

Abstract
Fluorescence spectroscopy and Raman spectroscopy are two major classes of spectroscopy methods in physical chemistry. Very recently stimulated Raman excited fluorescence (SREF) has been demonstrated (Xiong, H.; et al. Nature Photonics, 2019) as a new hybrid spectroscopy that combines the vibrational specificity of Raman spectroscopy with the superb sensitivity of fluorescence spectroscopy (down to single molecule level). However, this proof-of-concept study was limited by both the tunability of the commercial laser source and the availability of the excitable molecules in the near infrared. As a result, the generality of SREF spectroscopy remains unaddressed, and the understanding of the critical electronic pre-resonance condition is lacking. Herein we built a modified excitation source to explore SREF spectroscopy in the visible region. Harnessing a large palette of red dyes, we have systematically studied SREF spectroscopy on a dozen of different cases with a fine spectral interval of several nanometers. The results not only establish the generality of SREF spectroscopy for a wide range of molecules, but also reveal a tight window of proper electronic pre-resonance for the stimulated Raman pumping process. Our theoretical modeling and further experiments on newly synthesized dyes also support the obtained insights, which would be valuable in designing and optimizing future SREF experiments for single-molecule vibrational spectroscopy and super-multiplex vibrational imaging.
Graphical Abstract
The exquisite chemical specificity of Raman spectroscopy provides rich structure and dynamics information1. However, Raman scattering is intrinsically weak, with a cross-section of 10−30 cm−2 for typical chemical bonds2. Although several enchantment mechanisms (such as coherent Raman scattering and electronic resonance Raman) have been extensively exploited to amplify the Raman scattering cross sections3–11, it is still difficult to achieve all-far-field (without plasmonic resonance2,12–17) Raman spectroscopy at the ultimate single-molecule level. For example, electronic pre-resonance stimulated Raman scattering microscopy, which has combined coherent Raman scattering and electronic resonance Raman together under optical microscope, is still limited to about 50 molecules.
Another important school of strategies to enhance the Raman sensitivity is to couple vibrational information to more sensitive optical observables. Notably, the pioneering work of Laubereau et al. has employed fluorescence detection with time-resolved infrared spectroscopy to study vibrational relaxation dynamics19–21. Along this line of fluorescence-encoded infrared spectroscopy, Whaley-Mayda et. al have coupled this method with modern confocal microscope and reported an impressive sensitivity of nanomolar level22. Along the Raman direction, Wright proposed early in the 1980 that the fluorophore can be first pumped through stimulated Raman scattering (SRS) to vibrational excited state, which can then be brought up to fluorescence excited state by second excitation23 (Fig. 1a, which is related to but distinct from electronic resonance coherent anti-Stokes Raman scattering (CARS)24). This approach would have both the sharp lines characteristic of Raman spectroscopy and an increased sensitivity characteristic of fluorescence spectroscopy. In light of the revolutionary success of single-molecule fluorescence spectroscopy in the subsequent decades25–27, Winterhalder et al. were also optimistic and proposed in 2011 that this method might offer the single-molecule sensitivity of vibrational spectroscopy at optical far-field28. However, the first experimental attempt by Wright’s group in 1983 was hindered by overwhelming two-photon fluorescence background from perylene dye29. To the best of our knowledge, no further experimental studies have been published about this type of spectroscopy since 1983.
Fig. 1. New stimulated Raman excited fluorescence (SREF) laser microscopy system opening excitation band near 600-nm.

(a) Energy diagram for SREF process. (b) Wavelength of SHG of OPO idler (red) and corresponding resonance Raman shift (blue) as a function of OPO signal wavelength when OPO signal is set as Stoke beam and the SHG is set as pump beam. (c) System setup for SREF excitation band around 600-nm. PPLN, periodically poled Lithium Niobate; QWP, quarter-wave plate; DM, dichroic mirror; PBS, polarization beam splitter; BS, beam splitter; SPCM, single photon counting module; DAC, digital-to-analog converter; ADC, analog-to-digital converter; EOM, electro-optical modulator; APD, avalanched photodiode; PD, photodiode.
Very recently, our group has revisited this topic and reported stimulated Raman excited fluorescence (SREF) spectroscopy and imaging (Fig. 1a)30. By leveraging the joint advantage of chemical specificity and detection sensitivity, we have achieved single-molecule Raman sensitivity in all-far-field manner for the first time, and demonstrated multicolor SREF imaging of isotopologues to break the color barrier of fluorescence microscopy. We noted that the stimulated Raman pumping in the previous proposals and attempts was operated in the non-resonant condition. As a result, SREF can be easily buried by other competing process such as two-photon excited fluorescence, which was observed in both the 1983 study29 and our own experiment on the coumarin dye. Hence, we decided to harness electronic resonance to promote the stimulated Raman pumping process. Yet, we found in our pilot studies that bringing stimulated Raman pumping to rigorous electronic resonance gave us an overwhelming background largely from the anti-Stokes fluorescence background. We reasoned that this should be due to the fact that linear absorption cross section (which determines anti-Stokes fluorescence background) increases much faster than the resonance Raman cross section as the pump excitation approaches rigorous electronic resonance31. So we detuned stimulated Raman pumping empirically to some extent to electronic pre-resonance, and successfully achieved vibrational specificity and nanomolar sensitivity on two dyes (nitrile mode of Rh800 and C=C skeletal mode of ATTO 740).
