

Abstract
Background
Biliary tract cancers (BTCs) are a heterogeneous group of aggressive, rare malignancies with limited standard chemotherapeutic options for advanced disease. Recent studies have demonstrated potential novel biliary cancer targets and a possible role for immunotherapy in the treatment of patients with this disease. Intrahepatic cholangiocarcinoma (IHCC), extrahepatic cholangiocarcinoma (EHCC), and gallbladder carcinoma (GBC) are frequently grouped together in clinical trials despite differences in tumor biology.
Methods
To further investigate tumor biology differences, we profiled 1,502 BTCs using next-generation sequencing (NGS), immunohistochemistry, in situ hybridization, and RNA sequencing.
Results
IHCCs had higher rates of IDH1, BAP1, and PBRM1 mutations and FGFR2 fusions; EHCCs had higher rates of KRAS, CDKN2A, and BRCA1 mutations; and GBCs had higher rates of homologous recombination repair deficiency and Her2/neu overexpression and amplification. IHCCs and GBCs had higher rates of potential positive predictive biomarkers for immune checkpoint inhibition (PD-L1 expression, high microsatellite instability, and high tumor mutational burden) than EHCCs.
Conclusions
These findings support clinical molecular profiling of BTCs to inform potential therapeutic selection and clinical trial design based on the primary tumor’s site of origin within the biliary tree.
Keywords: Biliary cancer, intrahepatic cholangiocarcinoma (IHCC), extrahepatic cholangiocarcinoma (EHCC), gallbladder carcinoma (GBC)
Introduction
Biliary tract cancer (BTC) is an uncommon cancer type with an overall poor prognosis and limited therapeutic options (1). BTCs arise from epithelial cells from one of three distinct locations, producing a total of three BTC subtypes: intrahepatic cholangiocarcinoma (IHCC), extrahepatic cholangiocarcinoma (EHCC), and gallbladder carcinoma (GBC).
While BTC subtypes have varying clinical presentations, they are often treated similarly (2,3). Treatment relies on surgery for resectable disease; however, BTCs are frequently unresectable or recur following resection (4). For resectable BTCs, fluoropyrimidine- or gemcitabine-based chemotherapy remains the primary therapeutic option in the adjuvant setting, and there is a role for adjuvant chemoradiation with capecitabine in EHCC and GBC treatment (5-8).
For unresectable or metastatic BTC, the ABC-02 trial demonstrated that the combination of cisplatin and gemcitabine as first-line treatment prolonged overall survival over gemcitabine alone, and there were no significant differences in survival between IHCC, EHCC, and GBC subgroups (9). Despite being responsive to multiple chemotherapy agents, there is no established second-line treatment regimen for advanced BTC, and patients are strongly encouraged to enroll in clinical trials. This patient population is in dire need of novel, effective treatment options.
IHCCs, EHCCs, and GBCs have different molecular profiles (10), reflecting differences in underlying tumor etiology (11). In particular, Jusakul and colleagues carried out an integrated genomic, epigenomic, and transcriptomic analysis of BTCs, during which they identified 4 clusters, each with individual genetic, epigenetic, and clinical features (12). Cluster 1 is associated with increased Her2 amplification, Her2 gene expression, and CpG island hypermethylation; cluster 2 is associated with TP53 mutations and increased Her2 amplification and gene expression; cluster 3 is associated upregulated immune-related pathways (e.g., programmed cell death protein 1, PD-1); and cluster 4 is mostly IHCCs and is associated with BAP1 and IDH1/2 mutations, FGFR alterations, CpG shore methylation, and the best prognosis of the 4 clusters. These clusters are driven by different etiologies; for example, clusters 1 and 2 are frequently associated with liver fluke infections, whereas clusters 3 and 4 are not. A greater understanding of the genomic landscape of BTC has resulted in the identification of promising therapeutic targets including FGFR2 fusions (10), Her2/neu (13), and IDH1 (14), Many of these targets are being actively investigated in both basket and BTC-specific clinical trials.
Molecular profiling may also predict response to immunotherapy. The efficacy of immune checkpoint blockade across tumor types led to the first site-agnostic FDA approval of the anti-PD-1 antibody pembrolizumab for microsatellite instability high (MSI-H) and mismatch repair deficient (MMRd) cancers (15). In addition, high tumor mutational burden (TMB-H) and programmed death-ligand 1 (PD-L1) expression are potential positive predictive biomarkers for immune checkpoint blockade with anti-PD-1 and anti-PD-L1 antibodies (16-21).
We report the molecular characterization of a large cohort of BTCs, comparing IHCCs, EHCCs, and GBCs in order to explore potential therapeutic opportunities.
