

Abstract
Human alcoholic hepatitis (AH) carries a high mortality rate. AH is an acute-on-chronic form of liver injury characterized by hepatic steatosis, ballooned hepatocytes, neutrophil infiltration, and pericellular fibrosis. We aimed to study the pathogenesis of AH in an animal model which combines chronic hepatic fibrosis with intragastric alcohol administration. Adult male C57BL6/J mice were treated with CCl4 (0.2 ml/kg, 2×weekly by intraperitoneal injections for 6 weeks) to induce chronic liver fibrosis. Then, ethyl alcohol (up to 25 g/kg/day for 3 weeks) was administered continuously to mice via a gastric feeding tube, with or without one-half dose of CCl4. Liver and serum markers and liver transcriptome were evaluated to characterize acute-on-chronic-alcoholic liver disease in our model. CCl4 or alcohol treatment alone induced liver fibrosis or steatohepatitis, respectively, findings that were consistent with expected pathology. Combined treatment resulted in a marked exacerbation of liver injury, as evident by the development of inflammation, steatosis, and pericellular fibrosis, pathological features of human AH. E. coli and Candida were also detected in livers of mice cotreated with CCl4 and alcohol, indicating pathogen translocation from gut to liver, similar to human AH. Importantly, liver transcriptomic changes specific to combined treatment group demonstrated close concordance with pathways perturbed in patients with severe AH. Overall, mice treated with CCl4 and alcohol displayed key molecular and pathological characteristics of human AH—pericellular fibrosis, increased hepatic bacterial load, and dysregulation of the same molecular pathways. This model may be useful for developing therapeutics for AH.
Keywords: liver, systems toxicology, ethanol, transnational, fibrosis
Upon oral ingestion, ethyl alcohol diffuses readily through cell membranes and is metabolized by most tissues to acetaldehyde, a highly reactive molecule (Zakhari, 2006). Adverse effects of alcohol consumption in all tissues are a consequence formation of acetaldehyde, reactive oxygen and nitrogen species, depletion of cofactors (eg, NAD+), impairment in energy homeostasis, and activation of pro-inflammatory and other signaling pathways in many organs (Rusyn and Bataller, 2013). Alcohol metabolism and toxicity have most impact on the liver where adverse effects may range from steatosis to hepatitis, cirrhosis, and hepatocellular carcinoma (Altamirano et al., 2012).
Alcoholic hepatitis (AH) is an acute-on-chronic form of liver injury and represents the most severe form of alcoholic liver disease; it is associated with high mortality (Casanova and Bataller, 2014; Lucey et al., 2009; Michelena et al., 2015). Up to 40% of patients diagnosed with severe AH die within 6 months due to limited treatment options to supplement first-line therapy with prednisolone (Altamirano et al., 2012). Continuous alcohol consumption and loss of intestinal barrier are potential triggers of AH, which may be related to the immunosuppression effect of chronic alcohol consumption (Chiang et al., 2013; Monnig, 2017). Still, there does not appear to be a clear initiating event for AH in patients with pre-existing hepatic fibrosis who continue to consume alcohol.
Challenges with understanding the molecular and pathological drivers of AH in humans who usually present with an already advanced set of clinical signs and require immediate treatment are compounded by inadequate severity of alcohol-induced liver disease in commonly used animal models (Casanova and Bataller, 2014). Although several experimental models of alcohol-induced liver injury exist (Bertola et al., 2013; Ueno et al., 2012), they do not fully recapitulate key characteristics of clinical AH, such as pericellular fibrosis, elevated serum bilirubin, and increased hepatic bacterial load, as well as acute kidney injury (Prado et al., 2016). A number of studies in mice and rats combined a liquid diet alcohol feeding model and intraperitoneal injections of carbon tetrachloride (CCl4) to evaluate the molecular mechanisms of comorbidity (Chiang et al., 2013; Roychowdhury et al., 2014; Safer et al., 2015). These studies showed that alcohol exacerbates CCL4-induced liver fibrosis and identified several key pathogenic drivers, knowledge that can be used to identify targets for novel pharmacotherapy (Casanova and Bataller, 2014).
Our previous work demonstrated fibrosis- and alcohol-associated liver and kidney injury and identified key mechanistic pathways that drive acute kidney injury in response to alcohol in the mouse model of intragastric feeding (Furuya et al., 2016). We combined CCl4-induced fibrosis with chronic intragastric alcohol administration and observed that combined treatment with CCl4 and alcohol resulted in severe liver injury, more pronounced than using each treatment alone. This study details histopathological and molecular characterization of the liver effects in the model and provides a comparison of gene expression signatures with human AH. We demonstrate remarkable concordance with human AH, as evidenced by pericellular fibrosis, severe hepatic inflammation, profound transcriptome changes, and increased hepatic bacterial load, all key characteristics of human AH.
MATERIALS AND METHODS
Animals
Male mice (C57BL/6J, 20–25 g, 12 weeks of age) were obtained from the Jackson Laboratory (Bar Harbor, Maine) and housed in a temperature-controlled environment with a 12-h light-dark cycle and were given free access to regular laboratory chow diet and water. All studies were approved by the Institutional Animal Care and Use Committee at UNC-Chapel Hill.
Diets and treatment
CCl4 (≥ 99.5% pure) and olive oil vehicle were from Sigma (St. Louis, Missouri), ethyl alcohol (EtOH) (190 proof, Koptec) was from VWR (Radnor, Pennsylvania). Procedures for CCl4-induced liver fibrosis were as detailed elsewhere (Uehara et al., 2014). Mice were intraperitonealy injected (15 ml/kg) with CCl4 (0.2 ml/kg) or olive oil vehicle-alone 2 × week for 6 weeks (Supplementary Figure 1). After 6 weeks of CCl4 treatment, animals underwent surgical intragastric intubation (Kono et al., 2000). Following surgery, mice were housed in individual metabolic cages and allowed 1 week to recover with ad libitum access to food and water. Animals had free access to water and nonnutritious cellulose pellets throughout the remaining study. Alcohol groups received high-fat diet containing ethyl alcohol as detailed elsewhere (Kono et al., 2000). Alcohol was delivered continuously through the intragastric cannula initially at 16 g/kg/day and was gradually increased to 25 g/kg/day. All animals were given humane care in compliance with the National Institutes of Health guidelines and alcohol intoxication was assessed to evaluate the development of tolerance. Five experimental groups (n = 2–4 per group) comprised this study. Control (“Cont”) group are animals that received intraperitoneal injected injections (2×week) of olive oil vehicle for 6 weeks. “CCl4 (6w)” group are animals that received intraperitoneal injections (2×week) of CCl4 (0.2 ml/kg) for 6 weeks. “CCl4(9w)” are animals that received intraperitoneal injections (2×week) of CCl4 (0.2 ml/kg) for 6 weeks and then received intraperitoneal injections (2×week) of CCl4 (0.1 ml/kg) for 3 weeks. “EtOH” group are animals that received intraperitoneal injected injections (2×week) of olive oil vehicle for 6 weeks and then were administered alcohol intragastrically for 3 weeks. “CCl4(9w)+EtOH” group are animals that received intraperitoneal injections (2×week) of CCl4 (0.2 ml/kg) for 6 weeks and then were administered alcohol intragastrically with continuous intraperitoneal injected injections (2×week) with CCl4 (0.1 ml/kg). Animal survival was 100% in Cont, CCl4(6w) and CCL4(9w) groups, 70% in EtOH group, and 50% in CCl4(9w)+EtOH group.