Despite the excitement and promise, there are two critical (and also inter-related) limitations in this proof-of-concept study. First of all, the experiment is based on a commercial optical parametric oscillator (OPO) system, which output a fixed beam at 1031.2 nm and a tunable pump beam operated in near-IR region (i.e., the electronic resonance cannot be studied independent of the vibrational resonance). There are limited choices of bright fluorophores in the near-IR range. As a result, only two dyes have been demonstrated30. The generality of SREF spectroscopy remains unestablished beyond these two dyes. Second, the apparent “rare” success of SREF suggests that it is critical to achieve a proper electronic pre-resonance for the stimulated Raman pumping process: too much electronic resonance will make anti-Stokes fluorescence dominate SREF, and too little electronic resonance will make SREF too weak. Thus, the logical next step is to study the effect of electronic pre-resonance condition in a systematic way, in order to quantitatively characterize and understand this effect. This understanding is rather important as it will contribute to establishing the generality of SREF spectroscopy as well as providing practical guide for future design and application. Again, this task is difficult to perform with the commercial OPO system and the relative lack of dyes in this near-IR range.
In the present work, to address the two critical limitations related to SREF spectroscopy in our proof-of-concept paper, we will build a modified source to open up a new excitation range near 600-nm for SREF, which will allow us to test the generality of SREF spectroscopy on a number of visible dyes and to study the effect of electronic resonance condition in a systematic way. While a two-beam free tunable laser system might be more flexible to gain independent access to electronic resonance and vibrational resonance, such a laser system is rather difficult and expensive to obtain in the lab. Alternatively, we will adopt a simple approach by frequency doubling the idler beam from the commercial OPO laser source and then recombining it with the OPO signal beam as a new pair of pump and Stokes beams (Fig. 1, detail in methods) to open up a new SREF excitation range near 600-nm. Benefiting from a large variety of commercial fluorescence dyes in this visible range, the generality of SREF can be sufficiently tested. Moreover, we will select a rich list of dyes with slightly different (a few nm interval) absorption peak (λabs) in this range, which provides different electronic resonance condition for systematic evaluation.
Our new excitation source is based on an optical modification of the previous commercial OPO system (picoEmerald S, APE), which outputs the signal beam (λsignal, tunable from 790-nm to 960-nm) and the idler beam (λidler), with their wavelengths following a relationship of 1/λsignal +1/λidler = 1/515.6-nm. Hence, by tuning of λsignal, λidler will cover the wavelength range of 1484.4-nm to 1113.8-nm with the same pulse shape and laser linewidth (2-ps, 0.6-nm FWHM bandwidth). We then frequency doubled the idler beam by second harmonic generation (SHG) with a periodically poled Lithium Niobate (PPLN) crystal (Covesion, MSHG1420–0.5–5) (Fig. 1b, c). We then used the SHG (λidler/2) of the idler as the new pump beam (ωp), and the signal λsignal as the new Stokes beam (ωs). When tuning λsignal from 805-nm to 825-nm with the PPLN temperature matched simultaneously, the λidler/2 will sweep from 717.1-nm to 687.4-nm. As such, the energy difference between this new pair covers the Raman shift between 1523-cm−1 and 2426-cm−1 (Fig. 1b). The crystal length (5-mm) is chosen to be short enough to ensure neglectable group velocity mismatch, which well maintains of the ~2-ps pulse width for efficient SREF excitation30 (Fig. S1a).
With this new excitation source, the total excitation energy ((ωp - ωs) + ωp)) for a typical C=C skeletal mode (~1650 cm−1) of fluorescent dyes reaches ~638-nm. Therefore, C=C skeletal mode with dye absorption peak (λabs) around 638-nm can be potentially excited. Different from the previous near-IR region which lacks bright fluorophores, many commercial fluorophores with high photo-stability and large quantum yield, such as Alexa 610, ATTO Rho14, Nile Blue A, Alexa 633, ATTO 633, Alexa 647 etc., can be found with absorption peak around 638-nm (likely because of the famous He-Ne laser line at 633-nm). To test the generality of SREF spectroscopy and to study the critical electronic pre-resonance condition systematically, we select seven popular fluorescent dyes with λabs gradually shifting from 621-nm to 662-nm. With the additional help of solvatochromic effect in different solvents32, we are able to generate 11 different λabs within this range with a step size of several nanometers (Table 1). Since the total SREF excitation energy ((ωp - ωs) + ωp) of the C=C mode is fixed around 638-nm, these 11 λabs from 621-nm to 662-nm serve as a “sweep” to study the effect of electronic resonance condition in a systematic way. As shown in Table 1, we can divide these 11 cases into three categories depending on the energetic relationship between λabs and ((ωp - ωs) + ωp).