Methods
Biliary tract tumors profiled by Caris Life Sciences between 2009 and July 2017 were de-identified and retrospectively analyzed for molecular alterations. Tumor histology and diagnoses were taken from submitted pathology reports and confirmed by board certified pathologists.
Next-generation sequencing (NGS) using Miseq or NextSeq platforms (Illumina, Inc., San Diego, CA, USA) was performed on genomic DNA isolated from formalin-fixed, paraffin-embedded (FFPE) tumor samples, and no matched normal tissue was sequenced. A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA, USA). All variants were detected with >99% confidence based on allele frequency and amplicon coverage with an average sequencing depth of coverage of >500 times and an analytical sensitivity of 5%. Tumor enrichment was achieved by manual microdissection of harvested target tissue prior to molecular testing in all cases.
TMB was measured in each BTC by counting the number of non-synonymous, somatic mutations found per megabase (MB). The 592 genes sequenced comprised 1.4 MB of total genomic space. Tumors were considered to be TMB-H if they had greater than or equal to 17 mutations per megabase. This threshold had been previously established in colorectal cancer (CRC) studies: TMB was compared with MSI by fragment analysis, based on reports of TMB having concordance with MSI in CRC (22). MSI was examined at over 7,000 target microsatellite loci and compared to the reference genome hg19 from the UCSC Genome Browser database (23). Copy number variation (CNV) was tested by NGS and was determined by comparing the depth of sequencing of genomic loci to a diploid control as well as the known performance of these genomic loci. Calculated gains of 6 copies or greater were considered amplified.
For gene fusion detection, anchored multiplex polymerase chain reaction (PCR) was performed for targeted RNA sequencing using the ArcherDx fusion assay (Archer FusionPlex Solid Tumor panel). Unidirectional gene-specific primers were used to enrich for target regions, followed by NGS (Illumina MiSeq platform). Targets included 593 genes selected for known associations with various carcinomas (the complete panel of tested gene fusions is available at: https://www.carismolecularintelligence.com/tumor-profiling-menu/mi-profile-usa-excluding-new-york/). Fusions among the >11,000 fusions known to be found in normal tissues were excluded (24). The detection sensitivity of the assay allows for detection of a fusion that is present in at least 10% of the cells in the samples tested.
Immunohistochemistry (IHC) was performed on full FFPE sections of glass slides. Slides were stained using automated staining techniques per the manufacturer’s instructions and were optimized and validated per CLIA/CAP and ISO requirements. Staining was scored for intensity (0: no staining; 1+: weak staining; 2+: moderate staining; 3+: strong staining) and staining percentage (0–100%). Results were categorized as positive or negative by defined thresholds specific to each marker based on published clinical literature that associates biomarker status with patient response to therapeutic agents. A board-certified pathologist evaluated all IHC results independently. The primary antibodies used were PD-L1 (SP142), ERCC1 (8F1), RRM1 (polyclonal), TS (TS106), TOPO2A (3F6), Her2 (4B5), and cMET (SP44).
Fluorescence in situ hybridization (FISH) was performed to detect TOP2A (TOP2/CEP17 probe) gene amplification (Abbott Molecular/Vysis). Chromogenic in situ hybridization (CISH) was also used for Her2/neu (INFORM HER-2 Dual ISH DNA Probe Cocktail) and cMET (Ventana). HER2 amplification was defined as Her2/chr17 ratio ≥2.0; TOP2A amplification as TOP2A/CEP17 ratio ≥2.0, and cMET as >5 copies per tumor cells.
Human subjects had been de-identified prior to analysis. The study was deemed exempt by the Western Institutional Review Board (IRB registration number IRB00000533, WIRB work order #1-895778-1), and no informed consent was obtained for this retrospective analysis.
Results
Tumor characteristics
A total of 1,502 BTCs, including 825 IHCCs, 249 EHCCs, and 428 GBCs that underwent Caris molecular profiling were included in the analysis (Table 1). Three hundred and fifty-six BTCs were tested with NGS using the MiSeq platform, and 149 of these were also tested with RNA sequencing for genetic fusions. Of the 1,502 BTCs analyzed, 39.5% were from primary/localized tumors, 23.2% were from metastatic tumors, and 37.4% were from unknown sites (primary/localized or metastatic, Table 1).
