

Abstract
Environmental salinity is an important abiotic factor influencing normal physiological functions and productive performance in the sea cucumber Apostichopus japonicus. It is therefore important to understand how changes in salinity affect sea cucumbers in the face of global climate change. In this study, we investigated the responses to salinity stress in sea cucumbers using mRNA and miRNA sequencing. The regulatory network of mRNAs and miRNAs involved in salinity stress was examined, and the metabolic pathways enriched for differentially expressed miRNAs and target mRNAs were identified. The top 20 pathways were involved in carbohydrate metabolism, fatty acid metabolism, degradation, and elongation, amino acid metabolism, genetic information processing, metabolism of cofactors and vitamins, transport and catabolism, and environmental information processing. A total of 22 miRNAs showed differential expression during salinity acclimation. The predicted 134 target genes were enriched in functions consistent with the results of gene enrichment based on transcriptome analysis. These results suggested that sea cucumbers deal with salinity stress via changes in amino acid metabolism, ion channels, transporters, and aquaporins, under stimulation by environmental signals, and that this process requires energy from carbohydrate and fatty acid metabolism. Salinity challenge also induced miRNA expression. These results provide a valuable genomic resource that extends our understanding of the unique biological characteristics of this economically important species under conditions of salinity stress.
Electronic supplementary material
The online version of this article (10.1007/s12192-019-00996-y) contains supplementary material, which is available to authorized users.
Keywords: Apostichopus japonicus, Salinity stress, Differentially expressed genes, Different expression miRNA, mRNA and miRNA interaction
Introduction
Environmental salinity is an important abiotic factor affecting the normal physiology of aquatic animals. The health and productivity of sea cucumbers depend on optimal conditions, including a suitable salinity range. However, sea cucumbers are cultured in intertidal ponds where the water salinity fluctuates during monsoons or other periods of water exchange, evaporation, and natural precipitation disasters (Dong et al. 2008). Climate change threatens to further increase the frequency and intensity of salinity fluctuations, potentially leading to salinity stress in sea cucumbers. Previous studies indicated that salinity stress affected the behavior (Hamel et al. 2001), survival (Asha and Muthiah 2005), embryonic development (Kashenko 2000), growth (Li and Li 2010), energy budget (Yuan et al. 2010), reproduction (Talbot and Lawrence 2002), immune response (Wang et al. 2008), and metabolism in the sea cucumber Apostichopus japonicus (Dong et al. 2008), with potentially lethal effects. It is therefore necessary to understand the genetic differences and molecular mechanisms involved in the stress response, to aid the development of a long-term strategy for the selection of salinity-tolerant sea cucumbers, leading to increased productivity.
Stress-induced genes have previously been identified in Portunus trituberculatus, including genes related to immune function antioxidants, ion channels, ion cotransporter regulators, hormones, fatty acid and amino acid metabolism, gluconeogenesis, glycolysis, and glycan metabolism (Lv et al. 2013). Two genes encoding ecdysteroid-regulated protein and chitinase were protectively up-regulated under acute salinity stress, but were down-regulated under long-term salinity stress in white shrimp (Gao et al. 2012). Thirty-three salinity- and oxidative stress–induced genes, including LEA2, HSPs, GST, coagulation factor genes, and P450 enzyme, were differentially expressed in Haliotis discus, Eriocheir sinensis megalopae, and Crassostrea brasiliana (DeZoysa et al. 2009; Hui et al. 2014; Zacchi et al. 2017). The Na+/K+-ATPase (NKA) α-subunit, intracellular fatty acid-binding protein, and innexin 2 showed the highest gene expression levels under low-salinity stress in the shrimp Penaeus monodon (Shekhar et al. 2013). Heat shock protein genes were found to be key elements during salinity acclimation in the swimming crab (Xu and Liu 2011). Osmotic stress transcription factor 1, Na+/K+/2Cl− cotransporter (NKCC), cystic fibrosis transmembrane conductance regulator (CFTR), and Cl− channels were induced under salinity stress in fishes (Evans 2010; McCormick 2001), and potassium voltage-gated channel subfamily J member 9 (KCNJ9) and kinesin family member 5B (KINH) genes were considered important candidate genes in osmoregulation in tilapia (Gu et al. 2018; Zhang et al. 2018). Genes related to osmoregulation, signaling and interactions of the osmotic stress response, anti-apoptotic reactions, immune response, cell adhesion and communication, cytoskeleton, and cell cycle were affected by salinity stress in the Pacific oyster (Zhao et al. 2012), and ionic strength, the cytoskeleton, and MLC kinase were involved in the regulation of Na+–Cl− taurine transporter expression in Japanese eels (Chow et al. 2009). Growth hormone, insulin-like growth factor-1 (IGF-1), thyroid-stimulating hormone, and prolactin have also demonstrated important roles in osmoregulation among fish species (McCormick 2001; Sakamoto and McCormick 2006).
Salinity stress was recently shown to affect several cellular processes related to the immune response, cell cycle and differentiation, cytoskeleton rearrangements, and anti-apoptotic processes in Crassostrea gigas (Zhao et al. 2012; Meng et al. 2013). Hormone-mediated, cAMP-dependent, and osmotically mediated, and cAMP-independent pathways were shown to converge in a mechanism to activate CFTR and Cl− secretion, possibly through tyrosine phosphorylation of CFTR by focal adhesion kinase (FAK) in euryhaline killifish (Marshall et al. 2009). Chronic low-salinity stress could increase energy metabolism and amino acid and fatty acid metabolism, especially glycometabolism, as suggested by changes in the citrate cycle, oxidative phosphorylation, gluconeogenesis, glycolysis, and glycan metabolism in Litopenaeus vannamei (Xu et al. 2017). However, although changes in the expression levels of ribosomal proteins, mRNA–rRNA processing, and signal transduction might indicate metabolic and physiological changes in sea cucumbers under salinity stress (Kim and Jang 2002; Warner and McIntosh 2009); few studies have examined the molecular basis of tolerance to salinity stress in these organisms (Zhang et al. 2018).
miRNAs have been shown to play vital roles in environmental adaptation and stress in sea cucumbers (Huo et al. 2017), Crassostrea oysters (Zhao et al. 2016), and carp (Zhang et al. 2017). Twelve salinity-induced miRNAs in P. trituberculatus included two known (miR-2788a and miR-2788b) and 10 novel miRNAs. Thirty-four predicted corresponding target genes that were differentially expressed in the opposite way to the miRNAs were enriched in six important gene ontology (GO) biological processes related to osmoregulation, such as anion transport and chitin metabolism (Lv et al. 2016). The miR-8 family has been shown to be closely associated with zebrafish osmoregulation, and the family members miR-200a and miR-200b enabled precise control of ion transport by modulating the expression of Na+/H+ exchanger regulatory factor 1, a regulator of apical trafficking of transmembrane ion transporters in zebrafish (Flynt et al. 2009). Down-regulation of miR-429 in tilapia caused a substantial increase in the expression of OSTF1, which is responsible for osmosensory signal transduction (Yan et al. 2012a), and loss of function of miR-30c resulted in an inability to respond to osmotic stress in Nile tilapia (Yan et al. 2012b). IGF-1 is a target gene of miR-206 in tilapia, and IGF-1 treatment up-regulated the expression of transporters such as NKA and NKCC (Yan et al. 2013). Ten miRNA genes were identified in Gasterosteus aculeatus, suggesting a possible role for differential regulation of these miRNAs in salinity tolerance (Rastorguev et al. 2016).
However, despite research related to osmoregulation at the molecular, pathway, and hormone levels in many aquatic organisms, the effects of salinity stress on miRNAs and their predicted target genes in sea cucumbers remain unclear. In the present study, we determined the mRNA and miRNA expression profiles of A. japonicus under low-salinity stress using Illumina technology and identified the pathways enriched in the differentially expressed genes (DEGs). The interaction between miRNA and target genes was analyzed during salinity stress in sea cucumbers.
Materials and methods
Sea cucumbers, salinity stress, and sampling
Sea cucumbers were collected from Wafangdian Aquatic Farm, Dalian, P.R. China, in October 2016. All sea cucumbers were acclimated in tanks (90 × 75 × 60 cm) at the Institute of Dalian Ocean University, Key Laboratory of Mariculture & Stock Enhancement in North China’s Sea, Ministry of Agriculture. Half to two-thirds of the rearing water and sediment (feces and uneaten food) were changed daily during the acclimation period. During the experiment, sea cucumbers were fed once a day (3–5% body mass) with a laboratory-made formulated diet (equal parts Sargassum thunbergii, fish meal, and sea mud). Salinity and other water parameters were measured using an YSI multi-parameter water quality monitor (YSI incorporated, OH, USA). The temperature, pH, and dissolved oxygen were 24–25 °C, 7.92–8.29, and 3.76–5.56 mg/l, respectively, throughout the acclimation period. The sea cucumbers were then divided randomly into two groups (90 individuals per group) and transferred immediately into six tanks (30 individuals per tank): a control (CT) group maintained at 32 psu and a low-salinity stress (SS) group maintained at 18 psu. Low-salinity seawater was prepared by adding freshwater to a salinity of 18 psu. Complete salt-stress information was obtained by sampling three individuals at 1.5, 3, 6, 12, 24, 48, and 72 h, respectively, in the SS group. All samples were frozen immediately in liquid nitrogen prior to total RNA extraction for RNA library preparation, followed by subsequent mRNA and small RNA sequencing.
mRNA extraction and cDNA library construction and sequencing
mRNA was extracted from 21 samples (three replicates at seven time points) from the intestines, respiratory tree, and coelomic fluid of sea cucumbers in the SS group using an RNeasy mini kit including DNase treatment using an RNAprep pure Tissue Kit (TianGene, Beijing, China), according to the manufacturer’s protocol. Equal amounts of high-quality RNA were pooled to prepare the sequencing library. RNA purity was determined using a NanoPhotometer® (Implen, CA, USA). RNA concentration was measured using a Qubit® RNA Assay Kit and Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). RNA integrity was assessed using an RNA Nano 6000 Assay Kit (Agilent Bioanalyzer 2100 system; Agilent Technologies, CA, USA). Two cDNA libraries were constructed representing the CT and SS groups to assess the sea cucumber transcriptomes and create a quantitative and qualitative gene expression databases for salinity stress. Three samples were used to sequence two cDNA libraries representing the control and treated groups The libraries were subjected to Illumina paired-end sequencing to identify representative transcripts for a wide range of biological processes. A total of 1.5 μg of RNA per sample was used as the input material for RNA sample preparation. The sequencing libraries were generated using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (MA, USA), according to the manufacturer’s recommendations, and index codes were added to attribute the sequences to each sample. The library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, MA, USA) to preferentially select cDNA fragments of 150–200 bp in length. USER Enzyme (3 μl; NEB) was then used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before polymerase chain reaction (PCR). PCR was performed using Phusion High-Fidelity DNA Polymerase, universal PCR primers, and index (X) primer. The PCR products were purified (AMPure XP system), and the quality of the library was assessed using the Agilent Bioanalyzer 2100 system. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and paired-end reads were generated.
Bioinformatic analysis of RNA-seq data
RNA-seq clean data (clean reads) were obtained by removing reads containing adapters, poly-N, and low-quality reads (Q < 20) from the raw data. High-quality reads were assembled to contigs using the Trinity program (http://trinityrnaseq.sf.net) with min_kmer_cov set to 2 by default and all other parameters set to default values. The assembled unigene sequences were aligned for annotation by Blast-X search (cut-off E-value of 1e-10) in the public NCBI nonredundant (Nr) and nonredundant nucleotide (Nt) databases (http://www.ncbi.nlm.nih.gov/), Swiss-Prot (http://www.ebi.ac.uk/uniprot/), GO (http://www.geneontology.org/), Clusters of Orthologous Groups (COG) (http://www.ncbi.nlm.nih.gov/COG/), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/). GO terms at the second level were used for the GO annotation. GO enrichment analysis of the DEGs was implemented by Fisher’s exact test tool in Blast2GO. Term filter values were set to false discovery rate (FDR) ≤ 0.01 and P ≤ 0.005, with two-tailed tests. Finally, pathway assignments were generated using the KEGG database.
