma (28,29), approximately 50% of chondrosarcomas (30), and 6–10% of cases of acute myeloid leukemia (AML) (31-34). Mutations in IDH1 have also been observed in a subset of cases of CC, although the population incidence and prognostic impact of the mutation in this disease have not been established. Studies reporting on the frequency of mIDH1 in CC suggest that the incidence is higher in the ICC subtype (9,10). However, many of these studies have involved relatively small numbers of patients from single centers, which limits their interpretation.
The mutant IDH1 inhibitor ivosidenib has been approved by the USA Food and Drug Administration (FDA) for the treatment of adult patients with relapsed or refractory AML with a susceptible IDH1 mutation, as detected by an FDA-approved test, and has demonstrated activity in patients with mIDH1-positive CC in a large phase 1 trial (35). In patients with CC treated with ivosidenib, 6-month progression-free survival (PFS) was 38% and 12-month PFS was 20%; 56% of patients achieved stable disease and 5% achieved a partial response. A pivotal global phase 3 clinical trial is underway to determine the efficacy of ivosidenib compared to placebo after progression on standard therapies in advanced mIDH1 CC (ClincialTrials.gov NCT03173248).
The emergence of distinct molecular subgroups within CC along with the availability of targeted therapies, including ivosidenib, warrants further characterization of the prevalence and prognostic impact of the more frequently occurring subgroups. This systematic review was performed to investigate the frequency, clinical and pathologic covariates, and prognostic impact of activating mIDH1 in patients with CC.
Methods
The systematic review was conducted using a standardized approach, following Cochrane dual-reviewer methodology, and was in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses protocol (PRISMA-P) guidelines (36).
Search strategy and inclusion criteria
Searches were performed in Embase, MEDLINE via Ovid, the Cochrane Central Trials Register and Database of Systematic Reviews, and other Cochrane Library assets using terms for CC and mIDH1 with no date or language restrictions for articles published up to December 31, 2017. Searches were also performed for abstracts presented at the following conferences in 2016 and 2017: the American Society of Clinical Oncology (ASCO), ASCO-Gastrointestinal Cancers Symposium (ASCO-GI), the European Society for Medical Oncology (ESMO), and ESMO-Asia. In addition, the references of relevant systematic reviews were manually screened for relevant references not identified in the electronic searches.
The following search terms were used: CC; Klatskin tumor; biliary tract or biliary or bile duct and carcinoma or cancer or neoplasm or tumor or tumour; cholangiocellular carcinoma; hepatobiliary and carcinoma or cancer or neoplasm or tumor or tumour; cholangiolar carcinoma; hepatocholangiocarcinoma; isocitrate dehydrogenase 1; IDH1; IDH 1; IDH-1. The detailed search terms are provided as Table S1.
Screening and data extraction
Screening based on title and abstract was performed separately by two reviewers and cross-checked. Full papers were obtained for all references selected based on abstract and title and were screened for inclusion. The following data elements were extracted, where available, from the literature: full citation information, data source, country and region, publication type, study type and characteristics, quality of evidence, study sponsor, sample size, study population, interventions, treatment duration, length of follow-up, frequency/incidence rate of mIDH1, other genes analyzed for mutation, anatomic location of tumors, demographics (age, sex, and race), stage at diagnosis, number of previous lines of treatment, clinical covariates, clinical outcomes, and time from when endpoints were assessed. Data from all selected references were extracted into an agreed extraction grid by one researcher and reviewed by a second researcher. Any discrepancies between reviewers were discussed and uncertainties were resolved by a senior researcher.
Statistical analysis
Statistical significance for the differences in the frequency of mIDH1 in subgroups of patients according to tumor location and geographical location were assessed. Confidence intervals of proportions were calculated using GraphPad QuickCalcs (http://www.graphpad.com/quickcalcs/ConfInterval1.cfm) and odds ratios were calculated using MEDCALC Statistical Software (https://www.medcalc.org/calc/odds_ratio.php). Sensitivity analyses were conducted to assess the magnitude of impact of any potential double-counting.
Results
A total of 46 publications met the inclusion criteria and were investigated in this review (Figure 1, Table 1). Of these, 45 selected publications [29 full-length manuscripts and 16 conference abstracts (56-71)] reported the frequency of mIDH1; the other publication reported the results of a clinical trial in patients with mIDH1 CC (35). Because this study specifically selected for patients with mIDH1, it was not included in the mIDH1 prevalence assessment; however, it did contribute to the analysis of demographic characteristics of mIDH1-positive patients, mIDH1 subtypes, and clinical outcomes.
Figure 1.

PRISMA flow diagram for screening and selection of the literature search for CC and mutations in IDH1. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses; CC, cholangiocarcinoma; IDH1, isocitrate dehydrogenase 1; mIDH1, IDH1 mutation.
Table 1. Reported frequencies of mIDH1 in patients with ICC.
| Study (reference) | Country or region | ECC | ICC | |||||
|---|---|---|---|---|---|---|---|---|
| N | IDH1, n (%) | IDH1, female, n (%) | N | IDH1, n (%) | IDH1, female, n (%) | |||
| Akita et al. 2017 (11) | Japana | 32 | NR | NR | 47 | 4 (8.5) | NR | |