Table 1.
Spectral properties of commercial red SREF dyes experimented in this study.
| Dye1 | Solvent2 | λabs (nm) | C=C mode (cm−1) | (ωp - ωs) + ωp (nm) |
Concentration (nM) |
SNR3 |
|---|---|---|---|---|---|---|
| ATTO 610* | a | 621 | 1644 | 638.1 | 500 | undetectable |
| Alexa 633^ | a | 630 | 1651 | 637.6 | 100 | 2.5 |
| ATTO 610* | b | 631 | 1644 | 638.1 | 500 | 3.0 |
| ATTO Rho14* | a | 632 | 1652 &1680 | 637.5 & 635.7 | < 2000 | 7.5 & 2.1 |
| Nile Blue A | c | 635 | 1652 | 637.5 | 500 | 7.1 |
| ATTO 633* | a | 635 | 1640 | 638.4 | 500 | 3.1 |
| ATTO Rho14* | b | 638 | 1652 &1680 | 637.5 & 635.7 | 50 | 3.0 & 1.5 |
| Alexa 633^ | b | 640 | 1651 | 637.6 | 10 | 3.0 |
| ATTO 633* | b | 642 | 1640 | 638.4 | 500 | 1.6 |
| Alexa 647* | a | 654 | 1602 | 640.8 | 500 | undetectable |
| JF 646* | d | 662 | 1616 | 639.9 | 500 | undetectable |
We started from the high-energy side of the absorption peaks. ATTO 610 NHS ester in phosphate buffered saline (PBS, pH = 7.4) is the model dye in this category. As shown in Fig. 2, SRS spectrum clearly shows a Raman peak around 1638 cm−1 for its C=C skeletal mode. Note that, the electronic resonance is detuned substantially so that the total SREF excitation energy ((ωp - ωs) + ωp) is lying below the ensemble 0–0 transition energy (i.e. the cross point between the absorption spectrum and corresponding emission spectrum). As a result of this energetic gap, the pure SREF signal is observed to be very weak and barely detectable above the background (Fig. 2b, c). We reason that only a very small portion of molecules in the solution can be pumped to electronic excited state by SREF (likely because of inhomogeneous broadening). Therefore, the 0–0 transition energy can be set as the “red” side limit for the total SREF excitation energy. This limit would become more apparent after considering the complete “sweep” of the 11 cases shown below.
Fig. 2. SREF excitation with the total SREF excitation energy ((ωp - ωs) + ωp) below the 0–0 transition line.

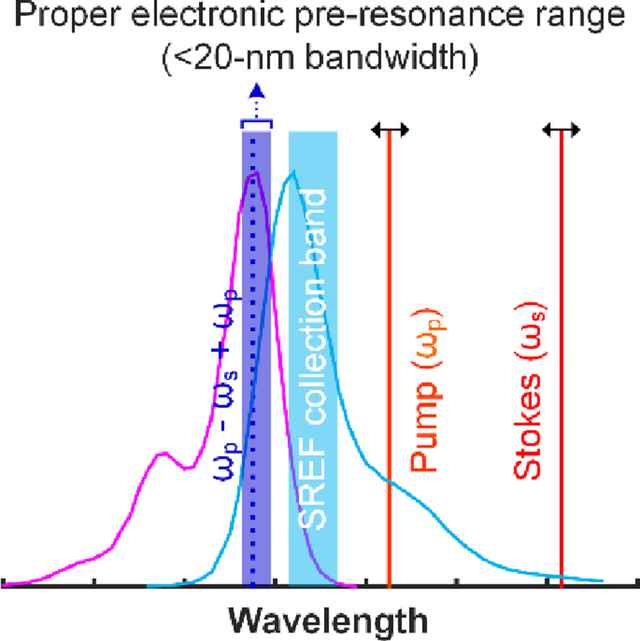
(a) show the SREF excitation and signal collection diagrams for ATTO 610 (NHS ester) in PBS (pH = 7.4). Purple curves and blue curves show the absorption and emission spectra; The dark red, light red and dash blue lines show the corresponding wavelength positions of Stokes beam (ωs), pump beam (ωp) and total excitation energy ((ωp - ωs) + ωp), respectively; and the blue band shows the fluorescence collection band of filter set (FF01-661/20, Semrock). All laser lines and CARS lines ((ωp - ωs) + ωp) are avoided. (b) and (c) show the SRS spectrum and the SREF spectrum of ATTO 610 (NHS ester), respectively. Corresponding concentrations and solvents are labeled on each panel. SRS spectrum was measured under 2.5-mW pump power and 13-mW Stokes power, and SREF spectrum was measured under 6-mW pump power and 13-mW Stokes power.