Sample collection
At the end of the study, mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and sacrificed via exsanguination through the vena cava, which was the site of blood collection. Tissues were excised and snap frozen in liquid nitrogen. Sections of the left and median liver lobes were fixed in formalin. Urine was collected using metabolic cages daily during alcohol administration and stored at −20°C until assayed.
Liver histopathological evaluation
Tissues were embedded in paraffin, sectioned at 5 µm, and stained with hematoxylin and eosin (H&E) or Sirius red. For Oil red O staining, tissues embedded in optimal cutting temperature compound were sectioned at 10 µm. Liver pathology was evaluated in a blind manner by two independent pathologists and scored as detailed elsewhere (Nanji et al., 1989). For Sirius red and oil red O staining, quantitative analysis was performed using NIH ImageJ at 100× magnification in 5 random fields.
Fluorescence in situ hybridization (FISH) and immunohistochemistry
Paraffin-embedded liver sections (5 μm) were stained with primary rabbit antimyeloperoxidase antibody (MPO; Abcam, Cambridge, Massachusetts; 1:50, 1 h, room temperature), goat IgG HRP-conjugated secondary antibody (Millipore, Billerica, Massachusetts; 1:2000, 1 h, room temperature), and Dako Liquid DAB+ Substrate chromogen System (Dako, Carpinteria, California). Quantitative analysis of staining was performed at 200× magnification in 5 random fields. FISH-enabled detection of bacteria in liver tissue was performed on the formalin-fixed paraffin-embedded liver tissue sections using a method detailed in Twedt et al. (2014). Nonspecific (non-EUB-TITC) and total bacteria-specific (EUB338-AlexaFluor® 555) FISH probes with sequences as detailed in Cassmann et al. (2016) were used. Slides were mounted with Gold Antifade reagent (with DAPI; P-36931 Invitrogen, Carlsbad, California) and imaged using LSM 780 NLO Confocal/multiphoton microscope equipped with an AxioObserver Z1 inverted microscope (Carl Zeiss AG, Oberkochen, Germany).
Biochemical measurements
Serum aminotransferase (ALT) levels and triglycerides in the liver tissue were determined spectrophotometrically with the infinity ALT liquid stable reagent (Thermo Electron, Melbourne, Australia) and L-type TG M kit (Wako, Richmond, Virginia), respectively.
Quantitative reverse-transcription PCR
Total liver RNA was extracted from mouse liver using the RNeasy Mini kit (Qiagen, Valencia, California). RNA concentrations were measured with ND-1000 spectrophotometer (NanoDrop, Wilmington, Delaware) and quality was verified using the Bio-Analyzer (Agilent, Santa Clara, California). Total liver RNA was reverse transcribed using random primers and the high-capacity cDNA archive kit (Applied Biosystems, Foster City, California). TaqMan gene expression assays (Life Technologies, Carlsbad, California) were used to query expression of DNA methyltransferases 1 (Dnmt1; Mm01151063_m1), 3a (Dnmt3a; Mm00432881_m1), and 3b (Dnmt3b; Mm01240113_m1), ubiquitin-like, containing PHD and RING finger domains, 1 (Uhrf1; Mm00477872_m1), and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; Mm99999915_g1). Reactions were performed in a 96-well assay format using a QuantStudio 7 Flex Real-Time PCR System (Life Technologies).
RNA sequencing
Libraries for mouse samples RNA-seq were prepared from total liver RNA using the Illumina TruSeq mRNA Sample Prep Kit (Illumina, San Diego, California). Pair-end (100 bp) sequencing was carried out using the Illumina HiSeq 2500 platform. RNA-seq reads were aligned to appropriate reference genomes (NCBI mm 10) using the “SNP-tolerant” GSNAP software. Gene expression data from mouse liver analyzes are available in Gene Expression Omnibus (study ID: GSE119470). Differential gene expression analysis was then performed on the complete list of 18 965 genes using R package DESeq2 (Love et al., 2014). A log2-fold change (compared with Control group) and false discovery rate (FDR)-adjusted p-value (q-value) cut-off at ±0.58 and < 0.1, respectively, were applied. For human liver, transcriptome profiling by RNA-Seq was performed using Illumina HiSeq2000 platform (San Diego, California) with liver samples provided by the InTeam Consortium Human Biorepository Core as described elsewhere (Brandl et al., 2018). A total of 51 patients with different phenotypes were selected: 12 patients with early alcoholic steatohepatitis, 18 patients with AH, and 10 normal control. The protocol was approved by the Institutional Board Review from the University of Pittsburgh. Human gene expression data are detailed in Argemi et al. (2018).
Pathway analyses
Gene Set Enrichment Analysis (GSEA 3.0) (Subramanian et al., 2007) and KEGG-annotated pathways (Kanehisa et al., 2017) were used. FDR q-value < 0.05 and FWER p-value < .05 were considered as threshold for the enrichment. Additional pathway analysis was conducted using Piano R package (Varemo et al., 2013) and mouse Reactome pathways (Fabregat et al., 2016). GGRNA tool was used to identify fibrosis, inflammation, hypoxia, and steatosis-associated transcripts (Naito and Bono, 2012).