| Arnold et al. 2015 (12) | Germany | 34 | NR | NR | 26 | 3 (11.5) | NR | |
| Borad et al. 2014 (21) | USA | NR | NR | NR | 6 | 0 (0.0) | NR | |
| Borger et al. 2012 (37) | USAa | 22 | NR | NR | 40 | 8 (20.0) | NR | |
| Borger et al. 2014 (38) | USA (screening cohort)a; China (validation cohort)a | NR | NR | NR | 31 38 |
11 (35.5) 4 (10.5) |
NR | |
| Chan-on et al. 2013 (39) | Romania, Singapore, Thailand | Liver fluke O. viverrini cohort: 46 | 1 (2.2) | 0 (0.0) | Liver fluke O. viverrini cohort: 62 | 1 (1.6) | 1 (100.0) | |
| Non-O. viverrini-related etiologies: 44 | 1 (2.3) | 0 (0.0) | Non-O. viverrini-related etiologies: 57 | 10 (17.5) | 7 (70.0) | |||
| Churi et al. 2014 (13) | USAa | 20 | NR | NR | 55 | 10 (18.2) | NR | |
| Doherty et al. 2016 (40)b | Canada | 19 | NR | NR | 21 | 2 (9.5) | NR | |
| Farshidfar et al. 2017 (41) | USA | TCGA analysis set: 4 | 1 (25.0) | NR | TCGA analysis set: 32 | 4 (12.5) | 3 (75.0) | |
| Additional sample set: 5 | 0 (0.0) | Additional sample set:10 | 2 (20.0) | 1 (50.0) | ||||
| Fujimoto et al. 2015 (42) | Japan | NR | NR | NR | 58 | 4 (6.9) | 3 (75.0) | |
| Goyal et al. 2015 (43) | USAa | NR | NR | NR | 104 | 26 (25.0) | 16 (61.5) | |
| Hess et al. 2015 (44)b | Central Europe | NR | NR | NR | 38 | 3 (7.9) | NR | |
| Holcombe et al. 2014 (45)b | USA | 115 | NR | NR | 291 | 52 (17.9) | NR | |
| Holcombe et al. 2015 (46)b | USA | 126 | NR | NR | 434 | 61 (14.1) | NR | |
| Javle et al. 2016 (9) | USA | 57 | NR | NR | 412 | 66 (16.0) | NR | |
| Jiao et al. 2013 (47) | Italy | NR | NR | NR | Discovery screen: 32 | Discovery screen: 4 (12.5) | Discovery screen: NR | |
| Prevalence screen: 32 | Prevalence screen: 5 (15.6) | Prevalence screen: 1 (20.0) | ||||||
| Jusakul et al. 2017 (10) | Brazil, China, France, Italy, Japan, Korea, Romania, Singapore, Taiwan, Thailand | 168 | 3 (1.8) | NR | 291 | 13 (4.5) | NR | |
| Kipp et al. 2012 (48) | USAa | 27 | 1 (3.7) | NR | 67 | 13 (19.4) | NR | |
| Lee et al. 2016 (14) | USA | 99 | NR | NR | NR | NR | NR | |
| Lee et al. 2016 (49) | South Korea | 24 | NR | NR | 17 | 1 (5.9) | 0 (0.0) | |
| Lee et al. 2017 (50) | South Korea | NR | NR | NR | 46 | 3 (6.5) | 1 (33.3) | |
| Liau et al. 2014 (51) | Taiwan | NR | NR | NR | 171 | 14 (8.2) | NR | |
| Lo et al. 2016 (52)b | USA | NR | NR | NR | 29 | 2 (6.9) | NR | |
| Lowery et al. 2016 (53)b | NR | NR | NR | NR | 30 | 9 (30.0) | NR | |
| Mattis et al. 2017 (54)b | USA | 12 | NR | NR | 9 | 5 (55.6) | NR | |
| Misumi et al. 2017 (55) | Japan | NR | NR | NR | 101 | 19 (18.8) | NR | |
| Nakamura et al. 2015 (15) | Japan | 86 | NR | NR | 145 | 8 (5.5) | NR | |
| Pak et al. 2017 (56)b | USA | NR | NR | NR | 66 | 15 (22.7) | NR | |
| Patel et al. 2017 (57)b,c | UK | 54 | NR | NR | NR | NR | NR | |
| Pawlick et al. 2014 (58)b | Europe, and USA | NR | NR | NR | 138 | 11 (8.0) | NR | |
| Putra et al. 2015 (16) | USA | 8 | NR | NR | 3 | 1 (33.3) | 1 (100.0) | |
| Reyes et al. 2017 (59)b | USA | 42 | NR | NR | 178 | 38 (21.3) | NR | |
| Ross et al. 2014 (60) | USA | NR | NR | NR | 28 | 9 (32.1) | 7 (77.8) | |
| Ruzzenente et al. 2016 (17) | Italya | 38 | 1 (2.6) | NR | 53 | 7 (13.2) | NR | |
| Sia et al. 2015 (61) | Italy, Spain, USA | NR | NR | NR | 107 | 11 (10.3) | NR | |
| Simbolo et al. 2014 (19) | Italy | 57 | NR | NR | 70 | 11 (15.7) | NR | |
| Tsokos et al. 2015 (62)b | USA | NR | NR | NR | 10 | 4 (40.0) | NR | |
| Ueno et al. 2017 (63)b | Japan | 27 | 1 (3.7) | NR | 31 | 3 (9.7) | NR | |
| Wachsmann et al. 2017 (64)b | USAa | 11 | NR | NR | 39 | 3 (7.7) | NR | |
| Wang et al. 2013 (65) | Chinaa; USA | NR | NR | NR | 326 | 23 (7.1) | NR | |
| Winter et al. 2017 (66)b | UK | NR | NR | NR | 4 | 1 (25.0) | NR | |
| Yasui et al. 2016 (67)b | Japan | NR | NR | NR | 49 | 5 (10.2) | NR | |
| Zhu et al. 2018 (68) | Chinaa | NR | NR | NR | 78 | 7 (9.0) | NR | |
| Zhu et al. 2014 (69) | China, Belgium, Romania, USA | NR | NR | NR | 200 | 31 (15.5) | NR | |
| Zou et al. 2014 (70) | Chinaa | NR | NR | NR | 102 | 5 (4.9) | NR | |
a, single center; b, conference abstract; c, anatomic location not specified. mIDH1, IDH1 mutation; IDH1, isocitrate dehydrogenase 1; ICC, intrahepatic cholangiocarcinoma; ECC, extrahepatic cholangiocarcinoma; NR, not reported; O. viverrini, Opisthorchis viverrini; TCGA: The Cancer Genome Atlas.
Most studies involved cohorts of patients from a single center. Publications include cohorts from centers in the USA (n=24), Europe (n=12, including France, Germany, Italy, Romania, Spain, and the United Kingdom), Asia (n=18, including China, Japan, Singapore, South Korea, Taiwan, and Thailand), and South America (n=1, Brazil) (Table 1) (some studies involved centers from more than one country).
Frequency of mIDH1
The 45 publications reporting the frequency of mIDH1 provided data for a total of 5,393 patients. Of these, 4,214 (78.1%) had ICC, 1,123 (20.8%) had ECC, and for 56 (1.0%) the anatomic location was not reported. Of the patients with ICC, 552 were mIDH1-positive (13.1%; 95% CI, 12.1–14.2%), as were nine patients with ECC (0.8%; 95% CI, 0.4–1.5%), and one patient for whom the anatomical location was not reported (1.8%; 95% CI, <0.0001–10.3%). The incidence in ICC was significantly enriched compared with ECC (P value <0.0001).
The frequency of mIDH1 in ICC was reported to range from 4.5% (13/291) for a study among patients from 10 different (primarily Asian) countries (10) to 55.6% (5/9) for a cohort of patients from a single USA center (63) (Figure 2). The equivalent data for the frequency of ECC are also shown in Figure 2.
Figure 2.
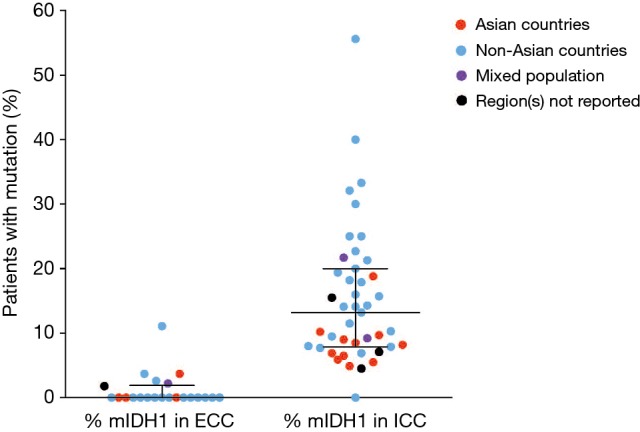
Frequency of mIDH1 according to tumor location. Dots represent individual studies where mIDH1 was assessed in each anatomic location. Black lines indicate median with interquartile range. mIDH1, isocitrate dehydrogenase 1 mutation; ECC, extrahepatic cholangiocarcinoma; ICC, intrahepatic cholangiocarcinoma.
