

SUMMARY
Gata4, Hand2, Mef2c, and Tbx5 (GHMT) can reprogram transduced fibroblasts into induced pacemaker-like myocytes (iPMs), but the underlying mechanisms remain obscure. Here, we explore the role of Hand2 in iPM formation by using a combination of transcriptome, genome, and biochemical as-says. We found many shared transcriptional signatures between iPMs and the endogenous sinoatrial node (SAN), yet key regulatory networks remain missing. We demonstrate that Hand2 augments chromatin accessibility at loci involved in sarcomere organization, electrical coupling, and membrane depolarization. Focusing on an established cardiac Hand2 cistrome, we observe selective reorganization of chromatin accessibility to promote pacemaker-specific gene expression. Moreover, we identify a Hand2 cardiac subtype diversity (CSD) domain through biochemical analysis of the N terminus. By integrating our RNA-seq and ATAC-seq datasets, we highlight desmosome organization as a hallmark feature of iPM formation. Collectively, our results illuminate Hand2-dependent mechanisms that may guide future efforts to rationally improve iPM formation.
Graphical Abstract
In Brief
Gata4, Hand2, Mef2c, and Tbx5 can reprogram fibroblasts into cardiomyocyte-like cells, including induced pacemakers (iPMs). Fernandez-Perez et al. show that Hand2 coordinates this process by influencing chromatin accessibility and gene expression in fibroblasts undergoing iPM lineage conversion. These insights could eventually inform the production of superior replacement cells.
INTRODUCTION
A growing body of evidence has established that cell identity is not completely fixed (Smith et al., 2016). After the identification of transcription factor (TF) cocktails capable of generating specific cell types (Ieda et al., 2010; Takahashi and Yamanaka, 2006; Vierbuchen et al., 2010), transcriptomic and epigenetic studies have elucidated key mechanisms by which direct reprogramming is accomplished (Buganim et al., 2012; Liu et al., 2017; Treutlein et al., 2016). Based on these insights, reprogramming cocktails have been rationally improved in many instances to optimize formation of functional cell types (Buganim et al., 2012; Liu et al., 2017; Wapinski et al., 2013, 2017). Intriguingly, there are even select cases in which detailed mechanistic insights into a particular reprogramming phenomenon have illuminated new developmental concepts (Mall et al., 2017; Pereira et al., 2016). Although these lineage conversion paradigms suggest plasticity in cell identity, many reprogramming systems use mouse embryonic fibroblasts (MEFs), which are inherently malleable.
Sinoatrial node (SAN) progenitors arise from the sinus horns to express Tbx5 and Tbx18 between E8.0 and E9.5 (Mommersteeg et al., 2007). Interestingly, these progenitors are negative for Nkx2–5, which directly antagonizes the pacemaker gene program. During subsequent SAN specification, which takes place between E9.5 and E18.5, Shox2, Tbx3, and Isl1 participate in downstream regulatory networks. Shox2 antagonizes Nkx2.5 and activates Tbx3 and Isl1 (Espinoza-Lewis et al., 2009). Tbx3 suppresses the gene expression program of neighboring atrial cardiomyocytes to demarcate the SAN boundary (Hoogaars et al., 2007). Finally, Isl1 directly activates the pacemaker gene program by cooperating with Shox2 and Tbx3 (Liang et al., 2015). Our previous attempt to leverage this knowledge to reprogram pacemaker cells was unsuccessful (Nam et al., 2014). One possible explanation for this observation is that at least one pioneer TF is typically required to achieve somatic cell lineage conversion (Morris, 2016), yet these four factors function near the bottom of the pacemaker developmental hierarchy.
The combination of Gata4, Mef2c, and Tbx5 converts fibroblasts into functionally induced cardiomyocyte-like myocytes (iCLMs) (Ieda et al., 2010). Addition of auxiliary factors (e.g., Hand2, Akt1, and Znf281) or manipulation of microRNAs (miRNAs), signaling pathways, culture conditions, or delivery strategies can further improve iCLM reprogramming (Abad et al., 2017; Ifkovits et al., 2014; Jayawardena et al., 2012; Miyamoto et al., 2018; Mohamed et al., 2017; Muraoka et al., 2014; Song et al., 2012; Yamakawa et al., 2015; Zhao et al., 2015; Zhou et al., 2015, 2017). We previously showed that Gata4, Hand2, Mef2c, and Tbx5 (GHMT) can generate a subset of induced pacemaker-like myocytes (iPM) based on gene expression, flow cytometry, immunocytochemistry, morphological, and electrical characteristics (Nam et al., 2014). Moreover, we found that iPMs do not pass through an Nkx2.5+ intermediate and exit the cell cycle rapidly, indicating that iPM formation is a direct reprogramming event (Nam et al., 2014).
Hand1 and Hand2 belong to the basic helix-loop-helix (bHLH) family of TFs and have essential roles during cardiac morphogenesis (George and Firulli, 2018; Wang and Baker, 2015). Hand2 is expressed in the embryonic right ventricle, whereas Hand1 is complementarily expressed in the left ventricle (George and Firulli, 2018), thus explaining the phenotypes of Hand1 and Hand2 knockout mice (Firulli et al., 1998; Srivastava et al., 1997). Hand2 also has specific and important functions in the neural crest, epicardium, and endocardium. However, a role for Hand2 in SAN specification has not been documented to date. Thus, our prior observation that GHMT mediates iPM for-mation was surprising (Nam et al., 2014), and the underlying mechanisms by which Hand2 facilitates iPM reprogramming remain unclear.
Here, we explored the role of Hand2 in iPM formation by using a combination of transcriptome, genome, and biochemical as-says. We observed many shared transcriptional signatures between iPMs and endogenous SAN tissue at various stages, although specific key TF networks are missing. Focusing on the established Hand2 cardiac cistrome, we observed selective reorganization of chromatin accessibility that promotes the activity of pacemaker-specific gene expression. We use fine-mapping of Hand2 protein domains to establish a cardiac subtype diversity (CSD) domain within the N terminus. Finally, we identify desmosome organization as a key feature of iPM formation by integrating the RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) datasets. Collectively, our results illuminate mechanisms by which Hand2 facilitates iPM formation and paves the way for future rational improvement of pacemaker formation.
RESULTS
Hand2 Facilitates Activation of Cardiac Conduction Genes with Gata4, Mef2c, and Tbx5
We first established an experimental system using Hcn4-GFP mice (Nam et al., 2014; Vedantham et al., 2015) to test mechanistic details of iPM reprogramming and a parallel pipeline for unbiased genome-wide analysis of iPMs at day 14 (Figure 1A). To confirm that Hand2 is specifically required for iPM reprogramming, we conducted a series of experiments in which a single factor was systematically omitted from GHMT. MEFs were infected with specific TF combinations and assessed by flow cytometry on day 10 for Hcn4-GFP and cardiac troponin T (cTnT) (Figure 1B). We observed that GHMT generated 4.85% Hcn4-GFP+/cTnT+ cells, which was not observed for any other combination. Importantly, our previous work indicates that double-positive Hcn4-GFP+/cTnT+ cells, rather than single-positive Hcn4-GFP−/cTnT+ or Hcn4-GFP+/cTnT− cells, most accurately reflect functional iPM formation (Nam et al., 2014).
Figure 1. Hand2 Facilitates Transcriptional Reprogramming of Induced Pacemaker (iPM) Cells.

(A) Experimental scheme for iPM reprogramming, which was analyzed in three independent workflows. Specific figure panels corresponding to each workflow are indicated to the right.
(B) MEFs were transduced with GHMT or each three-factor combination followed by flow cytometry analysis at day 10.
(C) Same experimental setup as in (B) but assayed by ICC and manual counting (means ± SD, n = 6) at day 14. All samples were compared with GMT to determine statistical significance.
(D) RNA-seq analysis was performed on control, GMT-infected, and GHMT-infected MEFs. Unsupervised hierarchical clustering of these genes highlights four distinct clusters (1–4).
(E) Gene expression for each cluster in (D) is shown for individual samples. p values for 1-way ANOVA testing across samples are shown within individual boxplots. Statistical testing for pairwise comparisons within a given cluster was performed by Tukey’s honestly significant difference (HSD) test between groups. All comparisons were significant, except for GHMT versus GMT in clusters 2 and 4. All numerical values for Tukey’s HSD testing are provided in Table S2.
(F) Gene ontology (GO) analysis for clusters 1 and 4.
(G) Gene-level view of RNA-seq data comprising “regulation of heart rate” and “sarcomere organization” GO terms across samples.
Statistical significance indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Non-significant comparisons indicated either by “ns” or no asterisk.
Next, we used a more rigorous immunocytochemistry (ICC) assay to evaluate sarcomere formation and subtype diversification (Fernandez-Perez and Munshi, 2017; Nam et al., 2014). We previously confirmed the cell type identity of Hcn4-GFP+/sarco-mere+ iPMs by direct, intracellular recordings (Nam et al., 2014). Nevertheless, Hcn4 (and Hcn4-GFP) can label other cardiomyocytes of the conduction system, aside from pacemaker cells, as well as certain non-myocytes, such as neurons, which do not express cardiac α-actinin or exhibit organized sarcomeres. Consistent with previous studies (Nam et al., 2014; Song et al., 2012), we found that Hand2 augments sarcomere formation and generation of all cardiac subtypes when added to Gata4, Mef2c, and Tbx5 (GMT) (Figure 1C). GHMT were all required for iPM formation, whereas Gata4 and Hand2 appear to have somewhat redundant roles during induced atrial myocyte (iAM) and induced ventricular myocyte (iVM) formation because GMT and Hand2, Mef2c, and Tbx5 (HMT) produced similar results in this reprogramming assay. Using an mCherry-Hand2 fusion construct, we conducted parallel studies of GHMT reprogramming that demonstrated that 80% of iCLMs and 100% of iPMs are Hand2+ (Figures S1A and S1B). Altogether, these results show that Hand2 is required for iPM reprogramming in the context of GHMT.
To gain a more comprehensive understanding of the transcriptional changes that underlie iPM reprogramming, we performed RNA-seq analysis on transduced MEFs on day 14. We were particularly interested in the unique ability of Hand2 to generate iPMs with GMT, so we profiled GFP+ cells after transduction of Hcn4-GFP MEFs with GMT or GHMT. In contrast to prior studies, we note that our transcriptome profiling occurs later in the reprogramming process, uses GFP-sorted cells, and directly compares GMT- and GHMT-transduced fibroblasts (Ieda et al., 2010; Song et al., 2012; Zhao et al., 2015). RNA-seq was performed in duplicate with good overall concordance among samples (Figure S1C). Unbiased analysis of the RNA-seq data revealed four major clusters (1–4) of gene expression patterns (Figure 1D). Analysis of each cluster in aggregate allowed better visualization of quantitative changes in gene expression among samples (Figure 1E).