There are eight different cases in the second category in which the total SREF excitation energy slightly exceeds that of the 0–0 transition line and lies in the vicinity of the dye absorption peak (λabs). Remarkably, robust SREF peaks were observed against the anti-stokes fluorescence background in all eight cases (Fig. 3). For several of these dyes such as ATTO Rho14 and Alexa 633 (Fig. 3f, g), high quality SREF spectra can be recorded with solution even below 50 nM (which corresponds to only a few molecules on average within our tight laser focal volume). We thus attribute this superb sensitivity to the nearly perfect match between ((ωp - ωs) + ωp) and λabs. Moreover, SREF spectra containing double vibrational peaks were recorded for ATTO Rho14 in both PBS and DMSO solutions (Fig. 3c, f), which showcases the accurate reflection of SREF on the vibrational dimension. This is an importance technical advance and generalization, as our proof-of-principle study only recorded a single/isolated vibrational peak in SREF spectrum.
Fig. 3. SREF excitation with the total SREF excitation energy ((ωp - ωs) + ωp) within the proper-electronic pre-resonance region.

Right column shows the SREF excitation and signal collection diagrams for Alexa 633 (Carboxy) in PBS (pH = 7.4), ATTO 610 (NHS ester) in DMSO, ATTO Rho14 (NHS ester) in PBS (pH = 7.4), Nile Blue A in ethanol, ATTO 633 (NHS ester) in PBS (pH = 7.4), ATTO Rho14 (NHS ester) in DMSO, Alexa 633 (Carboxy) in DMSO, ATTO 633 (NHS ester) in DMSO, respectively. In these panels, purple curves and blue curves show the absorption and emission spectra in corresponding solvents, respectively; The dark red, light red and dash blue lines show the corresponding wavelength positions of Stokes beam (ωs), pump beam (ωp) and total excitation energy ((ωp - ωs) + ωp), respectively; and the blue bands show the fluorescence collection band of filter set (FF01-661/20, Semrock). All laser lines and CARS lines ((ωp - ωs) + ωp) are avoided. Center column shows the SRS spectra of corresponding dyes. And all SRS spectra are measured under 2.5-mW pump power and 13-mW Stokes power. Left column shows the SREF spectra of corresponding dyes. All SREF spectra was measured under 6-mW pump power and 13-mW Stokes power. Concentrations and solvents are marked in corresponding panels, respectively. The concentration of the right panel of (c) is unknown because of the strong absorption of ATTO Rho14 (initially 2-μM in PBS) on the coverslip and spacer, which results to obvious decreasing of the concentration.
In the third and final category, we have two more cases in which the total SREF excitation energy exceeds that of the 0–0 transition line by 20 nm (approximately exceeding the dye absorption peak by 10-nm). As shown in Fig. 4, although SRS spectra display the Raman peaks around 1600 cm−1 from C=C skeletal mode for Alexa 647 in PBS and JF646 in 0.1% TFA ethanol, their vibrational features are largely buried by the anti-Stokes fluorescence background in the corresponding SREF spectra. Under this condition, the molecules are said to be over electronic pre-resonance and the SREF strategy for vibrational detection fails, which sets the “blue” side limit for the total SREF excitation energy.
Fig. 4. SREF excitation of two dyes with over electronic pre-resonance.

(a) and (d) show the SREF excitation and signal collection diagrams for Alexa 647 (NHS ester) and JF646 (NHS ester), respectively. In these panels, purple curves and blue curves show the absorption and emission spectra in corresponding solvents, respectively; The dark red, light red and dash blue lines show the corresponding wavelength positions of Stokes beam (ωs), pump beam (ωp) and total excitation energy ((ωp - ωs) + ωp), respectively; and the blue bands show the fluorescence collection band of filter set (FF01-661/20, Semrock). All laser lines and CARS lines ((ωp - ωs) + ωp) are avoided. (b) and (e) are the corresponding SRS spectra. And all SRS spectra are measured under 2.5-mW pump power and 13-mW Stokes power. (c) and (f) show the SREF spectra of corresponding dyes. All SREF spectra was measured under 6-mW pump power and 13-mW Stokes power. Concentrations and solvents are marked in corresponding panels, respectively.
This systematic SREF spectroscopy study on the C=C skeletal mode of a total of 11 cases distributed in three different categories has allowed us to gain quantitative insight into the generality and extendibility of SREF. We could draw an experimental conclusion that the “proper electronic pre-resonance” condition for successful SREF is within ~20-nm range between the absorption peak of fluorescence dye and the total SREF excitation energy: 0–0 transition energy sets the “red” limit for the total SREF excitation energy, and the 20-nm above the 0–0 transition energy sets the “blue” limit. We now have strong experimental data to show that, when under such electronic pre-resonance condition, SREF would be a general phenomenon for a large class of fluorophores with high sensitivity, as supported by all the eight cases shown in Fig. 3. Once outside this range, SREF signal could be either too weak or be buried in anti-Stokes fluorescence background, as shown in Fig. 2 and Fig. 4.