Both geographic location of the treatment center and mIDH1 status could be determined for 3,397 (80.6%) of the 4,214 patients with ICC and 955 (85.0%) of the 1,123 patients with ECC, allowing for comparisons of mIDH1 frequency rates across regions. Several multi-national studies that did not provide the geographic breakdown of their CC or IDH1-mutated CC patient populations could not be included in this calculation. Among patients with ICC, mIDH1 were reported in 399 (16.5%; 95% CI, 15.1–18.1%) of 2,416 patients treated at non-Asian centers and in 86 (8.8%; 95% CI, 7.2–10.7%) of 981 ICC patients treated at Asian centers (odds ratio, 2.06; 95% CI, 1.61–2.63, P<0.0001). The prevalence of mIDH1 reported among the subset of 1,904 ICC patients treated at USA centers was 18.0% (95% CI, 16.4–19.8%) (Table 2).
Table 2. Prevalence of mIDH1 in patients with ECC and patients with ICC.
| ECC | ICC | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| N (%) | Patients with ECC from Asian centers | Patients with ECC from non-Asian centers | Patients with ECC from USA centers | N (%) | Patients with ICC from Asian centers | Patients with ICC from non-Asian centers | Patients with ICC from USA centers | ||
| Patients with mIDH1 | 9 (0.8) | 2 (0.9) | 4 (0.6) | 2 (0.4) | 552 (13.1) | 86 (8.8) | 399 (16.5) | 343 (18.0) | |
| Median | 0.0 | 0.0 | 0.0 | 0.0 | 13.2 | 8.5 | 16.0 | 20.0 | |
| Range | 0.0–11.1 | 0.0–3.7 | 0.0–11.1 | 0.0–11.1 | 0.0–55.6 | 4.9–18.8 | 0.0–55.6 | 0.0–55.6 | |
| Studies | 22 | 5 | 17 | 12 | 43 | 13 | 29 | 20 | |
| Total patients | 1,123 | 230 | 725 | 548 | 4,214 | 981 | 2,416 | 1,904 | |
| Median [range] patients/study | 33 [8–168] | 32 [24–86] | 29 [8–126] | 24.5 [8–126] | 58 [3–434] | 58 [17–171] | 32 [3–434] | 32 [3–434] | |
mIDH1,isocitrate dehydrogenase 1 mutation; ECC, extrahepatic cholangiocarcinoma; ICC, intrahepatic cholangiocarcinoma.
No significant differences in mIDH1 rates by region were found for ECC, though interpretation is limited by the small patient numbers. Geographic location was available for six of the nine patients with ECC who were mIDH1-positive. Two of the 230 patients treated in Asian centers had mIDH1 (0.9%; 95% CI, 0.03–3.3%), as did four of 725 patients treated in non-Asian centers (0.6%; 95% CI, 0.2–1.5%). Of the 548 patients with ECC treated at USA centers, two were mIDH1-positive (0.4%; 95% CI, <0.0001–1.4%) (Table 2).
Two studies reported on the frequency of mIDH1 in patients with and without fluke infection, with the frequency being lower in fluke-infected patients (1.6–1.9% for fluke-infected patients vs. 4.2–10.9% for non-infected patients) (10,39). Chan-On et al. (39) provided further breakdown by anatomic location. Among the 108 fluke-infected patients in their study, mIDH1 were reported in one of 62 patients with ICC (1.6%) and one of 46 patients with ECC (2.2%). Among 101 non-infected patients, 10 of 57 patients with ICC had mIDH1 (17.5%) compared with one of 44 patients with ECC (2.3%).
One study reported on differences in the frequency of mIDH1 according to tumor subtypes, observing a lower frequency of mIDH1 in patients whose tumor cells resemble those of the large bile duct, compared with a second subtype in which the tumor cells resemble cholangiolar cells (4.5% vs. 16.9%) (46).
Demographic characteristics of patients with mIDH1 CC
Approximately half of the publications included demographic data for the patient population. However, in most cases this information related to the total study population and not specifically to patients with mIDH1 CC. In the eight studies that provided gender data by mIDH1 status, 86 (66.2%) of 130 patients with mIDH1 were female (95% CI, 57.7–73.7%). This was higher than the percentage of females in the overall CC population, as reported in 21 studies (796 of 1,792 patients, or 44.4%).
Seven studies reported the age of patients with mIDH1 (n=57), with the weighted average being 59.0 years (pooled standard deviation, 11.0) (16,39,41,44,45,53,55). Although the mIDH1 patient numbers were small, this was similar to the weighted average of 62.2 years reported for the overall CC populations reported in 18 studies (n=1,330) (11,13,14,16,17,21,37,39-42,44,45,50,51,53,64).
Characteristics of mIDH1 and co-mutations
Twenty of the 46 publications reported details of the specific mIDH1, representing 244 patients from the United States, Europe, and Asia (233 with ICC and 11 with ECC) (Figure 3). With the exception of two mutations, all of the mutations were observed on codon 132. R132C was the most frequently observed mutation across all publications reporting these data (n=155; 63.5% of patients). Other frequently reported mutations were R132L (n=35; 14.3% of patients) and R132G (n=29; 11.9% of patients). The other R132 mutations were R132S (n=9; 3.7% of patients), R132H (n=8; 3.3% of patients) and R132V (n=1; 0.6% of patients); for 5 patients, the specific change in R132 was not reported. The two other mutations (detected in one tumor each) were 199M and G97D.
Figure 3.
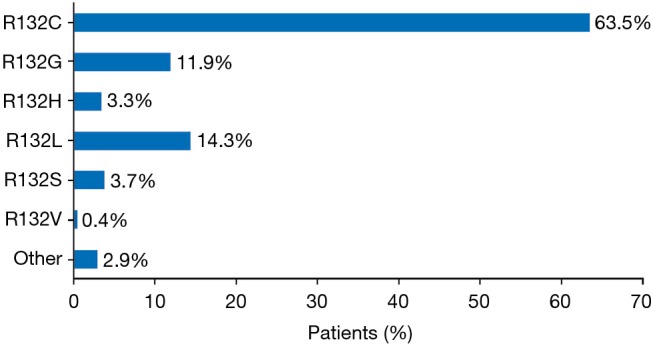
Percentage of patients with specific isocitrate dehydrogenase 1 mutations (across 20 studies and 244 patients).