Based on the coordinated changes in gene expression and the consistency of the data (Figures 1E and S1D), we focused on clusters 1 (C1: GHMT > GMT/control) and 4 (C4: control > GMT/GHMT) for gene ontology (GO) analysis. For GHMT > GMT/control (C1), we observed enrichment for the expected categories “regulation of cardiac muscle contraction” and “sarcomere organization” (Figure 1F). However, we also identified the GO terms “regulation of heart rate” and “bundle of His cell-Purkinje myocyte adhesion involved in cell communication,” suggesting activation of conduction system-specific genes (Figure 1F). For control > GMT/GHMT (C4), GMT and GHMT both suppressed genes involved in the inflammatory response and immune signaling pathways (Figure 1F), which were further repressed by the addition of Znf281 (Zhou et al., 2017).
Focusing on the GO term “sarcomere organization,” identified for GHMT > GMT/control (C1), we observed a nearly uniform upregulation of the genes comprising that category (Figures 1G and S1E). For the “regulation of heart rate” group, we also observed strong upregulation of individual genes specifically in GHMT-transduced iPMs, although the entire group was not uniformly activated, as seen for the “sarcomere organization” group (Figures 1G and S1E). Importantly, we note that reprogramming with Hand2 alone did not significantly increase gene expression in either of these groups, suggesting that Hand2 is not sufficient to activate pacemaker-and sarcomere-related genes. Taken together, these results demonstrate that Hand2 uniquely promotes iPM formation with GMT by activating genes necessary for myocyte contraction and cardiac conduction.
Comparative RNA-Seq Analysis Defines Shared Markers of iPMs and SAN
To independently evaluate the extent to which iPM transcriptional reprogramming resembles endogenous pacemaker cells, we sought to compare our RNA-seq data with previous datasets (Vedantham et al., 2015). In contrast to our RNA-seq data, which underwent standard library preparation, the prior datasets required extensive library amplification because of the limited amount of obtained tissue. Therefore, we were unable to make direct, quantitative comparisons between the two datasets. Using an alternative analytical approach, we qualitatively compared genes enriched in GHMT- versus GMT-transduced fibroblasts with those enriched in endogenous Hcn4-GFP+ SAN versus neighboring atrial tissue obtained at E14.5, P4, and P14 (Figure 2A).
Figure 2. Core Endogenous Pacemaker Marker Genes Are Enriched in iPMs.

(A) Analytical strategy for comparing iPM-specific genes with sinoatrial node (SAN)-specific genes in (B) and (C). Gene sets enriched in SAN compared with RA at14.5, P4, and P14 (red, green, or dark blue outline with yellow interior) were obtained from Vedantham et al. (2015). Gene sets enriched in GHMT- versus GMT- reprogrammed cells (light blue outline with yellow interior) were obtained from this study. Then, pairwise comparisons for overlapping genes at individual developmental time points were done in (B), whereas a four-way comparison for overlapping genes among all gene sets was performed in (C).
(B) Top: Venn diagrams demonstrating numbers of shared genes between iPMs and each developmental time point. Bottom: qRT-PCR for individual candidate genes in each group is shown.
(C) Four-way Venn diagram shows a core set of 12 pacemaker marker genes (dark red).
(D) RNA-seq data for pacemaker marker genes across reprogrammed MEF samples.
(E) qRT-PCR validation of a subset of pacemaker marker genes with micro-dissected mouse SAN tissue as a positive control.
(F) qRT-PCR time course of GMT- and GHMT-transduced MEFs.
From individual pairwise comparisons, we observed 41, 36, and 30 overlapping genes between iPMs and SAN at E14.5, P4, and P14, respectively, and enrichment of individual genes was confirmed by qRT-PCR analysis (Figure 2B). Based on this qualitative comparison, we conclude that iPMs share a modest degree of gene signatures with endogenous SAN tissue across developmental time points. An integrated four-way comparison identified a core set of 12 shared iPM-and SAN-specific marker genes (Figure 2C). Visualization of the core PM marker genes in our RNA-seq data revealed highly specific expression in GHMT-transduced, but not uninfected or GMT-transduced, fibroblasts (Figures 2D and S1E).
A subset of PM marker genes was independently validated at the RNA level by qRT-PCR analysis (Figure 2E). In total, we performed qRT-PCR validation on 7 of 12 pacemaker (PM) genes, 6 of which were successfully confirmed. Of the 7 core PM genes that were tested, qRT-PCR failed to robustly validate Tbx3 enrichment in iPMs. However, we emphasize that qRT-PCR validation was performed on unsorted cells, which may potentially dilute any signals of enrichment. Additional validation for Tbx3 at the protein level was obtained by ICC on GHMT-transduced fibroblasts (see Figure 3F). Interestingly, parallel experiments performed on GMT-reprogrammed iCLMs failed to stain for Tbx3 (Figure S1F), suggesting that iPMs are uniquely positive for Tbx3 protein.
Figure 3. Hand2 Reorganizes Chromatin Accessibility toward a Pacemaker-like State.

(A) ATAC-seq was performed on control, GMT-infected, and GHMT-infected MEFs, and P0 Hcn4-GFP+ endogenous pacemaker (P0 PM) cells were profiled for comparison. Unsupervised hierarchical clustering separated chromatin peaks into five clusters (1–5).
(B) Gene ontology (GO) term analysis of clusters 3 and 5.
(C) Known transcription factor motifs identified for clusters 3 and 5.
(D) RNA-seq enrichment of candidate transcription factors (TFs) for clusters 3 and 5.
(E) Cartoon diagram of the developmental pacemaker gene regulatory network (right) and associated RNA-seq data (left).
(F) Immunocytochemistry (ICC) on GHMT-reprogrammed cells for Tbx3 (top) and Shox2 (bottom). Scale bar, 20 mm.
To investigate the kinetics of pacemaker marker genes during iPM formation, we performed time-course qRT-PCR on GMT-and GHMT-infected fibroblasts (Figure 2F). Interestingly, we observed distinct gene expression dynamics for each pacemaker marker gene during iPM formation. Collectively, these data support the idea that direct reprogramming is distinct from developmental fate specification. In this regard, iPMs do not share gene expression signatures with endogenous SAN tissue at any specific developmental time point but, rather, seem to reflect a spectrum of developmental stages. Further evidence for that conclusion is derived from the observation that the early SAN developmental transcription factors Tbx3 and Tbx18 are activated in iPMs but Shox2 and Isl1 are not (see Figure 3E). Our results also identify a set of shared iPM and SAN markers that are activated with varying kinetics.
Hand2 Drives Chromatin Accessibility toward an Endogenous Pacemaker State
Although transcriptional signatures drive lineage specification, changes in chromatin accessibility can illuminate mechanisms by which gene expression is modulated (Long et al., 2016). Therefore, we used the assay for ATAC-seq to comprehensively profile chromatin accessibility (Buenrostro et al., 2013; Corces et al., 2017). ATAC-seq libraries were generated from GFP-sorted control, GMT-infected, and GHMT-infected fibroblasts. Sorted endogenous P0 Hcn4-GFP+ pacemaker cells (P0 PMs) were used to generate ATAC-seq libraries for comparison. Replicate ATAC-seq libraries showed a high degree of concordance, and the GMT and GHMT samples clustered together by similarity (Figure S2A). Unsupervised clustering analysis of chromatin accessibility peaks identified five unique clusters (1–5) representing distinct patterns of chromatin accessibility across control, GMT, GHMT, and P0 PM samples (Figures 3A and S2B).
For subsequent analyses, we focused on the behavior of chromatin accessibility peaks that were divergent between the starting cell population (MEFs) and the target cell population (P0 PMs). Therefore, we explored clusters 3 (C3: control > GMT/GHMT/P0 PM) and 5 (C5: P0 PM > GHMT > GMT > control) in more detail. In aggregate, chromatin accessibility diminished for control > GMT/GHMT/P0 PM (C3) and increased for P0 PM > GHMT > GMT > control (C5) when comparing GHMT to GMT (Figure S2B), suggesting that Hand2 promotes epigenome remodeling toward a more PM-like state. Interestingly, P0 PM > GHMT > GMT > control (C5) was relatively enriched for distal intergenic sites and depleted of proximal elements compared with control > GMT/GHMT/P0 PM (C3) (Figure S2C).
Next, we performed GO term analysis using the nearest annotated neighboring genes for individual chromatin accessibility peaks (Figures 3B and S2D). We found that the terms associated with control > GMT/GHMT/P0 PM (C3) chromatin included alternative fates (“mesonephric epithelium development”), fibroblast characteristics (“positive regulation of mesenchymal cell proliferation”), and signaling pathways (“platelet-derived growth factor receptor signaling pathway”). In contrast, terms associated with P0 PM > GHMT > GMT > control (C5) chromatin were similar to GO terms for iPM gene expression signatures (Figure 1F) and included the terms “regulation of cell communication by electrical coupling,” “regulation of sarcomere organization,” and “membrane depolarization during AV node cell action potential.” Altogether, these results strongly suggest that iPM chromatin accessibility mirrors observed changes in gene expression.
Changes in chromatin accessibility are typically orchestrated by coactivators, corepressors, and chromatin remodelers that are recruited to genomic DNA by sequence-specific TFs (Long et al., 2016). Therefore, we searched control > GMT/GHMT/P0 PM (C3) and P0 PM > GHMT > GMT > control (C5) chromatin accessibility peaks for recurrent motifs that bind known TFs (Figures 3C and S2E). For control > GMT/GHMT/P0 PM (C3) peaks, we observed a preponderance of binding motifs for the basic leucine zipper domain (bZIP) class of TFs, including Atf3, Fra1, and AP-1 (Fos/Jun). bZIP TFs have important roles in fibroblast gene expression, and their binding motifs are overrepresented in closing chromatin during iPS reprogramming (Li et al., 2017). In contrast, we identified Mef2c, Gata4, and Tbx5 motifs in P0 PM > GHMT > GMT > control (C5) chromatin, which would be expected for fibroblasts transduced with GHMT. Surprisingly, the Hand2 consensus motif was not detected in that analysis (see Figure 4 and Discussion). Intriguingly, however, our analysis did identify motifs for TEAD4 and Meis1, both of which have been implicated in heart development (Chen et al., 1994; Stankunas et al., 2008).
Figure 4. Modulation of Hand2 Cistrome Accessibility Contributes to iPM Reprogramming.

(A) Cardiac Hand2 ChIP-seq and iPM ATAC-seq peaks were intersected to establish a set of 1,684 iPM Hand2 cistrome peaks, which clustered into four groups (1–4).
(B) Gene ontology (GO) term analysis for clusters 2 and 3.
(C) Distribution of GMT binding motifs at 1,684 Hand2 genomic binding sites accessible in iPMs. Putative composite sites are boxed and labeled as follows: H-G-M, Hand2-Gata4-Mef2c; H-M-T, Hand2-Mef2c-Tbx5; H-G-T, Hand2-Gata4-Tbx5.
(G) Genome browser tracks for the Isl1, Shox2, Tbx3, and Tbx18 loci with associated ATAC-seq and RNA-seq tracks. Cardiac Hand2 ChIP-seq and E10.5 heart H3K27Ac ChIP-seq peaks are shown as a reference.
(H) Cardiac reprogramming of MEFs was assessed for sarcomere formation (left) and subtype specification (right) in the presence of the indicated PM gene regulatory TFs (means ± SD, n = 6). All samples were compared with GMT (left of dashed line) or GHMT (right of dashed line) to determine statistical significance. Additional pairwise comparisons are shown with horizontal lines at the top of each graph.