Table 1. Patient and tumor characteristics of age and gender by tumor location.
| Characteristic | IHCC (n=825) | EHCC (n=249) | GBC (n=428) | Total (n=1,502) |
|---|---|---|---|---|
| Tumors analyzed by NGS, n (%) | 200 (24.2) | 51 (20.5) | 105 (24.5) | 356 (23.7) |
| Age (years), mean [range] | 59.8 [20–90] | 63.0 [27–88] | 63.6 [32–90] | 61.4 [20–90] |
| Gender (female), % | 52.1 | 39.8 | 62.1 | 52.9 |
| Primary/local, n (%) | 387 (46.9) | 82 (32.9) | 124 (29.0) | 593 (39.5) |
| Metastatic, n (%) | 137 (16.6) | 61 (24.5) | 150 (35.0) | 348 (23.2) |
| Unknown (primary/local or metastatic), n (%) | 301 (36.5) | 106 (42.6) | 154 (36.0) | 561 (37.4) |
IHCC, intrahepatic cholangiocarcinoma; EHCC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma; NGS, next-generation sequencing.
Patient characteristics
The median age was similar across tumor subtypes (59.8 years in IHCC; 63.0 years in EHCC; and 63.6 years in GBC). GBC (62.1%) was significantly more prevalent in females compared to IHCC (52.1%, P=0.0008) or EHCC (39.8%, P<0.0001).
Molecular alterations
Among the 592 genes sequenced, 356 genes were mutated in at least one tumor (Figure 1). The most prevalent mutations were in TP53 (42.7%), ARID1A (21.7%), KRAS (15.7%), IDH1 (8.7%), CDKN2A (7.8%), BAP1 (6.7%), SMAD4 (6.5%), and PIK3CA (6.0%). Mutation rates in all other genes were seen in fewer than 5% of tumors. Of the 443 genes interrogated for CNVs, 119 genes were amplified in at least one tumor (Table 2). The highest amplification rates were in Her2/neu (4.7%), MYC (3.2%), MDM2 (3.2%), cMET (2.3%), CCND1 (2.0%), and CCNE1 (2.0%). Her2/neu amplifications were identified in 6.4% of tumors by CISH, 4.7% had CNVs by NGS read depth analysis, and overexpression was observed in 3.7% of tumors by IHC. cMET amplifications were seen in 1.8% of tumors by CISH, 2.3% of tumors by NGS, and was overexpressed in 40.9% of tumors by IHC. Protein expression of TS, ERCC1, and RRM1 by IHC was seen in 27.8%, 27.3%, and 14.2% of tumors, respectively.
Figure 1.
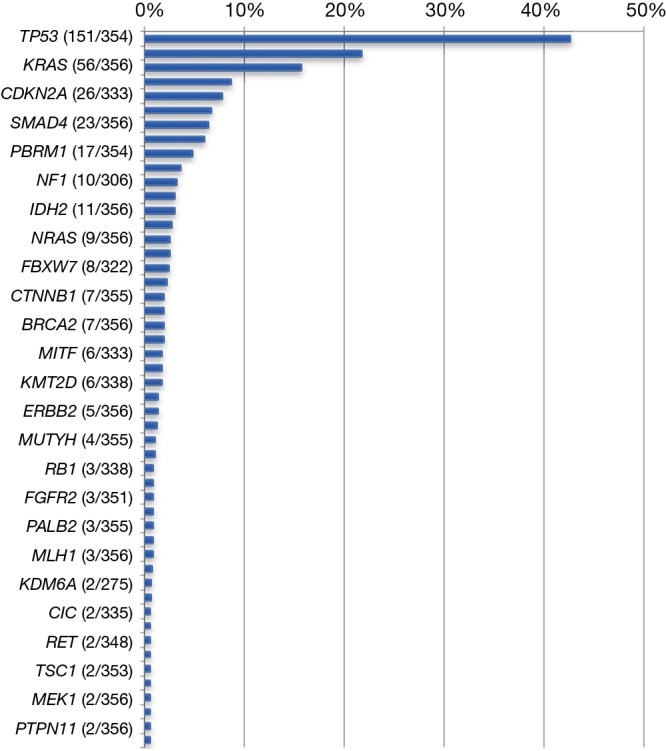
Gene mutation rates across all biliary cancers (n=356).
Table 2. Copy number variations in all biliary cancers.
| Gene | CNV+ | CNV total number | CNV, % |
|---|---|---|---|
| Her2/Neu | 16 | 342 | 4.7 |
| MYC | 11 | 342 | 3.2 |
| MDM2 | 11 | 342 | 3.2 |
| cMET | 8 | 342 | 2.3 |
| CCND1 | 7 | 342 | 2.0 |
| CCNE1 | 7 | 342 | 2.0 |
| HMGA2 | 6 | 325 | 1.8 |
| KRAS | 6 | 325 | 1.8 |
| FGF4 | 6 | 342 | 1.8 |
| FGF3 | 6 | 342 | 1.8 |
| WIF1 | 5 | 325 | 1.5 |
| FGF19 | 5 | 325 | 1.5 |
| MLLT6 | 5 | 325 | 1.5 |
| PDCD1 | 4 | 325 | 1.2 |
| RARA | 4 | 325 | 1.2 |
| MCL1 | 4 | 342 | 1.2 |
| CDK4 | 4 | 342 | 1.2 |
| FGFR3 | 4 | 342 | 1.2 |
CNV, copy number variation.