Eleven of the 46 publications reported on co-mutations present in patients with mIDH1 ICC (15,19,39-42,44,50,52,53,55). Figure 4 summarizes the mutations that were analyzed in ≥30 patients with ICC within each of the 11 studies. The three genes most frequently reported as co-mutations with mIDH1 were ARID1A (22.0%), BAP1 mutation or loss (15.5%), and PBRM1 (13.3%). Other mutations were reported in <8% of tumors analyzed. Insufficient data and small sample sizes prevented determination of whether the rates of these co-occurring mutations were significantly different from those seen in tumors with wild-type IDH1.
Figure 4.
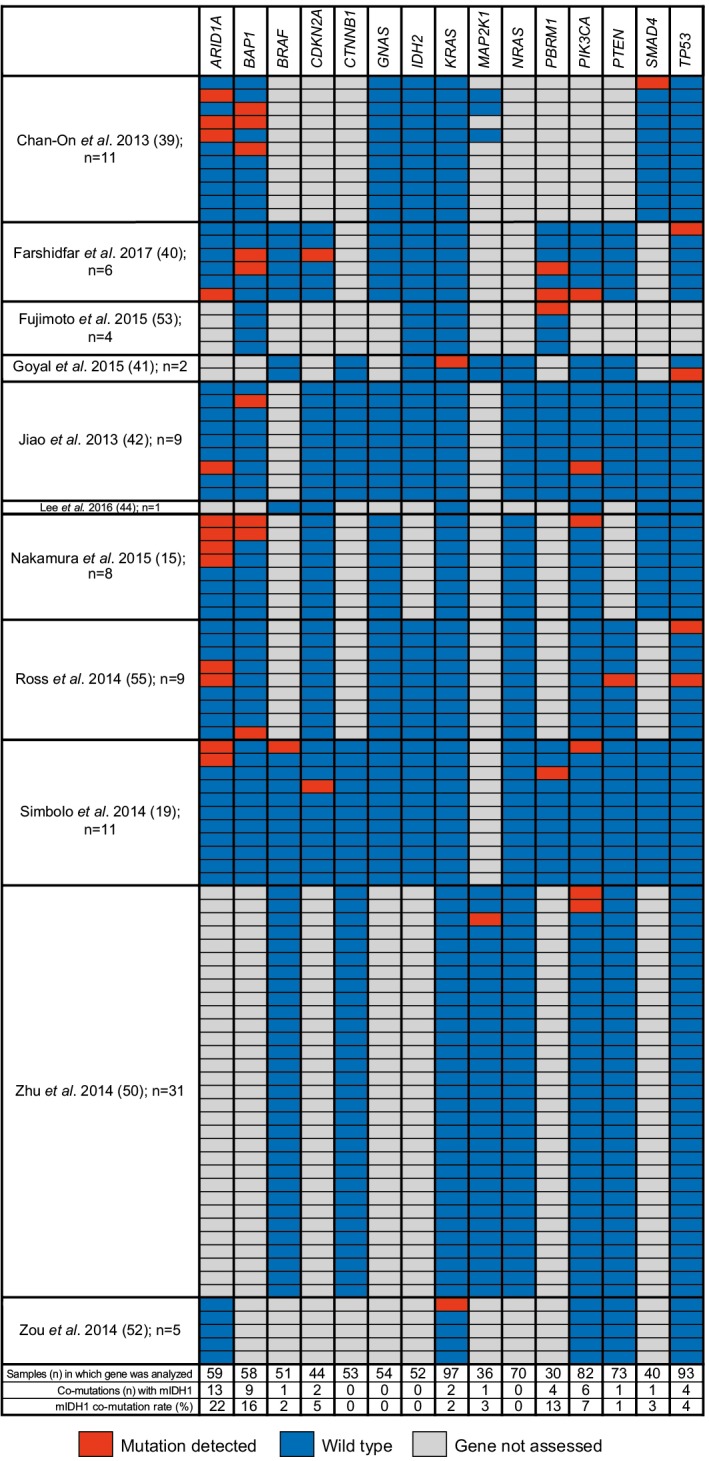
Representative co-mutations investigated in ≥30 patients with ICC having mIDH1. Only genes analyzed in ≥30 patients with ICC are presented. Values in pink boxes are the proportion of patients with mutations; lighter shading indicates lower proportions. ARID1A, AT-rich interactive domain-containing protein 1A ; BAP1, BRCA1-associated protein 1; BRAF, B-Raf proto-oncogene, serine/threonine kinase; CDKN2A, cyclin-dependent kinase inhibitor 2A; CTNNB1, catenin beta 1; GNAS, GNAS complex locus; IDH2, isocitrate dehydrogenase 2; KRAS, KRAS proto-oncogene, GTPase; MAP2K1, mitogen-activated protein kinase 1; NRAS, NRAS proto-oncogene, GTPase; PBRM1, polybromo 1; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PTEN, phosphatase and tensin homolog; SMAD4, SMAD family member 4; TP53, tumor protein 53; ICC, intrahepatic cholangiocarcinoma; mIDH1, IDH1 mutations.
Clinical outcomes in patients with mIDH1 CC
Eight of the 46 publications investigated the possible prognostic significance of mIDH1 in patients with ICC (Table 3) (9,13,17,49,50,62,64,66). These studies involved cohorts of 30 to 326 patients with ICC, with a prevalence of mIDH1 ranging from 7.1–30.0%. The number of patients with mIDH1 ICC ranged from 9 to 40. The duration of follow-up was reported in four studies; mean follow-up, reported for one study, was 28.3±25.8 months (17) and the median follow-up duration (reported for the other three studies) was 19 months (13), 23.2 months (50), and 11.0 (Chinese cohort) and 29.5 (USA cohort) months (49) (Table 3). None of the studies reported a statistically significant association between the presence of mIDH1 and clinical outcomes (OS, PFS, or time to progression). Given the low number of patients with mIDH1 ECC, no meaningful information could be extracted about clinical outcomes specific to these patients.
Table 3. Studies investigating the prognostic significance of mIDH1 in patients with ICC.
| Reference | Patients with ICC, N | Follow-up, months | Patients with mIDH1 and ICC, n (%) | Prognostic significance of mIDH1 |
|---|---|---|---|---|
| Churi et al. 2014 (13)a | 55 | Median, 19 | 10 (18.0) | Not associated with PFS or OS (not mentioned for mIDH2) |
| Javle et al. 2016 (9)a | 224 | NR | 40 (17.9) | Not prognostic for OS |
| Lowery et al. 2016 (53)a,b | 30 | NR | 9 (30.0) | Not associated with TTP in response to first line of chemotherapy in advanced disease (77% gemcitabine/platinum) |
| Pak et al. 2017 (56)a,b | 66 | NR | 15 (22.7) | Not prognostic for OS or DFS |
| Pawlik et al. 2014 (58)b | 138 | NR | 11 (7.8) | Not associated with survival |
| Ruzzenente et al. 2016 (17)a | 53 | Mean, 28.3±25.8 | 7 (13.2) | Not associated with OS |
| Wang et al. 2013 (65)a | 326 | Chinese cohort: median, 11.00 (range, 1–110.13); USA cohort: median, 29.5 (range, 0.67–153.43) |
23 (7.1) | mIDH2 but not mIDH1 associated with longer time to tumor recurrence after resection (P=0.021) |
| Zhu et al. 2014 (69)a | 200 | Median, 23.2 | 31 (15.5) | Not associated with OS |
a, studies with overall survival; b, conference abstract. mIDH1/2, isocitrate dehydrogenase 1/2; ICC, intrahepatic cholangiocarcinoma; NR, not reported; PFS, progression-free survival; OS, overall survival; TTP, time to progression; DFS, disease-free survival.