Statistical significance indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Non-significant comparisons indicated either by “ns” or no asterisk.
The close alignment of the GO terms and motifs enriched in iPMs and endogenous pacemaker cells was particularly impressive for the P0 PM > GHMT > GMT > control (C5) cluster. However, we wondered whether that was driven by the comparison with target cells or whether iPMs genuinely resembled endogenous pacemaker cells. To distinguish between those two possibilities, we reanalyzed the control, GMT, and GHMT datasets without the P0 PM samples (Figure S3). Unsupervised hierarchical clustering identified 5 clusters (Figures S3A and S3B). For cluster 3 (GHMT > GMT > control), which resembled cluster 5 (P0 PM > GHMT > GMT > control) in Figure 3, we identified GO terms related to cardiac conduction, such as “regulation of heart rate,” “regulation of heart rate by cardiac conduction,” and “cardiac conduction,” which were among the most enriched terms (Figure S3C). Furthermore, the list of motifs discovered in cluster 3 (GHMT > GMT > control) was highly concordant with the motifs identified for cluster 5 (P0 PM > GHMT > GMT > control) in Figure 3 (Figure S3D). Taken together, these observations strongly suggest that the alignment between iPM and P0 PM chromatin accessibility is not merely an artifact of our analytical method.
Given that specific binding motifs emerged from our ATAC-seq data, we next wished to evaluate whether the cognate TFs were expressed and/or enriched in GHMT-transduced iPMs. Therefore, we interrogated our companion RNA-seq datasets for TFs whose motifs were enriched in P0 PM > GHMT > GMT > control (C5) or control > GMT/GHMT/P0 PM (C3) chromatin (Figures 3D and S4A). For the motifs identified in control > GMT/GHMT/P0 PM (C3) chromatin, we observed that bZIP TFs were not specifically enriched or depleted in iPMs, whereas Tead1, Tead2, and Tead4 showed increased expression in GHMT-transduced fibroblasts compared with controls. For motifs obtained from P0 PM > GHMT > GMT > control (C5) chromatin, we reassuringly found increased expression of Gata4, Mef2c, Tbx5, and Hand2 in iPMs, whereas expression of Meis1/2 was less consistent. Collectively, these results demonstrate that global chromatin accessibility in GHMT-transduced iPMs moves toward a PM-like state. However, it remains unclear whether iPM reprogramming recapitulates key aspects of PM development.
Based on established gene regulatory networks that drive developmental specification of pacemaker cells (van Eif et al., 2018), we were particularly interested in assessing whether these networks were redeployed during iPM reprogramming (Figures 3E and S4A). Interestingly, we observed that Tbx3 and Tbx18 were upregulated in iPMs but Shox2 and Isl1 were not (Figures 3E and S4A). Expression of Notch1 and Id2, which regulate distinct aspects of cardiac conduction system development (van Eif et al., 2018), was not increased in iPMs, and expression of Nkx2.5, a negative regulator of pacemaker specification, was diminished (Figures 3E and S4A).
Expression levels for Tbx3, Tbx18, Shox2, and Isl1 were independently confirmed by qRT-PCR on separate samples (Figures 2E and S4B). Furthermore, protein levels of endogenous Tbx3 and Shox2 in iPMs were confirmed by ICC (Figure 3F), which was consistent with the RNA-seq and qRT-PCR results. Interestingly, we found little correlation between chromatin accessibility and gene expression for the PM regulatory TFs (Figure 3G). For example, chromatin was relatively accessible for all loci in fibroblasts and changed minimally in iPMs, but only Tbx3 and Tbx18 expression was detectable in iPMs (Figures 3E and 3G). Together, these observations suggest that substantial chromatin reorganization at developmentally important loci is not required for transcriptional activation during iPM reprogramming.
Analysis of the iPM ATAC-seq and RNA-seq datasets thus far demonstrated the following: (1) Shox2 and Isl1 are inactive members of the developmental pacemaker gene regulatory network, (2) opening loci are enriched for Meis1 and GMT motifs, and (3) closing loci are enriched for bZIP motifs. Based on these findings, we hypothesized that the addition of TFs from each of these categories would perturb iPM reprogramming either positively (Meis1/Isl1/Shox2) or negatively (bZIP). To test those possibilities, we performed iPM reprogramming with GMT and GHMT with or without single candidate factors.
When Tbx3 or Tbx18 were added to GMT or GHMT, we observed no detectable increase in either sarcomere+ iCLMs or iPMs (Figure S4C), which is consistent with the fact that GHMT already activates endogenous Tbx3 and Tbx18. In contrast, Isl1 and Shox2 each had interesting effects on cardiac reprogramming. Isl1 substituted for Hand2 to drive both sarcomere+ iCLMs and iPMs, whereas Isl1 added to GHMT further improved both iCLM and iPM reprogramming (Figure 3H). Shox2 had more subtle effects. When Shox2 was added in place of Hand2, overall sarcomere+ iCLM production was unaffected, but the resulting iCLMs were skewed toward iPMs (Figure 3H). In contrast, both iCLM and iPM generation were reduced when Shox2 was added to GHMT (Figure 3H). The addition of Meis1 or Atf3 to GHMT did not affect sarcomere+ iCLMs, but we observed a shift from iVMs to iAMs for Meis1 and suppression of iPMs by Atf3 (Figure S4C). We note that addition of Atf3 or Meis1 uniquely generated mixed iCLMs (Hcn4-GFP+/Myl2+), which we have not previously observed (Figure S4D). Interestingly, Meis1 plus GMT generated a small percentage of iPMs (Figure S4B). Taken together, our data demonstrate coordinated chromatin accessibility changes in GHMT-transduced iPMs toward an endogenous pacemaker-like state. Focusing on established PM gene regulatory networks, our results identify Isl1 and Shox2 addition as potential routes for rational improvement of iPM formation by GHMT.
Identification of Hand2-Dependent iPM cis-Regulatory Elements
Given that iPM reprogramming is dependent upon Hand2, we sought to determine how chromatin accessibility changes relative to established Hand2 genomic-binding sites. To address that question, we took advantage of a previously generated Hand2 ChIP-seq dataset derived from mouse embryonic heart (Laurent et al., 2017). Importantly, de novo motif discovery on that Hand2 ChIP-seq dataset showed that the consensus CATCTG Hand2 binding site was among the most common E-box motifs (Dai and Cserjesi, 2002; Laurent et al., 2017). We intersected that dataset (n = 12,101 peaks) with open iPM ATAC-seq peaks (n = 33,032 peaks) to identify a set of 1,684 Hand2 genomic binding sites that are accessible in iPMs (Figure 4A). Using that defined set of known Hand2 bindings sites, which we refer to as the iPM Hand2 cistrome, unbiased clustering was performed across all samples, including fibroblasts infected with Hand2 alone (Figure 4A). Interestingly, that analysis separated the iPM Hand2 cistrome into 4 distinct clusters. Clusters 1, 3, and 4 were accessible in both iPMs and P0 PMs, whereas cluster 2 had limited accessibility in P0 PMs.
We mapped the genomic location of individual clusters to gain a better appreciation for unique features of each group (Figure S5A). This analysis revealed that most of the Hand2 binding sites occurred at distal intergenic or intronic regulatory elements. Nevertheless, there were subtle differences among each cluster of Hand2 binding sites. For example, cluster 3 had a higher proportion of promoter-proximal Hand2 binding sites, whereas cluster 2 tended to have binding sites in the first intron (Figure S5A). GO terms for each iPM Hand2 cistrome cluster returned revealing gene categories (Figures 4B and S5B). The closing Hand2 genomic binding sites in cluster 2 (C2: control/Hand2 > GMT/GHMT > P0 PM) associated with generic terms related to developmental morphogenesis and actin filaments (Figure 4B). In contrast, the opening Hand2 genomic binding sites in cluster 3 (C3: P0 PM/GHMT > control/Hand2) returned terms related to the cardiac conduction system, including “cell-cell signaling involved in CCS” and “bundle of His development” (Figure 4B).
Next, we searched for recurrent TF co-binding motifs in each cluster of Hand2 genomic binding sites (Figure S5C). As expected, motifs for GATA, MEF2, and T-box families of TFs were prevalent in the opening clusters (1, 3, and 4), whereas bZIP and TEAD motifs were identified in the closing cluster 2 (Figure S5C). Because Gata4, Hand2, Mef2c, and Tbx5 are known to function cooperatively (Dai and Cserjesi, 2002; Garg et al., 2003; Morin et al., 2000; Zang et al., 2004), we identified genomic locations for GATA, MEF2, and T-box family binding motifs to assess potential co-occupancy of GHMT across the iPM Hand2 cistrome (Figure 4C). Interestingly, we found a large number of potential Hand2-Gata4-Mef2c (H-G-M), Hand2-Mef2c-Tbx5 (H-M-T), and Hand2-Gata4-Tbx5 (H-G-T) composite binding sites dispersed across the iPM Hand2 cistrome. Given the flexibility of the E-box motif (CANNTG), we also tested the hypothesis that subtle alterations in the Hand2 binding motif may account for the distinct chromatin accessibility profiles of clusters 1–4. However, we found no evidence that specific E boxes segregated with a particular cluster (Figure S5D). Taken together, these observations suggest that GHMTs cooperate to activate the iPM Hand2 cistrome.
The Hand2 N Terminus Harbors a CSD Domain
To explore the biochemical mechanisms by which Hand2 regulates iPM formation, we performed an extensive structure-function analysis. To address whether Hand2 functions as an activator or repressor during cardiac reprogramming, we created Hand2-VP16 and Hand2-EnR fusion proteins and tested their ability to support cardiac reprogramming with GMT (Figures S6A and S6B). We found that Hand2-VP16 slightly improved and Hand2-EnR strongly repressed cardiac reprogramming, suggesting that Hand2 functions as an activator in this context. Using a previously established dimerization-defective Hand2 mutant (Hand2F119P) (McFadden et al., 2002), we also demonstrated that Hand2 dimerization is required for optimal cardiac reprogramming (Figure S6C). Taken together, these data show that Hand2 functions as a dimeric transcriptional activator during cardiac reprogramming.
The Hand2 protein sequence can be partitioned into unstructured N- and C-termini and a central bHLH domain, which regulates DNA binding, dimerization, and protein-protein interactions (Conway et al., 2010). To analyze the function of each domain, we performed cardiac reprogramming experiments in which fibroblasts were infected with GMT plus Hand2 or one of the variants shown in Figure 5A. Based on the presumption that Hand2 DNA-binding is required for efficient cardiac reprogramming, we were encouraged to find that transduction of the Hand2EDE mutant, which cannot bind DNA (McFadden et al., 2002), led to reduced iPM formation (Figure 5B). Interestingly, neither the N-terminal nor C-terminal deletion Hand2 mutants could support optimal cardiac reprogramming with GMT, suggesting that each domain is functionally required.
Figure 5. The Hand2 N Terminus Encodes a Cardiac Subtype Diversification (CSD) Domain.

(A) Schematic for the Hand2 mutants used in (B). Unstructured N and C termini are shown in blue and orange, respectively, whereas the known transactivation domain (TAD) is shown in black. Beta helix-loop-helix (bHLH) domain is shown in gray, and mutations in DNA-contacting basic residues in the RRR109–111EDE construct are depicted as red asterisks.