Bacterial DNA detection in liver using quantitative reverse-transcription PCR
DNA from liver tissue was isolated with DNeasy Blood&Tissue kits (Qiagen). PCR was performed using Taqman (Life Technologies) gene expression assay probe for Rps18 (Mm02601778_g1), or custom-made probes for E.coli, pan-candida and specific Candida species (C. albicans, C. glabrata, C. tropicalis, and C. dubliniensis) (Supplementary Table 1). Expression Rps18 was used as the housekeeping gene. Reactions were performed in a 96-well assay format using Roche 480 instrument (Roche Applied Science, Indianapolis, Indiana).
Western blotting analysis of histone modifications
Western blot analysis was conducted using primary antibodies against histone 3 lysine 4 trimethylation (H3K4me3; Cell Signaling Technology, Danvers, Massachusetts; 1:1000, cat. 9727), histone 3 lysine 9 trimethylation (H3K9me3; Millipore Corporation, Billerica, Massachusetts; 1:1000, cat. 07-442), histone 3 lysine 27 trimethylation (H3K27me3; Millipore; 1:1000, cat. 07-449), histone 4 lysine 20 trimethylation (H4K20me3; Millipore; 1:1000, cat. 07-463), histone 3 lysine 27 acetylation (H3K27ac; Millipore; 1:1000, cat. 07-360), histone 3 lysine 9 acetylation (H3K9ac; Abcam; 1:1000, cat. ab10812), and histone 3 lysine 56 acetylation (H3K56ac; Abcam; 1:1000, ab76307). IRDye 800 conjugated secondary antirabbit antibody (LI-COR Biosciences, Lincoln, Nebraska; 1:15 000) was used for visualization. Blots were scanned and analyzed using the Odyssey CLx Infrared Imaging System (LI-COR Biosciences). Protein loading was confirmed by immunostaining against total histone 3 (H3K27ac; Millipore; cat. 17-10046).
Statistical analyses
Statistical significance for data other than RNA sequencing was determined using Prism5 (GraphPad, San Diego, California). Quantitative values are expressed as mean ± SD unless otherwise noted and statistical significance was assessed using 1-way ANOVA within each time point followed by the Tukey’s post hoc test with significance cut-off p < .05.
RESULTS
Combined Treatment With CCl4 and Alcohol Results in Severe Liver Injury
Animals maintained on alcohol alone or CCl4(9w) + EtOH presented with marked liver steatosis or steatohepatitis, respectively, as evident in quantitative histopathological analyses, Oil RedO staining, and biochemical measures of ALT and triglycerides (Figs. 1 and 2). Liver injury was more severe in the combined treatment group, CCl4(9w) + EtOH, as compared with EtOH group. Bridging fibrosis of increased severity was observed in CCl4(6w) and CCl4(9w) groups as evidenced by the characteristic increases in Sirius red staining, which were accompanied by progressive neutrophil infiltration (MPO staining) in liver and especially their localization to the areas of fibrosis. In CCl4(9w) + EtOH group, Sirius red staining revealed bridging fibrosis accompanied by massive pericellular collagen deposition (Figure 1B) and increased, yet generally diffuse neutrophil infiltration (Figure 1C). Thus, although neutrophil infiltration was a pronounced feature of liver injury elicited by CCl4 with, or without coadministration of alcohol (Figure 2F), the patterns of neutrophil deposition in liver were markedly different (Figure 1C). Overall, these data confirmed that animals in this study exhibited fibrosis- and alcohol-related disease phenotypes, and that combined treatment group showed more severe liver injury compared with mice treated with CCl4 or alcohol alone, consistent with histopathological findings in human AH. Specifically, administration of EtOH to mice with chronic injury resulted in a “chicken-wire” pattern of liver fibrosis similar to that described in patients with AH (ie, pericellular fibrosis) (Bataller and Gao, 2015).
Figure 1.

Histological evaluation of liver injury in alcohol- and fibrosis-associated mouse model. Representative photomicrographs of liver sections are shown. (A) Images of H&E, Oil RedO, Sirius red, and myeloperoxidase (MPO) staining. Magnification 100–200×, scale bar = 200 μm. Higher-resolution images of Sirius red (B) and MPO (C) staining in select groups. Magnification 400×, scale bar = 50 μm.
Figure 2.

Quantitative analysis of liver injury in alcohol- and fibrosis-associated mouse model. (A) Serum aminotransferase levels. (B) Liver injury score (see Materials and Methods). (C) Quantitative analysis of Oil RedO staining (5 random fields at 200× magnification). (D) Triglyceride levels in liver tissue. (E) Quantitative analysis of Sirius red staining (5 random fields at 200× magnification). (F) Quantitative analysis of MPO-positive cell counts (5 random fields at 200× magnification). All data are presented as mean ± SD. Asterisks denote statistical significance as follows: a, p < .05, compared with control group; b, p < .05, compared with CCl4(6w) group; c, p < .5, compared with CCl4(9w) group; d, p < .05, compared with EtOH group.
Transcriptome Analysis of Molecular Drivers of Fibrosis- and Alcohol-Associated Liver Injury in the Mouse
To further elucidate the mechanism of fibrosis and alcohol-associated liver injury, transcriptome and pathway analyses were performed on liver tissue. Liver transcriptional effects of CCL4 were time dependent with progressively greater effect was observed at 9 weeks (Figs. 3A and 3B). The predominant effect was transcriptional induction of pathways involved in fibrogenesis, cell proliferation and cytokine cascades (Supplementary Table 2). EtOH treatment had a robust effect on the liver transcriptome with many transcripts induced 30-fold or above (Figure 3C); these were enriched for upregulation of metabolism (eg, lipid metabolism, cytochrome P450s, etc.), and downregulation of immune responses (Supplementary Table 2). Remarkably, CCl4(9w) + EtOH treatment group had the greatest effect on the liver transcriptome (Figure 3D); it comprised of upregulation of apoptosis, cell cycle, DNA damage, inflammation, and bacterial infection, and downregulation of lipid accumulation and immune response (Supplementary Table 2). Most of the transcriptional changes elicited by EtOH were nonoverlapping with those of CCL4(9w) or CCl4(9w) + EtOH (Figs. 3E–G); however, the effects were highly concordant (r2 = 0.63) between CCL4(9w) and CCl4(9w) + EtOH groups (Figure 3H).
Figure 3.