The present review also identified a publication by Lowery et al. (35) reporting the results of a phase 1 trial of ivosidenib in patients with previously treated mIDH1 CC. This dose-escalation study involved 73 patients with CC, of which 65 had ICC. A partial response was observed in 5% of patients, and 56% achieved stable disease. Six-month PFS was 38% and 12-month PFS was 20%. The reported results did not distinguish patients with ICC from those with ECC.
Discussion
The results of this systematic review confirm the findings of previous individual studies demonstrating that, in patients with CC, mIDH1 is largely confined to ICC tumors. Indeed, in all the studies identified in this review, which included both patients with ICC and with ECC, the proportion of patients with mIDH1 tumors was substantially greater in patients with ICC compared with those with ECC. Based on all the patients included in the identified studies, the overall frequency of mIDH1 was 13.1% in patients with ICC versus 0.8% in patients with ECC (P value <0.0001).
Further characterization of the frequency of mIDH1 in ICC across studies reveals possible differences according to geographical location, with generally lower frequencies being observed in Asian centers (8.8%; 95% CI, 7.2–10.7%) compared with non-Asian centers (16.5%; 95% CI, 15.1–18.1%) and particularly USA centers (18.0%; 95% CI, 16.4–19.8%). This may reflect differences in the distribution of risk factors. Recognized risk factors for ICC include chronic inflammation of the bile ducts due to infection with liver fluke, and chronic viral hepatitis associated with hepatitis B or C infections, both of which are more prevalent in Asian countries. One study identified in our review reported mIDH1 ICC to occur at a lower frequency in patients with fluke infection (1.6% vs. 4.2%) (10); this has also been observed in a study reporting on the combined frequency of IDH1 and mIDH2 in a cohort of patients from Thailand with fluke infection, and in a cohort of patients from Singapore without fluke infection (3.2% vs. 22.2%; P=0.029) (39). Two Japanese studies provide evidence to suggest that the frequency of mIDH1 is lower in patients with chronic hepatitis. Both of these studies considered the combined frequency of IDH1 or IDH2: Fujimoto et al. (53) reported a significantly higher frequency of IDH1/2 in patients without hepatitis (20% vs. 2%; P<0.01), while Yasui et al. (71) reported that none of the patients in their cohort who had hepatitis had mIDH1/2. Ascertainment bias due to geographical differences in access to comprehensive clinical tumor sequencing could also have contributed to the differences in observed frequency. Any conclusions, however, need to be cautious, given the range of frequencies reported for studies within a particular geographical region, the relatively small number of patients included in some studies, and the lack of rigorous inclusion criteria for any of the studies.
Given that mIDH1 appears to be present in a clinically relevant proportion of patients with ICC tumors, it is of interest whether the patient characteristics, and the course of disease progression in patients with these tumors, differ from those of patients with tumors with wild-type IDH1. Although some of the studies identified in this review provided information on the demographic characteristics of patients with ICC, most did not provide data specifically for the mIDH1 subtype. Based on a small subset of studies, we found the mIDH1 appears to occur more frequently in women than in men, though this finding requires validation in a larger cohort with symmetric demographic information available across subjects. There was insufficient information to definitively determine whether mIDH1 CC occurs more frequently in older or younger patients or in a particular racial group compared with tumors with wild-type IDH1. However, one study has suggested that mIDH1 tumors may show a different morphology to wild-type IDH1 tumors (46), and a Japanese study distinguished two ICC subtypes and found that mIDH1/2 were confined to type 2 tumors (frequency, 40% vs. 0%); type 2 tumors were associated with a better recurrence-free survival and OS compared with type 1 tumors (71).
Various genomic profiling studies have identified a range of oncogenic mutations present in CC tumors. In this study, we have summarized the most frequently reported co-mutations in ICC mIDH1 tumors. The three most frequently observed co-mutations were ARID1A, BAP1 (loss or mutation), and PBRM1. The frequency of mutations in ARID1A was numerically higher when mIDH1 was present compared with the overall study sample, although the statistical significance could not be determined. BAP1 mutations and losses could not be distinguished, owing to variability in testing and reporting across studies. This is a noteworthy limitation precluding the delineation of mutation versus loss of protein expression due to epigenetic dysregulation according to mIDH1 status (10,47,72).
Although some clinical differences have been noted between patients with mIDH1 versus wild-type ICC tumors, evidence to date suggests that the presence of mIDH1 does not significantly affect the prognosis of patients with ICC. This was the conclusion from eight studies that have sought to assess the possible prognostic implications of the mIDH1 in ICC tumors (9,13,17,49,50,62,64,66). While these studies have been small, involving no more than 40 patients with mIDH1 tumors in each study, the consistency of results across the eight studies suggests that the presence of mIDH1 does not significantly affect the natural history of ICC or the outcomes of current chemotherapy. These results further indicate that survival outcomes reported for ICC as a whole can be used as a benchmark against which the effects of investigational mIDH1-targeted therapies are measured.
The findings of this review bring together significant information on the frequency, clinical characteristics, and genetic characteristics of ICC tumors harboring mIDH1. This adds to the growing literature on the role of mIDH1 in other tumors, such as AML, glioma, and chondrosarcoma. As with these other tumors, almost all the mIDH1 reported involved changes in the arginine codon, R132. However, the most common substitutions appear to differ between tumors. In gliomas, the most common mutation is substitution of arginine by histidine, while less common mutations include R132C, R132S, R132G, and R132L (73). In contrast, the findings of this review suggest that R132C is the most common mutation in ICC, followed by R132L and R132G. All of these mutations are understood to result in gain of function activity, resulting in the accumulation of very high levels of the oncogenic metabolite 2-HG. This leads to a cellular hypermethylation profile that is believed to contribute to tumorigenesis through epigenetic changes that lead to inhibition of cellular differentiation (73). Further recent research suggests that mIDH1 is associated with increased methylation, and hence reduced expression, of the genes encoding the immunosuppressive molecules (PD-L1) and programmed cell death protein 1 (PD-1) (notably, being associated with a lack of response to checkpoint inhibitors in gliomas), and an increase in tumor-infiltrating lymphocytes (74). Other studies have shown that T-cell uptake of 2-HG results in suppression of T-cell activity (75) and reduced accumulation of T cells in tumor sites (76). These results indicate that 2-HG may also exert tumorigenic effects by acting as an immunosuppressant. Thus, while tumorigenesis associated with mIDH1 is likely to be mediated through changes in gene expression as with most other tumors, uniquely, the changes in gene transcription appear to be brought about through the effects of the oncometabolite 2-HG, rather than the modulation of cell signaling pathways. The precise role of mIDH1 in tumorigenesis is not understood and may differ across tumor types. Characterization of the genetic changes present in mIDH1 ICC tumors and the clinical course of the tumor may help understand the role of mIDH1 in ICC and hence its potential as a therapeutic target.