(B) Cardiac reprogramming with GMT alone or with the indicated Hand2 mutants was assessed for sarcomere organization (left) and subtype diversification (right) (means ± SD, n = 8). All samples were compared with GMT to determine statistical significance.
(C) Cardiac reprogramming was conducted with GMT plus Hand2 or Hand1, and iCLMs were assessed for sarcomere organization (left) and subtype diversity (right). The degree of primary amino acid conservation between Hand2 and Hand1 for each protein domain is indicated at the top (means ± SD, n = 6). All samples were compared with GMT to determine statistical significance.
(D) Cartoon diagram of Hand2 predicted secondary structure with low-complexity regions (LCRs) highlighted in green and bHLH region in blue cylinders and red line (top). The primary sequence of the N terminus is shown below with the indicated LCRs.
(E) Schematic for the Hand2 N-terminal mutants used for reprogramming in (F).
(F) Cardiac reprogramming was performed with GMT alone or combined with the indicated Hand2 N-terminal mutants. iCLMs were assessed for sarcomere organization (left) and subtype diversity (right) (means ± SD, n = 6). All samples were compared with GMT to determine statistical significance.
Statistical significance indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Non-significant comparisons indicated either by “ns” or no asterisk.
Because Hand1 and Hand2 exhibit complementary expression patterns and a high degree of amino acid conservation (Figure 5C), they may function similarly within distinct anatomical compartments. However, gene replacement studies and in vitro binding-site selection assays favor the notion that each is functionally distinct (Dai and Cserjesi, 2002; Firulli et al., 2010; Hollenberg et al., 1995). To test these alternatives in the context of cardiac reprogramming, we compared the outcome of infecting fibroblasts with GMT + Hand2 versus GMT + Hand1. Interestingly, we found that Hand1 could not support optimal cardiac reprogramming as compared with Hand2 (Figure 5C). Thus, we conclude that Hand2 uniquely augments cardiac reprogramming in the context of GMT.
Although each domain of Hand2 appears to be functionally important, we chose to focus on the N terminus for four reasons. First, the N- and C-termini remain functionally underexplored. Second, amino acid conservation between Hand1 and Hand2 is lowest for the N terminus (Figure 5C), implying that differences in that region are likely to account for functional disparities between the two proteins. Third, a parallel experiment in which a VP16 activation domain was fused to Hand2D1-90 failed to restore cardiac reprogramming efficiency in combination with GMT (data not shown), suggesting that the N terminus contains more than just the previously established transactivation domain (Dai and Cserjesi, 2002). Fourth, the Hand2 N terminus harbors three predicted low-complexity regions, including a stretch of poly-alanines (Figure 5D), which have been implicated in several human diseases associated with TF gene mutations (Albrecht and Mundlos, 2005).
To rigorously evaluate the function of the Hand2 N terminus, we constructed a series of N-terminal deletion mutants (Figure 5E) and subjected them to several quality-control measures. ICC of infected MEFs confirmed nuclear localization of all Hand2 constructs (Figure S6D). Western blotting demonstrated production of full-length Hand2 proteins (Figure S6E). Transient transfection assays with an alpha myosin heavy chain (aMHC) reporter construct and Hand2 mutant VP16 fusions suggested intact DNA binding ability (Figure S6F). Finally, co-immunoprecipitation assays confirmed that Hand2 mutants retained the ability to interact with known binding partners (Figure S6G). Taken together, these results demonstrate that Hand2 N-terminal mutants maintain proper nuclear localization, protein expression, DNA binding, and protein-protein interactions.
Using the collection of Hand2 N-terminal mutants, we systematically tested their ability to mediate cardiac reprogramming of MEFs along with GMT (Figure 5F). These studies led to two interesting observations. First, we found that truncation to aa 75 had no major effect on overall cardiac reprogramming because the number of sarcomere+ cells remained unaffected (Figure 5F). Consistent with the presence of the previously described transactivation domain between aa 50 and 90, however, we observed that cardiac reprogramming diminished substantially when the Hand2Δ1–90 mutant was used (Figure 5F). Second, we noted profound changes in the proportion of iPMs generated by specific Hand2 N-terminal deletion mutants. In particular, Hand2 mutants in which the transactivation domain remained intact, yet any portion of the poly-alanine stretch was compromised (e.g., Hand2Δ1–25, Hand2Δ1–32, and Hand2Δ1–50), led to dramatically increased production of iPMs at the expense of iAMs and iVMs (Figure 5F). The iPMs generated by the poly-alanine mutant Hand2 constructs appeared morphologically indistinguishable from those generated by GHMT (Figure S6H). Based on these experiments, we conclude that the Hand2 N terminus harbors both a transactivation domain and a CSD domain.
Integrated Analysis Uncovers Desmosome Gene Activation in iPMs
Encouraged by the insights provided by our RNA-seq and ATAC-seq data analysis, we sought to identify potential milestones on the path toward generating pacemaker cells. Therefore, we performed integrated analysis of the differences between GHMT- and GMT-transduced fibroblasts to illuminate Hand2-dependent gene regulatory networks that reflect acquisition of a PM-like transcriptional state. To accomplish that objective, we simultaneously compared ATAC-seq and RNA-seq datasets derived from GHMT- and GMT-infected fibroblasts. We plotted RNA-seq and ATAC-seq peaks as a fold change between the GHMT and GMT samples. In this manner, peaks that appear in the right upper quadrant represent genes whose loci become more accessible and whose expression increases. Likewise, peaks in the left lower quadrant represent genes whose loci become less accessible and whose expression decreases. Confirming the overall feasibility of this approach, we found that genes comprising the “sarcomeric organization” GO term (Figure 1G) and pacemaker marker genes (Figure 2D) tended to appear in the right upper quadrant (Figure S7A).
Focusing on genes in the right upper quadrant, we identified a group of desmosomal genes (Figure 6A). We observed concerted upregulation of the desmosomal gene battery specifically in iPMs (Figures 6B and S7B). Desmosomes are cellular structures that allow tight connections between cells and consist of several individual protein components (Figure 6C). Desmosomes are critical for intercellular communication, and human mutations in desmosomal genes can cause familial arrhythmia syndromes (Corrado et al., 2017). Furthermore, mouse knockout studies have clearly demonstrated that the desmosomal protein desmoplakin is required for normal sinus node function (Mezzano et al., 2016).
Figure 6. Hand2 Coordinates Activation of the Desmosome Machinery.

(A) Integrated analysis of RNA-seq (x axis) and ATAC-seq (y axis) data between GHMT- and GMT-transduced MEFs. Open and closed circles denote open and closed chromatin, whereas upward and downward pointing arrows indicate increased and decreased gene expression. Gene loci are labeled with colored symbols and corresponding gene names.
(B) RNA-seq heatmap for genes that comprise the desmosome machinery in control, GMT-infected, and GHMT-infected MEFs.
(C) Cartoon diagram of the desmosome complex.
(D) Genome browser tracks for Dsp and Pkp2 loci with corresponding ATAC-seq tracks derived from control, GMT, GHMT, and P0 PM samples. Hand2 and E10.5 heart H3K27 ChIP-seq peaks are provided as a reference.
(E) qRT-PCR confirmation that desmosome genes are enriched in iPMs.
(F) ICC validation of Pkp2 protein expression in GHMT, but not GMT, transduced MEFs. Scale bar, 20 mm.
(G) Kinetics of desmosome gene expression during iPM formation demonstrated by qRT-PCR analysis.
Consistent with the integrated analysis, we observed that chromatin accessibility at the Dsp1 locus increased substantially between GMT and GHMT samples, whereas accessibility at the Pkp2 locus increased less dramatically (Figure 6D). Interestingly, we note that multiple Hand2 ChIP-seq peaks underlie regions of increased chromatin accessibility, suggesting that Hand2 is a primary driver of that process. To confirm the RNA-seq data, we performed qRT-PCR analysis on a separate set of reprogrammed fibroblasts (Figure 6E). As expected, we found increased expression of desmosomal components Dsp, Pkp2, Dsc2, Jup, and Dsg2 in GHMT- versus GMT-infected fibroblasts. Moreover, we observed that endogenous Pkp2 was expressed at the protein level in GHMT-infected, but not GMT-infected, iCLMs (Figure 6F).
To evaluate desmosome gene-expression dynamics, we performed qRT-PCR on GHMT-infected fibroblasts at various time points after transduction (Figure 6G). Although all tested genes increased expression after GHMT transduction, each desmosomal component exhibited unique expression dynamics and maximal levels. A recent study suggested that Pkp2 can modulate gene expression (Cerrone et al., 2017), so we tested that possibility in the context of iPM reprogramming (Figure S7C). However, we found no evidence that Pkp2 could either substitute for Hand2 or augment GHMT to improve iPM formation. Taken together, these results identify a Hand2-dependent desmosome gene battery that undergoes coordinated chromatin reorganization and gene activation during iPM reprogramming.
DISCUSSION
In this study, we conducted a detailed genomic and biochemical analysis of the mechanisms by which Hand2, in cooperation with Gata4, Mef2c, and Tbx5, coordinates iPM formation. Hand2 or- chestrates the activation of a panel of endogenous pacemaker marker genes that span embryonic and postnatal SAN development. Interestingly, we found that Shox2 and Isl1, which participate in the developmental pacemaker gene regulatory network, are not activated in iPMs, and they can augment iPM formation to some extent when added exogenously. Furthermore, we demonstrated that Hand2 cistrome accessibility undergoes coordinated changes to open loci of cardiac conduction genes, including Hcn4. Through structure-function analysis of Hand2, we discovered a CSD domain residing within the unstructured N terminus. Finally, we found that a battery of desmosomal genes is specifically activated in the presence of Hand2 via concerted chromatin reorganization (Figure 7).
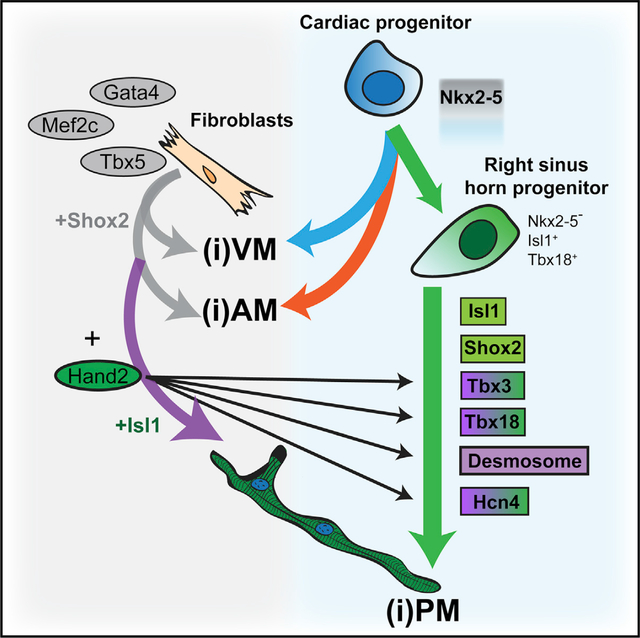
Figure 7. Model for Hand2-Dependent Mechanisms That Regulate iPM Formation by GHMT.