Differential expression analysis
Gene expression values were calculated as reads aligned to gene per kilobase of exon per million mapped reads. Differential expression analysis between the two samples was performed using the DEGseq (2010) R package. P values were adjusted using the Q value (Storey and Tibshirani 2003). A Q value < 0.005 and |log2(foldchange)|> 1 were set as the threshold for significantly differential expression. A FDR < 0.05 was used as the threshold P value in multiple tests to measure significantly differential gene expression. Genes were only considered differently expressed in a given library when P < 0.05 and a greater than twofold change (absolute value of log2 ratio > 1) in expression across libraries was observed.
miRNA extraction and cDNA library construction and sequencing
We assessed miRNA expression and obtained a quantitative and qualitative miRNA expression databases for low-salinity stress in sea cucumbers by constructing two cDNA libraries representing the CT and SS groups. A total of 1.5 μg total RNA per sample was used as input material for the small RNA library. Sequencing libraries were generated using NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB) following the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. PCR amplification was performed using LongAmp Taq 2× Master Mix, SR Primer for Illumina, and index (X) primer. PCR products were purified on an 8% polyacrylamide gel (100 V, 80 min). DNA fragments corresponding to 140–160 bp (length of small noncoding RNA plus 3′ and 5′ adaptors) were recovered and dissolved in an 8-L elution buffer. Library quality was assessed using an Agilent Bioanalyzer 2100 with DNA High Sensitivity Chips. Clustering of the index-coded samples was performed using a cBot Cluster Generation System using TruSeq SR Cluster Kit v3-cBot-HS according to the manufacturer’s instructions. After cluster generation, library preparations were sequenced on an Illumina Hiseq 2500/2000 platform and 50-bp single-end reads were generated.
miRNA analysis and miRNA identification
Raw data (raw reads) in the fastq format were first processed using custom Perl and Python scripts (Novogene Bioinformatics Institute, Beijing, China). Clean data (clean reads) were processed by getting rid of low-quality reads, and reads containing poly-N, with 5′ primer contaminants, without 3′ primers or the insert tag, and with poly A or T or G or C from the raw data. The Q20, Q30, and GC-content of the raw data were calculated. Small RNA tags within a certain length range were then used for subsequent analyses. All the clean reads were searched against the Rfam (http://rfam.sanger.ac.uk/) database for annotation, and reads annotated as tRNA, rRNA, snoRNA, and snRNA were discarded. The remainder of the small RNA reads were mapped to the sea cucumber genome (http://www.genedatabase.cn/aja_genome_20161129.html) using Bowtie (Langmead et al. 2009) without mismatch to analyze their expression and distribution on the reference. Mapped small RNA tags were used to search for known miRNAs. Because miRBase21.0 does not include miRNA information for sea cucumbers, we used miRBase20.0 (http://www.mirbase.org/) as a reference, with modified software mirdeep2 (Friedlander et al. 2011) and srna-tools-cli to identify conserved miRNAs. Only perfect matches were considered conserved miRNAs. Reads that were not aligned to the miRBase database were used to predict novel miRNAs. miREvo (Wen et al. 2012) and mirdeep2 (Friedlander et al. 2011) software were integrated to predict novel miRNAs by exploring the secondary structure, the Dicer cleavage site.
Differential expression of miRNAs
miRNA expression levels were estimated as transcript per million using the following criteria (Zhou et al. 2010). Differential expression analysis of the CT and SS samples was performed using the DEGseq (2010) R package. P values were adjusted using the Q value (Storey and Tibshirani 2003). A Q value < 0.01 and |log2(foldchange)|> 1 were set as the threshold for significantly differential expression.
miRNA target prediction and regulatory network construction
miRNA target genes among the DEGs were predicted using the miRanda 4.0 algorithm (microrna.sanger.ac.uk/targets/v4) and TargetScan (www.targetscan.org), based on the complementary region between miRNAs and mRNAs and the thermodynamic stability of the miRNA–mRNA duplex. All the miRNA-target mRNAs were calculated and clustered by GO terms and KEGG pathway annotations. Regulatory network predictions for mRNA and miRNA were performed using Cytoscape 3.3.0 (http://www.cytoscape.org/).
Real-time-PCR confirmation of Illumina sequencing data
To validate our Illumina sequencing data, nine DEGs were selected for quantitative RT-PCR (RT-qPCR) analysis. The primers were designed using Primer 5 software (Premier Biosoft International, CA, US). The β-actin gene was used as an internal control for target gene qPCR analysis. The primers were designed according to the contig sequences (Table 1). RNA (500 ng) from each sample of coelomic fluid was measured and treated with RQ1 RNase-Free DNase (Promega, Beijing, China) to remove any genomic DNA. The cDNA was synthesized from the treated RNA using a reverse transcriptase reagent kit (PrimeScript™ RT reagent Kit, TaKara, Beijing, China). RT-qPCR was carried out on an ABI 7500 Real-Time PCR system using SYBR® Premix Ex Taq™ (Tli RNaseH Plus) (TaKaRa). Each reaction was prepared in 20 μl containing 10 μl SYBR® Premix Ex Taq (2×), 10 μmol/l each of target PCR forward and reverse primers 0.8 μl, 0.4 μl ROX reference dye II, 2 μl of 1:5 diluted cDNA, and 6 μl ultra-pure water. Melting curve analysis was performed at the end of each PCR to confirm the specificity of the reaction. The PCR reaction program was 95 °C for 30 s, then 40 cycles of 95 °C for 5 s, and 60 °C for 32 s, followed by one cycle of 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s, and 60 °C for 15 s.
Table 1.
Primer list of selected DEGs and DEMs in RT-qPCR verification experiments
| Genes/miRNA | Forward/reverse primer (5′-3′) |
|---|---|
| Calcium-transporting ATPase (Ca-ATP) | F: 5′CGGTACTGGTGACAATAGAA3′ |
| R: 5′GAGGATGACAAAGTGGAGC3′ | |
| V-TYpe H+-transporting ATPase subunit alpha (H-ATP) | F: 5′AGAATATCCGAGTCCACG3′ |
| R: 5′ACAAGACCACATCCCAAC3′ | |
| Nicotinic acetylcholine receptor subunit alpha-9 (CHRNA9) | F: 5′ACTCCCTCTACAGATGCG3′ |
| R: 5′TGGTGCCACTAAGGTGAA3′ | |
| Sodium- and chloride-dependent GABA transporter 2 (SLC6a8) | F: 5′GACCAAAGTTACTGCTCCAC3′ |
| R: 5′TTACCGTTTACCCGTGCC3′ | |
| Sodium-/potassium-transporting ATPase subunit alpha (NKA) | F: 5′GTCCAACAGGGCATGAGT3′ |
| R: 5′TGAGTGGGTACATACGAAGT3′ | |
| Calcium-activated chloride channel regulator 4-like (CLCA4) | F: 5′CGTATGTTCGTATTTCTCCCTC3′ |
| R: 5′ATGGCTACCATCCGGTCT3′ | |
| Glycine receptor subunit alpha-4 | F: 5′AACCCGTGGTAGTGGTGG3′ |
| (Glra4a) | R: 5′TTCCCTGCTGGTCCTCAT3′ |
| Potassium channel tetramerization domain 6 | F: 5′TGTCGGCGGTAACTTCTA3′ |
| (KCTD6) | R: 5′TTGTGGGTACGATGAGCT3′ |
| NADH dehydrogenase (Ubiquinone) 1 ą Subcomplex, 1 (NDUFA1) | F: 5′TGCCTCTGAACGGGGAAAC3′ |
| R: 5′CCAGCCAGCATAGTACCTGTAA3′ | |
|
Aquaporin-9 (AQP9) |
5′AGGCACCGACTACAGAAC3′ |
| 5′CACCTCTTAATCCAGCAC3′ | |
| β-actin | F: 5′CGGCTGTGGTGGTGAAGGAGTA3′ |
| R: 5′TCATGGACTCAGGAGACGGTGTG3′ | |
| miR-2010 | GGGGTTACTGTTGATGTCAGCCCCTT |
| miR-2011 | GGGACCAAGGTGTGCTAGTGATGAC |
| miR-2013 | CGTGCAGCATGATGTAGTGGTGT |
| miR-10 | GCGAACCCTGTAGATCCGAATTTGTG |
| miR-92b-3p | TATTGCACTTGTCCCGGCCT |
|
miR-278-3p U6 |
GTCGGTGGGACTTTCGTTCGATT |
| ACGCAAATTCGTGAAGCGTT |
Six differentially expressed miRNAs (DERs) were selected for RT-qPCR analysis. Total RNAs extracted from the coelomic fluid were reverse transcribed using a stem-looped antisense primer. The resultant cDNA was submitted to amplification of mature miRNAs using a miRNA-specific primer and a universal primer. The U6 snRNA gene was employed as an endogenous control (Table 1). qPCR was conducted in 15-μl reaction volumes containing 300 nM of each primer and cDNA derived from 0.1 μg of total RNA. Cycling parameters were 95 °C for 1 min, followed by 40 cycles of 95 °C for 35 s and 60 °C for 30 s. All reactions were run in triplicate. The expression levels of miRNAs were normalized against U6 snRNA levels. All data were analyzed using the 2−ΔΔCt method. Significant differences were examined by paired t tests and a P value < 0.05 was considered to be statistically significant.
Results
Assessment of differential gene expression induced by salt stress in sea cucumbers
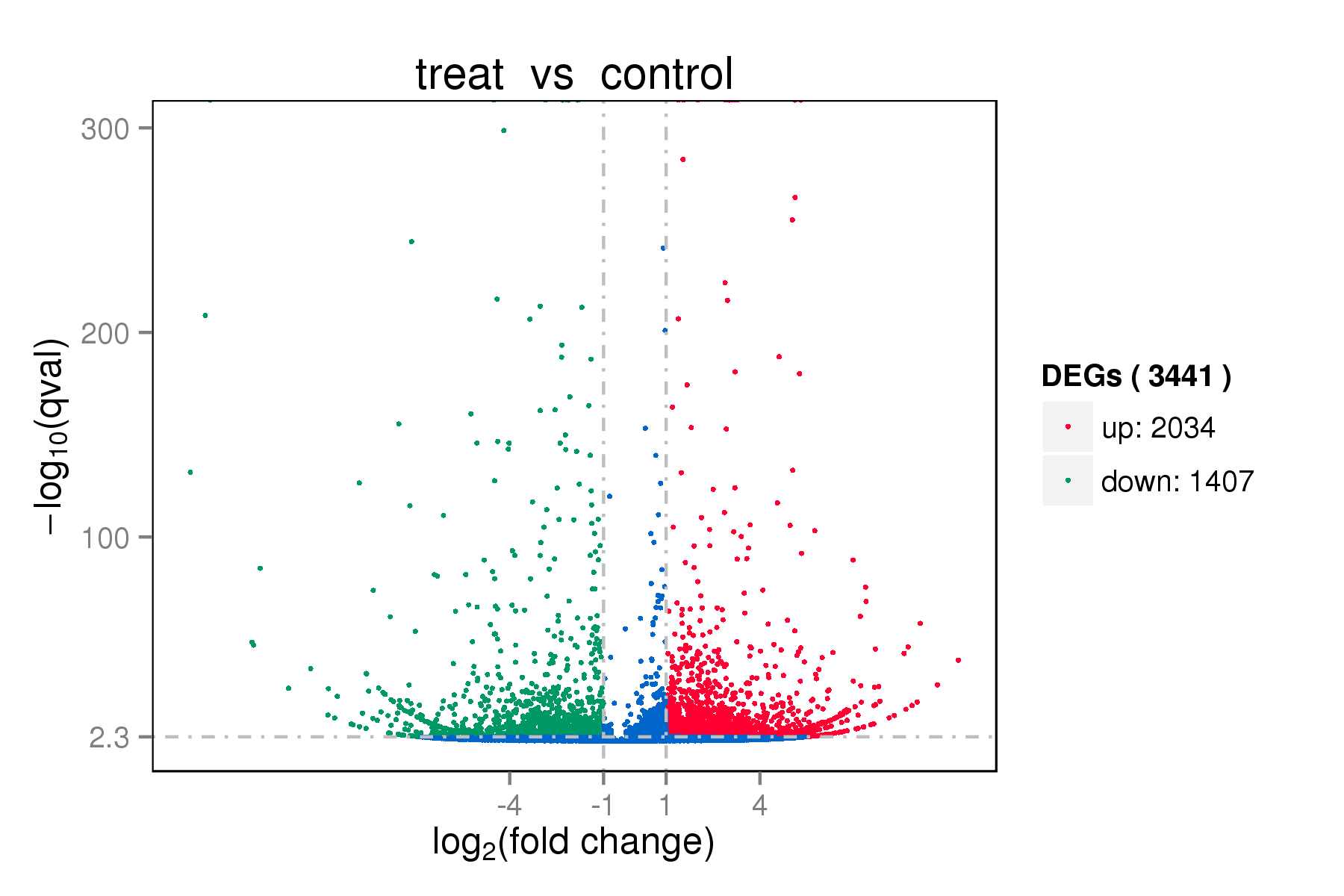
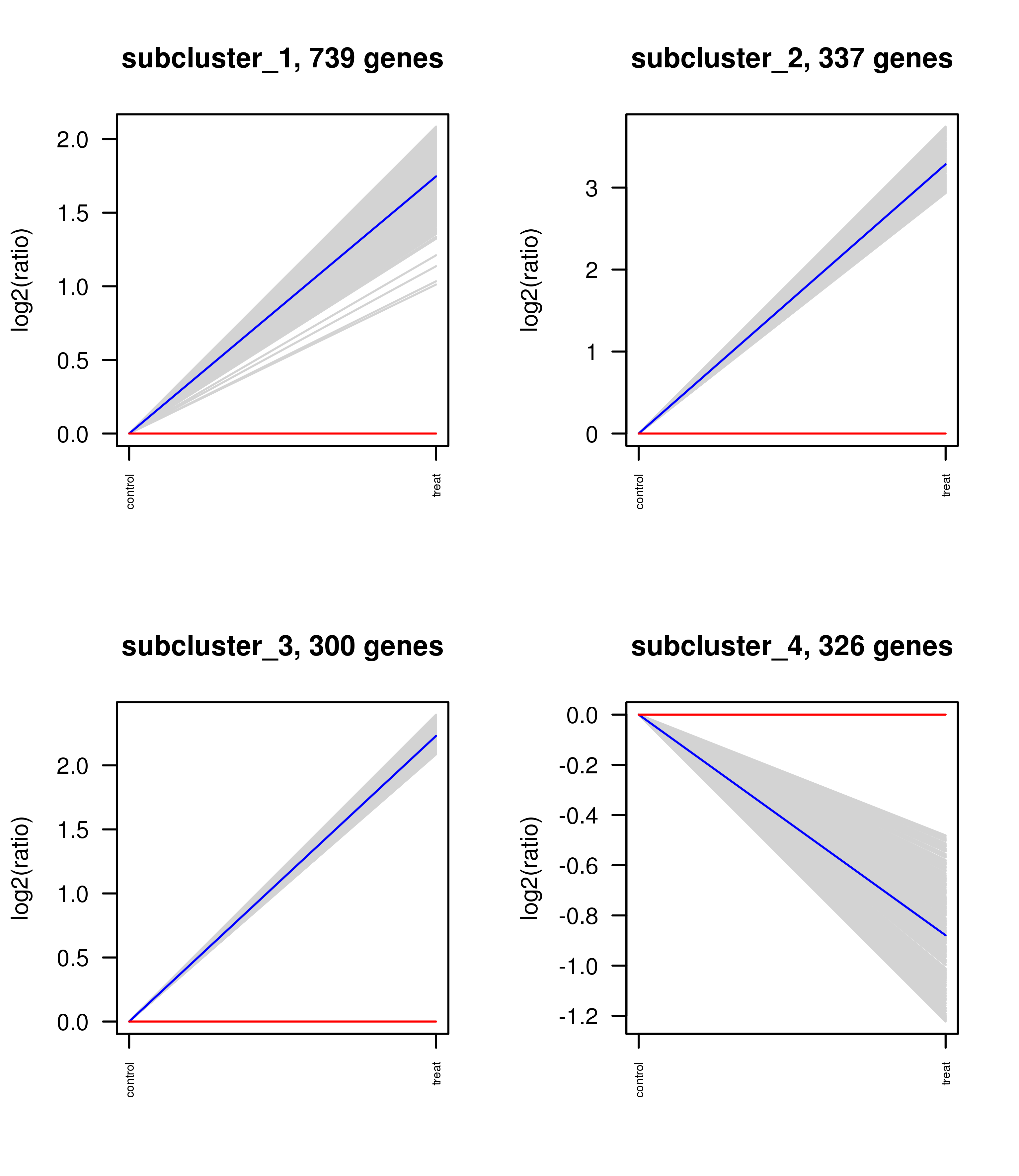
RNA Illumina sequencing identified 90,589,942-bp paired-end reads and 84,740,294 clean reads in sea cucumbers (Table 2). High-quality reads were mapped to sea cucumber databases (Table 3). A total of 34,837 unigenes were obtained, of which 3441 unigenes were differentially expressed after low-salinity challenge, including 2034 up-regulated and 1407 down-regulated unigenes that were differentially expressed between the CT and SS groups (Supplementary Information, Figs. S1 and S2 and Table S1). The top four sub-clusters are presented in Fig. S3 (Supplementary Information), of which sub-Clusters 1, 2, 3, and 4 were composed of 739, 739, 300, and 326 transcripts, respectively.