Global transcriptomic analysis of liver injury in alcohol- and fibrosis-associated mouse model. (A–D) Scatter plots of gene expression changes, relative to control group, in treatment groups as indicated. Red dots indicate significant (FDR-adjusted q-value < 0.1, log2-fold change ±1.5-fold) differentially expressed genes. (E) Venn diagrams of significant differentially expressed genes among indicated groups. (F–H) Pair-wise correlation plots of gene expression changes among CCl4(9w), EtOH, or CCl4(9w)+EtOH groups. Dotted lines are indicating ±1.5-fold change, colored dots represent significant (FDR-adjusted q-value <0.1) genes. Differentially expressed genes are color coded by whether they were changed by one (orange or red), or both (purple) treatments. Pathways for gene expression changes in panels B–D are listed in Supplementary Table 2.
Clinically, severe AH is commonly attributed to pre-existing alcohol-induced hepatic fibrosis and inflammation which are preceding the onset of acute-on-chronic alcohol-induced severe liver injury. Thus, we have examined transcriptional effects on fibrosis-, inflammation-, hypoxia-, and steatosis-related genes in this mouse model (Figure 4). Unsupervised hierarchical clustering analysis of the transcripts that are assigned into these broad categories using GGRNA tool (Naito and Bono, 2012) showed clear separation of CCl4(9w) and CCl4(9w) + EtOH groups from CCl4(6w) and EtOH groups for fibrosis-, inflammation-, and hypoxia-related transcripts (Figs. 4A–C). In contrast, no clear separation among groups was observed for steatosis-related transcripts, most of the effects were in the EtOH group (Figure 4D). These results at the transcriptome level strongly suggest that the existence of underlying fibrosis sensitizes the liver to the deleterious effects of EtOH.
Figure 4.
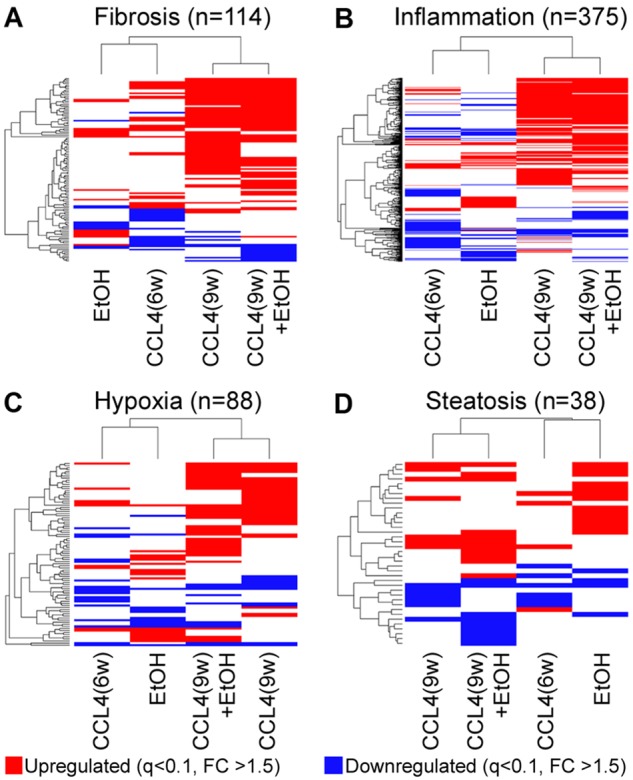
Pathway-specific analysis of liver injury in alcohol- and fibrosis-associated mouse model. Supervised clustering diagrams of 114 (A), 375 (B), 88 (C), and 38 (D) transcripts identified as related to fibrosis, inflammation, hypoxia, and steatosis, respectively, by GGRNA (Naito and Bono, 2012). Group averages are shown [sample number per group were Cont = 4, CCl4(6w) = 2, CCl4(9w)-2, EtOH = 4, CCl4(9w) + EtOH = 3]. Transcripts are color coded if they have been significantly (FDR-adjusted q-value < 0.1) affected with ±1.5-fold change (red = upregulated, blue = downregulated). Ranked lists of transcripts in this figure are included in Supplementary Table 3. Raw gene expression data are available in GEO (accession ID GSE119470).
Mouse-Human Comparison of the Transcriptomic Signature of Fibrosis, Steatohepatitis and Alcoholic Hepatitis
Molecular signatures of human cirrhosis, steatohepatitis, and AH have been previously characterized (Affo et al., 2013). Therefore, we explored how well the study groups in the mouse model match various disease phenotypes in human liver with respect to gene expression. Figure 5 shows molecular pathway plots for the mouse (Figs. 5A–C) and humans (Figs. 5D–F). There is little overlap between mouse steatohepatitis induced by intragastric feeding of alcohol alone (EtOH group) and human early alcoholic steatohepatitis. Only downregulation of the immune system pathway was in common, confirming the challenges with using traditional mouse models to study human alcohol-induced injury (Casanova and Bataller, 2014; Gao and Bataller, 2011; Prado et al., 2016). Transcriptomic signatures of mouse liver fibrosis [CCl4(9w) group] and human liver cirrhosis were much more concordant; these included upregulation of immune system, cell cycle and mitosis, and signal transduction cascades. CCl4 treatment led to downregulation of drug and lipid metabolism pathways in the mouse, a known competitive inhibition effects (Weber et al., 2003), whereas in human cirrhosis the metabolism pathways were prominently induced. Most interesting is the observation that transcriptional signature of CCl4(9w)+EtOH group was nearly identical to that of human severe AH. Most of the significantly up- and downregulated pathways overlapped; cell cycle and mitosis, signal transduction, and fibrogenesis pathways were induced, whereas metabolism-associated pathways were repressed.
Figure 5.

Species concordance in the effects of alcohol, fibrosis, or alcoholic hepatitis on the liver transcriptome. Reactome (Fabregat et al., 2016) pathways (gene set size > 25) that were significantly (FDR-adjusted q-value < 0.05) up- (red) or down-(blue) regulated in each condition as indicated were visualized using Piano R package. Ranked lists of pathways in this figure are included in Supplementary Table 4.