On the right, the pathway from cardiac progenitor cell to Tbx18+/Isl1+/Nkx2–5− right sinus horn progenitor to endogenous pacemaker cell is shown. Previous work has established that Isl1, Shox2, Tbx3, and Tbx18 have instructive roles during developmental pacemaker formation. Cardiac progenitor cells can alternatively give rise to atrial myocytes (AM) and ventricular myocytes (VM) as depicted by orange and blue arrows, respectively. On the left is a proposed pathway for iPM reprogramming by GHMT. GMT alone is capable of converting fibroblasts into iAMs and iVMs (gray arrows), whereas GHMT augments iAM and iVM production and uniquely promotes iPM formation (purple arrow). In this study, we show that Hand2 partially activates the developmental pacemaker gene regulatory network via Tbx3 and Tbx8 (purple-green shading). However, Isl1 and Shox2 remain inactive after GHMT infection of fibroblasts (green shading), and exogenous Isl1 and Shox2 can each promote iPM formation. Hand2 also coordinates activation of various core pacemaker marker genes, such as Hcn4 (purple-green shading), and we uncovered an iPM desmosome gene battery (purple shading).
To gain a sense for the degree of similarity observed between iPMs and SAN (Figure 2), we note that, in the original comparison of SAN and right atrial (RA) RNA-seq datasets (Vedantham et al., 2015), there were 142, 166, and 128 overlapping genes between the SAN-enriched gene list for E14.5 versus P4, P4 versus P14, and E14.5 versus P14, respectively. Thus, we consider the degree of overlap between iPMs and SAN tissue (Figure 2B) to be at least moderate. Furthermore, the degree of observed overlap likely represents an underestimate for the following three reasons. First, the RNA amplification required for the SAN-enriched dataset (Vedantham et al., 2015) would lead to overrepresentation of certain genes and skewing of the comparison with the iPM dataset, which was generated without amplification. Second, the SAN-enriched RNA-seq dataset was generated by laser capture, such that both cardiomyocytes (CMs) and non-CM (as well as some Hcn4-GFP−) tissue may have been obtained, thus leading to a more heterogeneous sample composition and a dilution of the similarity with our iPM dataset. Third, iPMs are engineered cells that approach a transcriptional signature of SAN tissue, yet they remain short of the target population.
Because Isl1 is known to regulate pacemaker formation (Liang et al., 2015), we were surprised that Isl1 transcripts were not increased in GHMT-reprogrammed cells (Figure 3E). We surmised that the Isl1 transcriptional network failed to be engaged by GHMT, which motivated our decision to test it as a candidate reprogramming factor. Consistent with that hypothesis, we found that addition of Isl1 to GHMT improved iPM reprogramming, but Isl1 could also partially substitute for Hand2 (Figure 3H). Interestingly, the Isl1 motif did appear in the hypergeometric optimization of motif enrichment (HOMER) analysis of cluster 5 (Table S3), but it was only enriched 1.1-fold above background, so we did not consider that level of enrichment meaningful. To reconcile these observations, we posit that Hand2 and Isl1 activate a set of common transcriptional targets requiring each TF alone and a separate set of targets requiring both. Presumably, the latter category of target genes involves cooperative binding of a Hand2-Isl1 complex. This proposed mechanistic model is consistent with either direct or indirect gene regulation, and distinguishing between those possibilities represents an important area of future investigation.
Surprisingly, the consensus Hand2 motif (CATCTG) did not emerge from our HOMER analysis of the iPM ATAC-seq data (Figure 3C). However, careful review of the raw analysis (Table S3) reveals motifs for other bHLH family members in clusters 3–5 using less-stringent criteria for motif discovery. Overall, none of the identified bHLH motifs enrich substantially above background, suggesting that Hand2 does not bind to a strong E-box consensus sequence during iPM reprogramming. Furthermore, a strong E-box motif did not emerge from our analysis of iPM ATAC-seq peaks that overlap with known Hand2 binding sites as determined by ChIP-seq (Figures S5C and S5D). We think that context-dependent alterations in Hand2 DNA binding affinity help to explain these seemingly contradictory observations.
The original Hand2 consensus motif derived from in vitro-binding site-selection assays is CATCTG(G) with very little flexibility (Dai and Cserjesi, 2002). Interestingly, the derived Hand2 consensus based on the embryonic limb Hand2 ChIP-seq dataset is CRTCTG(G) with “A” being the preferred nucleotide at the “R” position (Osterwalder et al., 2014). This consensus is almost identical to the one identified in vitro, suggesting that Hand2 by itself is capable of binding to these motifs in the limb. In contrast, the Hand2 consensus based on the embryonic heart Hand2 chromatin immunoprecipitation sequencing (ChIP-seq) dataset is CAKCTG(B) (Laurent et al., 2017). Here, the “K” and “B” positions are evenly divided among all nucleotide possibilities and are not strongly specified overall, suggesting that the consensus is much more flexible in the heart. We believe that this reflects different binding modes in limb versus heart and iPMs, which require cooperative binding of Hand2 with GMT. Consistent with that idea, a Hand2 DNA-binding-deficient mutant can rescue heart, but not limb, development (Liu et al., 2009), although we note that this mutant cannot completely rescue efficient iPM formation (Figure 5B).
Given that Hand2 coordinates activation of cardiac conduction genes, we hypothesize that Hand2 may also regulate developmental pacemaker specification. Two observations provide intriguing support for this idea. First, Hand2 is unique among GHMT to activate pacemaker genes. Second, several known cardiac genomic Hand2 binding sites coincide with chromatin peaks specifically opened when Hand2 functions with GMT. Why has a potential role for Hand2 in pacemaker specification remained obscure? Because Hand2 knockout mice are embryonically lethal (Srivastava et al., 1997), pacemaker phenotypes would have been difficult to detect. However, re-examination of the Hand2 knockout phenotype should resolve this question, and the tools now exist to delete Hand2 in pacemaker cells. We speculate that Hand2 functions to establish the competence of pacemaker progenitor cells to undergo subsequent lineage specification.
Cardiac reprogramming provides a unique opportunity to perform structure-function analysis and to uncover important mechanistic insights, some of which may also apply to development. We found that the Hand2 N terminus harbors a protein domain that ensures cardiac subtype diversification during cardiac reprogramming by GHMT. We envision two general models for how the CSD domain functions. First, the CSD domain regulates the binding specificity of Hand2 to promote cardiac subtype-specific gene expression signatures. Second, the CSD domain mediates key protein-protein interactions that are required for subtype diversification. To address the first possibility, we performed ATAC-seq using Hand2D1-32 with GMT and found no significant differences in chromatin accessibility between full-length and mutant Hand2 (data not shown). Thus, we currently favor the second model. We also found that Hand2 coordinates desmosome gene activation in iPMs. Interestingly, desmosomes are important for coordinating the assembly and function of gap junction proteins, which are essential for intercellular electrical communication (Corrado et al., 2017). Whether the insights gleaned from our studies have developmental importance remains an important question that now appears testable in parallel experimental systems for developmental and reprogramming cell-type specification.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nikhil Munshi (nikhil.munshi@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments described in this study were conducted under the oversight of the University of Texas Southwestern Institutional Animal Care and Use Committee. Hcn4-GFP reporter mice were generated by bacterial artificial chromosome (BAC) transgenesis using a ~237 kb BAC clone (RP23-281H22) that includes the Hcn4 gene and nearby regulatory regions as previously described (Gong et al., 2003). Equal ratios of male and female mice were harvested at E12.5 for this study.
METHOD DETAILS
Isolation of mouse embryonic fibroblasts (MEFs)
Hcn4-GFP E12.5 reporter mouse embryos were harvested and removed of the head and internal organs. The remaining tissue was finely minced and digested in 1 mL of 0.25% trypsin-EDTA per embryo for 15 min in a 37°C water bath. Cells were resuspended in 2 mL per embryo of fibroblast medium (DMEM/Hi glucose supplemented with 10% FBS and 1% [v/v] penicillin/streptomycin) and plated on a 15-cm dish. In 24 h, cells were harvested and stored for future use. All animal experiments described in this study were conducted under the oversight of the University of Texas Southwestern Institutional Animal Care and Use Committee.
DNA Constructs
The GFP-Hand2 fusion expression constructs were created by Phusion High-Fidelity PCR amplification of the appropriate fragment using primers containing BamHI, ClaI, and SalI linkers. Digested fragments were cloned into pBabe vector. For Hand2 domains, and N terminus variants, the desired fragments were amplified using primers that contained the desired truncation size. All constructs were confirmed via sequencing, expression via western blotting, and localization via immunofluorescence.
Generation of retroviruses, viral infection, and cardiac reprogramming
Two micrograms of retroviral plasmid DNA mix expressing mouse Gata4, Mef2c, Tbx5, Hand2, or Hand2 variants, were transfected using FuGENE 6 into Platinum E cells plated on 6-well plate at a density of 1 × 106 cell per well 24h post-plating (Song et al., 2012). In parallel, Hcn4-GFP MEFs were seeded into culture dishes or plates that were precoated with SureCoat (Cellutron) for 2 h at a density of 6 × 104 per square centimeter. 24 h post-transfection, viral supernatant was collected and filtered through a 0.45 μm cellulose filter. The viral medium was mixed with hexadimethrine bromide (polybrene) at a final concentration of 8 μg/mL and added to the culture plate with MEFs. Platinum E cells were replenished with growth medium. 24 h later, MEFs were infected with a second round of viral supernatant, and Platinum E cells were properly discarded. 48 h post-infection, the medium was replaced with induction medium composed of DMEM/199 (4:1), 10% FBS, 5% HI Horse serum. 1% penicillin/streptomycin, 1% non-essential amino acids, 1% essential amino acids, 1% B27, 1% insulin-selenium-transferrin, 1% vitamin mixture, 1% sodium pyruvate (Invitrogen). The medium was replaced every 2–3 days until the end of the experiment.
Immunocytochemistry
Cells were washed two times with PBS and fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 at room temperature. Samples were blocked with universal blocking buffer (BioGenex) for 10 mins at RT and then incubated with primary antibodies against α-actinin (Sigma), GFP (Invitrogen), Nppa (Abgent), or Myl2 (ProteinTech) diluted in 0.5X universal blocking buffer overnight at 4°C. The next day, samples were washed three times with 0.1% Triton X-100, and incubated with goat anti-mouse Alexa Fluor 555 (Invitrogen), goat anti-chicken Alexa Fluor 488 (Invitrogen), and donkey anti-rabbit Alexa Fluor 647 (Invitrogen) for 1 h at room temperature. Cells were washed three times with 0.1% Triton X-100, and mounted with ProLong Glass antifade mountant with NucBlue (Invitrogen). Images where capture with either a Zeiss LSM700 or Nikon A1R+ confocal microscope.
We previously identified a set of minimal immunostaining markers that distinguish endogenous atrial, ventricular, and pacemaker myocytes. This analysis demonstrated that endogenous pacemaker cells are sarcomere+/Hcn4-GFP+/Nppa−/Myl2−. We have also shown that this combination of immunostaining and morphological criteria prospectively identifies reprogrammed iPMs as confirmed by patch clamp recordings (Nam et al., 2014). Thus, we feel confident that our parsimonious choice of markers accurately identifies functional pacemaker cells.