Table 2.
Summary of RNA Illumina sequencing results
| Sample name | Raw reads | Clean reads | Clean bases | Error rate (%) | Q20 (%) | Q30 (%) | GC content (%) |
|---|---|---|---|---|---|---|---|
| CTT | 44,010,986 | 40,396,842 | 6.06G | 0.01 | 97.23 | 93.35 | 40.07 |
| SST | 46,578,956 | 44,343,452 | 6.65G | 0.01 | 97.12 | 93.1 | 40.59 |
Table 3.
Summary of high-quality reads mapped to sea cucumbers databases
| Sample name | CTT | SST |
|---|---|---|
| Total reads | 40,396,842 | 44,343,452 |
| Total mapped | 23,471,851 (58.1%) | 28,080,373 (63.32%) |
| Multiple mapped | 1,665,590 (4.12%) | 1,933,177 (4.36%) |
| Uniquely mapped | 21,806,261 (53.98%) | 26,147,196 (58.97%) |
| Read-1 | 10,992,580 (27.21%) | 13,194,150 (29.75%) |
| Read-2 | 10,813,681 (26.77%) | 12,953,046 (29.21%) |
| Reads map to “+” | 10,880,625 (26.93%) | 13,052,960 (29.44%) |
| Reads map to “−“ | 10,925,636 (27.05%) | 13,094,236 (29.53%) |
| Non-splice reads | 14,164,807 (35.06%) | 16,348,015 (36.87%) |
| Splice reads | 7,641,454 (18.92%) | 9,799,181 (22.1%) |
GO enrichment of DEGs
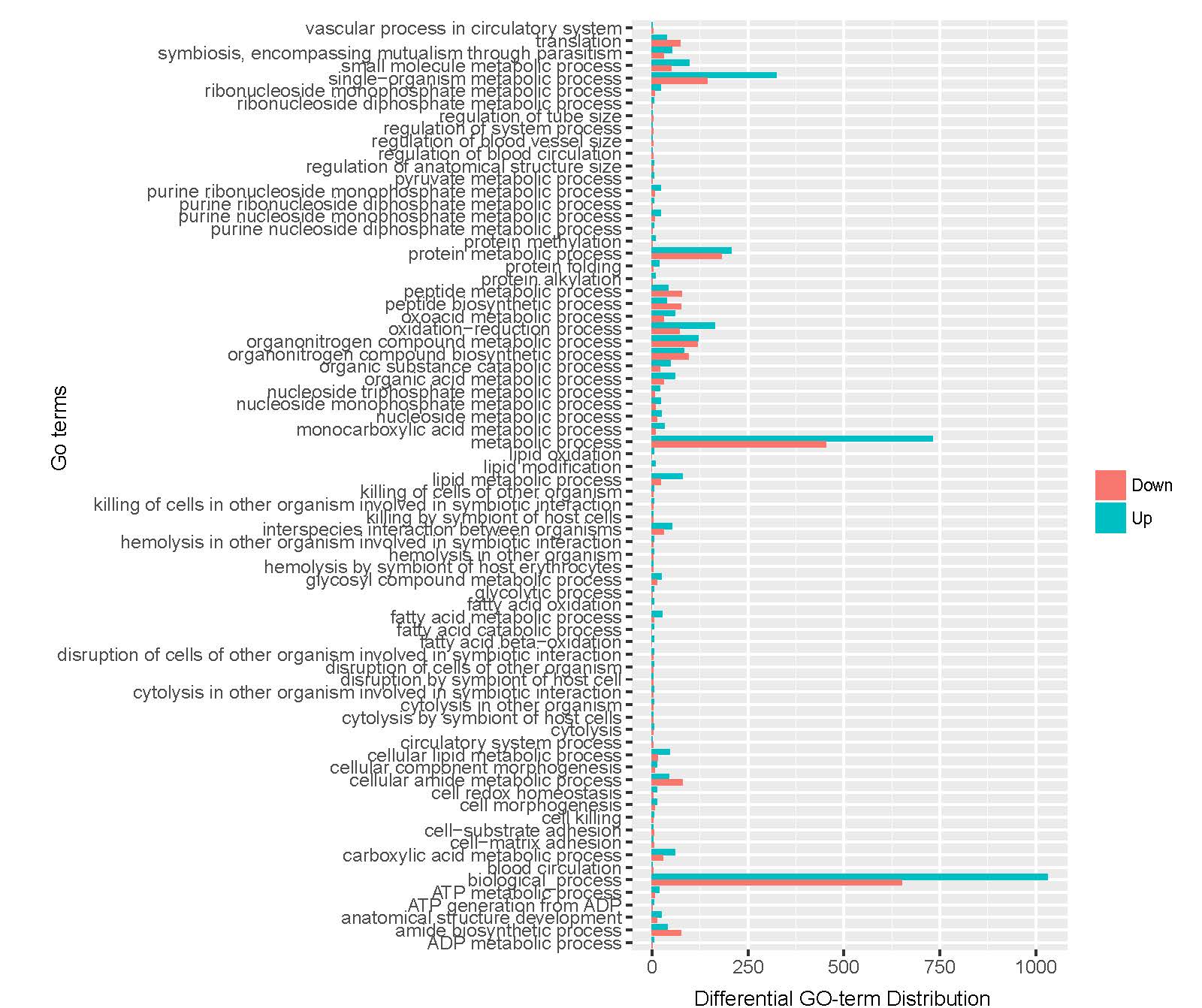
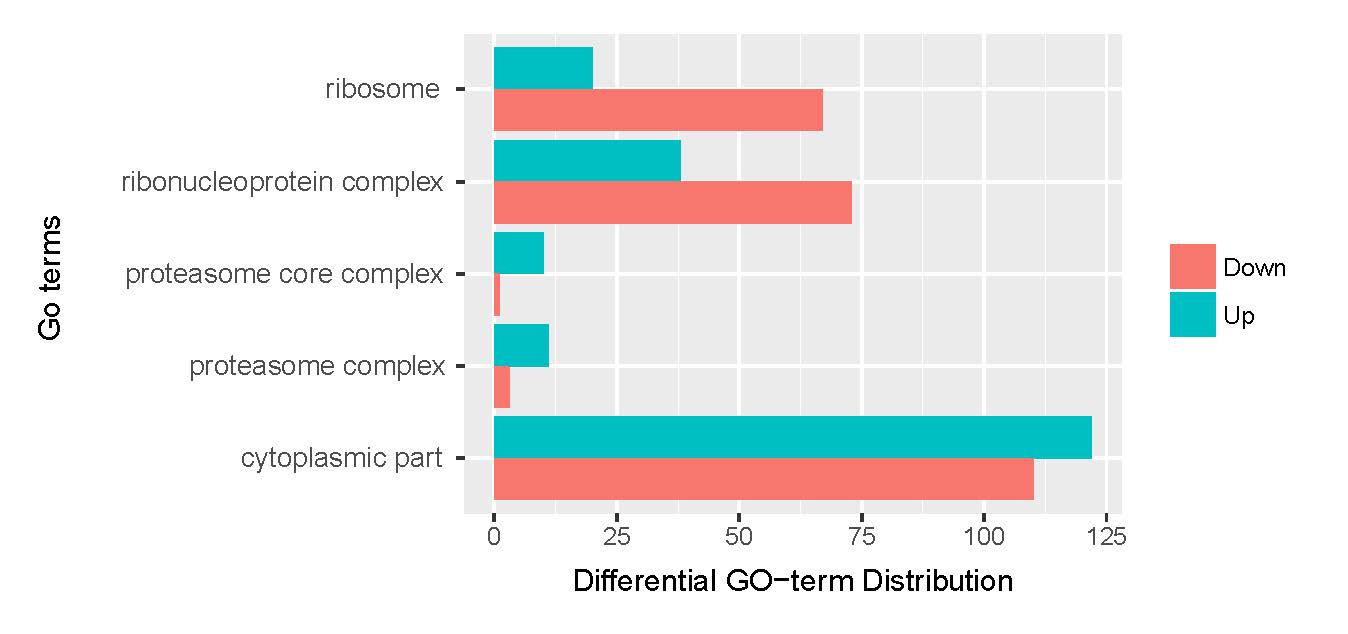
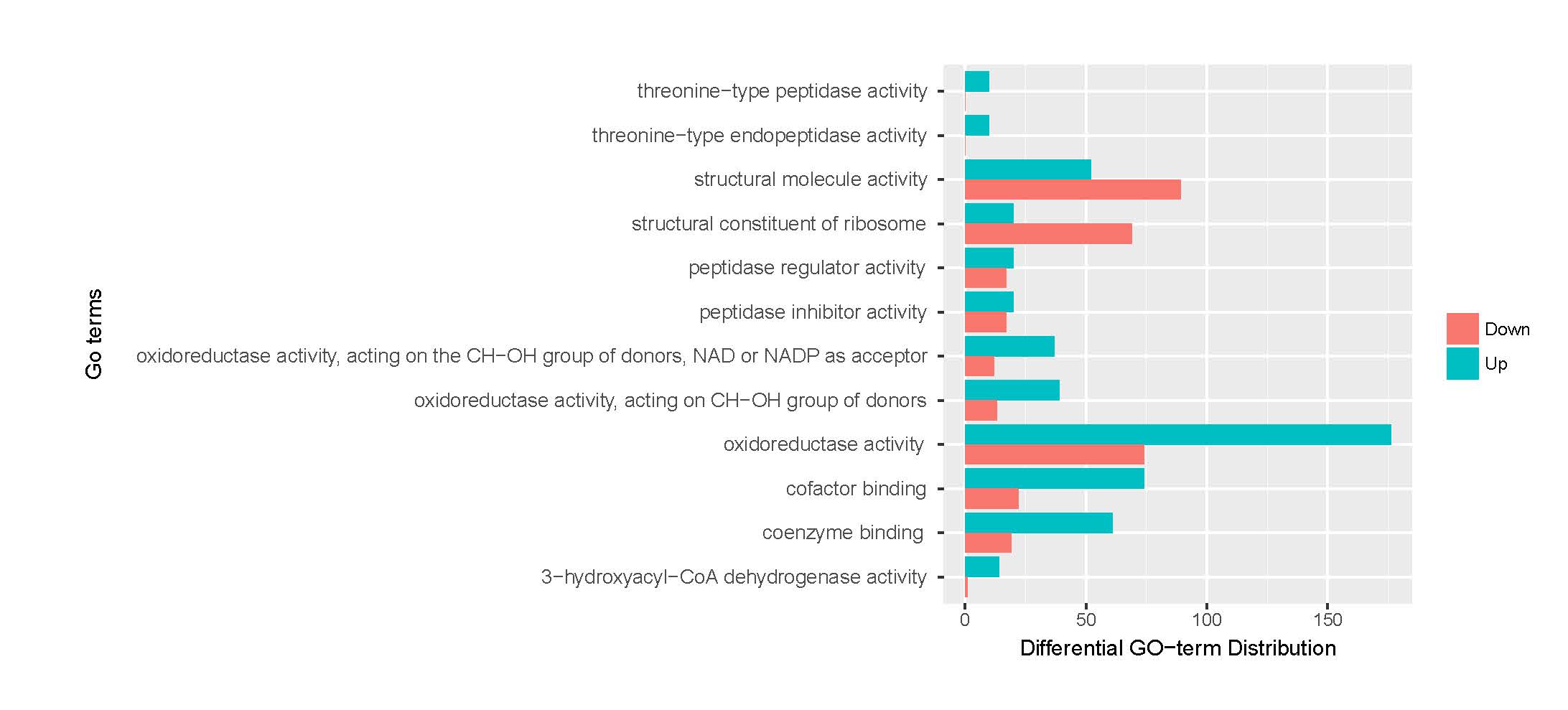
A total of 21,910 unigenes were annotated by GO analysis, with one or more GO terms. Of these, 17,968 unigenes were annotated with molecular functions, 14,777 with biological processes, and 9930 with cellular components. A total of 21,910 unigenes were enriched 3117 GO terms (Supplementary Information Fig. S4–6, Table S1). To retrieve a more accurate range of GO terms, we applied a term mode value of P < 0.05, resulting in 46 GO terms.