To the best of our knowledge, the robust methodology employed in this systematic review has ensured the identification of all relevant published data relating to the frequency, mutations, co-mutations and clinical outcomes for patients with mIDH1 ICC. However, the conclusions from this review are necessarily cautious, given the limitations of the available data and the individual studies, which are only partly overcome by considering the whole body of literature. Firstly, reporting of mutation status is influenced by the availability and cost of the required genetic tests, which are not generally available in some centers and geographical locations. Secondly, there is likely to be selection bias, with tumors that are more amenable to biopsy and those without substantial stromal contamination, being more likely to be genetically analyzed. Furthermore, some studies have not distinguished between tumors with mIDH1 and mIDH2, although these are two distinct enzymes. In addition, many studies focused on genetic characterization of the tumors rather than the clinical course of disease or the characteristics of patients developing ICC tumors. An additional limitation is the possibility of overlapping patients across several of the publications, owing to shared datasets. We excluded publications with overt overlap, but there remained several that could have had partial overlap due to shared authorship and institution (37,38,41,56). In accordance with systematic review guidelines, it would have been inappropriate to exclude any of these studies from our analysis (77). Sensitivity analyses were conducted to assess the magnitude of impact of any potential double-counting by, for example, keeping only the largest of a set of potentially overlapping studies or only the ones with the highest or lowest mIDH1 rates; these did not materially impact the overall findings.
In conclusion, this review has identified a growing body of literature relating to ICC tumors harboring mIDH1. These studies substantiate the early clinical data suggesting that mIDH1 is largely confined to ICC tumors and extremely rare in ECC. The studies also provide preliminary evidence that the frequency of mIDH1 in ICC may be lower in geographic regions where specific risk factors, such as fluke infection or chronic hepatitis, contribute significantly to the occurrence of CC. mIDH1 is a recognized therapeutic target for a number of different tumors including AML. The development of targeted therapies against mIDH1 will help to characterize these tumors and to refine our understanding of the role of mIDH1 in tumorigenesis. There is an urgent need for better treatment options for patients with CC, and understanding the genetic basis of this disease is a key step in developing new treatments.
Table S1. Terms employed for database searches.
| Search number | Search terms | Hits |
|---|---|---|
| Embase | ||
| 1 | exp bile duct carcinoma/ | 19,619 |
| 2 | (cholangiocarcinoma or hepatocholangiocarcinoma).mp | 14,543 |
| 3 | exp bile duct/ | 23,896 |
| 4 | ((bile duct or cholangiocellular or klatskin or biliary or hepatobiliary or cholangiolar) and (cancer* or carcinoma* or neoplasm* or tumor* or tumour*)).mp. | 53,832 |
| 5 | or/1−4 | 70,319 |
| 6 | exp isocitrate dehydrogenase 1/ | 2,649 |
| 7 | (isocitrate dehydrogenase 1 or IDH1 or IDH-1).mp. | 5,973 |
| 8 | 6 or 7 | 5,973 |
| 9 | 5 and 8 | 176 |
| 10 | limit 9 to English language | 175 |
| MEDLINE via Ovid | ||
| 1 | exp Adenoma, Bile Duct/ or exp Cholangiocarcinoma/ or exp Bile Duct Neoplasms/ | 20,176 |
| 2 | (Cholangiocarcinoma or hepatocholangiocarcinoma).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] | 12,364 |
| 3 | exp Bile Ducts/ | 48,387 |
| 4 | ((bile duct or cholangiocellular or klatskin or biliary or hepatobiliary or cholangiolar) and (cancer* or carcinoma* or neoplasm* or tumor* or tumour*)).mp. | 41,358 |
| 5 | or/1−4 | 77,568 |
| 6 | (isocitrate dehydrogenase 1 or IDH1 or IDH-1).mp. | 2,601 |
| 7 | 5 and 6 | 81 |
| 8 | limit 7 to English language | 81 |
| Cochrane Library | ||
| 1 | MeSH descriptor: [Adenoma, Bile Duct] explode all trees | 6 |
| 2 | MeSH descriptor: [Cholangiocarcinoma] explode all trees | 82 |
| 3 | MeSH descriptor: [Bile Ducts] explode all trees | 493 |
| 4 | Cholangiocarcinoma or hepatocholangiocarcinoma | 333 |
| 5 | ((bile duct or cholangiocellular or klatskin or biliary or hepatobiliary or cholangiolar) and (cancer* or carcinoma* or neoplasm* or tumor* or tumour*)) | 2,160 |
| 6 | #1 or #2 or #3 or #4 or #5 | 2,541 |
| 7 | MeSH descriptor: [Isocitrate Dehydrogenase] explode all trees | 24 |
| 8 | (isocitrate dehydrogenase 1 or IDH1 or IDH-1) | 145 |
| 9 | #7 or #8 | 153 |
| 10 | #6 and #9 | 11 |
| PubMed | ||
| 1 | ((bile duct carcinoma[MeSH Terms]) OR bile duct adenoma[MeSH Terms]) OR cholangiocarcinoma[MeSH Terms] | 18,209 |
| 2 | bile duct[MeSH Terms] | 44,347 |
| 3 | (bile duct or cholangiocellular or klatskin or biliary or hepatobiliary or cholangiolar | 158,094 |
| 4 | ((((cancer*) OR carcinoma*) OR neoplasm*) OR tumor*) OR tumour* | 3,668,071 |
| 5 | #3 and #4 | 51,211 |
| 6 | Cholangiocarcinoma or hepatocholangiocarcinoma | 11,521 |
| 7 | #1 or #2 or #5 or #6 | 83,317 |
| 8 | (((isocitrate dehydrogenase i[MeSH Terms]) OR isocitrate dehydrogenase 1) OR IDH1) OR IDH-1 | 7,408 |
| 9 | #7 and #8 | 91 |
| 10 | #9 Filters: English | 90 |
| 11 | #10 Filters: Publication date from 2016/01/01 to 2017/12/31 | 41 |
Acknowledgments
Medical writing assistance was provided by Rowena Hughes of Envision Pharma Group and funded by Agios Pharmaceuticals Inc. Editorial assistance was also provided by Excel Medical Affairs, Horsham, UK, and supported by Agios.
Ethical Statement: Ethics approval was not required and informed consent from patients was not obtained as this was a literature-based study.