Real-time qPCR
Total RNA was extracted using RNeasy Mini kit (QIAGEN) according to manufacturer’s instructions. One microgram of RNA was reverse-transcribed using SuperScript III (Invitrogen). AzuraQuant (Azura Genomics) RT-qPCR was performed in triplicate using an ABI PRISM real-time PCR machine (Applied Biosystems) and analyzed with SDS2.45 Software (Applied Biosystems). mRNA levels were normalized to Gapdh. Primer sequences can be found in Table S6.
Western blotting and co-immunoprecipitation
Platinum-E cells were transiently transfected with myc-epitope or GFP fusion Hand2, or Hand2 variants. 24 and 48 h post-transfection, 10T1/2 cells were infected with viral supernatant as previously described. 48 h post-infection 10T1/2 were collected and protein lysate prepared using three freeze-thaw cycles in RIPA buffer (50 mM HEPES-KOH, Ph 7.5, 1 mM EDTA, pH 8.0, 1%[v/v] NP-40 Igepal CA-630, 0.7% Sodium deoxycholate, 0.5M Lithium Chloride, 1X protein inhibitor). Cell debris was cleared by centrifugation at 2000x g for 5 min at 4°C. Supernatant was collected and used for downstream applications. Twenty micrograms of total protein lysate (as determined by Bradford assay) were resolved by SDS/PAGE, transferred to PDVF membrane (BioRad), probe with mouse anti-myc (Thermo Fisher Scientific), or rabbit anti-GFP (Invitrogen), and detected with HRP-conjugated anti-mouse/rabbit. Membranes were incubated with Clarity MAX western ECL blotting substrates (BioRAD) according to manufactures instruction and developed using Li-Cor’s Odyssey Fc imaging system. For Co-IP experiments, PE cells were co-transfected with HA-Tbx5 and GFP-Hand2 variants for 48hrs. Cell lysates were incubated overnight with 1 mg of rabbit polyclonal anti-GFP (Thermo Fisher Scientific) with end-over-end rotation at 4°C. The cell lysates were pulled down using magnetic Protein G Dynabeads (Invitrogen), and then the GFP-Hand2 complex was eluted with 100 mM Glycine, pH 2, for 10 min at 55°C. Solution was neutralized with 10 mL of 1M Tris, pH 8.0. Eluted and input samples were analyzed by SDS-PAGE western blot using a mouse anti-HA or a rabbit anti-GFP polyclonal antibody.
Flow cytometry
For isolation of GFP-Hand2+Hcn4-GFP+ populations after 14 days of reprogramming, cells were trypsinized and washed twice with FACS buffer (PBS, 0.5%BSA, 0.1% NaN3), resuspended at 1 × 106 cells per mL in FACS buffer and filtered with a 100 μm cell strainer. Cells were sorted using a FACSAria 2 (BD Biosciences). For ATAC-seq samples, 12.5k GFP-Hand2+Hcn4-GFP+ cells were isolated per biological replicates, and ~40k cells for RNA samples. Separate plates were considered as biological samples and were not pooled.
RNA-seq library preparation and sequencing
RNA-seq was performed in biological duplicates. For RNA-seq, total RNA was extracted from sorted GFP-Hand2+Hcn4-GFP+ cells using the RNeasy Mini kit (QIAGEN). 100 ng of total RNA was used for the RNA-seq. Library preparation was performed using TruSeq Stranded mRNA with Ribo-Zero human/mouse/rat assay (Illumina) following the manufacture’s protocol. Optimal library size was confirmed using Agilent 2200 TapeStation using D1000 screen tape. Samples were then paired-end sequenced using an Illumina HiSeq 500 High Output (400M) at the University of Texas Southwestern Microarray Core Facility.
RNA-seq analysis
Raw FASTQ files were analyzed using FastQC v0.11.2 (Andrews, 2010) and FastQ Screen v0.4.4 (Wingett, 2011), and reads were quality-trimmed using fastq-mcf. (ea-utils/1.1.2–806) (Aronesty, 2013). The trimmed reads were mapped to the hg19 assembly of the human genome (the University of California, Santa Cruz, version from igenomes) using Tophat (Kim et al., 2013). Duplicated reads were marked using Picard tools (v1.127; https://broadinstitute.github.io/picard/), the RNA counts generated from FeatureCounts (Liao et al., 2014) were TMM normalized (Robinson and Oshlack, 2010), and differential expression analysis was performed using edgeR (Robinson et al., 2010). For differential expression analysis, statistical cutoffs of FDR ≤ 0.05 and log2CPM ≥ 0 and were used to identify statistically significant and possibly biologically relevant differentially regulated genes. Mouse sinoatrial node transcriptome raw data was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65658 (P4, E14, E14.5 for SAN and RA samples) and counting and normalization was done as above. Genes common to each cluster from the RNASEQ heat- map and also significantly differentially expressed between SAN versus RA for E14.5, E14 and P4 datasets were plotted in venn charts generated using Intervene (Khan and Mathelier, 2017).
Pathway and network analysis were conducted using the Database for Annotation, Visualization, and Integrated Discovery (DAVIDv.6.8) (Huang et al., 2009) software. The threshold was set as modified Fisher Exact P value (EASE score) % 0.05. Differentially expressed gene heatmaps were clustered by hierarchical clustering using R (http://www.R-project.org) and generated using the heatmap.2 function within the gplots package (Warnes et al., 2016) and ComplexHeatmap package (Gu et al., 2016).
Gene ontology terms were generated using the web-based PANTHER Gene Ontology (GO) tool using nearest neighbor gene for common peaks within replicates. GO clusters and mini RNA-seq heatmaps were generated using the ggplot2 R package (https://ggplot2.tidyverse.org/).
ATAC-seq library preparation and sequencing
ATAC-seq was performed in biological duplicates. To profile low amounts of open chromatin in our samples we used a modified ATAC-seq protocol previously published (Corces et al., 2017). In brief, 12.5k GFP-Hand2+Hcn4-GFP+ cells were sorted and pelleted by centrifugation at 500 × g at 4°C for 10 min. Cells were resuspended in 50 μL of cold RBS (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 in H20) with 0.1%NP40, 0.1%Tween-20, and 0.01% Digitonin, and incubated on ice for 3 min. The isolated nuclei was washed one time with 1mL of cold RSB containing 0.1% Tween-20 but not NP40 or Digitonin. Nuclei were pelleted by centrifugation at 500x g at 4°C for 10 min before the supernatant was carefully discarded. The isolated nuclei was resuspended in 50 mL of transposition mixture (1xTD Buffer, 100nM Tn5, PBS, 0.01% Digitonin, 0.1% Tween-20, H20) and incubated at 37°C for 30 min in a thermomixer at 1000 RPM mixing. The DNA was cleaned with Zymo DNA clean and concentrator-5 (Zymo Research), and eluted in 20 mL of Elution buffer. Illumina adaptor Indexes were added to the libraries using KAPA HiFi enzyme with manufacture’s PCR conditions (Kapa Biosystems) and pre-amplified for 5 cycles. One mL of this reaction was run on the Kapa library quantification kit (Kapa Biosystems) to estimate leftover cycles for a final concentration of 20 nM per library. Amplified DNA was cleanup twice using AMPure XP Beads (Beckman Coulter); the first using a 0.5X volume to remove large DNA fragments (> 1kb), and the second with 1.2X to clean up the remaining small fragments. The final clean fragments were resuspended in 20 mL of Elution buffer. DNA was quantify using a Qubit fluorometer (life technologies), and library sizes were determined with a TapeStation (Agilent Technologies). Sequencing was performed using the Illumina HiSeq 500 High Output (400M) to obtain an average of 50 million paired-end 75bp reads per sample.
ATAC-seq analysis
Raw FASTQ files were analyzed using FastQC v0.11.5 (Andrews, 2010) and FastQ Screen v0.11.4 (Wingett, 2011), and reads were quality-trimmed using fastq-mcf. (ea-utils/1.1.2–806) (Aronesty, 2013). The trimmed reads were mapped to the mm10 assembly of the mouse genome (the University of California, Santa Cruz, version from igenomes) with bowtie2 (version 2.3.3.1) (Langmead and Salzberg, 2012). The duplicates were marked using picard-tools (v2.2.1) and blacklist regions were removed using bedtools (v2.7.1). TN5 shifting of bam files was performed using the open-source Perl script “ATAC_BAM_shiftrt_gappedAlign.pl” (Ahmed and Ucar, 2017). The ATAC-Seq peaks were called using MACS2 (version 2.1.0.20160309) (Zhang et al., 2008), with a q-value threshold of 0.05 and using the random background. Differential binding analysis was performed using the DiffBind package (Stark and Brown, 2011).
Furthermore, the fragments were divided as nucleosome free (sub-nucleosomal, < 100 bp) and as nucleosome occupied or bound (> 100 bp). Overlapping peaks from replicates were merged using bedtools (v2.7.1) merge to yield a peak set for each sample. We used bedtools (v2.7.1) intersect to get common regions between peak files of replicates of the same experimental group and subsequently filtered for all peaks which were consistently and significantly different (FDR < 0.05) between at least two conditions. The annotation of peaks, genome-wide distribution of ATAC-Seq regions on promoters, exons, introns, and intergenic regions in each of the samples was done using ChIPseeker (Yu et al., 2015).
For ATAC-seq in model organisms, the peak file (NAME_peaks.narrowPeak) can be uploaded directly to the UCSC genome browser for generating the browser tracks and ENCODE peaks were added by configuring the UCSC Genome Browser Track to import public track hubs, in this case ENCODE ChIP-Seq dataset from mouse limb https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE55707 (GSM1447340 – (ChIPseq), GSM1447341- (Input)) and also mouse heart https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73368. The mm9 genome based wig files were converted to mm10 genome bigwig files using CrossMap. For genome browser tracks bigWig files were hosted in Cyverse{Merchant, 2016 #600}. Web-based PANTHER Gene Ontology (GO) tool was used for generating GO terms for nearest neighbor genes for peaks that were common to 2 replicates of each ATAC-seq sample. We generated Pearson correlation graphs as well as RPKM normalized bigwig files (bamCoverage module), PCA plots, heatmaps and profile plots for the ATAC-Seq using deepTools2 (Ramírez et al., 2016). For ATAC-seq profile plots, ATAC-seq counts were first normalized by RPKM and then z-scores were calculated on a per-peak basis across samples. RPKM in this case is Reads per Kilobase per Million mapped reads.
The method mentioned in Laurent et al. (2017) for motif analysis was followed in the ATAC-seq dataset. De-novo and known motif over-representation analysis in the selected merged peaksets was done using the findMotifsGenome.pl script in the HOMER collection (Heinz et al., 2010). Raw HOMER known motif lists for Figures 3A and S3A are provided in Tables S3 and S4, respectively. A fixed length of 7 nucleotides was used for de-novo motif finding in ± 150 bp region around the summit of each peak. We used FIMO (Grant et al., 2011) from the MEME suite to confirm the discovery of de-novo motifs. The top 5 over-represented motifs with all possible alternative sequences were used to scan the region for high-affinity matches with p value threshold of 1e-4.