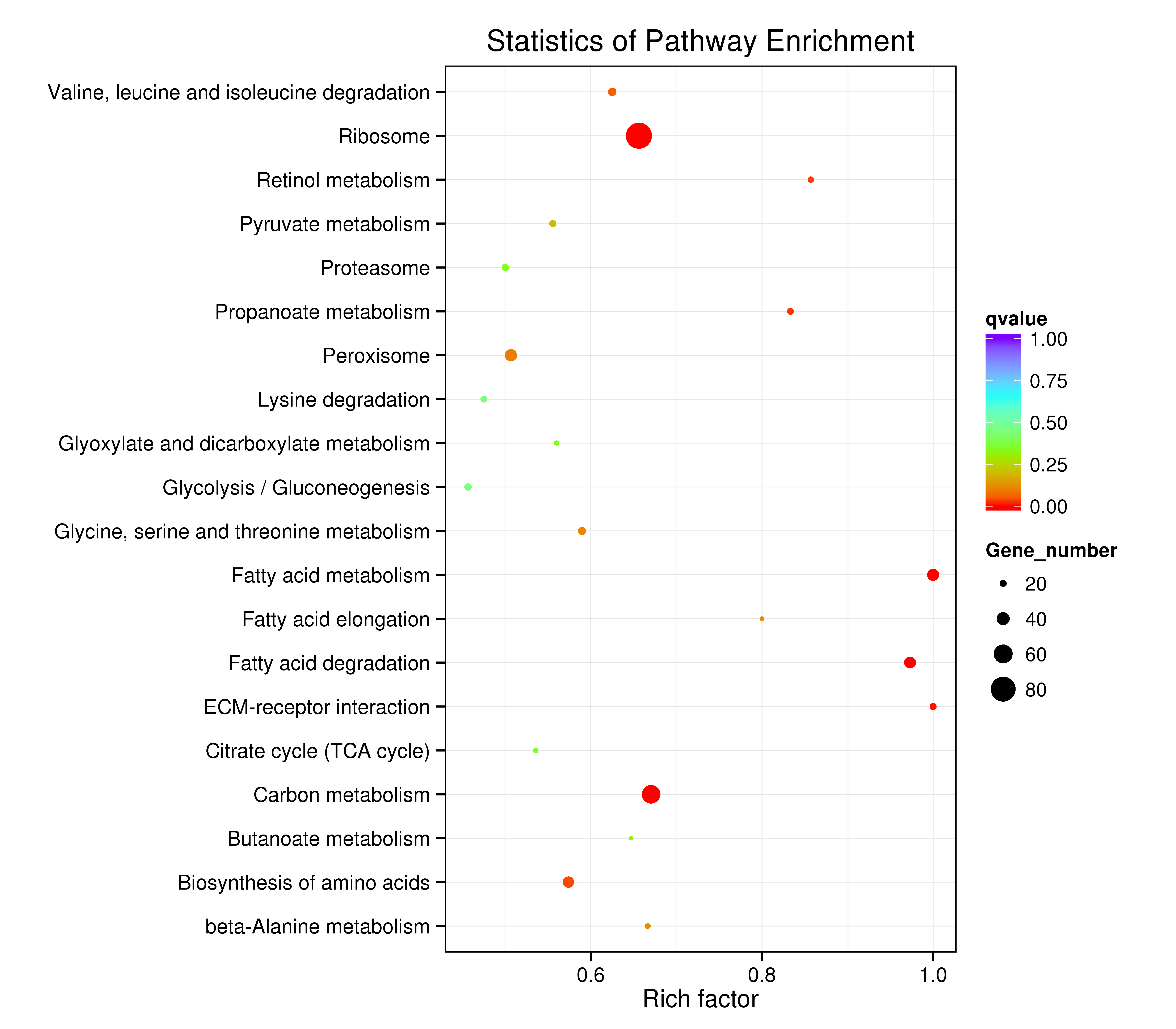
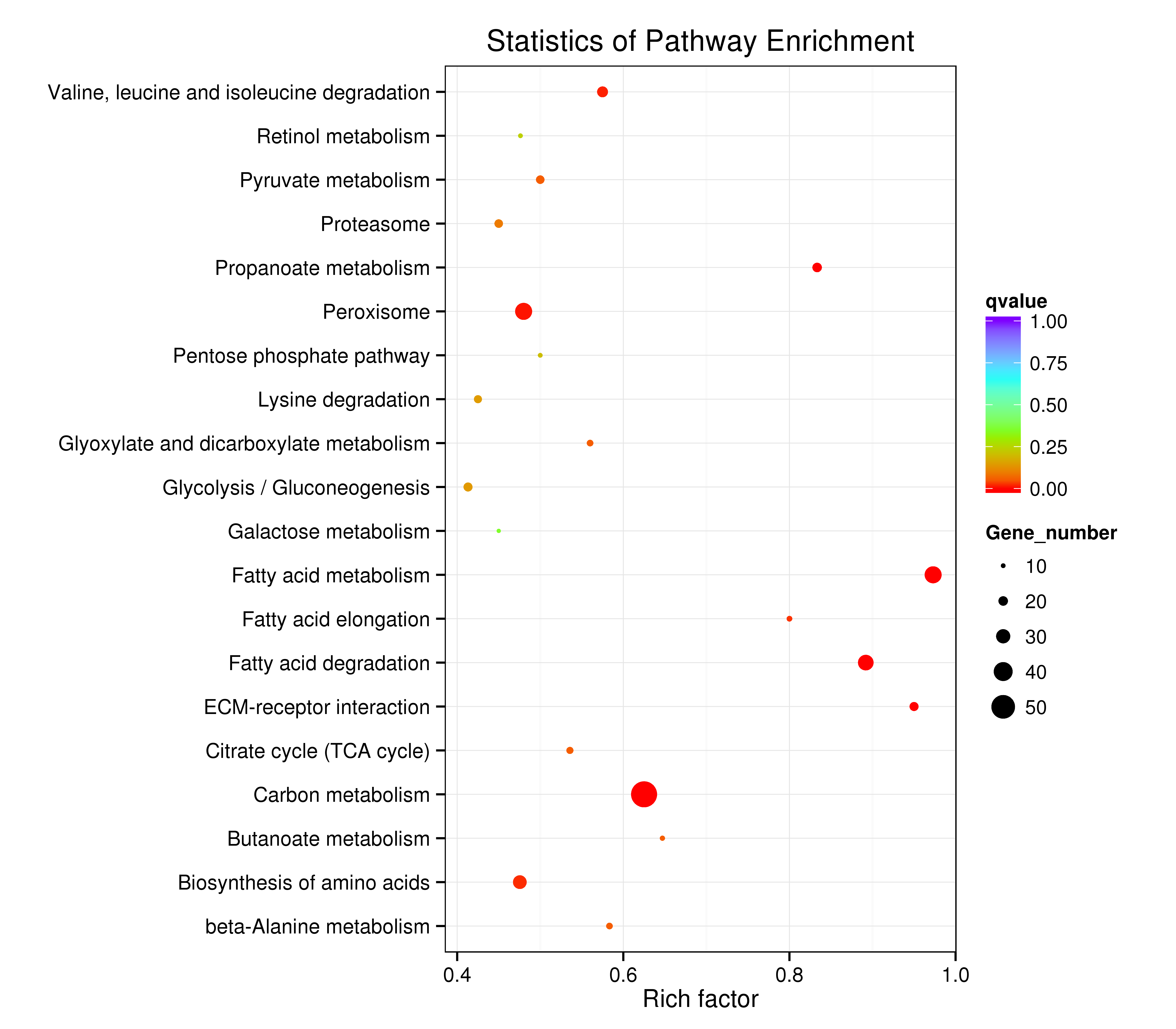
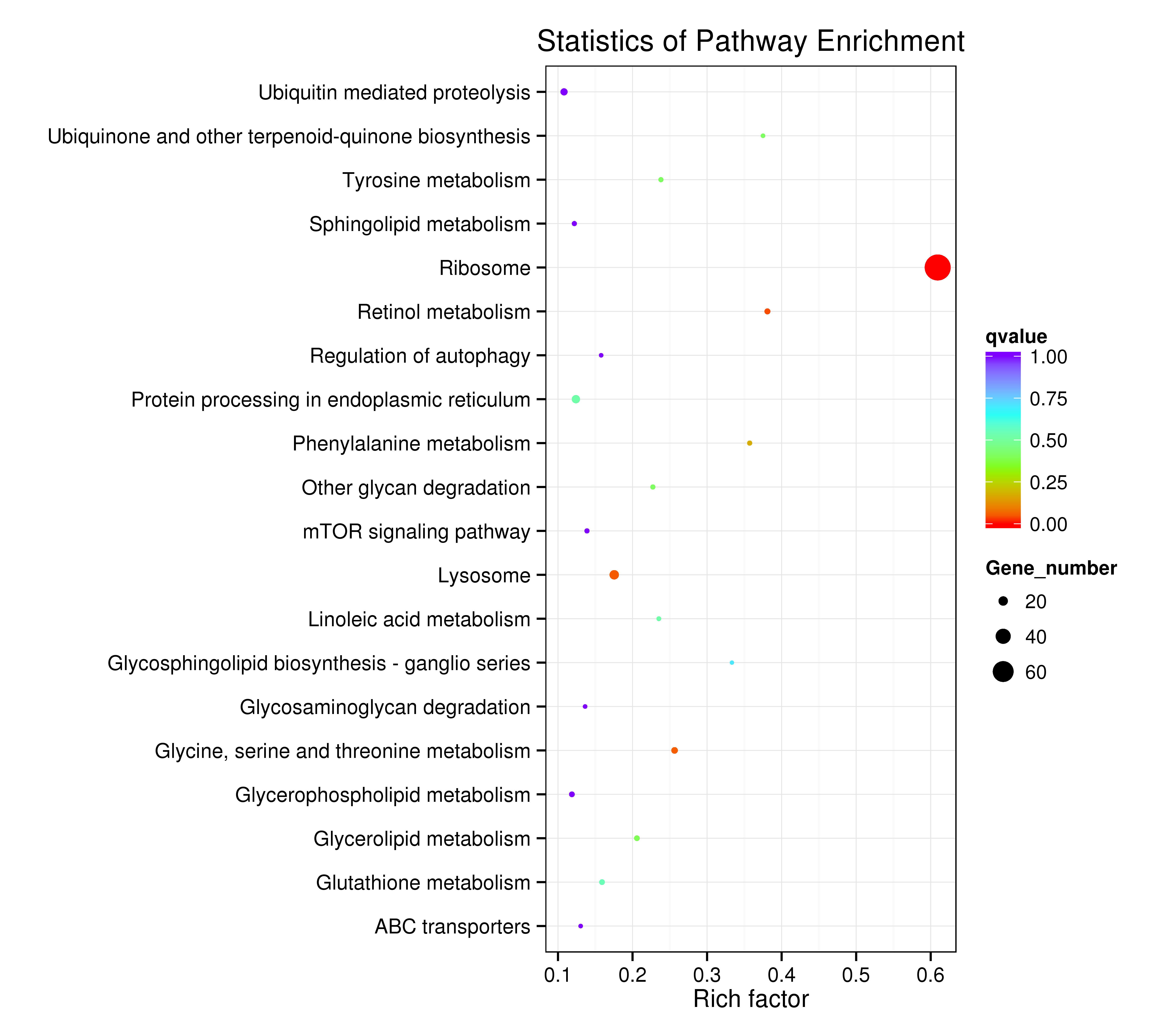
The assembled unigenes were also subjected to KEGG pathway analysis to identify the biochemical pathways operating in sea cucumbers. All differentially expressed unigenes were classified into 106 different pathways, of which the top 20 pathways were presented (Supplementary Information, Fig. S7). Up-regulated unigenes were mainly classified into 97 different pathways, of which the top 20 pathways were involved in carbohydrate metabolism, fatty acid metabolism, degradation, and elongation, amino acid metabolism, genetic information processing, metabolism of cofactors and vitamins, transport and catabolism, and environmental information processing (Supplementary Information, Fig. S8, Table 4). A total of 171 sequences were classified into carbohydrate metabolism, 83 into amino acid metabolism, and 81 sequences into lipid metabolism pathways. Thirty-six genes were involved in the peroxisome sub-pathway, which contributes to many crucial metabolic processes such as fatty acid oxidation, biosynthesis of ether lipids, and free radical detoxification, and 18 genes belonged to the proteasome sub-pathway, which is involved in signal transduction pathways, antigen processing for appropriate immune responses, stress signaling, inflammatory responses, and apoptosis.