RNA sequencing was performed on 149 tumors in total (Table 3), of which 10 had a detectable fusion (6.7%). FGFR2 fusions were seen in 7 tumors (all with different fusion partners), one EHCC, one GBC, and 5 IHCCs. A single FGFR3 fusion was seen in one GBC (FGFR3-TACC3). BRAF fusions were identified in one GBC (CUX1-BRAF) and one IHCC (SND1-BRAF).
Table 3. Fusion genes detected by RNA sequencing.
| Gene | Fusions | Total tested | Frequency, % | IHCC fusions | EHCC fusions | GBC fusions |
|---|---|---|---|---|---|---|
| BRAF | 2 | 131 | 1.5 | SND1-BRAF | – | CUX1-BRAF |
| FGFR2 | 7 | 149 | 4.7 | FGFR2-AHCYL1 FGFR2-AXDND1 FGFR2-FMR1 FGFR2-PAWR FGFR2-MYLK | FGFR2-ADAM9 | FGFR2-BICC1 |
| FGFR3 | 1 | 149 | 0.7 | – | – | FGFR3-TACC3 |
IHCC, intrahepatic cholangiocarcinoma; EHCC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma.
Comparison of molecular profiles in IHCC, EHCC and GBC
Significant differences were seen when comparing gene mutation rates between the 3 different BTC subtypes (Figure 2A). IHCCs had significantly higher IDH1 (14.5%), BAP1 (9.5%), and PBRM1 (7.5%) mutation rates than GBCs (P<0.05). On the other hand, EHCCs were characterized by significantly more frequent mutations in KRAS (37.3%), CDKN2A (22.0%), and BRCA1 (3.9%, all P<0.05). GBCs were characterized by significantly higher Her2/neu overexpression (9.2%) and amplification (14.4%), as well as high TOP2A expression (78.3%) and amplification (25.0%, all P<0.05). EHCCs and GBCs carry some similar molecular features when compared to IHCCs, including higher mutation rates of TP53, SMAD4, APC, and ERBB2. Grouping the mutations in homologous repair genes together (ATM, ATRX, BRCA1, BRCA2, CHEK2, PALB2, PTEN, and NBN), 13.8% of EHCCs, 19.1% of GBCs, and 7.4% of IHCCs demonstrated evidence of homologous repair deficiency (Figure 2B). Looking at mutations or rearrangements in BRAF and receptor tyrosine kinases HER2 and FGFR, 15.7% of EHCCs, 23.8% of GBCs, and 12.0% of IHCCs had potentially therapeutic targets.
Figure 2.

Difference in mutation rates between extrahepatic cholangiocarcinoma, intrahepatic cholangiocarcinoma, and gallbladder carcinoma. (A) Significant differences in a range of gene mutation rates between extrahepatic cholangiocarcinoma, intrahepatic cholangiocarcinoma, and gallbladder carcinoma. Relative gene mutational frequencies across subtypes of biliary tract cancer. Horizontal brackets indicate significant differences by Chi-squared tests (P<0.05); (B) rates of homologous recombination repair deficiency, receptor tyrosine kinase mutations, and other significant actionable mutations across tumor subtypes. IHCC, intrahepatic cholangiocarcinoma; EHCC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma; CISH, chromogenic in situ hybridization; FISH, fluorescent in situ hybridization; IHC, immunohistochemistry.
Immune checkpoint inhibitor-associated biomarkers
PD-L1 overexpression was seen on tumor cells from 63 of 798 (7.9%) tumors using IHC. Increased microsatellite instability (MSI-H) was seen in 7 out of 352 tumors (2.0%) using NGS (Table 4). Four percent of all BTCs (all subtypes) were TMB-H based on a cutoff of 17 somatic missense mutations per MB of targeted sequence. Of note, gene amplification of CD274 (the PD-L1-encoding gene) was only seen in one of 325 tumors. Overall, when all 3 markers (PD-L1, MSI-H, and TMB-H) were considered, 11.4% of tumors carried at least one favorable biomarker for immune checkpoint blockade. When the 3 BTC subtypes were investigated individually, IHCCs and GBCs had a similar prevalence of predictive markers to immune checkpoint blockade (13.0% and 12.0%, respectively), whereas EHCCs had a prevalence of only 6.9% (P>0.05).