Evidence for Pathogenic Microorganism Translocation as a Mechanism of Fibrosis- and Alcohol-Associated Liver Injury in the Mouse
Translocation of the pathogenic microorganisms from the gut to liver is one fundamental mechanism of the pathogenesis of alcohol- and cirrhosis-associated liver diseases in animals and humans (Starkel and Schnabl, 2016). Moreover, patients with severe AH suffer from candida infections due to immune deficiency (Lahmer et al., 2014; Wernlund et al., 2014). A recent study suggest that translocation of Candida and other fungal species could play a pathogenic role in the development of AH (Yang et al., 2017). Indeed, bacterial infection-associated pathways were upregulated in both CCl4(9w) + EtOH group and in human severe AH. Next, we tested whether livers in mice affected by both fibrosis and alcohol exhibit higher load of pathogenic microorganisms. First, we performed FISH analysis of E. coli in mouse livers and found increased load of bacterial DNA fragments only in CCl4(9w) + EtOH group (Figure 6A). Then, we evaluated E. coli and candida presence in livers using PCR (Figure 6A). We found that the abundance of E. coli- and candida-specific DNA was significantly increased in CCl4(9w)+EtOH group.
Figure 6.
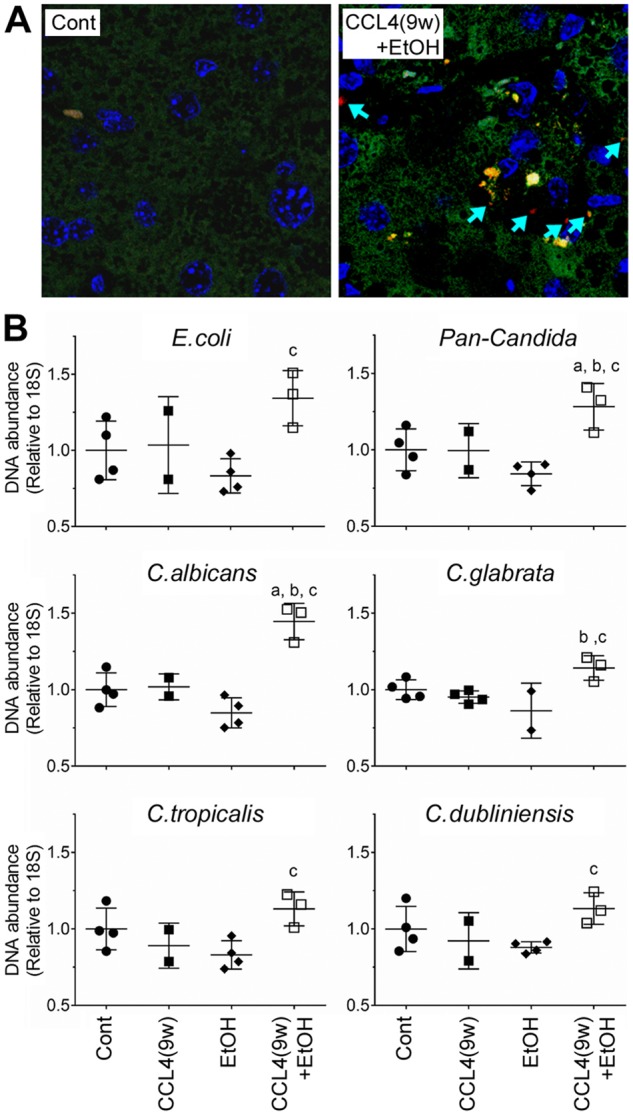
Pathogenic microorganism analysis in the liver in alcohol- and fibrosis-associated mouse model. (A) Presence of E. coli DNA in liver sections was evaluated using FISH. Representative images of merged liver sections (nuclei = blue, nonspecific staining = green, bacteria = red) from Cont and CCL4(9w) + EtOH groups are shown. Arrows indicate E. coli in liver tissue. (B) Presence of E. coli and candida (subspecies are indicated) DNA in the liver tissue was assessed by qPCR. Scatter plots show the individual values (dots), group medians (line) and standard deviations (whiskers). Asterisks denote statistical significance as follows: a, p < .05, compared with control group; b, p < .05, compared with CCl4(6w); c, p < .05, compared with CCl4(9w).
Epigenetic Mechanisms of Fibrosis- and Alcohol-Associated Liver Injury in the Mouse
Increasing evidence indicates that molecular pathways to liver fibrosis and alcohol-induced liver injury include epigenetic mechanisms (Mandrekar, 2011; Pirola and Sookoian, 2016). Thus, we sought to explore effects of alcohol, fibrosis, and combined treatment on liver epigenome by exploring transcriptional changes in DNA methyltransferases (Figure 7A) and histone modifications (Figure 7B). Expression of 3 active DNA methyltransferases (Dnmt1, 3a, and 3b) and methyltransferase-regulating gene Uhrf1 was evaluated. EtOH had a repressive effect on expression of Dnmt3b, whereas fibrosis groups showed upregulation of Dnmt1 and Uhrf1. In CCl4(9w) + EtOH group, Dnmt3b was downregulated, and Dnmt1 and Uhrf1 were prominently upregulated, the latter effect was about 2.5-fold higher than the effect of CCl4-induced fibrosis alone.
Figure 7.

Analysis of the epigenetic effects in the liver in alcohol- and fibrosis-associated mouse model. (A) Quantitative analysis of expression of DNA methylation-associated genes, as indicated on each graph, was performed by qRT-PCR. Gapdh was used as a loading control. (B) Western blot analysis of histone modifications, as indicated on each graph. Scatter plots show the individual values (dots), group medians (line), and standard deviations (whiskers). Values in Cont group were set to 1 and ratios were derived as compared with Cont. Asterisks denote statistical significance as follows: a, p < .05, compared with control group; b, p < .05, compared with CCl4(6w) group; c, p < .05, compared with CCl4(9w) group; d, p < .05, compared with EtOH group. Images of the individual Western blots are shown in Supplementary Figure 2.
H3K27ac, H3K9me3, and H3K4me3 are marks of actively transcribed chromatin, whereas H4K20me3 is a histone mark found in regions of heterochromatin (Barski et al., 2007). Indeed, concordant with the transcriptional responses to various treatments in this mouse model (Figs. 3–5), we found that most pronounced effects on histone modifications were in the CCl4(9w) + EtOH group (Figure 7). An increase in H3K27ac, H3K9me3, and H3K4me3 marks and a decrease of H4K20me3 were significant in the combined treatment group, whereas EtOH alone only affected H4K20me3 (Figure 7B and Supplementary Figure 2).