Table 4.
All DEG regulated unigenes involved in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway for sea cucumbers
| KEGG pathway | Sub-pathway (Up unigene number) | Sub-pathway (Down unigene number) |
|---|---|---|
| Amino acid metabolism |
beta-Alanine metabolism (14) Biosynthesis of amino acids (29) Lysine degradation (17) Valine, leucine, and isoleucine degradation (23) |
Glycine, serine, and threonine metabolism (10) Phenylalanine metabolism (5) Tyrosine metabolism (5) Glutathione metabolism (7) |
| Carbohydrate metabolism |
Butanoate metabolism (11) Carbon metabolism (55) Citrate cycle (TCA cycle) (15) Glycolysis/gluconeogenesis (19) Glyoxylate and dicarboxylate metabolism(14) Pentose phosphate pathway (10) Propanoate metabolism (20) Pyruvate metabolism (18) Galactose metabolism (9) |
Other glycan degradation (5) Glycosphingolipid biosynthesis (2) Glycosaminoglycan degradation (3) |
| Lipid metabolism |
Fatty acid degradation (33) Fatty acid elongation (12) Fatty acid metabolism (36) |
Glycerolipid metabolism (7) Linoleic acid metabolism (4) Glycerophospholipid metabolism (7) Sphingolipid metabolism (5) |
| Genetic information processing | Proteasome (18) |
Ribosome (78) Protein processing in endoplasmic reticulum (16) Ubiquitin mediated proteolysis (12) |
| Metabolism of cofactors and vitamins | Retinol metabolism (10) |
Retinol metabolism (8) Ubiquinone and other terpenoid -quinone biosynthesis (3) |
| Transport and catabolism | Peroxisome (36) |
Lysosome (20) Regulation of autophagy (3) |
| Environmental information processing | ECM-receptor interaction (19) |
mTOR signaling pathway (5) ABC transporters (3) |
All differentially expressed down-regulated unigenes were classified into 80 different pathways. The top 20 pathways were mainly consistent with the up-regulated pathways (Table 4, Supplementary Information, Fig. S9). A total of 27 sequences were classified into amino acid metabolism, 23 sequences into lipid metabolism, and 23 into lysosome and regulation of autophagy cellular processes.
Analysis of small RNA library datasets and small RNA profiles
We constructed two small RNA libraries (CTR and SSR) from sea cucumbers using Illumina Solexa high-throughput sequencing technology to identify the miRNAs involved in salt stress. A total of 8,133,756 and 7,650,042 raw reads were obtained from the CTR and SSR libraries, respectively (Table 5), which produced 7,928,444 (99.04%) and 7,188,397 (95.96%) small RNA reads, respectively, after trimming the adapter sequences. Of these, 94.91% and 96.68% were mapped to the reference. There were 411 and 283 mapped novel miRNAs, 360 and 306 mapped known miRNAs, 48 and 41 mapped mature novel miRNAs, and 32 and 31 mapped mature known miRNAs in the CTR and SSR libraries, respectively.
Table 5.
Mapping statistics for sea cucumber miRNA types from Illumina sequencing reads
| Type | Control (CTR) | % of total | Salt stress (SSR) | % of total |
|---|---|---|---|---|
| Raw reads | 8,133,756 | 100.00% | 7,650,042 | 100.00% |
| Clean reads | 8,005,507 | 98.42% | 7,491,540 | 97.93% |
| Small RNA reads after length filter | 7,928,444 | 99.04% | 7,188,397 | 95.96% |
| Total mapped small RNA | 7,525,099 | 94.91% | 6,950,055 | 96.68% |
| Mapped novel miRNA | 411 | 0.0055 | 283 | 0.0041 |
| Mapped mature | 48 | 0.0006 | 41 | 0.0006 |
| Mapped hairpin | 56 | 0.0007 | 51 | 0.0007 |
| Mapped uniq sRNA | 411 | 0.0055 | 283 | 0.0041 |
| Mapped known miRNA | 360 | 0.0048 | 306 | 0.0044 |
| Mapped mature | 32 | 0.0004 | 31 | 0.0004 |
| Mapped hairpin | 32 | 0.0004 | 30 | 0.0004 |
Differential expression profiles of miRNAs
In the present study, we aimed to identify miRNAs involved in salinity acclimation in sea cucumbers. A total of 22 miRNAs were identified that showed differential expression during salinity acclimation, including seven miRNAs that were up-regulated and 15 that were down-regulated between the CT and SS groups (Table 6).
Table 6.
All differentially expressed miRNAs (DER) in CCT (control) vs SST (treat) of sea cucumbers during salinity acclimation
| sRNA | Treat | Control | log2.fold_change | P value | Up/down |
|---|---|---|---|---|---|
| miR-153-3p | 3759.01 | 10,661.84 | − 1.504 | 0 | Down |
| miR-2011 | 32,165.77 | 15,990.37 | 1.0083 | 0 | Up |
| miR-278-3p | 1820.50 | 350.83 | 2.3755 | 3.92E-259 | Up |
| miR-2008 | 285.54 | 1168.72 | − 2.0332 | 2.05E-114 | Down |
| miR-2005 | 607.87 | 156.20 | 1.9604 | 1.13E-70 | Up |
| miR-29b | 32.60 | 271.87 | − 3.0601 | 6.63E-45 | Down |
| miR-10 | 116.55 | 351.78 | − 1.5938 | 2.72E-25 | Down |
| miR-2007 | 46.25 | 180.10 | − 1.9611 | 3.41E-18 | Down |
| miR-92a | 79.94 | 179.33 | − 1.1656 | 1.25E-08 | Down |
| miR-2013 | 7.83 | 37.85 | − 2.2733 | 1.02E-05 | Down |
| miR-2010 | 2.19 | 15.10 | − 2.789 | 0.0016061 | Down |
| miR-124 | 11.29 | 33.27 | − 1.5589 | 0.0016931 | Down |
| novel-miR-3 | 148,776.03 | 297,808.92 | − 1.0012 | 0 | Down |
| novel-miR-4 | 17,134.09 | 81,004.73 | − 2.2411 | 0 | Down |
| novel-miR-14 | 1253.98 | 3614.17 | − 1.5272 | 9.92E-228 | Down |
| novel-miR-12 | 3191.03 | 1311.92 | 1.2823 | 3.83E-208 | Up |
| novel-miR-20 | 2108.23 | 629.19 | 1.7445 | 4.53E-208 | Up |
| novel-miR-16 | 909.61 | 2772.57 | − 1.6079 | 5.87E-190 | Down |
| novel-miR-15 | 572.72 | 2032.68 | − 1.8275 | 1.19E-169 | Down |
| novel-miR-91 | 378.78 | 147.60 | 1.3597 | 2.45E-28 | Up |
| novel-miR-27 | 270.97 | 626.51 | − 1.2092 | 3.97E-28 | Down |
| novel-miR-51 | 24.40 | 8.22 | 1.5696 | 0.002008 | Up |
The heatmap of 22 DERs in the CT and SS groups is shown in Fig. S10 (Supplementary Information). Four of the miRNAs clustered in salinity stress conditions, including novel-miR-3, novel-miR-4, miR-153-3p, and miR-2011, indicating that these miRNAs had similar expression profiles. Five novel miRNAs (novel-miRNAs 12, 14, 15, 16, and 20) clustered with a known miRNA-278-3p, suggesting that these novel miRNAs had similar expression profiles to the known miRNA-278-3p. This heatmap imaging of the DERs between salinity-stressed and control sea cucumbers contributes to our understanding of the functions of novel miRNAs in salt stress.
GO and pathway enrichment analysis for miRNA target genes
The physioregulatory properties of the significantly DERs (P < 0.05) in response to salinity stress were elucidated by predicting their target genes using miRanda. A total of 134 target genes of 22 DERs were found (Supplementary Information, Table S2) and were further categorized by GO annotation and KEGG pathway analysis. The 2-oxocarboxylic acid metabolism, nicotinate and nicotinamide metabolism, basal transcription factors, mucin-type O-glycan biosynthesis, fatty acid biosynthesis, spliceosome, and biosynthesis of amino acids pathways were enriched in KEGG (Supplementary Information, Table S3). We finally constructed a network of DERs and their target mRNAs using Cytoscape 3.3.0 (Fig. 1).
Fig. 1.

The network of DEM and their target mRNAs was constructed using Cytoscape 3.3.0. Yellow rectangular box miRNAs and white oval target genes
Validation of RNA-seq data by qPCR
After comprehensive RT-qPCR validation, the expression levels of nine selected DEGs and six DERs were compared with the expression profiles from RNA-seq analysis (Figs. 2 and 3). The expression trends of the DEGs and DERs predicted by RNA-seq were consistent with the results of RT-qPCR (Figs. 2 and 3). However, the magnitude of expression levels of some miRNA from RT-qPCR varied with the RNA-Seq results, and this may be attributed to differences in sensitivity and algorithms of the two techniques. A hypothetical model depicting the components involved in sea cucumber salt-responsive networks is shown in Fig. 4.
Fig. 2.
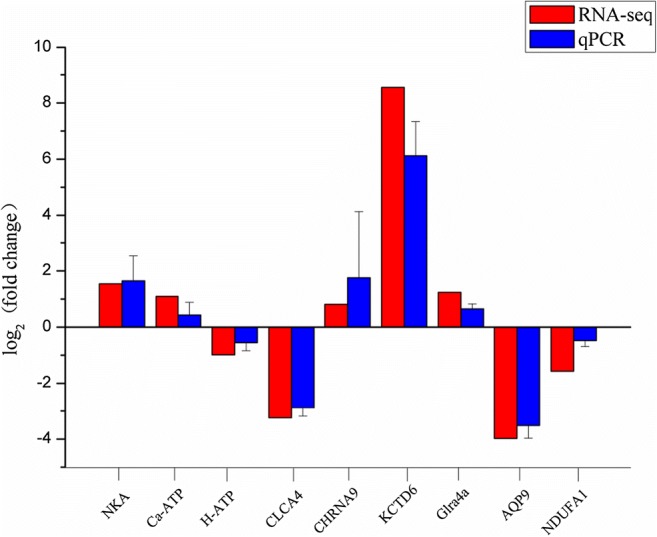
Analysis of selected nine differentially expressed genes (DEGs) in salinity-stressed and non-salinity-stressed sea cucumbers by sequencing and RT-qPCR methods. The results revealed that expression of these selected differentially expressed genes (DEGs) by RTqPCR were in agreement with the sequencing data
Fig. 3.
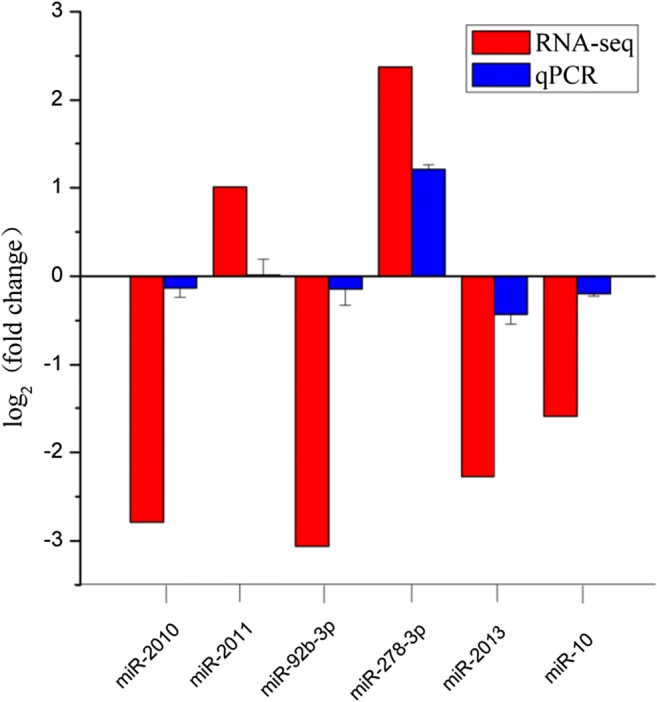
Analysis of selected six differentially expressed miRNA (DEMs) in salinity-stressed and non-salinity-stressed sea cucumbers by sequencing and RT-qPCR methods
Fig. 4.