ChIP-seq and ATAC-seq integration analysis
1684 regions from merged ATAC-seq data (for the figure (ATACSEQ final heatmap) that overlapped with ChIP-seq Peaks from Laurent et al. (2017) were found using bedtools (v2.7.1) intersect.
De-novo motif finding was performed in these regions as above and FIMO was used to scan motifs for GATA factors 1, 2, 3, 4 and 6; TBX factors 2, 3 and 5 as well as MEF2 factors A, C and D. Hierarchically clustered heatmaps representing the patterns of these TF motifs as also all possible E-box motif (CANNTG) combinations against the ChIP-seq and ATAC-seq integrated regions was plotted using the ComplexHeatmap package (Gu et al., 2016).
RNA-seq and ATAC-seq integration analysis
Fold changes (log to the base 2) of significant differentially expressed genes (FDR < 0.05) were calculated for RNASEQ and corresponding ATACSEQ peaks annotated closest to the TSS of these genes by ChIPseeker for GHMT versus GMT conditions. A scatterplot was generated using this dataset with base R (http://www.R-project.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as mean ± SD and have at least n = 3 per group (refer to figure legend to detailed information). P values were calculated with either unpaired t test or one-way ANOVA except where noted in the Figure Legend. Statistical analysis were run using GraphPad Prism 7.04 software package. P < 0.05 was considered significant in all cases. P values are depicted using the GP style with one to four asterisks (< 0.05, < 0.01, < 0.001, < 0.0001). Raw Pearson correlation values for Figures S1C and S2A are provided in Table S1.
DATA AND SOFTWARE AVAILABILITY
The accession number for the ATAC-seq, and RNA-seq data in this paper is GEO: GSE124338.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Troponin T, Cardiac Isoform Ab-1 | Thermo Scientific | Cat#MS-295-P; RRID:AB_61806 |
| Rabbit GFP polyclonal antibody | Thermo Scientific | Cat#A-11122; RRID:AB_221569 |
| Chicken IgY GFP polyclonal antibody | Thermo Scientific | Cat#A10262; RRID:AB_2534023 |
| Mouse anti-HA.11 Epitope tag monoclonal antibody clone 16B12 | Bio Legend | Cat#MMS-101R; RRID:AB_291262 |
| Rabbit myosin light chain 2 polyclonal antibody | Proteintech | Cat#10906-1-AP; RRID:AB_2147453 |
| Rabbit NPPA polyclonal antibody (N-term) | Abgent | Cat#AP8534a; RRID:AB_10552945 |
| Mouse monoclonal anti-alpha-Actinin clone EA-53 | Sigma | Cat#A7811; RRID:AB_476766 |
| Rabbit anti-Tbx3 polyclonal antibody | Abcam | Cat#ab99302; RRID:AB_10861059 |
| Rabbit anti-Shox2 polyclonal antibody | Abcam | Cat#ab82918; RRID:AB_1860605 |
| Rabbit RFP polyclonal antibody | Rockland | Cat#600-401-379; RRID:AB_2209751 |
| Rabbit c-myc tag polyclonal antibody | Thermo Scientific | Cat#PA1-981; RRID:AB_325961 |
| Mouse beta-actin (8H10D10) monoclonal | Cell Signaling Technology | Cat#3700; RRID:AB_2242334 |
| Goat anti-Mouse IgG (H+L) antibody, Alexa Fluor 555 | Thermo Scientific | Cat#A-21422; RRID:AB_141822 |
| Goat anti-Rabbit IgG (H+L) antibody, Alexa Fluor 488 | Thermo Scientific | Cat#A-11034; RRID:AB_2576217 |
| Donkey anti-Rabbit IgG (H+L) antibody, Alexa Fluor 647 | Thermo Scientific | Cat#A-31573; RRID:AB_2536183 |
| Donkey anti-Mouse IgG (H+L) antibody, Alexa Fluor 647 | Thermo Scientific | Cat#A-31571; RRID:AB_162542 |
| Goat anti-Chicken IgY (H+L) antibody, Alexa Fluor 488 | Abcam | Cat#ab150169; RRID:AB_2636803 |
| Bacterial and Virus Strains | ||
| NEB Turbo Competent E. coli (High Efficiency) | NEB | Cat#C2984l |
| Biological Samples | ||
| E12.5 Hcn4-GFP mouse embryonic fibroblasts | This paper | N/A |
| P0 Hcn4-GFP+ pacemaker myocytes | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SureCoat | Cellutron | Cat#SC-9035 |
| Polybrene | Millipore | Cat#TR-1003-G |
| Dulbecco’s Modified Eagle Medium (DMEM) | Sigma | Cat#D5796 |
| Medium 199, Earle’s Salts | Thermo Fisher | Cat#11150059 |
| Fetal bovine serum (FBS) | Sigma | Cat#F2442 |
| Insulin-Transferrin-Selenium G | Thermo Fisher | Cat#41400-045 |
| MEM vitamin solution | Thermo Fisher | Cat#11120-052 |
| MEM amino acids | Thermo Fisher | Cat#1601149 |
| Non-Essential amino acids | Thermo Fisher | Cat#11140-050 |
| Antibiotic-Antimycotics | Thermo Fisher | Cat# 15240062 |
| B-27 supplement | Thermo Fisher | Cat#17504044 |
| Heat-Inactivated Horse Serum | Thermo Fisher | Cat#26050-088 |
| NaPyruvate | Thermo Fisher | Cat#11360-70 |
| Penicillin/Streptomycin | Thermo Fisher | Cat#1514022 |
| Puromycin | Thermo Fisher | Cat#A11139-03 |
| Blasticidin | Gemini Bio-Products | Cat#400-128P |
| Glutamax | Thermo Fisher | Cat#35050-061 |
| FuGENE 6 transfection Reagent | Promega | Cat#E2692 |
| Tn5 | Gift by Gary Hon lab | N/A |
| Clarity Max western blot ECL substrate | BIO-RAD | Cat#1705062S |
| Azuraquant Green Fast QPCR Mix Hi-Rox | Phenix Research Products | Cat#RA-2005 |
| Luciferase Assay System | Promega | Cat#E1500 |
| Power block 10X | BioGenex | Cat#HK083-50K |
| 12mm #1.5 Coverslip with Fibronectin | NeuVitro Corporation | Cat#GG-12-15-fibronectin |
| Trypsin 0.25% EDTA | Thermo Fisher | Cat#25200056 |
| Dynabeads Protein G | Thermo Fisher | Cat#10003D |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| AMPure XP Beads | Beckman Coulter | Cat#A63881 |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold Set B | Illumina | Cat#RS-122-2302 |
| Zymo DNA clean and concentrator | ZYMO RESEARCH | Cat#D4014 |
| KAPA library quantification kit | Kapa Biosystems | Cat#KK4828 |
| Agilent DNA1000 kit | Agilent Technologies | Cat#5067-1504 |
| Qubit dsDNA HS Assay kit | Thermo Fisher | Cat#Q328251 |
| Deposited Data | ||
| Sorted RNA-seq, sorted ATAC-seq data | This paper | Gene Expression Ommibus (GEO): GSE124338 |
| Hand2 ChIP-seq data | Laurent et al., 2017 | Gene Expression Ommibus (GEO): GSE73368 |
| H3K27ac ChIP-seq on embryonic 10.5 mouse heart | ENCODE | https://www.encodeproject.org/experiments/ENCSR582SPN/ |
| Hcn4-GFP RNA-seq | Vedantham et al., 2015 | Gene Expression Ommibus (GEO): GSE65658 |
| Experimental Models: Cell Lines | ||
| Platinum-E Retroviral Packaging Cell line, Ecotropic | Cell Biolabs | Cat#RV-101 |
| C3H/10T1/2, Clone 8 | ATCC | Cat#CCL-226 |
| Experimental Models: Organisms/Strains | ||
| Tg(Hcn4-EGFP)JK158Gsat | MMRRC | RRID:MMRRC_030842-UCD |
| Oligonucleotides | ||
| ClaI_Hand2_aa20-aa216_5: gcgcATCGATGCCGCAGCCGCCGCC | This paper | N/A |
| ClaI_Hand2_aa25-aa216_5: gcgcATCGATGCTGCCGCCGCCGCC | This paper | N/A |
| ClaI_Hand2_aa32-aa216_5: gcgcATCGATAGCCGCTGCAGCCACGAG | This paper | N/A |
| ClaI_Hand2_aa50-aa216_5: gcgcATCGATGAGATGTCGCCCCCCGAC | This paper | N/A |
| ClaI_Hand2_aa75-aa216_5: gcgcATCGATGGTGCCGCGGGCCTG | This paper | N/A |
| ClaI_Hand2_aa90_5: gcgcATCGATGTGA AGCGCCGGGGC | This paper | N/A |
| ClaI_Hand2_5: gcgcATCGATATGAGTCT GGTGGGGGGC | This paper | N/A |
| SalI_Hand2_stop_3: gcgcGTCGACTCACTG CTTGAGCTCCAGGG | This paper | N/A |
| EcoRI_stop_Hand2_aa154_3: gcgcGAATTCt caCAGATCCATGAGGTAGGCGATGTAGC | This paper | N/A |
| Recombinant DNA | ||
| pBabemycHand2 | Song et al., 2012 | N/A |
| pBabemycGata4 | Song et al., 2012 | N/A |
| pBabeMef2cmyc | Song et al., 2012 | N/A |
| pBabemycTbx5 | Song et al., 2012 | N/A |
| pBabemycHand1 | This paper | N/A |
| pBabemycHand2F119P | This paper | N/A |
| pBabemycHand2VP16 | This paper | N/A |
| pBabemycHand2Engrailed | This paper | N/A |
| pBabemycIsl1 | This paper | N/A |
| pBabeShox2 | This paper | N/A |
| pBabemycPkp2 | This paper | N/A |
| pBabeTbx3 | This paper | N/A |
| pBabeTbx18 | This paper | N/A |
| pBabeMeis1 | This paper | N/A |
| pBabemycAtf3 | This paper | N/A |
| pBabemycHand2Δ1-20 | This paper | N/A |
| pBabemycHand2Δ1-25 | This paper | N/A |
| pBabemycHand2Δ1-35 | This paper | N/A |
| pBabemycHand2Δ1-50 | This paper | N/A |
| pBabemycHand2Δ1-75 | This paper | N/A |
| pBabemycHand2Δ1-90 | This paper | N/A |
| pBabeGFPHand2Δ1-20 | This paper | N/A |
| pBabeGFPHand2Δ1-25 | This paper | N/A |
| pBabeGFPHand2Δ1-35 | This paper | N/A |
| pBabeGFPHand2Δ1-50 | This paper | N/A |
| pBabeGFPHand2Δ1-75 | This paper | N/A |
| pBabeGFPHand2Δ1-90 | This paper | N/A |
| Software and Algorithms | ||
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| Fiji | NIH | https://fiji.sc/ |
| NIS-Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/viewer |
| Zeiss LSM ZEN Lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| FlowJo Software | FlowJo, LLC | https://www.flowjo.com/ |
| GraphPad Prism 7.04 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Li-Cor Image Studio Lite | Li-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| UCSC Genome Browser | N/A | http://genome.ucsc.edu/ |
| DAVID | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| PANTHER Gene Ontology | N/A | http://geneontology.org/ |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/index.php |
| R Development Core | N/A | https://www.r-project.org |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/motif/ |
| FIMO (MEME suite 5.0.3) | Grant et al., 2011 | http://meme-suite.org/tools/fimo |
| FastQC | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| FastQ Screen | Wingett, 2011 | http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen |
| fastq-mcf. | Aronesty, 2013 | https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md |
| deepTools | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/ |
| TopHat | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Picard tools | N/A | https://broadinstitute.github.io/picard/ |
| featureCounts | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| TMM | Robinson and Oshlack, 2010 | N/A |
| edgeR | Robinson et al., 2010 | N/A |
| ComplexHeatmap | Gu et al., 2016 | N/A |
| ChIPseeker | Yu etal., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| DiffBind package | Stark and Brown, 2011 | N/A |
| CyVerse | NSF | https://www.cyverse.org/ |
| Other | ||
Highlights.