To further test if our experimental insight obtained above is extendable to other electronic coupled Raman modes, we have synthesized two new fluorophores33–34 (Fig. 5a, e; synthesis detail in supporting materials) with nitrile group installed on their conjugation systems. For both these two fluorophores, strong electronic coupled nitrile modes are observed with Raman resonance around 2200 cm−1 (Fig. 5c, g). For fluorophore A which is designed with λabs = 635-nm in PBS (pH = 7.4) (Fig. 5a), the total SREF excitation energy for nitrile mode reaches 600.4-nm, meaning the SREF excitation is far beyond the blue side limit of proper electronic pre-resonance (Fig. 5b). Consistent with our insight obtained above, an overwhelming anti-Stokes fluorescence background was observed, and no sharp vibrational feature can be found on the fluorescence excitation spectrum (Fig. 5c, d). In contrast, for fluorophore B which is designed with λabs = 595-nm (Fig. 5e), the total SREF excitation energy of nitrile mode (resonance at 2250 cm−1) reaches 599.6-nm, which makes it well within the proper electronic pre-resonance (Fig. 5f). Indeed, an obvious SREF peak was detected above the anti-Stokes fluorescence background at the exact Raman resonance of the nitrile mode (Fig. 5g, h). Therefore, our newly synthesized dyes on different Raman modes also support the obtained insights. Note that the fluorophore A experiences a strong quenching effect in many solvents (including in PBS, pH = 7.4), mainly due to the ionization dynamics that led to the failure of the push-pull electronic conjugation system. Together with the low fluorescence collection efficiency of the emission filter (Fig. 5f, blue band), only a moderate detection sensitivity of ~700-nM is achieved. Further engineering of less-quenchable fluorophores will significantly improve the sensitivity up to single molecule level30.
Fig. 5. SREF excitation of nitrile mode on newly synthesized fluorophores.

(a) and (e) show the newly synthesized dyes with electronic-transition-coupled nitrile mode. (b) and (f) show the energy diagrams for nitrile mode SREF excitation. purple curves and blue curves show the absorption and emission spectra in corresponding solvents, respectively; The dark red, light red and dash blue lines show the corresponding wavelength positions of Stokes beam (ωs), pump beam (ωp) and total excitation energy ((ωp - ωs) + ωp), respectively; and the blue bands show the fluorescence collection band of filter set (FF01-661/20, Semrock). (c) and (g) show the SRS spectra of corresponding dyes. (d) and (h) show the SREF spectra of corresponding dyes. All SRL and SREF spectra was measured under 2.5-mW pump power and 13-mW Stokes power. Concentrations and solvents are marked in corresponding panels, respectively.
We then seek for a theoretical explanation for our experimental insight obtained above by modeling the detuning trend of SREF signal and anti-Stokes fluorescence background (Fig. 6, details in Supporting materials, Fig. S2–4). In our experiments, the anti-Stokes fluorescence background can be modeled by Boltzmann statistics35–36 (Fig. S4). On the other hand, the modeling of SREF signal will depend on whether the total excitation energy ((ωp - ωs) + ωp) is above the 0–0 transition line. If so, the SREF signal is mainly determined by the SRS pumping rate which can be modeled by the Albrecht A-term9, 37, as our pump pulse can easily saturate the transitions from ground-state vibrational-excited states to the first electronic excited state30. If not, SREF transition rate will be further modulated by the profile of the absorption tail (Fig. S3). Fig. 6 shows the evolving trend of the ratio between the SREF signal and anti-Stokes fluorescence background, if the competition between different processes can be ignored. Obviously, when the total excitation energy ((ωp - ωs) + ωp) is above 0–0 transition line, the anti-Stokes fluorescence increases much faster than SREF. As a result, when the total excitation energy is above 0–0 transition line by ~20-nm, the ratio between SREF and anti-Stokes fluorescence decreases by more than two times. Note that this ratio can be further decreased due to competition between different processes, such as ground state depletion, etc. Hence this trend can explain our observed “blue” limit (Fig. 4). When the total excitation energy ((ωp - ωs) + ωp) is below 0–0 transition line, the absolute SREF signal drops very fast (Fig. S3b), which can explain our observed “red” limit for successful SREF detection. Besides offering theoretical support to the blue and red limits for the resonance condition of the total excitation energy, this simple model also suggests that SREF to anti-Stokes fluorescence ratio would reach the maximum near the 0–0 transition line, which is indeed observed in the SREF spectra of ATTO 633 and ATTO Rho14 in PBS and DMSO (Fig. 3c & f, e &h).