Table 4. Biomarkers associated with response to immune checkpoint inhibitors.
| BTC subtype | PD-L1 by IHC | MSI-H | TMB-H | Overall percent positive |
|---|---|---|---|---|
| All BTCs | 7.9% (63/798) | 2.0% (7/352) | 4.0% (14/352) | 11.4% (39/342) |
| IHCC | 8.1% (36/444) | 2.5% (5/198) | 3.5% (7/198) | 13.0% (25/193) |
| EHCC | 6.9% (8/116) | 2.0% (1/50) | 2.0% (1/50) | 6.9% (2/29) |
| GBC | 8.0% (19/238) | 1.0% (1/104) | 5.8% (6/104) | 12.0% (12/100) |
Overall percent positive is calculated in the subgroups of tumors with results from all 3 available biomarkers. PD-L1, programmed death-ligand 1; MSI-H, microsatellite instability high; TMB-H, high tumor mutational burden; BTC, biliary tract cancer; IHCC, intrahepatic cholangiocarcinoma; EHCC, extrahepatic cholangiocarcinoma; GBC, gallbladder carcinoma.
Discussion
BTCs are an uncommon group of cancers with a generally aggressive clinical course and poor prognosis. Molecular characterization of BTCs provides potentially actionable clinical information, especially because EHCC, IHCC, and GBC are anatomically close and histologically similar cancer entities (13). In order to improve patient outcomes for BTC, it is imperative to better tailor therapies for patients, selecting who will benefit from immune checkpoint inhibitors and specific targeted therapies. BTCs are a good candidate for molecular analysis because they are rich in therapeutic targets and are highly heterogeneous (13,14). The targetable alterations in BTCs vary significantly with etiology, patient geographical origin, and different anatomical locations despite similar histology (12).
Chemotherapy resistance and sensitivity
High expression of TS, RRM1, and ERCC1 has been shown to confer resistance to fluoropyrimidines, gemcitabine, and platinum-based agents, respectively (25-30). We found that over one-quarter of all BTCs had increased expression of at least one of these markers. The high prevalence of TS, RRM1, and ERCC1 may indicate resistance to standard chemotherapies (de novo resistance) or reflect previous exposure to those cytotoxic agents (acquired resistance). TOP2A overexpression was also very common, especially in GBCs (78.3% positive). TOP2A is often associated sensitivity to anthracyclines, more so in breast cancer than in hepatocellular carcinoma (31), and its predictive power in BTCs is unknown. Anthracyclines have been previously used in salvage regimens in chemorefractory BTCs (32-35), although their use has declined due to their associated cardiotoxicity and effective alternative agents. However, anthracyclines may be still appropriate in a specific subset of patients, conceivably those with TOP2A amplification by FISH (9.0% of our cohort), although this should be further evaluated in prospective clinical trials.
Homologous recombination repair deficiency
An increasing number of malignancies have been associated with defects in the homologous recombination DNA repair pathway including mutations in BRCA1, BRCA2, PALB2, and others (see above). Although many of these genes are mutated relatively infrequently, they are a sizable subset when grouped together and were seen in 19.1% of GBCs in our study. This finding has potential therapeutic implications, as tumors driven by homologous recombination repair deficiency may be more sensitive to poly ADP-ribose polymerase (PARP) inhibitors and platinum-based chemotherapy (36-39).
cMET
In our cohort, cMET expression was seen in 40.9% of BTCs, with no significant difference between subgroups. Although cMET expression has been associated with an unfavorable prognostic marker in some studies, conflicting results have also been reported (40-42). Agents that specifically target cMET have shown promise in various cancer types, and although BTC-specific trials are lacking, early preclinical studies in cholangiocarcinoma cell-lines that overexpress cMET have shown promising results (43). In contrast to the high cMET overexpression rate observed in BTCs (40.9%; Figure 3), cMET amplification was seen in only 1.8% of tumors, and unsurprisingly, all 6 cMET amplified tumors showed strong positive IHC staining. While cMET inhibitors are being investigated in various solid tumors, these data suggest that BTCs should be included. The best predictive biomarker (overexpression by IHC vs. amplification by CNV vs. other) for cMET-targeted therapies in BTC remains undetermined and warrants further investigation.
Figure 3.

IHC and CISH protein overexpression and amplification rates across all biliary cancers. IHC, immunohistochemistry; CISH, chromogenic in situ hybridization.