Footnotes
Conflicts of Interest: AN Boscoe is an employee and shareholder of Agios Pharmaceuticals Inc. C Rolland is an employee of Envision Pharma Group, paid consultants to Agios Pharmaceuticals Inc in connection with this study. RK Kelley receives support (to institution) for conduct of clinical trials from: Agios, Astra Zeneca, Bayer, Bristol-Myers Squibb, Eli Lilly, Exelixis, MedImmune, Merck, QED, Novartis, Taiho; she also receives consulting fees (to individual) for advisory board/IDMC membership from Genentech/Roche and Target Pharma Solutions.
References
- 1.Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15:95-111. 10.1038/nrclinonc.2017.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiao MS, Chiablaem K, Charoensawan V, et al. Emergence of intrahepatic cholangiocarcinoma: how high-throughput technologies expedite the solutions for a rare cancer type. Front Genet 2018;9:309. 10.3389/fgene.2018.00309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan SA, Emadossadaty S, Ladep NG, et al. Rising trends in cholangiocarcinoma: is the ICD classification system misleading us? J Hepatol 2012;56:848-54. 10.1016/j.jhep.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 4.Elfaki DH, Gossard AA, Lindor KD. Cholangiocarcinoma: expanding the spectrum of risk factors. J Gastrointest Cancer 2008;39:114-7. 10.1007/s12029-008-9040-0 [DOI] [PubMed] [Google Scholar]
- 5.Petrick JL, Thistle JE, Zeleniuch-Jacquotte A, et al. Body mass index, diabetes and intrahepatic cholangiocarcinoma risk: The liver cancer pooling project and meta-analysis. Am J Gastroenterol 2018;113:1494-505. 10.1038/s41395-018-0207-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget 2016;7:46750-67. 10.18632/oncotarget.8775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathan H, Pawlik TM, Wolfgang CL, et al. Trends in survival after surgery for cholangiocarcinoma: a 30-year population-based SEER database analysis. J Gastrointest Surg 2007;11:1488-96; discussion 1496-7. 10.1007/s11605-007-0282-0 [DOI] [PubMed] [Google Scholar]
- 8.Mavros MN, Economopoulos KP, Alexiou VG, et al. Treatment and prognosis for patients with intrahepatic cholangiocarcinoma: systematic review and meta-analysis. JAMA Surg 2014;149:565-74. 10.1001/jamasurg.2013.5137 [DOI] [PubMed] [Google Scholar]
- 9.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 2016;122:3838-47. 10.1002/cncr.30254 [DOI] [PubMed] [Google Scholar]
- 10.Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116-35. 10.1158/2159-8290.CD-17-0368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akita M, Fujikura K, Ajiki T, et al. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod Pathol 2017;30:986-97. 10.1038/modpathol.2017.22 [DOI] [PubMed] [Google Scholar]
- 12.Arnold A, Bahra M, Lenze D, et al. Genome wide DNA copy number analysis in cholangiocarcinoma using high resolution molecular inversion probe single nucleotide polymorphism assay. Exp Mol Pathol 2015;99:344-53. 10.1016/j.yexmp.2015.08.003 [DOI] [PubMed] [Google Scholar]
- 13.Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One 2014;9:e115383. 10.1371/journal.pone.0115383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H, Wang K, Johnson A, et al. Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J Clin Pathol 2016;69:403-8. 10.1136/jclinpath-2015-203394 [DOI] [PubMed] [Google Scholar]
- 15.Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10. 10.1038/ng.3375 [DOI] [PubMed] [Google Scholar]
- 16.Putra J, de Abreu FB, Peterson JD, et al. Molecular profiling of intrahepatic and extrahepatic cholangiocarcinoma using next generation sequencing. Exp Mol Pathol 2015;99:240-4. 10.1016/j.yexmp.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruzzenente A, Fassan M, Conci S, et al. Cholangiocarcinoma heterogeneity revealed by multigene mutational profiling: clinical and prognostic relevance in surgically resected patients. Ann Surg Oncol 2016;23:1699-707. 10.1245/s10434-015-5046-6 [DOI] [PubMed] [Google Scholar]
- 18.Schlitter AM, Jang KT, Klöppel G, et al. Intraductal tubulopapillary neoplasms of the bile ducts: clinicopathologic, immunohistochemical, and molecular analysis of 20 cases. Mod Pathol 2015;28:1249-64. 10.1038/modpathol.2015.61 [DOI] [PubMed] [Google Scholar]
- 19.Simbolo M, Fassan M, Ruzzenente A, et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014;5:2839-52. 10.18632/oncotarget.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Ding X, Wang S, et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett 2016;380:163-73. 10.1016/j.canlet.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014;10:e1004135. 10.1371/journal.pgen.1004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726-36. 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silkin SV, Startsev SS, Krasnova ME, et al. Complete clinical response of BRAF-mutated cholangiocarcinoma to vemurafenib, panitumumab, and irinotecan. J Gastrointest Cancer 2016;47:502-5. 10.1007/s12029-015-9792-2 [DOI] [PubMed] [Google Scholar]
- 24.Czink E, Kloor M, Goeppert B, et al. Successful immune checkpoint blockade in a patient with advanced stage microsatellite-unstable biliary tract cancer. Cold Spring Harb Mol Case Stud 2017;3:a001974. 10.1101/mcs.a001974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739-44. 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Ye D, Guan KL, et al. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 2012;18:5562-71. 10.1158/1078-0432.CCR-12-1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network , Brat DJ, Verhaak RG, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015;372:2481-98. 10.1056/NEJMoa1402121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765-73. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011;224:334-43. 10.1002/path.2913 [DOI] [PubMed] [Google Scholar]
- 31.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058-66. 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225-34. 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel KP, Ravandi F, Ma D, et al. Acute myeloid leukemia with IDH1 or IDH2 mutation: frequency and clinicopathologic features. Am J Clin Pathol 2011;135:35-45. 10.1309/AJCPD7NR2RMNQDVF [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol 2015;90:732-6. 10.1002/ajh.24072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol 2017;35:abstr 4015.