Fig. 6. Theoretical modeling of the SREF signal and anti-Stokes fluorescence background as a function of pump detuning rate.

Assume the ensemble 0–0 transition energy of the molecule is fixed at 638-nm. Yellow line for anti-Stokes fluorescence; red curve for SREF. Blue curve is the ratio between SREF and anti-Stokes fluorescence. Red dash lines in (a) and (b) show the position of 713-nm, while the total excitation energy ((ωp - ωs) + ωp) equals the 0–0 transition line, which we defined as the “red” limit for “proper” electronic pre-resonance.
In summary, through constructing a new excitation source and exploring a large palette of popular dyes, we have established the generality of SREF spectroscopy on a wide variety of visible dyes with nanomolar-level sensitivity. This has put the validity of SREF spectroscopy on firm experimental ground, which is a significant result given that only two dyes were successfully detected since the proposal back in 1980. Moreover, by systematic studying how the SREF performance depends on the electronic resonance, we have revealed the experimental rule for the critical electronic pre-resonance condition. Specially, the total excitation energy ((ωp - ωs) + ωp) should lie within the 20-nm range around the absorption peak λabs of fluorophores above the 0–0 transition energy. Our theoretical modeling (Fig. 6) also lends support to this insight. On one hand, such a relatively narrow range might appear stringent and thus partly explain why experimental progress on this line has been slow during the past three decades29. On the other hand, the successful SREF detection of all 8 cases falling within the proper electronic pre-resonance window strongly supports the generality and robustness of SREF spectroscopy (Table 1). Furthermore, as we have demonstrated here for the novel nitrile-containing fluorophores (Fig. 5), the insights obtained here can serve as a valuable guide to design SREF-optimized dyes for advanced application such as super-multiplexed biomedical imaging18, 31, 38–40. Beside optimized laser excitation configuration, successful single-molecule SREF spectroscopy would require high quantum yield and good photo-stability of fluorophores in the environment, which would be a topic for further study.
Experimental methods
The OPO system we used (APE picoEmerald S) has an idler beam output up to 350-mW, signal beam output up to 800-mW, both with 0.6-nm HWFM bandwidth, 80-MHz repetition rate, and ~2-ps pulse width. We weakly focus (focus length f = 75-mm) the idler beam (~1-mm beam diameter) into a 5-mm PPLN crystal (Covesion, MSHG1420–0.5–5) for SHG generation. And a home-build software based on LabVIEW is used to synchronize the OPO tuning and the temperature of the oven (Covesion, PV10 and OC2) that hosts the crystal. A power up to 120-mW can be achieved across the whole tuning range of the crystal. The benefit of using PPLN for frequency doubling is that the output beam shares exactly the same polarization and propagation direction as the input idler beam, which ensures the spatial overlap between pump beam and Stokes beam during laser tuning process. A feedback power control loop based on a photodiode (Thorlabs, PDA36A) and a halfwave plate mount on a motorized rotation stage (Thorlabs, PRM1Z8) has been built to precisely adjust the power within 2% fluctuation range during laser tuning for high accurate excitation spectrum recording (Fig. 1c and Fig. S1). Then, the SHG of idler was set as pump beam, and OPO signal was set as Stokes beam. They were expanded, collimated, and sent to a home-build galvanometer-based (GVSM002, Thorlabs) two-dimension laser scanning microscopy. The scan lens (Thorlabs, SL50-CLS2) and tube lens (Thorlabs, TL200-CLS2) enable an additional 4× beam expansion. At last, the back pupil of the objective (UPLSAPO, 1.2 N.A., Olympus) were overfilled by both two beams to approach diffraction limited focus. A delay line on SHG of idler was used to control the delay between pump beam and Stokes beam. An electro-optic modulator (Thorlabs, EO-PM-R-20-C1) was used on the Stokes beam for all SRL measurements. The laser itself provide a 20-MHz driving signal, it was amplified by a power amplifier (Mini-Circuits, ZHL-1–2W+) before sent to the modulator to achieve a modulation depth more than 90%. For all SREF measurements, a shortpass dichroic mirror (Chroma, T690spxxr) was used to reflect the pump and Stokes beam but pass the backward fluorescence, and two bandpass filters (FF01-661/20, Semrock) were used to totally block pump, Stokes laser lines and CARS lines. A 75-mm doublet was used to relay the fluorescence emitted in the objective focus to a 100-μm diameter small area avalanched photodiode (SPCM-NIR-14-FC, 70-cps dark counts, Excelitas) worked at single-photon-counting mode to form serious confocal detection. For all the SRL and SREF signal acquisition, samples were prepared by sandwich solutions with standard 1-mm thick glass slide and 0.17-mm coverslip, separated by 0.12-mm thick imaging spacers (20-mm diameter, Sigma, GBL654006). For nanomolar-concentration solutions, to avoid dye absorption on glass interface, both slides and coverslips were cleaned in Piranha solution41 (H2SO4: H2O2 solution = 3:1 v/v) at 90 degrees Celsius overnight, and further rinsed by 30-min ultrasonic cleaning in deionized water for more than 4 times. Data points were all collected with 1-ms dwell time while laser focus was driven to scan in the solution to avoid obvious photo-bleaching at a single point. And each data point on the spectra represents the mean value of 200 independent measurements, the corresponding error bar represents 95% confidence intervals of the mean values. All data collection and laser-scan control were achieved by a NI card (PCI-6259, NI) driven by our home-written LabVIEW program. The details about the detection of SRL signal can be found in other reference30. For all the commercial dyes, they are directly used without further purification. For the dyes synthesized by us, their details about synthesis and characterization can be found in the supporting material (Synthesis of fluorophore A and B).