A hypothetical model depicting the components involved in the sea cucumbers salt-responsive networks was established. The purple rectangular box of the figure represents the pathway involved after salt stress; the gray rectangular box represents the possible three salinity adaption modes, and the orange rectangular box represents the related miRNAs
Discussion
Salinity-stress responses can be mediated via multiple processes, including the induction of molecular chaperones, rapid clearance of damaged macromolecules, and activation of certain gene expression programs (Yang and Guo 2018). Numerous studies had investigated salinity acclimation in fish species at a large-scale data level (Yan et al. 2013), but previous studies of salinity acclimation in sea cucumbers are still based on few candidate genes and miRNAs (Zhang et al. 2017). However, the rapid development and decreasing cost of RNA sequencing technologies meant that we were able to identify 34,837 unigenes and 1360 miRNAs in sea cucumbers.
Salinity can affect aquatic animals in ecological systems via complex biological effects. However, the mechanisms by which sea cucumbers adapt to changes in salinity remain unclear. Some research indicated that sea cucumbers were stenohaline because they lacked an obvious osmoregulatory organ, but other studies (Diehl 1986) suggested that most echinoderms may maintain some ion concentrations at slightly different levels from environmental seawater. Vidolin et al. (2002) reported that the gray sea cucumber (Holothuria grisea) could temporally regulate the osmotic pressure of its coelomic fluids. Sea cucumbers remain in continuous contact with environmental water and are thus constantly challenged to adapt their plasma ion balance within the defined range necessary for proper cell function. Furthermore, acute changes in salinity might induce the expression of a series of genes to initiate various physiological functions.
DEGs identified in this study were mainly enriched in pathways related to amino acid metabolism, carbohydrate metabolism, lipid metabolism, genetic information processing, metabolism of cofactors and vitamins, transport and catabolism, and environmental information processing, suggesting that these processes are involved in regulating the response to low-salinity stress in sea cucumbers. These results in sea cucumbers were similar to those seen in oysters, Zhikong scallop, Manila clam, and tilapia (Zhang et al. 2012; Shi et al. 2013; Milan et al. 2011; Zhang et al. 2014a, b, c; Nie et al. 2017). The most representative KEGG pathways affected by salinity stress also included lipid, glycerophospholipid, and glycerolipid metabolism, vitamin digestion and absorption, and fat digestion (Zhang et al. 2017), also implicating these processes in the adaptation to low-salinity stress in sea cucumbers.
Aquaporin-9 (AQP9), as a target gene of miRNA-2005, was induced by salinity stress and is involved in transport and catabolism pathways. AQP9 expression, as a coordinator of water and ion transport, was significantly down-regulated, consistent with the current transcriptomic results. Down-regulation of AQP9 has previously been recorded in the pufferfish Takifugu obscurus (Jeong et al. 2014), Salmo salar (Tipsmark et al. 2010), A. japonicus (Tse et al. 2006), and Oreochromis mossambicus (Breves et al. 2010). Results in oysters indicated the aquaporins were important salt-responsive effectors in establishing water homeostasis (Berger and Kharazova 1997). Transcriptome analysis revealed that long-term stress reduced the activity of water channels to protect against water currents and swelling and shrinkage of gill cells in oysters (Meng et al. 2013). The results indicated that AQP9 might form a broad-specificity channel able to mediate the passage of a wide variety of non-charged solutes to achieve salinity adaption in sea cucumbers. Reduced expression of AQP genes could be explained by increased protein phosphorylation mediated by stress (Tournaire-Roux et al. 2003; Santoni et al. 2003).
Ion channels (including Na+, K+, Ca2+, and Cl− channels) and osmolyte transporters, which control ion and osmolyte balances, were activated by environmental stimulation. In sea cucumbers, NKA, Ca2+-transporting ATPase (Ca-ATP), V-type H+-transporting ATPase subunit alpha (H-ATP), nicotinic acetylcholine receptor subunit alpha-9 (CHRNA9), glycine receptor subunit alpha-4 (Glra4a), potassium channel tetramerization domain-containing 6 (KCTD6), and chloride channel accessory 4 (CLCA4) were all induced under salinity stress and were all involved in transport and catabolism pathways. Hypo-osmoregulation usually involves Cl−- and Na+-excretion-related genes, such as NKA, NKCC, CFTR anion channels, and claudin transmembrane proteins, which provide a driving force for ion transport systems in osmoregulatory epithelia (Evans et al. 2005; Tipsmark et al. 2008;Choe et al. 2007 Evans and Claiborne 2006). NKA has been shown to play an important role during salinity adaptation in many species, including Sarotherodon melanotheron (Ouattara et al. 2009), O. mossambicus (Fiess et al. 2007), and Cyprinus carpio (Salati et al. 2011). The abundance of NKA in sea cucumbers was consistent with the above results, suggesting that NKA also plays an important role in salt stress in sea cucumbers. Rengmark et al. (2007) indicated that the Ca2+-transporting plasma membrane ATPase was involved in the response to salinity exposure in Nile tilapia. Ca-ATP was similarly up-regulated in oysters under salt stress (Meng et al. 2013). Ca-ATP was also up-regulated in sea cucumbers under salinity stress, indicating that it was also an important regulatory molecule for maintaining ion balance. In the current study, H+-ATPase was down-regulated by salinity stress in sea cucumbers, suggesting that the osmolality tolerance of sea cucumbers may be influenced by acidification conditions, proton transport, and ATP hydrolysis. H+-ATPase supported the breakdown of macromolecules by up to 40 types of acid hydrolases (Beyenbach and Wieczorek 2006). Moreover, V-type H+-ATPase influences CO3− and Cl− exchange and aids the uptake of cations such as Na+, Ca2+, and Cd2+ via H+-driven antiport by regulating cytosolic pH (Dietz et al. 2001). V-type H+-ATPase provided H+ required by neurotransmitter transport via vesicular solute-linked carrier (SLC) family (Hediger et al. 2004; Parsons et al. 2000). Overall, H+-ATPase appeared to play a vital role in dealing with salinity stress in sea cucumbers. NKA, Ca-ATP, H-ATP, Glra4a, KCTD6, CLCA4, and CHRNA9 genes were either up- or down-regulated, supporting the participation of this transport and catabolism pathway in salinity adaptation in sea cucumbers. These ion transport genes play important roles in salinity adaptation and are involved in complex networks with other factors, such as the SLC family, pH, protein hydrolysis, neurotransmitters and their receptors, free amino acids (glycine, taurine, and beta-alanine), glucose and other sugars, bile salts and organic acids, metal ions, and amine compounds.
Free amino acids, such as glycine, taurine, and alanine, can activate neurotransmitter-gated chloride channels and neurotransmitter membrane receptors (Glra4a, CHRNA9, CLCA4) (Lustig et al. 2002). Moreover, lysine is metabolized in eukaryotes to yield acetyl-CoA via acetylation (Starai et al. 2002; Schwer et al. 2006). Acetyl-CoA participates in osmoregulation as an intermediate metabolite commonly produced from amino acid, lipid, and carbohydrate metabolism. In our study, gene expression of 3-hydroxyacyl-CoA dehydrogenase, which can produce acetyl-CoA, was significantly up-regulated by salinity stress, resulting in more acetyl-CoA entering the citrate cycle for energy production. Similar results were found in Ruditapes philippinarum (Nie et al. 2017), C. gigas (Meng et al. 2013), Oreochromis niloticus (Yan et al. 2012a, b, c), and the Pacific white shrimp (Chen et al. 2015). Ubiquitin-mediated proteolysis and ubiquinone and other terpenoid–quinone biosynthesis contribute to protein transport and turnover.
Many DEGs were enriched in carbohydrate metabolism pathways, suggesting that stored energy is important for stress tolerance and that the metabolic response to salinity stress in sea cucumbers depends on carbohydrate catabolism. NDUFA1 encodes an essential component of complex I of the respiratory chain, which transfers electrons from NADH to ubiquinone for ATP synthesis by chemiosmotic coupling. In this study, NDUFA1 was down-regulated under salinity stress in sea cucumbers, indicating that energy reserves were reduced in sea cucumbers subjected to salinity stress, as also seen in oysters under stressors such as virus infections and cadmium (Tamayo et al. 2014; Ivanina et al. 2011). ATPGD1 is involved in metabolic processes and catalyzes the degradation of beta-alanine in oysters according to KEGG, to maintain osmotic equilibrium under hypo-osmotic stress. Genes related to the respiratory chain complex were also regulated, including those encoding 3-demethylubiquinone-93-methyltransferase to produce ubiquinone under hyposaline conditions (Zhang et al. 2014a, b, c). Other carbohydrate metabolism pathways, including phosphoenolpyruvate carboxykinase and adenosine kinase (Chaney and Gracey 2011) and malate dehydrogenase and fructose 1,6-biphosphatase (Corporeau et al. 2014) pathways, were also shown to be affected by stress. Notably, carbohydrate metabolism was the main and direct energy source utilized to deal with the energy demands associated with salinity stress.
Lipid metabolism was shown to be enriched in sea cucumbers under low-salinity stress by KEGG pathway analysis in this study, consistent with previous research (Zhang et al. 2018). The 18 top KEGG pathways involved in salinity stress in L. vannamei were significantly involved in physiological responses, particularly lipid metabolism, including fatty acid biosynthesis, arachidonic acid metabolism, and glycosphingolipid and glycosaminoglycan metabolism (Chen et al. 2015). The authors suggested that lipids or fatty acids might reduce osmotic stress in L. vannamei by providing additional energy or changing the membrane structure to allow osmoregulation in relevant organs. These pathways were also identified in Eriocheir sinensis under osmotic stress (Li et al. 2014). If carbohydrates are scarce, energy must be obtained from the breakdown of fatty acids from body tissue instead of from glucose (Ranallo and Rhodes 1998; Bradbury 2006), and it is reasonable to assume that sea cucumbers may use long-chain fatty acids for energy supplementation (Palacios et al. 2004a, b). The up-regulated long-chain-specific acyl-CoA dehydrogenase and phytanoyl-CoA dioxygenase were both involved in fatty acid oxidation, and the final product of fatty acid β-oxidation is acetyl-CoA. Moreover, acetyl-CoA can be completely oxidized into carbon dioxide and water via the tricarboxylic acid cycle to generate energy. These results implied that lipid metabolites are important substances for energy storage and supply in sea cucumbers under salinity stress and may also act as regulators of many ion channels.
In this study, seven miRNAs were up-regulated and 15 were down-regulated in sea cucumbers exposed to salinity stress, suggesting that miRNAs are required to deal with environmental changes in salinity by sea cucumbers. Previous studies found that miRNAs were involved in the regulation of salinity in other species. miRNA-429 expression decreased under osmotic stress and miRNA-30c inhibition led to a loss of osmotic stress tolerance in tilapia (Yan et al. 2012a, b). The miRNA-8 family binds to the Na+/H+ exchanger regulatory factor 1 in zebrafish, suggesting a functional role for this pathway in ionocyte-dependent osmoregulation (Flynt et al. 2009). A series of studies in Nile tilapia showed that osmotic stimuli regulated gill and kidney miRNAs and were functionally involved in mediating molecular and organismal level responses to maintain homeostasis under osmotic stress (Yan et al. 2012a, b; Zhao et al. 2016).