DISCUSSION
The only approved specific therapy for AH (ie, prednisolone), a form of acute-on-chronic liver injury, was proposed in 1971 (Porter et al., 1971). Since then, no new targeted therapies have been effectively developed, mostly due to the lack of suitable animal drug efficacy testing models that recapitulate the main histological and molecular features of AH in humans. An ideal animal model should reproduce the clinical events, histological findings, and molecular changes that occur in humans. Clinically, AH usually develops in patients with underlying cirrhosis/advanced fibrosis who consume large amounts of alcohol. Several research groups have proposed rodent models that combine liver fibrosis and alcohol treatment. In studies with mice, both drinking water (Han et al., 2017) and liquid diet (Chiang et al., 2013; Roychowdhury et al., 2014) methods of administering alcohol have been used. Regardless of whether alcohol was administered before, together with, or after CCl4, liver fibrosis was exacerbated through pathways that involved oxidative stress, inflammation, stellate cell activation, and angiogenesis. Similar effects were reported in studies in rats (Bingul et al., 2016; Safer et al., 2015). However, a detailed comparison with the histopathological and molecular signatures of human AH has not been performed because only recently the human disease has been well characterized.
In our recent study in which we developed a histological scoring system for patients with AH, we found that the vast majority of patients had underlying cirrhosis (Altamirano et al., 2014). This fact prompted us to seek a model of liver injury where severe fibrosis exists before animals are exposed to large amounts of EtOH. The second requisite was to induce histological changes similar to those described in patients with AH. In our previous human studies (Bataller and Gao, 2015), we found that the pattern of fibrosis was pericellular (“chicken wire”). Reproducing this particular type of fibrosis was an important finding in our model. Finally, the animal model should partially reproduce the main cellular and molecular events that occurs in humans including hepatocellular damage, neutrophil infiltration, passage of gut bacterial and fungi through the portal circulation to the liver, and profound changes in liver transcriptome with an inflammatory and fibrogenic profile (Affo et al., 2013; Yang et al., 2017). Unfortunately, the existing animal models of alcohol-induced liver injury are characterized by only mild changes in liver histology and lack severe pericellular fibrosis (Bertola et al., 2013; Tsukamoto et al., 1985, 1986; Wilkin et al., 2016). Based on the design of the animal model presented herein (EtOH exposure on a chronically damage liver), the histological findings and the transcriptome and epigenetic changes, we conclude that the current model reasonably recapitulates key aspects of human AH. Of course, our model uses a toxicant different from alcohol (ie, CCl4) to induce underlying fibrosis, but this approach was needed, as shown by a number of previous studies (Chiang et al., 2013; Han et al., 2017; Roychowdhury et al., 2014), because rodents do not develop significant fibrosis after chronic exposure to alcohol alone. Despite this limitation, our model represents a new tool to perform preclinical studies to test potential new drugs for patients with AH.
The main consequences of acute-on-chronic liver injury in the setting of AH include systemic inflammatory response (Michelena et al., 2015), hepatocellular failure with a reactive inefficient ductular reaction (Sancho-Bru et al., 2012), as well as neutrophil infiltration and pericellular fibrosis. Although the mechanisms are not well understood, a role for oxidative stress and bacterial and fungal translocation has been proposed (Yang et al., 2017). The existing animal models that employ different protocols of alcohol administration reproduce only few of these changes (Mandrekar et al., 2016). In our study, combined treatment group showed more severe liver injury and difference of inflammation type compared with single-treatment groups. In particular, we found neutrophil infiltration which closely reproduces typical findings in humans (Gao and Tsukamoto, 2016). One of the most important findings of our study is that the combination of CCl4 and EtOH resulted in pericellular type of liver fibrosis, which is the pattern found in patients with AH (Bataller and Gao, 2015). This is important given that fibrosis is a key determinant of the outcome in these patients and that targeting fibrosis is an important goal for novel molecular therapies in AH (Lackner et al., 2017). Recently, several studies reported that different types of inflammatory cells, including neutrophils, accumulate within portal tracts and correlate with liver fibrosis and ductular reaction in human NAFLD (Gadd et al., 2014).
Besides histological similarities, a striking finding of our study is the fact that the combination model (CCl4-induced fibrosis plus alcohol) resulted in liver transcriptome changes that resemble human disease. The overall transcriptome analysis revealed that alcohol and CCl4 are synergistic in deregulating hepatic gene expression. Pathway analysis of gene expression of mice treated with CCl4 + alcohol showed profound changes in terms of inflammation, fibrosis, and hypoxia, which could reflect prolongation of CCl4-induced hepatic stellate cell activation and immune cell infiltration in our model. Compared with CCl4-treated mice, hepatic transcriptome of mice treated with CCl4 + alcohol showed higher number of differentially expressed genes more closely related to human AH liver transcriptome. Interestingly, the enrichment of amino acid and lipid metabolism among downregulated gene sets in our model could reflect the initial stage of hepatocellular failure. This feature has been difficult to reproduce in other mouse models, due to the characteristic resilience of mice to develop liver failure. These results strongly suggest that preexisting live fibrosis sensitizes the liver to the deleterious effects of alcohol. In contrast with these findings, mice treated with alcohol alone showed only mild transcriptome changes, which involved upregulation of genes related to fat metabolism, suggesting that even this intragastric model of alcohol feeding, a model considered to be more severe than others (Ueno et al., 2012), only exhibits early mild disease stage. As we show, the trancriptome of human livers with AH includes, instead, an important downregulation of liver anabolic functions, including lipid metabolism.
We also note that our finding of the presence of E. coli and Candida species in livers of mice treated with CCl4+ alcohol is another feature of this mouse model that is concordant with human AH. Lipopolysaccharide (LPS), an essential component of the outer membrane of gram-negative bacteria, is known to play an important role in the pathogenesis and complications of human AH (Bataller and Mandrekar, 2015). Resident Kuppfer cells are activated by LPS and LPS serum levels are increased in the portal, as well as in the systemic, circulation after excessive alcohol intake (Petrasek et al., 2010; Uesugi et al., 2001). Moreover, we recently showed that LPS mediates ductular cell expansion and that LPS levels predict multiorgan failure and death in patients with AH (Michelena et al., 2015). Concomitantly, the presence of most common Candida species DNA in livers of CCl4 + alcohol mice, parallels recent evidence of fungi translocation in patients with AH. In a recent study by our NIAAA-funded consortium InTeam, a role for Candida in the pathogenesis of AH was proposed (Yang et al., 2017). Mice exposed to alcohol exhibited increased translocation of fungal β-glucan into systemic circulation and β-glucans-activated Kupffer cells via CLEC7A. In that study, microbiome analysis in alcoholic patients showed reduced intestinal fungal diversity and Candida overgrowth. The model reported in this manuscript represents an experimental setting to further explore the role of fungal overgrowth in alcoholic-induced liver injury.