Supplementary Material
Acknowledgements
W.M. acknowledges support from National Institutes of Health (NIH) R01 grant (GM128214) and R01 grant (GM132860).
Footnotes
Supporting materials
Fig. S1
Synthesis of fluorophore A and B
Theoretical modeling of SREF and Anti-Stoke fluorescence background
Fig. S2
Fig. S3
Fig. S4
Reference
- 1.Herzberg G, Infrared and Raman spectra of polyatomic molecules; D. Van Nostrand Company: New York, 1945; Vol. 2. [Google Scholar]
- 2.Nie S; Emory SR, Probing single molecules and single nanoparticles by surface-enhanced Raman scattering. Science 1997, 275, 1102–1106. [DOI] [PubMed] [Google Scholar]
- 3.Min W; Freudiger CW; Lu S; Xie XS, Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annual review of physical chemistry 2011, 62, 507–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carey P, Biochemical applications of Raman and resonance Raman spectroscopes Elsevier: 2012. [Google Scholar]
- 5.Kukura P; McCamant DW; Mathies RA, Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem 2007, 58, 461–488. [DOI] [PubMed] [Google Scholar]
- 6.Shim S; Stuart CM; Mathies RA, Resonance Raman cross-sections and vibronic analysis of Rhodamine 6G from broadband stimulated Raman spectroscopy. ChemPhysChem 2008, 9, 697–699. [DOI] [PubMed] [Google Scholar]
- 7.Spiro TG; Strekas TC, Resonance Raman spectra of hemoglobin and cytochrome c: inverse polarization and vibronic scattering. Proceedings of the National Academy of Sciences 1972, 69, 2622–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson BB; Peticolas WL, The resonant Raman effect. Annual Review of Physical Chemistry 1976, 27, 465–521. [Google Scholar]
- 9.Asher SA, UV resonance Raman studies of molecular structure and dynamics: applications in physical and biophysical chemistry. Annual review of physical chemistry 1988, 39, 537–588. [DOI] [PubMed] [Google Scholar]
- 10.Efremov EV; Ariese F; Gooijer C, Achievements in resonance Raman spectroscopy: Review of a technique with a distinct analytical chemistry potential. Analytica chimica acta 2008, 606, 119–134. [DOI] [PubMed] [Google Scholar]
- 11.Prince RC; Frontiera RR; Potma EO, Stimulated Raman scattering: from bulk to nano. Chemical reviews 2016, 117, 5070–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kneipp K; Wang Y; Kneipp H; Perelman LT; Itzkan I; Dasari RR; Feld MS, Single molecule detection using surface-enhanced Raman scattering (SERS). Physical review letters 1997, 78, 1667–1670. [Google Scholar]
- 13.Zhang W; Yeo BS; Schmid T; Zenobi R, Single molecule tip-enhanced Raman spectroscopy with silver tips. The Journal of Physical Chemistry C 2007, 111, 1733–1738. [Google Scholar]
- 14.Le Ru EC; Etchegoin PG, Single-molecule surface-enhanced Raman spectroscopy. Annual review of physical chemistry 2012, 63, 65–87. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y; Zhen Y-R; Neumann O; Day JK; Nordlander P; Halas NJ, Coherent anti-Stokes Raman scattering with single-molecule sensitivity using a plasmonic Fano resonance. Nature communications 2014, 5, 4424. [DOI] [PubMed] [Google Scholar]
- 16.Yampolsky S; Fishman DA; Dey S; Hulkko E; Banik M; Potma EO; Apkarian VA, Seeing a single molecule vibrate through time-resolved coherent anti-Stokes Raman scattering. Nature Photonics 2014, 8, 650–656. [Google Scholar]
- 17.Zrimsek A; Chiang N; Mattei M; Zaleski S; McAnally M; Chapman C; Henry A; Schatz G; Van Duyne R, Single-molecule chemistry with surface-and tip-enhanced Raman spectroscopy. Chemical reviews 2017, 117, 7583–7613. [DOI] [PubMed] [Google Scholar]
- 18.Wei L; Chen Z; Shi L; Long R; Anzalone AV; Zhang L; Hu F; Yuste R; Cornish VW; Min W, Super-multiplex vibrational imaging. Nature 2017, 544, 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laubereau A; Seilmeier A; Kaiser W, A new technique to measure ultrashort vibrational relaxation times in liquid systems. Chemical Physics Letters 1975, 36, 232–237. [Google Scholar]