Her2/neu
Her2 overexpression and amplification were seen in 4–6% of BTCs profiled. Consistent with previous observations, the extent of Her2 overexpression and amplification in GBCs (9–14%) is greater than in EHCCs (4–8%), which is greater than in IHCCs (1%). Large, randomized, controlled trials of Her2-targeted therapies have demonstrated benefit for patients with Her2-positive breast and gastroesophageal cancers, but similar trials in BTCs have not been carried out. This is because the prevalence of Her2 overexpression and amplification is more common in breast and gastroesophageal cancers. However, case reports and series have shown Her2 to be an effective target in GBCs (44,45). The MyPathway basket trial included 7 patients with Her2 amplified or overexpressed BTC treated with Her2-targeted therapy (trastuzumab and pertuzumab); 2 patients had a partial response and another 3 patients had stable disease beyond 120 days (46). In addition, the SUMMIT trial using the pan-Her kinase inhibitor neratinib included 9 patients with Her2-mutated BTC; 2 patients had a partial response and a third patient had stable disease (47). Currently, a clinical trial (NCT03093870) is underway to investigate the use of varlitinib, a poly-tyrosine kinase inhibitor (including epidermal growth factor receptor (EGFR), HER2, and HER4), in BTCs.
EHCCs and GBCs had higher rates of ERBB2 and TP53 mutations than IHCCs. Jusakul and colleagues observed a similar tendency for ERBB2 and TP53 mutations in EHCCs (clusters 1 and 2) (12). Their study found these mutations were associated with fluke infection, O. viverrini and C. cinensis, which is relatively uncommon in the United States. In general, Her2-targeted therapy may be the best hope for EHCCs since they tend to lack other viable targets.
FGFR
Gene fusions are often oncogenic driver mutations. However, only about 10% of our cohort had fusions assayed, and they were found in all 3 tumor-subtypes. FGFR2 fusions were seen most often in IHCCs. This is noteworthy because of ongoing clinical trials of FGFR inhibitors in FGFR2 fusion-positive cholangiocarcinomas (NCT02150967; NCT03230318; NCT02924376) and various tumors with FGFR2-related abnormalities (basket trials; NCT02052778; NCT01948297).
IDH1
We found a high frequency of chromatin-remodeling gene mutations in our patient cohort, which is consistent with other reports identifying mutations in IDH1, BAP1, ARID1A, and PBRM1 (12,14,44,48). IHCCs are frequently not associated with liver fluke infection (cluster 4), and mutations in these chromatin modifying genes often serve as the primary epigenetic driver mutation leading to DNA hypermethylation and tumor growth (12). IDH1 is particularly significant because there are several ongoing clinical trials (NCT02073994; NCT02989857; NCT02746081; NCT03212274) that are investigating small molecule IDH1 inhibitors in solid tumors, including cholangiocarcinomas.
RNF43
RNF43 was rarely mutated in our study, seen in 2.0% of EHCC, 1.0% of GBCs, and 1.5% of IHCCs. Ring finger protein 43 (RNF43) mutations can promote Wnt/β-catenin pathway signaling which causes tumorigenesis, and novel Wnt inhibitors are currently being studies in ongoing clinical trials (NCT02675946, NCT025221844, NCT01351103). Therefore, although this is a rare subset of BTCs, patients with RNF43-mutated tumors should be referred to these ongoing clinical trials as appropriate.
Immune checkpoint inhibition
Although only a single gene amplification of CD274 was observed in the 352 tumors analyzed for CD274 CNV by NGS, 7.9% of all tumors overexpressed PD-L1 by IHC. Abundant somatic mutations in solid tumors have been associated with a greater responsiveness to immune checkpoint inhibitors (20,21,49). TMB-H and MSI-H are potentially useful for assessing neoantigen presentation and viability of immune checkpoint inhibition. In this cohort, 11.4% of all BTCs had a predictive biomarker for immune checkpoint inhibitors, particularly in IHCC and GBC types. Our findings support the notion that immune checkpoint blockade may be used to treat BTCs, and this is currently being tested in multiple ongoing clinical trials (NCT03267940, NCT02703714, NCT03101566, NCT03111732).
Limitations to our study include the lack of clinical outcome information and the use of diagnostic testing platforms that are commonly used for clinical purposes. Also, tumors did not undergo identical diagnostic testing (many underwent analysis by IHC only). In addition, we do not have data on previous exposure of patients to liver flukes, incidence of primary sclerosing cholangitis, or incidence of choledochal cysts, all of which would help elucidate the etiology of individual BTCs.
Conclusions
Our characterization of 1,502 BTCs has led to insights into molecular identities of IHCCs, EHCCs, and GBCs, which may be helpful in understanding treatment and improving outcomes. Patients with IHCCs may benefit from enrollment in clinical trials targeting FGFR2 and IDH1. GBCs in particular may be suitable for agents targeting homologous recombination repair deficient tumors (e.g., PARP inhibitors). IHCCs and GBCs may warrant immunotherapeutic approaches due to increased levels of PD-L1 expression, TMB-H, and MSI-H. EHCCs might have the fewest prospects of future treatments based on current clinical trials and the molecular targets assessed by our study. While no targeted therapies or immunotherapies (except for MSI-H tumors) are currently FDA approved for BTC, several promising agents are in development, and comprehensive molecular profiling will enable optimal therapeutic selection for patients.