Another important finding of our model is the profound deregulation of epigenetic regulators in mice expose to acute-on-chronic alcohol injury. The study of RNA expression of epigenetic regulator genes confirmed the potential relevance in DNA and chromatin modifications in and alcoholic liver disease-induced liver fibrosis (Massey et al., 2017). In accordance with previous studies (Komatsu et al., 2012), mice treated with CCL4 for 6 weeks showed a decrease in the expression of type 3 DNA methyltransferases (DNMT) genes. To the contrary, long-term treatment with CCL4 (9 weeks), alone or with alcohol administration, increased the levels of DNMT1 and restored levels of DNMT3 mRNA. Interestingly, the increase of DNMTs mRNA levels in mice treated with CCl4 (9 weeks) with or without EtOH were accompanied with higher global levels of histone markers of active transcription, such as H3K27Ac and repressive markers like H3K9me, indicating intense epigenetic remodeling upon liver damage, data concordant with major transcriptional effects we observed in this model. Interestingly, our model of AH showed significant effects of alcohol compared with CCl4(9w)-treated mice. First, the expression of DNMT3b gene remained inhibited, probably revealing an inhibitory effect of alcohol on DNA methylation, seen also in only alcohol-treated mice. Second, the levels of Uhrf1 were higher, suggesting that methylation-related mechanisms are possibly compensating for alcohol-mediated DNMT3b inhibition. Further studies assessing specific gene promoter/enhancers along with RNA-seq data are warranted to uncover the genes which transcription is activated or inhibited, and their potential role of these modulations in development of AH.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
DISCLOSURE
The authors have no conflicts to disclose. The views expressed in this manuscript do not necessarily represent those of the U.S. Food and Drug Administration.
FUNDING
National Institute on Alcohol Abuse and Alcoholism (U01 AA021908).
Supplementary Material
REFERENCES
- Affo S., Dominguez M., Lozano J. J., Sancho-Bru P., Rodrigo-Torres D., Morales-Ibanez O., Moreno M., Millan C., Loaeza-del-Castillo A., Altamirano J. (2013). Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 62, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamirano J., Fagundes C., Dominguez M., Garcia E., Michelena J., Cardenas A., Guevara M., Pereira G., Torres-Vigil K., Arroyo V., et al. (2012). Acute kidney injury is an early predictor of mortality for patients with alcoholic hepatitis. Clin. Gastroenterol. Hepatol. 10, 65–71. [DOI] [PubMed] [Google Scholar]
- Altamirano J., Miquel R., Katoonizadeh A., Abraldes J. G., Duarte-Rojo A., Louvet A., Augustin S., Mookerjee R. P., Michelena J., Smyrk T. C., et al. (2014). A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 146, 1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argemi J., Latasa M. U., Atkinson S. R., Blokhin I., Massey V., Gue J. P., Cabezas J., Lozano J. J., Van Booven D., Bell A., et al. (2018). Defective HNF4a-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat. Commun., in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Bataller R., Gao B. (2015). Liver fibrosis in alcoholic liver disease. Semin. Liver Dis. 35, 146–156. [DOI] [PubMed] [Google Scholar]
- Bataller R., Mandrekar P. (2015). Identifying molecular targets to improve immune function in alcoholic hepatitis. Gastroenterology 148, 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A., Mathews S., Ki S. H., Wang H., Gao B. (2013). Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingul I., Basaran-Kucukgergin C., Aydin A. F., Coban J., Dogan-Ekici I., Dogru-Abbasoglu S., Uysal M. (2016). Betaine treatment decreased oxidative stress, inflammation, and stellate cell activation in rats with alcoholic liver fibrosis. Environ. Toxicol. Pharmacol. 45, 170–178. [DOI] [PubMed] [Google Scholar]
- Brandl K., Hartmann P., Jih L. J., Pizzo D. P., Argemi J., Ventura-Cots M., Coulter S., Liddle C., Ling L., Rossi S. J., et al. (2018). Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 69, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J., Bataller R. (2014). Alcoholic hepatitis: Prognosis and treatment. Gastroenterol. Hepatol. 37, 262–268. [DOI] [PubMed] [Google Scholar]
- Cassmann E., White R., Atherly T., Wang C., Sun Y., Khoda S., Mosher C., Ackermann M., Jergens A. (2016). Alterations of the ileal and colonic mucosal microbiota in canine chronic enteropathies. PLoS One 11, e0147321.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang D. J., Roychowdhury S., Bush K., McMullen M. R., Pisano S., Niese K., Olman M. A., Pritchard M. T., Nagy L. E. (2013). Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One 8, e69114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., et al. (2016). The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya S., Chappell G.A., Iwata Y., Uehara T., Kato Y., Kono H., Bataller R., Rusyn I., et al. (2016). A mouse model of alcoholic liver fibrosis-associated acute kidney injury identifies key molecular pathways. Toxicol. Appl. Pharmacol. 310, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd V. L., Skoien R., Powell E. E., Fagan K. J., Winterford C., Horsfall L., Irvine K., Clouston A. D. (2014). The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 59, 1393–1405. [DOI] [PubMed] [Google Scholar]
- Gao B., Bataller R. (2011). Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B., Tsukamoto H. (2016). Inflammation in alcoholic and nonalcoholic fatty liver disease: Friend or foe? Gastroenterology 150, 1704–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Y., Hu J. N., Wang Z., Wei S. N., Zheng S. W., Wang Y. P., Li W. (2017). 5-HMF attenuates liver fibrosis in CCl4-plus-alcohol-induced mice by suppression of oxidative stress. J. Nutr. Sci. Vitaminol. 63, 35–43. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45(D1), D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y., Waku T., Iwasaki N., Ono W., Yamaguchi C., Yanagisawa J. (2012). Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med. Genomics 5, 5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H., Bradford B. U., Rusyn I., Fujii H., Matsumoto Y., Yin M., Thurman R. G. (2000). Development of an intragastric enteral model in the mouse: Studies of alcohol-induced liver disease using knockout technology. J. Hepatobiliary Pancreat. Surg. 7, 395–400. [DOI] [PubMed] [Google Scholar]