- 36.Shamseer L, Moher D, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ 2015;350:g7647. 10.1136/bmj.g7647 [DOI] [PubMed] [Google Scholar]
- 37.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012;17:72-9. 10.1634/theoncologist.2011-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borger DR, Goyal L, Yau T, et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res 2014;20:1884-90. 10.1158/1078-0432.CCR-13-2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan-On W, Nairismägi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke–related and non-infection-related bile duct cancers. Nat Genet 2013;45:1474-8. 10.1038/ng.2806 [DOI] [PubMed] [Google Scholar]
- 40.Doherty M, Chiu JWY, McNamara MG, et al. Molecular profiling of advanced biliary cancer: Lost in translation from bench to bedside. J Clin Oncol 2016;34:283 10.1200/jco.2016.34.4_suppl.283 [DOI] [Google Scholar]
- 41.Farshidfar F, Zheng S, Gingras MC, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 2017;18:2780-94. 10.1016/j.celrep.2017.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujimoto A, Furuta M, Shiraishi Y, et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun 2015;6:6120. 10.1038/ncomms7120 [DOI] [PubMed] [Google Scholar]
- 43.Goyal L, Govindan A, Sheth RA, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015;20:1019-27. 10.1634/theoncologist.2015-0210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hess T, Bertrand C, Chan C. Comprehensive genomic characterization of intrahepatic cholangiocarcinoma. Med Genet 2015;27:abstr pp 79-196. [Google Scholar]
- 45.Holcombe RF, Xiu J, Gatalica Z, et al. Molecular profiling of bile duct and gallbladder cancer to identify different therapeutic options. J Clin Oncol 2014;32:4097 10.1200/jco.2014.32.15_suppl.4097 [DOI] [Google Scholar]
- 46.Holcombe RF, Xiu J, Pishvaian MJ, et al. Tumor profiling of biliary tract carcinomas to reveal distinct molecular alterations and potential therapeutic targets. J Clin Oncol 2015;33:285 10.1200/jco.2015.33.3_suppl.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3. 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kipp BR, Voss JS, Kerr SE, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol 2012;43:1552-8. 10.1016/j.humpath.2011.12.007 [DOI] [PubMed] [Google Scholar]
- 49.Lee CH, Wang HE, Seo SY, et al. Cancer related gene alterations can be detected with next-generation sequencing analysis of bile in diffusely infiltrating type cholangiocarcinoma. Exp Mol Pathol 2016;101:150-6. 10.1016/j.yexmp.2016.07.010 [DOI] [PubMed] [Google Scholar]
- 50.Lee JH, Shin DH, Park WY, et al. IDH1 R132C mutation is detected in clear cell hepatocellular carcinoma by pyrosequencing. World J Surg Oncol 2017;15:82. 10.1186/s12957-017-1144-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liau JY, Tsai JH, Yuan RH, et al. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod Pathol 2014;27:1163-73. 10.1038/modpathol.2013.241 [DOI] [PubMed] [Google Scholar]
- 52.Lo AA, Costa HA, Zehnder J, et al. Comparative study of isocitrate dehydrogenase 1 mutations in intraheptatic cholangiocarcinoma by immunohistochemistry and pyrosequencing. Lab Invest 2016;96: abstr 1758 443–4A.
- 53.Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling and analysis of mutual exclusivity of genetic aberrations (MEGA) of intra- and extrahepatic cholangiocarcinomas (IHC and EHC) evaluation of prognostic features and potential targets for intervention. J Clin Oncol 2016;34:abstr 4088.
- 54.Mattis DM, Joseph NM, Yeh I, et al. Deep Sequencing of Intrahepatic Cholangiocarcinoma, Extrahepatic Cholangiocarcinoma and Klatskin Tumors. San Antonio, TX: United States and Canadian Academy of Pathology Annual Meeting; 2017:Abstract 748. [Google Scholar]
- 55.Misumi K, Hayashi A, Shibahara J, et al. Intrahepatic cholangiocarcinoma frequently shows loss of BAP1 and PBRM1 expression, and demonstrates specific clinicopathological and genetic characteristics with BAP1 loss. Histopathology 2017;70:766-74. 10.1111/his.13127 [DOI] [PubMed] [Google Scholar]
- 56.Pak LM, Goldman D, Gonen M, et al. Mutational profiling of resected intrahepatic cholangiocarcinoma. J Clin Oncol 2017;35:e15675 10.1200/JCO.2017.35.15_suppl.e15675 [DOI] [Google Scholar]
- 57.Patel D, Hamoudi R, Khan S, et al. Genetic and epigenetic alterations in biliary tree cancers: combined FISH, IHC and NGS approach. J Pathol 2017;243:S33. [Google Scholar]
- 58.Pawlik TM, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: Refining prognostic determinants and identifying therapeutic targets. J Clin Oncol 2014;32:abstr 210. [DOI] [PMC free article] [PubMed]
- 59.Reyes S, Horick N, Clark J, et al. Frequency and impact of tumor genotyping in clinical practice of patients with advanced biliary tract cancers (ABTCs). Ann Oncol 2017;28:mdx262.020.
- 60.Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014;19:235-42. 10.1634/theoncologist.2013-0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087. 10.1038/ncomms7087 [DOI] [PubMed] [Google Scholar]
- 62.Tsokos C, Liu X, Solomon D. Telomerase reverse transcriptase promoter mutations (mTERTp) in combined hepatocellular(HCC)-cholangiocarcinoma(CC; CHCC-CC) support clonal and HCC-like origin for both components. Lab Invest 2015;95:abstr 1697 425A.
- 63.Ueno M, Morizane C, Kawamoto Y, et al. The nationwide cancer genome screening project in Japan, SCRUM-Japan GI-screen: efficient identification of cancer genome alterations in advanced biliary tract cancer. Ann Oncol 2017;28:v209-68. 10.1093/annonc/mdx369.100 [DOI] [Google Scholar]
- 64.Wachsmann M, Pinho D, Ram R. Isocitrate dehydrogenase gene mutations in cholangiocarcinoma. Lab Invest 2017;97:452A-3A. [Google Scholar]
- 65.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091-100. 10.1038/onc.2012.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winter H, Kaisaki P, Harvey J, et al. Circulating tumour DNA and oncometabolites in patients with intrahepatic cholangiocarcinoma. Ann Oncol 2017;28:mdx508.024.
- 67.Yasui K, Nagasaka T, Umeda Y, et al. Comprehensive analysis of intrahepatic cholangiocarcinoma based on viral infections and mutational status in the IDH1/2 and KRAS genes. Cancer Res 2016;76:abstr 3172.
- 68.Zhu Y, Chen J, Kong W, et al. Predicting IDH mutation status of intrahepatic cholangiocarcinomas based on contrast-enhanced CT features. Eur Radiol 2018;28:159-69. 10.1007/s00330-017-4957-y [DOI] [PubMed] [Google Scholar]
- 69.Zhu AX, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: Refining prognosis and identifying therapeutic targets. Ann Surg Oncol 2014;21:3827-34. 10.1245/s10434-014-3828-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zou S, Li J, Zhou H, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696. 10.1038/ncomms6696 [DOI] [PubMed] [Google Scholar]
- 71.Hayashi A, Misumi K, Shibahara J, et al. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am J Surg Pathol 2016;40:1021-30. 10.1097/PAS.0000000000000670 [DOI] [PubMed] [Google Scholar]
- 72.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474-8. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romanidou O, Kotoula V, Fountzilas G. Bridging cancer biology with the clinic: comprehending and exploiting IDH gene mutations in gliomas. Cancer Genomics Proteomics 2018;15:421-36. 10.21873/cgp.20101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berghoff AS, Kiesel B, Widhalm G, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol 2017;19:1460-8. 10.1093/neuonc/nox054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bunse L, Pusch S, Bunse T, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 2018;24:1192-203. 10.1038/s41591-018-0095-6 [DOI] [PubMed] [Google Scholar]
- 76.Kohanbash G, Carrera DA, Shrivastav S, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 2017;127:1425-37. 10.1172/JCI90644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ 2009;339:b2700. 10.1136/bmj.b2700 [DOI] [PMC free article] [PubMed] [Google Scholar]