In this study, miR-10 expression was reduced under salinity stress, indicating its involvement in adaptation to these conditions. TBC1D5 (TBC1 domain family member 5), which is involved in the process of glucose metabolism, was a predicted target gene of miR-10. Some reports demonstrated glucose achieved by trafficking of the key nutrient transporter, GLUT1/SLC2A1 in response to diverse metabolic stresses. TBC1D5 facilitated retromer-dependent GLUT1 trafficking by shuttling during metabolic stress (Roy et al. 2017). We therefore inferred that down-regulation of miR-10 and consequent up-regulation of its target gene TBC1D5 may help to meet the need for glucose to generate energy under low-salt stress in sea cucumbers. Further investigations are required to explore the molecular mechanisms responsible for the regulation of the target gene TBC1D5 by miR-10 during osmotic adaptation in sea cucumbers.
miR-2008 and miR-92a were also down-regulated under low-salinity stress. The predicted target genes of miR-2008 and miR-92a were pleckstrin homology domain-containing family A member 3 (PLEKHA3/FAPP1) and phospholipid scramblase 2 (PLSCR2), respectively. PLSCR2 mediated accelerated ATP-independent bidirectional trans-bilayer migration of phospholipids upon binding of calcium ions and was involved in lipid metabolism (Greenberg and McDaniel 2002; Wiedmer et al. 2004). PLEKHA3/FAPP1 bound to phosphatidylinositol 4-phosphate and was involved in cholesterol and sphingolipid transport/transport from Golgi and endoplasmic reticulum to the apical membrane (Godi et al. 2004). These results suggest that miR-2008 and miR-92a may be involved in salinity adaptation in sea cucumbers by targeting PLEKHA3/FAPP1 and PLSCR2. Moreover, these results were consistent with the results of transcriptome sequencing, confirming the participation of lipid metabolism in response to salinity stress in sea cucumbers.
miR-2007 was down-regulated by salinity stress. Its predicted target gene was solute carrier family 22 member 15 (SLC22a15), which acts as an organic ion transporter to transport various medically and physiologically important compounds, including pharmaceuticals, toxins, hormones, neurotransmitters, and cellular metabolites (Mihaljevic et al. 2016; Koepsell 2013). SLC22a15 is also involved in the transport of glucose and other sugars, bile salts and organic acids, metal ions, and amine compounds. The current results indicated that miR-2007 and its target gene SLC22a15 participated in salinity adaptation in sea cucumbers by transporting glucose and other sugars to fulfill the energy requirements imposed by salinity stress. The predicted target genes of miR-2013 were transient receptor potential cation channel subfamily A member 1 (TRPA1), neuronal acetylcholine receptor subunit alpha-7 (CHRNA7), and Rho guanine nucleotide exchange factor 3 (ARHGEF3). These three target genes were involved in the processes of lipid metabolism and transport. TRPA1 is a member of the transient receptor potential superfamily of ion channels gated by temperature and functions as a temperature transducer and is involved in binding the inositol polyphosphates that are required for optimal channel activity (Paulsen et al. 2015). CHRNA7 binds to acetylcholine resulting in conformational changes leading to the opening of ion-conducting channels across the plasma membrane (Lansdell et al. 2005). ARHGEF3 accelerate the GTPase activity of Rho GTPases by catalyzing the release of bound GDP. Overall, these results indicated that low-molecular-weight crystalline materials, such as SLC22a15, TRPA1, CHRNA7, PLSCR2, and PLEKHA3, participated in the adaptation of sea cucumbers to environmental salinity changes under the miRNA regulation, and that carbohydrate metabolism-related factors, such as ARHGEF3 and TBC1D5, were involved in providing energy for the adaptive changes. The pathways enriched for the miRNA-predicted target genes were consistent with the pathways identified by transcriptome analysis.
Based on the above experimental results, we established a hypothetical model of the components involved in salt-responsive networks in sea cucumbers. Pathways included environmental information processing, including extracellular matrix–receptor interaction (19 genes up-regulated), mTOR signaling pathway (5 genes down-regulated), and ABC transporters (3 genes down-regulated), in accordance with previous studies by Zhao et al. (2016). Environmental signals could thus stimulate sea cucumbers to adapt to salinity stress through amino acid metabolism, ion channels, transporters, and aquaporins. Amino acid metabolites participate in the process of colloidal osmotic regulation through free amino acids, while ion channels and transporters are responsible for crystal osmotic regulation, and aquaporin is responsible for regulating water channels. Acetyl-coA, as a metabolite from amino acid, lipid, and carbohydrate metabolism, and hormones and neurotransmitters are also involved in this salinity adaptation process. Sea cucumbers thus adapt to salinity stress by cooperation among these three aspects, together with miRNA regulation.
Conclusions
We have constructed a comprehensive mRNA and miRNA dataset for sea cucumbers. Based on these results, we inferred that sea cucumbers adapt to salinity stress via changes in amino acid metabolism, ion channels, transporters, and aquaporins following stimulation by environmental signals. This process requires energy from carbohydrate and fatty acid metabolism. Moreover, the salinity adaptation process involves regulation by miRNAs. The information on enriched genes and miRNA expression profile data generated in this study will support further environmental, molecular, and physiological studies of sea cucumbers. The results thus provide valuable genomic resources that extend our understanding of the unique biological characteristics of this economically important species under conditions of salinity stress.
Electronic supplementary material
{kind=link}
(PNG 24 kb)
{kind=link}
(PNG 78 kb)
{kind=link}
(PNG 310 kb)
{kind=link}
(JPG 259 kb)
{kind=link}
(JPG 49 kb)
{kind=link}
(JPG 102 kb)
{kind=link}
(PNG 595 kb)
{kind=link}
(PNG 596 kb)
{kind=link}
(PNG 639 kb)
{kind=link}
(PNG 162 kb)
(XLSX 388 kb)
(XLSX 517 kb)
(XLSX 16.4 kb)
Funding information
The authors are thankful to the National Key Research and Development Program of China (2018YFD0901601), Department of Education of Liaoning Province (L201620) for their financial support.
Compliance with ethical standards
All the experimental procedures involving animals were approved by the Institutional Animal Ethics Committee (IAEC).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Asha PS, Muthiah P. Effects of temperature, salinity and pH on larval growth, survival and development of the sea cucumber Holothuria spinifera Theel. Aquaculture. 2005;250(3–4):823–829. [Google Scholar]
- Berger VJ, Kharazova AD. Mechanisms of salinity adaptations in marine molluscs. In interactions and adaptation strategies of marine organisms. Dordrecht: Springer; 1997. pp. 115–126. [Google Scholar]
- Beyenbach KW, Wieczorek H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol. 2006;209(4):577–589. doi: 10.1242/jeb.02014. [DOI] [PubMed] [Google Scholar]
- Bradbury MW. Lipid metabolism and liver inflammation. I Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol-Gastr L. 2006;290(2):194–198. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- Breves JP, Fox BK, Pierce AL, Hirano T, Grau EG. Gene expression of growth hormone family and glucocorticoid receptors, osmosensors, and ion transporters in the gill during seawater acclimation of Mozambique tilapia, Oreochromis mossambicus. J Exp Zool A. 2010;313(7):432–441. doi: 10.1002/jez.613. [DOI] [PubMed] [Google Scholar]
- Chaney ML, Gracey AY. Mass mortality in Pacific oysters is associated with a specific gene expression signature. Mol Ecol. 2011;20(14):2942–2954. doi: 10.1111/j.1365-294X.2011.05152.x. [DOI] [PubMed] [Google Scholar]
- Chen K, Li E, Li T, Xu C, Wang X, Lin H, Qin JG, Chen LQ. Transcriptome and molecular pathway analysis of the hepatopancreas in the Pacific white shrimp Litopenaeus vannamei under chronic low-salinity stress. PLoS One. 2015;10:e0131503. doi: 10.1371/journal.pone.0131503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow SC, Ching LY, Wong AMF, Wong CK. Cloning and regulation of expression of the Na+–Cl––taurine transporter in gill cells of freshwater Japanese eels. J Exp Biol. 2009;212(20):3205–3210. doi: 10.1242/jeb.031302. [DOI] [PubMed] [Google Scholar]
- Corporeau C, Tamayo D, Pernet F, Quéré C, Madec S. Proteomic signatures of the oyster metabolic response to herpesvirus OsHV-1μVar infection. J Proteome. 2014;109:176–187. doi: 10.1016/j.jprot.2014.06.030. [DOI] [PubMed] [Google Scholar]
- DeZoysa M, Whang I, Lee Y, Lee S, Lee JS, Lee J. Transcriptional analysis of antioxidant and immune defense genes in disk abalone (Haliotis discus discus) during thermal, low-salinity and hypoxic stress. Comp Biochem Physiol B. 2009;154(4):387–395. doi: 10.1016/j.cbpb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Diehl WJ. Osmoregulation in echinoderms. Comp Biochem Phys A. 1986;84(2):199–205. [Google Scholar]
- Dietz KJ, Tavakoli N, Kluge C, Mimura T, Sharma SS, Harris GC, Chardonnens AN, Golldack D. Significance of the V-type ATPase for the adaptation to stressful growth conditions and its regulation on the molecular and biochemical level. J Exp Bot. 2001;52(363):1969–1980. doi: 10.1093/jexbot/52.363.1969. [DOI] [PubMed] [Google Scholar]
- Dong Y, Dong S, Meng X. Effects of thermal and osmotic stress on growth, osmoregulation and Hsp70 in sea cucumber (Apostichopus japonicus Selenka) Aquaculture. 2008;276:179–186. [Google Scholar]
- Evans DH, Piermarini PM, Choe KP. The multifunctional fish gill: dominant site of gas exchange osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol Rev. 2005;85(1):97–177. doi: 10.1152/physrev.00050.2003. [DOI] [PubMed] [Google Scholar]
- Evans DH, Claiborne JB (2006) The physiology of fishes.CRC
- Evans TG. Co-ordination of osmotic stress responses through osmosensing and signal transduction events in fishes. J Fish Biol. 2010;76(8):1903–1925. doi: 10.1111/j.1095-8649.2010.02590.x. [DOI] [PubMed] [Google Scholar]
- Fiess JC, Kunkel-Patterson A, Mathias L, Riley LG, Yancey PH, Hirano T, Grau EG. Effects of environmental salinity and temperature on osmoregulatory ability, organic osmolytes, and plasma hormone profiles in the Mozambique tilapia (Oreochromis mossambicus) Comp Biochem Phys A. 2007;146(2):252–264. doi: 10.1016/j.cbpa.2006.10.027. [DOI] [PubMed] [Google Scholar]
- Flynt AS, Thatcher EJ, Burkewitz K, Li N, Liu Y, Patton JG. miR-8 microRNAs regulate the response to osmotic stress in zebrafish embryos. J Cell Boil. 2009;185(1):115–127. doi: 10.1083/jcb.200807026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, et al. (2011) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. J Nucleic Acids Res 40(1):37–52 [DOI] [PMC free article] [PubMed]
- Gao W, Tan B, Mai K, Chi S, Liu H, Dong X, Yang Q. Profiling of differentially expressed genes in hepatopancreas of white shrimp (Litopenaeus vannamei) exposed to long-term low salinity stress. Aquaculture. 2012;364:186–191. [Google Scholar]
- Godi A, DiCampli A, Konstantakopoulos A, DiTullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, DeMatteis MA. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns (4) P. Nat Cell Boil. 2004;6(5):393. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- Greenberg AS, McDaniel ML. Identifying the links between obesity, insulin resistance and β-cell function: potential role of adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur J Clin Investig. 2002;32:24–34. doi: 10.1046/j.1365-2362.32.s3.4.x. [DOI] [PubMed] [Google Scholar]
- Gu XH, Jiang DL, Huang Y, Li BJ, Chen CH, Lin HR, Xia JH (2018) The sea cucumber Holothuria scabra in Nile Tilapia using QTL-Seq. Mar Biotechnol:1–10 [DOI] [PubMed]
- Hamel JF, Conand C, Pawson DL, Mercier A (2001) The sea cucumber Holothuria scabra (Holothuroidea: Echinodermata): its biology and exploitation as beche-de-mer:129–223
- Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch. 2004;447(5):465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- Hui M, Liu Y, Song C, Li Y, Shi G, Cui Z. Transcriptome changes in Eriocheir sinensis megalopae after desalination provide insights into osmoregulation and stress adaption in larvae. PLoS One. 2014;9(12):e114187. doi: 10.1371/journal.pone.0114187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo D, Sun L, Li X, Ru X, Liu S, Zhang L, Yang H. Differential expression of miRNAs in the respiratory tree of the sea cucumber Apostichopus japonicus under hypoxia stress. G3. 2017;7(11):3681–3692. doi: 10.1534/g3.117.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanina AV, Froelich B, Williams T, Sokolov EP, Oliver JD, Sokolova IM. Interactive effects of cadmium and hypoxia on metabolic responses and bacterial loads of eastern oysters Crassostrea virginica Gmelin. Chemosphere. 2011;82(3):377–389. doi: 10.1016/j.chemosphere.2010.09.075. [DOI] [PubMed] [Google Scholar]
- Jeong SY, Kim JH, Lee WO, Dahms HU, Han KN. Salinity changes in the anadromous river pufferfish, Takifugu obscurus, mediate gene regulation. Fish Physiol Biochem. 2014;40(1):205–219. doi: 10.1007/s10695-013-9837-z. [DOI] [PubMed] [Google Scholar]
- Kashenko SD. Acclimation of the sea cucumber Apostichopus japonicus to decreased salinity at the blastula and gastrula stages: its effect on the desalination resistance of larvae at subsequent stages of development. Russ J Mar Biol. 2000;26(6):422–426. [Google Scholar]