- Lackner C., Spindelboeck W., Haybaeck J., Douschan P., Rainer F., Terracciano L., Haas J., Berghold A., Bataller R., Stauber R. E. (2017). Histological parameters and alcohol abstinence determine long-term prognosis in patients with alcoholic liver disease. J. Hepatol. 66, 610–618. [DOI] [PubMed] [Google Scholar]
- Lahmer T., Messer M., Schwerdtfeger C., Rasch S., Lee M., Saugel B., Schmid R. M., Huber W. (2014). Invasive mycosis in medical intensive care unit patients with severe alcoholic hepatitis. Mycopathologia 177, 193–197. [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey M. R., Mathurin P., Morgan T. R. (2009). Alcoholic hepatitis. N. Engl. J. Med. 360, 2758–2769. [DOI] [PubMed] [Google Scholar]
- Mandrekar P. (2011). Epigenetic regulation in alcoholic liver disease. World J. Gastroenterol. 17, 2456–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar P., Bataller R., Tsukamoto H., Gao B. (2016). Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology 64, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey V., Cabezas J., Bataller R. (2017). Epigenetics in liver fibrosis. Semin. Liver Dis. 37, 219–230. [DOI] [PubMed] [Google Scholar]
- Michelena J., Altamirano J., Abraldes J. G., Affo S., Morales-Ibanez O., Sancho-Bru P., Dominguez M., Garcia-Pagan J. C., Fernandez J., Arroyo V., et al. (2015). Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology 62, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnig M. A. (2017). Immune activation and neuroinflammation in alcohol use and HIV infection: Evidence for shared mechanisms. Am. J. Drug Alcohol. Abuse 43(1), 7–23. doi: 10.1080/00952990.2016.1211667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y., Bono H. (2012). GGRNA: An ultrafast, transcript-oriented search engine for genes and transcripts. Nucleic Acids Res. 40(Web Server issue), W592–W596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanji A. A., Mendenhall C. L., French S. W. (1989). Beef fat prevents alcoholic liver disease in the rat. Alcohol Clin. Exp. Res. 13, 15–19. [DOI] [PubMed] [Google Scholar]
- Petrasek J., Mandrekar P., Szabo G. (2010). Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol. Res. Pract. 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola C. J., Sookoian S. (2016). The modulation of liver methylome in liver diseases. J. Hepatol. 64, 987–988. [DOI] [PubMed] [Google Scholar]
- Porter H. P., Simon F. R., Pope C. E., Volwiler W., Fenster L. F. (1971). Corticosteroid therapy in severe alcoholic hepatitis. A double-blind drug trial. N. Engl. J. Med. 284, 1350–1355. [DOI] [PubMed] [Google Scholar]
- Prado V., Caballeria J., Vargas V., Bataller R., Altamirano J. (2016). Alcoholic hepatitis: How far are we and where are we going? Ann. Hepatol. 15, 463–473. [PubMed] [Google Scholar]
- Roychowdhury S., Chiang D. J., McMullen M. R., Nagy L. E. (2014). Moderate, chronic ethanol feeding exacerbates carbon-tetrachloride-induced hepatic fibrosis via hepatocyte-specific hypoxia inducible factor 1alpha. Pharmacol. Res. Perspect. 2, e00061.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I., Bataller R. (2013). Alcohol and toxicity. J. Hepatol. 59, 387–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safer A. M., Afzal M., Hanafy N., Mousa S. (2015). Green tea extract therapy diminishes hepatic fibrosis mediated by dual exposure to carbon tetrachloride and ethanol: A histopathological study. Exp. Ther. Med. 9, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Bru P., Altamirano J., Rodrigo-Torres D., Coll M., Millán C., José Lozano J., Miquel R., Arroyo V., Caballería J., Ginès P., et al. (2012). Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 55, 1931–1941. [DOI] [PubMed] [Google Scholar]
- Starkel P., Schnabl B. (2016). Bidirectional communication between liver and gut during alcoholic liver disease. Semin. Liver Dis. 36, 331–339. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Kuehn H., Gould J., Tamayo P., Mesirov J. P. (2007). GSEA-P: A desktop application for gene set enrichment analysis. Bioinformatics 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H., French S. W., Benson N., Delgado G., Rao G. A., Larkin E. C., Largman C. (1985). Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 5, 224–232. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H., Towner S. J., Ciofalo L. M., French S. W. (1986). Ethanol-induced liver fibrosis in rats fed high fat diet. Hepatology 6, 814–822. [DOI] [PubMed] [Google Scholar]
- Twedt D. C., Cullen J., McCord K., Janeczko S., Dudak J., Simpson K. (2014). Evaluation of fluorescence in situ hybridization for the detection of bacteria in feline inflammatory liver disease. J. Feline Med. Surg. 16, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T., Pogribny I. P., Rusyn I. (2014). The DEN and CCl4 -induced mouse model of fibrosis and inflammation-associated hepatocellular Carcinoma. Curr. Protoc. Pharmacol. 66, 14 30 1–14 30 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno A., Lazaro R., Wang P. Y., Higashiyama R., Machida K., Tsukamoto H. (2012). Mouse intragastric infusion (iG) model. Nat. Protoc. 7, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesugi T., Froh M., Arteel G. E., Bradford B. U., Thurman R. G. (2001). Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 34, 101–108. [DOI] [PubMed] [Google Scholar]
- Varemo L., Nielsen J., Nookaew I. (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L. W., Boll M., Stampfl A. (2003). Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 33, 105–136. [DOI] [PubMed] [Google Scholar]
- Wernlund P. G., Stoy S., Lemming L., Vilstrup H., Sandahl T. D. (2014). Blood culture-positive infections in patients with alcoholic hepatitis. Scand. J. Infect. Dis. 46, 902–905. [DOI] [PubMed] [Google Scholar]
- Wilkin R. J., Lalor P. F., Parker R., Newsome P. N. (2016). Murine models of acute alcoholic hepatitis and their relevance to human disease. Am. J. Pathol. 186, 748–760. [DOI] [PubMed] [Google Scholar]
- Yang A. M., Inamine T., Hochrath K., Chen P., Wang L., Llorente C., Bluemel S., Hartmann P., Xu J., Koyama Y., et al. (2017). Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 127, 2829–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhari S. (2006). Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 29, 245–254. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.