Hand2 orchestrates reprogramming of fibroblasts toward pacemaker cell fate
Induced pacemaker (iPM) gene expression resembles endogenous sinoatrial node tissue
Hand2 reorganizes chromatin accessibility toward a pacemaker-like state
The Hand2 N terminus diversifies cardiomyocyte subtype formation
ACKNOWLEDGMENTS
We thank the UT Southwestern Medical Center Flow Cytometry, Histology Core, Shared Molecular Biology Confocal Microscopy, and Next Generation Sequencing Facilities for use of resources and expert technical assistance. We gratefully acknowledge Gary Hon, Ning Liu, and Beverly Rothermel for critical review of the manuscript and members of the Olson, Hon, and Munshi labs for technical advice, reagents, and scientific discussions. This work was supported by the NSF (grant 2015165336 to A.F.-P.), NIH (HL136604, HL133642, and HL135217 to N.V.M.), the Burroughs Wellcome Fund (1009838 to N.V.M.), and the March of Dimes Foundation (5-FY13–203 to N.V.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.077.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abad M, Hashimoto H, Zhou H, Morales MG, Chen B, Bassel-Duby R, and Olson EN (2017). Notch inhibition enhances cardiac reprogramming by increasing MEF2C transcriptional activity. Stem Cell Reports 8, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, and Ucar D (2017). I-ATAC: interactive pipeline for the management and pre-processing of ATAC-seq samples. PeerJ 5, e4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht A, and Mundlos S (2005). The other trinucleotide repeat: polyalanine expansion disorders. Curr. Opin. Genet. Dev 15, 285–293. [DOI] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data.https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Aronesty E (2013). Comparison of sequencing utility programs. Open Bioinform. J 7, 1–8. [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, and Jaenisch R (2012). Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrone M, Montnach J, Lin X, Zhao YT, Zhang M, Agullo-Pascual E, Leo-Macias A, Alvarado FJ, Dolgalev I, Karathanos TV, et al. (2017). Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun 8, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Friedrich GA, and Soriano P (1994). Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev. 8, 2293–2301. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Firulli B, and Firulli AB (2010). A bHLH code for cardiac morphogenesis. Pediatr. Cardiol 31, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado D, Basso C, and Judge DP (2017). Arrhythmogenic Cardiomyopathy. Circ. Res. 121, 784–802. [DOI] [PubMed] [Google Scholar]
- Dai YS, and Cserjesi P (2002). The basic helix-loop-helix factor, HAND2, functions as a transcriptional activator by binding to E-boxes as a heterodimer.J. Biol. Chem 277, 12604–12612. [DOI] [PubMed] [Google Scholar]
- Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, and Chen Y (2009). Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2–5. Dev. Biol 327, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Perez A, and Munshi NV (2017). Assessing cardiomyocyte subtypes following transcription factor-mediated reprogramming of mouse embryonic fibroblasts. J. Vis. Exp (121), e55456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli AB, McFadden DG, Lin Q, Srivastava D, and Olson EN (1998). Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat. Genet 18, 266–270. [DOI] [PubMed] [Google Scholar]
- Firulli AB, Firulli BA, Wang J, Rogers RH, and Conway SJ (2010). Gene replacement strategies to test the functional redundancy of basic helix-loophelix transcription factor. Pediatr. Cardiol 31, 438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447. [DOI] [PubMed] [Google Scholar]
- George RM, and Firulli AB (2018). Hand factors in cardiac development. Anat. Rec. (Hoboken). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, and Heintz N (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineagedetermining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, and Weintraub H (1995). Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol. Cell. Biol 15, 3813–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogaars WM, Engel A, Brons JF, Verkerk AO, de Lange FJ, Wong LY, Bakker ML, Clout DE, Wakker V, Barnett P, et al. (2007). Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 21, 1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifkovits JL, Addis RC, Epstein JA, and Gearhart JD (2014). Inhibition of TGFb signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 9, e89678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, and Dzau VJ (2012). MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res 110, 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, and Mathelier A (2017). Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics 18, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent F, Girdziusaite A, Gamart J, Barozzi I, Osterwalder M, Akiyama JA, Lincoln J, Lopez-Rios J, Visel A, Zuniga A, and Zeller R (2017). HAND2 target gene regulatory networks control atrioventricular canal and cardiac valve development. Cell Rep. 19, 1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, et al. (2017). Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21, 819–833.e816. [DOI] [PubMed] [Google Scholar]
- Liang X, Zhang Q, Cattaneo P, Zhuang S, Gong X, Spann NJ, Jiang C, Cao X, Zhao X, Zhang X, et al. (2015). Transcription factor ISL1 is essential for pacemaker development and function. J. Clin. Invest 125, 3256–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu N, Barbosa AC, Chapman SL, Bezprozvannaya S, Qi X, Richardson JA, Yanagisawa H, and Olson EN (2009). DNA binding-dependent and -independent functions of the Hand2 transcription factor during mouse embryogenesis. Development 136, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. (2017). Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Kareta MS, Chanda S, Ahlenius H, Perotti N, Zhou B, Grieder SD, Ge X, Drake S, Euong Ang C, et al. (2017). Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, McAnally J, Richardson JA, Charité J, and Olson EN (2002). Misexpression of dHAND induces ectopic digits in the developing limb bud in the absence of direct DNA binding. Development 129, 3077–3088. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnology 28, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano V, Liang Y, Wright AT, Lyon RC, Pfeiffer E, Song MY, Gu Y, Dalton ND, Scheinman M, Peterson KL, et al. (2016). Desmosomal junctions are necessary for adult sinus node function. Cardiovasc. Res 111, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K, Akiyama M, Tamura F, Isomi M, Yamakawa H, Sadahiro T, Muraoka N, Kojima H, Haginiwa S, Kurotsu S, et al. (2018). Direct in vivo reprogramming with Sendai virus vectors improves cardiac function after myocardial infarction. Cell Stem Cell 22, 91–103.e105. [DOI] [PubMed] [Google Scholar]
- Mohamed TM, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, Ang YS, Yu P, Wang H, Tang S, et al. (2017). Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. Circulation 135, 978–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mommersteeg MT, Hoogaars WM, Prall OW, de Gier-de Vries C, Wiese C, Clout DE, Papaioannou VE, Brown NA, Harvey RP, Moorman AF, and Christoffels VM (2007). Molecular pathway for the localized formation of the sinoatrial node. Circ. Res 100, 354–362. [DOI] [PubMed] [Google Scholar]
- Morin S, Charron F, Robitaille L, and Nemer M (2000). GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 19, 2046–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA (2016). Direct lineage reprogramming via pioneer factors; a detour through developmental gene regulatory networks. Development 143, 2696–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, et al. (2014). MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 33, 1565–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam YJ, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, Bassel-Duby R, Olson EN, and Munshi NV (2014). Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development 141, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder M, Speziale D, Shoukry M, Mohan R, Ivanek R, Kohler M, Beisel C, Wen X, Scales SJ, Christoffels VM, et al. (2014). HAND2 targets define a network of transcriptional regulators that compartmentalize the early limb bud mesenchyme. Dev. Cell 31, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Gomes A, Bernitz J, Papatsenko D, Niu X, Swiers G, Azzoni E, de Bruijn MF, Schaniel C, et al. (2016). Hematopoietic reprogramming in vitro informs in vivo identification of hemogenic precursors to definitive hematopoietic stem cells. Dev. Cell 36, 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Sindhu C, and Meissner A (2016). Molecular features of cellular reprogramming and development. Nat. Rev. Mol. Cell Biol. 17, 139–154. [DOI] [PubMed] [Google Scholar]
- Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, and Olson EN (1997). Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat. Genet 16, 154–160. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Shang C, Twu KY, Kao SC, Jenkins NA, Copeland NG, Sanyal M, Selleri L, Cleary ML, and Chang CP (2008). Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res 103, 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind: differential binding analysis of ChIP-Seq peak data (R package version 100.). http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf.
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SA, Sim S, Neff NF, Skotheim JM, Wernig M, and Quake SR (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eif VWW, Devalla HD, Boink GJJ, and Christoffels VM (2018). Transcriptional regulation of the cardiac conduction system. Nat. Rev. Cardiol 15, 617–630. [DOI] [PubMed] [Google Scholar]
- Vedantham V, Galang G, Evangelista M, Deo RC, and Srivastava D (2015). RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circ. Res 116, 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, and Baker NE (2015). E proteins and ID proteins: helix-loop-helix partners in development and disease. Dev. Cell 35, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Lee QY, Chen AC, Li R, Corces MR, Ang CE, Treutlein B, Xiang C, Baubet V, Suchy FP, et al. (2017). Rapid Chromatin Switch in the Direct Reprogramming of Fibroblasts to Neurons. Cell Rep. 20, 3236–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, and Schwartz M (2016). gplots: various R programming tools for plotting data (R package version3.0. 1.). https://rdrr.io/cran/gplots/. [Google Scholar]
- Wingett S (2011). FastQ Screen: a quality control tool to screen a library of sequences in FastQ format against a set of sequence databases. (Babraham Bioinformatics). [Google Scholar]
- Yamakawa H, Muraoka N, Miyamoto K, Sadahiro T, Isomi M, Haginiwa S, Kojima H, Umei T, Akiyama M, Kuishi Y, et al. (2015). Fibroblast growth factors and vascular endothelial growth factor promote cardiac reprogramming under defined conditions. Stem Cell Reports 5, 1128–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zang MX, Li Y, Wang H, Wang JB, and Jia HT (2004). Cooperative interaction between the basic helix-loop-helix transcription factor dHAND and myocyte enhancer factor 2C regulates myocardial gene expression.J. Biol. Chem 279, 54258–54263. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, et al. (2015). High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of profibrotic signalling. Nat. Commun 6, 8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Dickson ME, Kim MS, Bassel-Duby R, and Olson EN (2015). Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA 112, 11864–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Morales MG, Hashimoto H, Dickson ME, Song K, Ye W, Kim MS, Niederstrasser H, Wang Z, Chen B, et al. (2017). ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 31, 1770–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the ATAC-seq, and RNA-seq data in this paper is GEO: GSE124338.