- 20.Seilmeier A; Kaiser W; Laubereau A; Fischer S, A novel spectroscopy using ultrafast two-pulse excitation of large polyatomic molecules. Chemical Physics Letters 1978, 58, 225–229. [Google Scholar]
- 21.Hübner H-J; Wörner M; Kaiser W; Seilmeier A, Subpicosecond vibrational relaxation of skeletal modes in polyatomic molecules. Chemical physics letters 1991, 182, 315–320. [Google Scholar]
- 22.Whaley-Mayda L; Penwell SB; Tokmakoff A, Fluorescence encoded infrared spectroscopy: ultrafast vibrational spectroscopy on small ensembles of molecules in solution. The journal of physical chemistry letters 2019. [DOI] [PubMed] [Google Scholar]
- 23.Wright JC, Double resonance excitation of fluorescence in the condensed phase—an alternative to infrared, Raman, and fluorescence spectroscopy. Applied Spectroscopy 1980, 34, 151–157. [Google Scholar]
- 24.Min W; Lu S; Holtom GR; Xie XS, Triple-resonance coherent anti-Stokes Raman scattering microspectroscopy. ChemPhysChem 2009, 10, 344–347. [DOI] [PubMed] [Google Scholar]
- 25.Orrit M; Bernard J, Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Physical review letters 1990, 65, 2716. [DOI] [PubMed] [Google Scholar]
- 26.Nie S; Chiu DT; Zare RN, Probing individual molecules with confocal fluorescence microscopy. Science 1994, 1018–1018. [DOI] [PubMed] [Google Scholar]
- 27.Moerner W; Orrit M, Illuminating single molecules in condensed matter. Science 1999, 283, 1670–1676. [DOI] [PubMed] [Google Scholar]
- 28.Winterhalder M; Zumbusch A; Lippitz M; Orrit M, Toward far-field vibrational spectroscopy of single molecules at room temperature. The Journal of Physical Chemistry B 2011, 115 (18), 5425–5430. [DOI] [PubMed] [Google Scholar]
- 29.Lee S; Nguyen D; Wright J, Double resonance excitation of fluorescence by stimulated Raman scattering. Applied Spectroscopy 1983, 37, 472–474. [Google Scholar]
- 30.Xiong H; Shi L; Wei L; Shen Y; Long R; Zhao Z; Min W, Stimulated Raman excited fluorescence spectroscopy and imaging. Nature Photonics 2019, 13, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei L; Min W, Electronic preresonance stimulated Raman scattering microscopy. The Journal of Physical Chemistry Letters 2018, 9, 4294–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reichardt C, Solvatochromic dyes as solvent polarity indicators. Chemical Reviews 1994, 94, 2319–2358. [Google Scholar]
- 33.Shi J; Zhang X; Neckers DC, Xanthenes: fluorone derivatives. 1. The Journal of Organic Chemistry 1992, 57, 4418–4421. [Google Scholar]
- 34.Shi J; Zhang X-P; Neckers DC, Xanthenes: flourone derivatives II. Tetrahedron letters 1993, 34, 6013–6016. [Google Scholar]
- 35.Erickson L, On anti-Stokes luminescence from Rhodamine 6G in ethanol solutions. Journal of Luminescence 1972, 5, 1–13. [Google Scholar]
- 36.Zander C; Drexhage KH, Cooling of a dye solution by anti-Stokes fluorescence. Advances in photochemistry 1995, 20, 59–78. [Google Scholar]
- 37.Albrecht A; Hutley M, On the dependence of vibrational Raman intensity on the wavelength of incident light. The Journal of Chemical Physics 1971, 55, 4438–4443. [Google Scholar]
- 38.Shi L; Xiong H; Shen Y; Long R; Wei L; Min W, Electronic resonant stimulated raman scattering micro-spectroscopy. The Journal of Physical Chemistry B 2018, 122, 9218–9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu F; Zeng C; Long R; Miao Y; Wei L; Xu Q; Min W, Supermultiplexed optical imaging and barcoding with engineered polyynes. Nature methods 2018, 15, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu F; Brucks SD; Lambert TH; Campos LM; Min W, Stimulated Raman scattering of polymer nanoparticles for multiplexed live-cell imaging. Chemical Communications 2017, 53, 6187–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X; Wu Z; Nie H; Liu Z; He Y; Yeung E, Single DNA molecules as probes for interrogating silica surfaces after various chemical treatments. Analytica chimica acta 2007, 602, 229–235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.