- Kim YK, Jang SK. Continuous heat shock enhances translational initiation directed by internal ribosomal entry site. Biochem Biophys Res Commun. 2002;297(2):224–231. doi: 10.1016/s0006-291x(02)02154-x. [DOI] [PubMed] [Google Scholar]
- Koepsell H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Asp Med. 2013;34(2–3):413–435. doi: 10.1016/j.mam.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. J Gen Biol 10(3):25 [DOI] [PMC free article] [PubMed]
- Lansdell SJ, Gee VJ, Harkness PC, Doward AI, Baker ER, Gibb AJ, Millar NS (2005) RIC-3 enhances functional expression of multiple nicotinic acetylcholine receptor subtypes in mammalian cells. Mol Pharmacol [DOI] [PubMed]
- Li E, Wang S, Li C, Wang X, Chen K, Chen L. Transcriptome sequencing revealed the genes and pathways involved in salinity stress of Chinese mitten crab, Eriocheir sinensis. Physiol Genomics. 2014;46(5):177–190. doi: 10.1152/physiolgenomics.00191.2013. [DOI] [PubMed] [Google Scholar]
- Li L, Li Q. Effects of stocking density temperature, and salinity on larval survival and growth of the red race of the sea cucumber Apostichopus japonicus (Selenka) Aquac Int. 2010;18(3):447–460. [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22(4):1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Liu P, Gao B, Li J. The identification and characteristics of salinity-related microRNAs in gills of Portunus trituberculatus. Cell Stress Chaperones. 2016;21(1):63–74. doi: 10.1007/s12192-015-0641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Liu P, Wang Y, Gao B, Chen P, Li J. Transcriptome analysis of Portunus trituberculatus in response to salinity stress provides insights into the molecular basis of osmoregulation. PLoS One. 2013;8(12):e82155. doi: 10.1371/journal.pone.0082155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WS, Watters KD, Hovdestad LR, Cozzi RR, Katoh F. CFTR Cl–channel functional regulation by phosphorylation of focal adhesion kinase at tyrosine 407 in osmosensitive ion transporting mitochondria rich cells of euryhaline killifish. J Exp Biol. 2009;212(15):2365–2377. doi: 10.1242/jeb.030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick SD. Endocrine control of osmoregulation in teleost fish. Am Zool. 2001;41(4):781–794. [Google Scholar]
- Meng J, Zhu Q, Zhang L, Li C, Li L, She Z, Zhang G. Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS One. 2013;8(3):e58563. doi: 10.1371/journal.pone.0058563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaljevic I, Popovic M, Zaja R, Smital T. Phylogenetic, syntenic, and tissue expression analysis of slc22 genes in zebrafish (Danio rerio) BMC Genomics. 2016;17(1):626. doi: 10.1186/s12864-016-2981-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan M, Coppe A, Reinhardt R, Cancela LM, Leite RB, Saavedra C, Bargelloni L. Transcriptome sequencing and microarray development for the Manila clam, Ruditapes philippinarum: genomic tools for environmental monitoring. BMC Genomics. 2011;12(1):234. doi: 10.1186/1471-2164-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Jiang L, Chen P, Huo Z, Yang F, Yan X. High throughput sequencing of RNA transcriptomes in Ruditapes philippinarum identifies genes involved in osmotic stress response. Sci Rep-UK. 2017;7(1):4953. doi: 10.1038/s41598-017-05397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouattara NG, Bodinier C, Nègre-Sadargues G, D'Cotta H, Messad S, Charmantier G, Panfili J, Baroiller JF. Changes in gill ionocyte morphology and function following transfer from fresh to hypersaline waters in the tilapia Sarotherodon melanotheron. Aquaculture. 2009;290(1–2):155–164. [Google Scholar]
- Palacios E, Bonilla A, Luna D, Racotta IS. Survival, Na+/K+-ATPase and lipid responses to salinity challenge in fed and starved white pacific shrimp (Litopenaeus vannamei) postlarvae. Aquaculture. 2004;234(1–4):497–511. [Google Scholar]
- Palacios E, Bonilla A, Pérez A, Racotta IS, Civera R. Influence of highly unsaturated fatty acids on the responses of white shrimp (Litopenaeus vannamei) postlarvae to low salinity. J Exp Mar Biol Ecol. 2004;299(2):201–215. [Google Scholar]
- Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19(49):5606. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520(7548):511. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo RF, Rhodes EC. Lipid metabolism during exercise. Sports Med. 1998;26(1):29–42. doi: 10.2165/00007256-199826010-00003. [DOI] [PubMed] [Google Scholar]
- Rastorguev SM, Nedoluzhko AV, Sharko FS, Boulygina ES, Sokolov AS, Gruzdeva NM, Prokhortchouk EB. Identification of novel microRNA genes in freshwater and marine ecotypes of the three-spined stickleback (Gasterosteus aculeatus) Mol Ecol Resour. 2016;16(6):1491–1498. doi: 10.1111/1755-0998.12545. [DOI] [PubMed] [Google Scholar]
- Rengmark AH, Slettan A, Lee WJ, Lie Ø, Lingaas F. Identification and mapping of genes associated with salt tolerance in tilapia. J Fish Biol. 2007;71:409–422. [Google Scholar]
- Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell. 2017;67(1):84–95. doi: 10.1016/j.molcel.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, McCormick SD. Prolactin and growth hormone in fish osmoregulation. Gen Comp Endocrinol. 2006;147(1):24–30. doi: 10.1016/j.ygcen.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Salati AP, Baghbanzadeh A, Soltani M, Peyghan R, Riazi G. Effect of different levels of salinity on gill and kidney function in common carp Cyprinus carpio (Pisces: Cyprinidae) Ital J Zool. 2011;78(3):298–303. [Google Scholar]
- Santoni V, Joëlle V, Pflieger D, Sommerer N, Maurel C. A proteomic study reveals novel insights into the diversity of aquaporin forms expressed in the plasma membrane of plant roots. Bio Chem J. 2003;373(1):289–296. doi: 10.1042/BJ20030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2P. Proc Natl Acad Sci USA. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar MS, Kiruthika J, Ponniah AG. Identification and expression analysis of differentially expressed genes from shrimp (Penaeus monodon) in response to low salinity stress. Fish Shellfish Immunol. 2013;35(6):1957–1968. doi: 10.1016/j.fsi.2013.09.038. [DOI] [PubMed] [Google Scholar]
- Shi M, Lin Y, Xu G, Xie L, Hu X, Bao Z, Zhang R. Characterization of the Zhikong scallop (Chlamys farreri) mantle transcriptome and identification of biomineralization-related genes. Mar Biotechnol. 2013;15(6):706–715. doi: 10.1007/s10126-013-9517-0. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298(5602):2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. J Proc Natl Acad Sci 100(16): 9440–9445 [DOI] [PMC free article] [PubMed]
- Talbot TD, Lawrence JM. The effect of salinity on respiration excretion regeneration and production in Ophiophragmus filograneus (Echinodermata: Ophiuroidea) J Exp Mar Biol Ecol. 2002;275(1):1–14. [Google Scholar]
- Tamayo D, Corporeau C, Petton B, Quere C, Pernet F. Physiological changes in Pacific oyster Crassostrea gigas exposed to the herpesvirus OsHV-1 μVar. Aquaculture. 2014;432:304–310. [Google Scholar]
- Tipsmark CK, Kiilerich P, Nilsen TO, Ebbesson LO, Stefansson SO, Madsen SS. Branchial expression patterns of claudin isoforms in Atlantic salmon during seawater acclimation and smoltification. Am J Physiol-RegI. 2008;294(5):R1563–R1574. doi: 10.1152/ajpregu.00915.2007. [DOI] [PubMed] [Google Scholar]
- Tipsmark CK, Sørensen KJ, Madsen SS. Aquaporin expression dynamics in osmoregulatory tissues of Atlantic salmon during smoltification and seawater acclimation. J Exp Biol. 2010;213(3):368–379. doi: 10.1242/jeb.034785. [DOI] [PubMed] [Google Scholar]
- Tournaire-Roux C, Sutka M, Javot H, Gout E, Gerbeau P, Luu DT. Cytosolic pH regulates root water transport during anoxic stress through gating of aquaporins. Nature. 2003;425(6956):393. doi: 10.1038/nature01853. [DOI] [PubMed] [Google Scholar]
- Tse WP, Che CT, Liu K, Lin ZX. Evaluation of the anti-proliferative properties of selected psoriasis-treating Chinese medicines on cultured HaCaT cells. J Ethnopharmacol. 2006;108(1):133–141. doi: 10.1016/j.jep.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Vidolin D, Santos-Gouvea IA, Freire CA. Osmotic stability of the coelomic fluids of a sea-cucumber (Holothuria grisea) and a starfish (Asterina stellifera) (Echinodermata) exposed to the air during low tide: a field study. Acta Biol Paranaense. 2002;31:113–121. [Google Scholar]
- Wang F, Yang H, Gao F, Liu G. Effects of acute temperature or salinity stress on the immune response in sea cucumber Apostichopus japonicas. Comp Biochem Phys A. 2008;151(4):491–498. doi: 10.1016/j.cbpa.2008.06.024. [DOI] [PubMed] [Google Scholar]
- Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34(1):3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen M, Shen Y, Shi S, et al. (2012) miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC bioinformatics 13(1):140 [DOI] [PMC free article] [PubMed]
- Wiedmer T, Zhao J, Li L, Zhou Q, Hevener A, Olefsky JM. Adiposity dyslipidemia and insulin resistance in mice with targeted deletion of phospholipid scramblase 3 (PLSCR3) Proc Natl Acad Sci. 2004;101(36):13296–13301. doi: 10.1073/pnas.0405354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Li E, Liu Y, Wang X, Qin JG, Chen L. Comparative proteome analysis of the hepatopancreas from the Pacific white shrimp Litopenaeus vannamei under long-term low salinity stress. J Proteome. 2017;162:1–10. doi: 10.1016/j.jprot.2017.04.013. [DOI] [PubMed] [Google Scholar]
- Xu Q, Liu Y. Gene expression profiles of the swimming crab Portunus trituberculatus exposed to salinity stress. Mar Biol. 2011;158(10):2161. [Google Scholar]
- Yan B, Guo JT, Zhao LH, Zhao JL. MiR-30c: a novel regulator of salt tolerance in tilapia. Biochem Biophys Res Commun. 2012;425(2):315–320. doi: 10.1016/j.bbrc.2012.07.088. [DOI] [PubMed] [Google Scholar]
- Yan B, Wang ZH, Zhao JL. Mechanism of osmoregulatory adaptation in tilapia. Mol Biol Rep. 2013;40(2):925–931. doi: 10.1007/s11033-012-2133-7. [DOI] [PubMed] [Google Scholar]
- Yan B, Zhao LH, Guo JT, Zhao JL. miR-429 regulation of osmotic stress transcription factor 1 (OSTF1) in tilapia during osmotic stress. Biochem Biophys Res Commun. 2012;426(3):294–298. doi: 10.1016/j.bbrc.2012.08.029. [DOI] [PubMed] [Google Scholar]
- Yan B, Zhao L, Guo J, Zhao J (2012c) miR-206 regulates the growth of the teleost tilapia (Oreochromis niloticus) through the modulation of IGF-1 gene expression. J Exp Biol:079590 [DOI] [PubMed]
- Yang Y, Guo Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018;217(2):523–539. doi: 10.1111/nph.14920. [DOI] [PubMed] [Google Scholar]
- Yuan X, Yang H, Wang L, Zhou Y, Gabr HR. Effects of salinity on energy budget in pond-cultured sea cucumber Apostichopus japonicus (Selenka) (Echinodermata: Holothuroidea) Aquaculture. 2010;306(1–4):348–351. [Google Scholar]
- Zacchi FL, de Lima D, Flores-Nunes F, Mattos JJ, Luechmann KH, de Miranda Gomes CHA, Bainy ACD. Transcriptional changes in oysters Crassostrea brasiliana exposed to phenanthrene at different salinities. Aquat Toxicol. 2017;183:94–103. doi: 10.1016/j.aquatox.2016.12.016. [DOI] [PubMed] [Google Scholar]
- Zhang BC, Zhang J, Sun L. In-depth profiling and analysis of host and viral microRNAs in Japanese flounder (Paralichthys olivaceus) infected with megalocyti virus reveal involvement of microRNAs in host-virus interaction in teleost fish. BMC Genomics. 2014;15(1):878. doi: 10.1186/1471-2164-15-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Tong C, Tian F, Zhao K. Integrated mRNA and microRNA transcriptome analyses reveal regulation of thermal acclimation in Gymnocypris przewalskii: a case study in Tibetan Schizothoracine fish. PLoS One. 2017;12(10):e0186433. doi: 10.1371/journal.pone.0186433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Fang X, Guo X, Li L, Luo R, Xu F. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490(7418):49. doi: 10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- Zhang L, Feng Q, Sun L, Ding K, Huo D, Fang Y. Differential gene expression in the intestine of sea cucumber (Apostichopus japonicus) under low and high salinity conditions. Comp Biochem Phys D. 2018;25:34–41. doi: 10.1016/j.cbd.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li L, Zhu Y, Zhang G, Guo X. Transcriptome analysis reveals a rich gene set related to innate immunity in the Eastern oyster (Crassostrea virginica) Mar Biotechnol. 2014;16(1):17–33. doi: 10.1007/s10126-013-9526-z. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sun J, Mu H, Li J, Zhang Y, Xu F. Proteomic basis of stress responses in the gills of the pacific oyster Crassostrea gigas. J Proteome Res. 2014;14(1):304–317. doi: 10.1021/pr500940s. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yu H, Kong L, Liu S, Li Q. High throughput sequencing of small RNAs transcriptomes in two Crassostrea oysters identifies microRNAs involved in osmotic stress response. Sci Rep- UK. 2016;6:22687. doi: 10.1038/srep22687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yu H, Kong L, Li Q. Transcriptomic responses to salinity stress in the Pacific oyster Crassostrea gigas. PLoS One. 2012;7(9):e46244. doi: 10.1371/journal.pone.0046244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P, Wang B, Wang G, Jia Z, Pu P. Zhang W, Kang C (2010) Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Invest. 90:144–155 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 24 kb)
(PNG 78 kb)
(PNG 310 kb)
(JPG 259 kb)
(JPG 49 kb)
(JPG 102 kb)
(PNG 595 kb)
(PNG 596 kb)
(PNG 639 kb)
(PNG 162 kb)
(XLSX 388 kb)
(XLSX 517 kb)
(XLSX 16.4 kb)