Acknowledgments
We would like to thank Marion L. Hartley, PhD, for her edits and suggestions during the composition of this manuscript.
Ethical Statement: The study was deemed exempt by the Western Institutional Review Board (IRB registration number IRB 00000533, WIRB work order #1-895778-1), and no informed consent was obtained for this retrospective analysis.
Footnotes
Conflicts of Interest: Drs. Xiu and Poorman are employed by Caris Life Sciences. The other authors have no conflicts of interest to declare.
References
- 1.Noel MS, Hezel AF. New and emerging treatment options for biliary tract cancer. Onco Targets Ther 2013;6:1545-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horgan AM, Amir E, Walter T, et al. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934-40. 10.1200/JCO.2011.40.5381 [DOI] [PubMed] [Google Scholar]
- 3.Ma N, Cheng H, Qin B, et al. Adjuvant therapy in the treatment of gallbladder cancer: a meta-analysis. BMC Cancer 2015;15:615. 10.1186/s12885-015-1617-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarnagin WR, Shoup M. Surgical management of cholangiocarcinoma. Semin Liver Dis 2004;24:189-99. 10.1055/s-2004-828895 [DOI] [PubMed] [Google Scholar]
- 5.Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: The BILCAP randomized study. J Clin Oncol 2017;35:Abstr4006.
- 6.Ben-Josef E, Guthrie KA, El-Khoueiry AB, et al. SWOG S0809: A Phase II Intergroup Trial of Adjuvant Capecitabine and Gemcitabine Followed by Radiotherapy and Concurrent Capecitabine in Extrahepatic Cholangiocarcinoma and Gallbladder Carcinoma. J Clin Oncol 2015;33:2617-22. 10.1200/JCO.2014.60.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigt J, Malfertheiner P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev Gastroenterol Hepatol 2010;4:395-7. 10.1586/egh.10.45 [DOI] [PubMed] [Google Scholar]
- 8.Lamarca A, Hubner RA, David Ryder W, et al. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328-38. 10.1093/annonc/mdu162 [DOI] [PubMed] [Google Scholar]
- 9.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273-81. 10.1056/NEJMoa0908721 [DOI] [PubMed] [Google Scholar]
- 10.Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10. 10.1038/ng.3375 [DOI] [PubMed] [Google Scholar]
- 11.Cardinale V, Carpino G, Reid L, et al. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J Gastrointest Oncol 2012;4:94-102. 10.4251/wjgo.v4.i5.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jusakul A, Cutcutache I, Yong CH, et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov 2017;7:1116-35. 10.1158/2159-8290.CD-17-0368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo KH, Kim NK, Kwon WI, et al. Genomic Alterations in Biliary Tract Cancer Using Targeted Sequencing. Transl Oncol 2016;9:173-8. 10.1016/j.tranon.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain A, Javle M. Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol 2016;7:797-803. 10.21037/jgo.2016.09.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.FDA. Keytruda. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125514s024lbl.pdf. Accessed November 21 2017.
- 16.Colli LM, Machiela MJ, Myers TA, et al. Burden of Nonsynonymous Mutations among TCGA Cancers and Candidate Immune Checkpoint Inhibitor Responses. Cancer Res 2016;76:3767-72. 10.1158/0008-5472.CAN-16-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodman AM, Kato S, Bazhenova L, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther 2017;16:2598-608. 10.1158/1535-7163.MCT-17-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson DB, Frampton GM, Rioth MJ, et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol Res 2016;4:959-67. 10.1158/2326-6066.CIR-16-0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463-9. 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377:2500-1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadler ZK, Battaglin F, Middha S, et al. Reliable Detection of Mismatch Repair Deficiency in Colorectal Cancers Using Mutational Load in Next-Generation Sequencing Panels. J Clin Oncol 2016;34:2141-7. 10.1200/JCO.2015.65.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderwalde A, Spetzler D, Xiao N, et al. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018;7:746-56. 10.1002/cam4.1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babiceanu M, Qin F, Xie Z, et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res 2016;44:2859-72. 10.1093/nar/gkw032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao Y, Cui J, Xi H, et al. Association of thymidylate synthase expression and clinical outcomes of gastric cancer patients treated with fluoropyrimidine-based chemotherapy: a meta-analysis. Onco Targets Ther 2016;9:1339-50. 10.2147/OTT.S98540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CY, Chang YL, Shih JY, et al. Thymidylate synthase and dihydrofolate reductase expression in non-small cell lung carcinoma: the association with treatment efficacy of pemetrexed. Lung Cancer 2011;74:132-8. 10.1016/j.lungcan.2011.01.024 [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, Sun J, Zhen J, et al. Thymidylate synthase predicts for clinical outcome in invasive breast cancer. Histol Histopathol 2005;20:871-8. [DOI] [PubMed] [Google Scholar]
- 28.Lee SJ, Choi YL, Park YH, et al. Thymidylate synthase and thymidine phosphorylase as predictive markers of capecitabine monotherapy in patients with anthracycline- and taxane-pretreated metastatic breast cancer. Cancer Chemother Pharmacol 2011;68:743-51. 10.1007/s00280-010-1545-0 [DOI] [PubMed] [Google Scholar]
- 29.Minami K, Shinsato Y, Yamamoto M, et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol Sci 2015;127:319-25. 10.1016/j.jphs.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 30.Li P, Fang YJ, Li F, et al. ERCC1, defective mismatch repair status as predictive biomarkers of survival for stage III colon cancer patients receiving oxaliplatin-based adjuvant chemotherapy. Br J Cancer 2013;108:1238-44. 10.1038/bjc.2013.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao S, Beckman RA, Riazi S, et al. Quantification and expert evaluation of evidence for chemopredictive biomarkers to personalize cancer treatment. Oncotarget 2017;8:37923-34. 10.18632/oncotarget.13544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim KH, Han SW, Oh DY, et al. Outcome of infusional 5-fluorouracil, doxorubicin, and mitomycin-C (iFAM) chemotherapy and analysis of prognostic factors in patients with refractory advanced biliary tract cancer. Oncology 2012;83:57-66. 10.1159/000338795 [DOI] [PubMed] [Google Scholar]
- 33.Lee S, Oh SY, Kim BG, et al. Second-line treatment with a combination of continuous 5-fluorouracil, doxorubicin, and mitomycin-C (conti-FAM) in gemcitabine-pretreated pancreatic and biliary tract cancer. Am J Clin Oncol 2009;32:348-52. 10.1097/COC.0b013e31818c08ff [DOI] [PubMed] [Google Scholar]
- 34.Furuse J, Okusaka T, Funakoshi A, et al. Early phase II study of uracil-tegafur plus doxorubicin in patients with unresectable advanced biliary tract cancer. Jpn J Clin Oncol 2006;36:552-6. 10.1093/jjco/hyl075 [DOI] [PubMed] [Google Scholar]
- 35.Park SH, Park YH, Lee JN, et al. Phase II study of epirubicin, cisplatin, and capecitabine for advanced biliary tract adenocarcinoma. Cancer 2006;106:361-5. 10.1002/cncr.21621 [DOI] [PubMed] [Google Scholar]
- 36.Somlo G, Frankel PH, Arun BK, et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin Cancer Res 2017;23:4066-76. 10.1158/1078-0432.CCR-16-2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244-50. 10.1200/JCO.2014.56.2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bendell J, O'Reilly EM, Middleton MR, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol 2015;26:804-11. 10.1093/annonc/mdu581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Bono J, Ramanathan RK, Mina L, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 2017;7:620-9. 10.1158/2159-8290.CD-16-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blumenschein GR, Jr, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol 2012;30:3287-96. 10.1200/JCO.2011.40.3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heo MH, Kim HK, Lee H, et al. The Clinical Impact of c-MET Over-Expression in Advanced Biliary Tract Cancer (BTC). J Cancer 2017;8:1395-9. 10.7150/jca.17898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyamoto M, Ojima H, Iwasaki M, et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer 2011;105:131-8. 10.1038/bjc.2011.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barat S, Bozko P, Chen X, et al. Targeting c-MET by LY2801653 for treatment of cholangiocarcinoma. Mol Carcinog 2016;55:2037-50. 10.1002/mc.22449 [DOI] [PubMed] [Google Scholar]
- 44.Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58. 10.1186/s13045-015-0155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Law LY. Dramatic response to trastuzumab and paclitaxel in a patient with human epidermal growth factor receptor 2-positive metastatic cholangiocarcinoma. J Clin Oncol 2012;30:e271-3. 10.1200/JCO.2012.42.3061 [DOI] [PubMed] [Google Scholar]
- 46.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol 2018;36:536-42. 10.1200/JCO.2017.75.3780 [DOI] [PubMed] [Google Scholar]
- 47.Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189-94. 10.1038/nature25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3. 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gatalica Z, Snyder C, Maney T, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev 2014;23:2965-70. 10.1158/1055-9965.EPI-14-0654 [DOI] [PubMed] [Google Scholar]