

Abstract
Evolutionary history is typically portrayed as a branching phylogenetic tree, yet not all evolution proceeds in a purely bifurcating manner. Introgressive hybridization is one process that results in reticulate evolution. Most known examples of genome-wide introgression occur among closely related species with relatively recent common ancestry; however, we present evidence for ancient hybridization and genome-wide introgression between major stem lineages of darters, a species-rich clade of North American freshwater fishes. Previous attempts to resolve the relationships of darters have been confounded by the uncertain phylogenetic resolution of the lineage Allohistium. In this study, we investigate the phylogenomics of darters, specifically the relationships of Allohistium, through analyses of approximately 30,000 RADseq loci sampled from 112 species. Our phylogenetic inferences are based on traditional approaches in combination with strategies that accommodate reticulate evolution. These analyses result in a novel phylogenetic hypothesis for darters that includes ancient introgression between Allohistium and other two major darter lineages, minimally occurring 20 million years ago. Darters offer a compelling case for the necessity of incorporating phylogenetic networks in reconstructing the evolutionary history of diversification in species-rich lineages. We anticipate that the growing wealth of genomic data for clades of non-model organisms will reveal more examples of ancient hybridization, eventually requiring a re-evaluation of how evolutionary history is visualized and utilized in macroevolutonary investigations.
Keywords: Ancient hybridization, darters, Etheostomatinae, phylogenetic networks, phylogenomics, RADseq
One of the primary goals in systematic biology is to reconstruct the evolutionary history of life. With genomic data increasingly accessible for non-model organisms (e.g., Lemmon and Lemmon 2013; McCormack and Faircloth 2013; McCormack et al. 2013), thousands of loci are easily generated for many lineages, potentially providing unprecedented power to infer phylogenetic relationships (Dunn et al. 2008; Faircloth et al. 2013; Jarvis et al. 2014; Prum et al. 2015; Dornburg et al. 2017; Near et al. 2018). There is considerable excitement for the advent of phylogenomics (e.g., Blaimer et al. 2015), yet there are appropriate misgivings (e.g., Hahn and Nakhleh 2015). It is often assumed that simply analyzing more data will reveal a genome-wide central evolutionary trend, providing solutions for the most difficult phylogenetic questions (Wolf et al. 2002; Rokas et al. 2003). While large molecular data sets have certainly helped resolve contentious relationships, they also reveal patterns of substantial gene tree discordance (e.g., Jarvis et al. 2014; Arcila et al. 2016; Shen et al. 2017; Hughes et al. 2018). Genomic data have power to not only resolve phylogenetic relationships, but also to uncover genealogical discordance that would otherwise go undetected.
With thousands of loci providing a window into evolutionary history, we now have an unprecedented ability to both detect phylogenetic incongruence and investigate the causes of discordance. Some phylogenetic incongruence may be the result of incomplete lineage sorting (ILS) that can be accounted for with the multispecies coalescent model in a “species tree” framework (Edwards 2009). ILS can produce a high degree of gene tree incongruence in phylogenomic data sets (e.g., Pollard et al. 2006; Salichos and Rokas 2013). For instance, none of the gene trees inferred from introns, exons, or ultra-conserved elements match the species tree topology inferred from 48 bird genomes (Jarvis et al. 2014). In extreme cases, ILS can result in a mismatch between the most probable gene tree and the underlying species tree (Degnan and Rosenberg 2006).
However, ILS is not the only potential source of gene tree discordance. Hybridization and introgression can also lead to phylogenetic incongruence between loci. While ILS is frequently accounted for in phylogenetic inference using multispecies coalescent models, models that account for horizontal gene flow has seen less widespread use. Even when phylogenetic studies detect hybridization and introgression, they are often limited to cases involving recently diverged taxa. Examples in animals include swordtail fishes (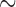 4–6 million years ago [Ma], Cui et al. 2013; Schumer et al. 2016), east African cichlids (
4–6 million years ago [Ma], Cui et al. 2013; Schumer et al. 2016), east African cichlids (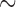 2.8 Ma, Gante et al. 2016), Mexican highland jays (
2.8 Ma, Gante et al. 2016), Mexican highland jays (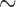 5 Ma, Zarza et al. 2016), Hawaiian ducks (
5 Ma, Zarza et al. 2016), Hawaiian ducks (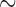 0.4–1.2 Ma, Lavretsky et al. 2015), Campephilus and melanerpine woodpecker (
0.4–1.2 Ma, Lavretsky et al. 2015), Campephilus and melanerpine woodpecker ( 5–11 Ma, Fuchs et al. 2013), equids (
5–11 Ma, Fuchs et al. 2013), equids ( 4.5 Ma, Jónsson et al. 2014), macaques (
4.5 Ma, Jónsson et al. 2014), macaques ( 5 Ma, Fan et al. 2014), humans and Neanderthals (
5 Ma, Fan et al. 2014), humans and Neanderthals ( 0.5 Ma, Green et al. 2010), bears (
0.5 Ma, Green et al. 2010), bears ( 1–2 Ma, Kutschera et al. 2014), felids (
1–2 Ma, Kutschera et al. 2014), felids ( 2–3 Ma, Li et al. 2016), Anopheles mosquitoes (
2–3 Ma, Li et al. 2016), Anopheles mosquitoes ( 0.5–1.85 Ma, Wen et al. 2016), and Heliconius butterflies (
0.5–1.85 Ma, Wen et al. 2016), and Heliconius butterflies ( 2 Ma, Zhang et al. 2016). Most of these examples of gene flow occurred between sister species or recently diverged taxa. However, older gene flow events are also likely to decrease the accuracy of phylogenetic inference (Huson and Bryant 2006; Degnan and Rosenberg 2009). Unfortunately, signals of widespread genomic introgression may erode or obscure the true history of speciation, making detection of old hybridization events challenging (Eaton et al. 2015; Schumer et al. 2016). Thus, the potential impact of genome-wide introgression on deeper time scales remains relatively unknown in animals.
2 Ma, Zhang et al. 2016). Most of these examples of gene flow occurred between sister species or recently diverged taxa. However, older gene flow events are also likely to decrease the accuracy of phylogenetic inference (Huson and Bryant 2006; Degnan and Rosenberg 2009). Unfortunately, signals of widespread genomic introgression may erode or obscure the true history of speciation, making detection of old hybridization events challenging (Eaton et al. 2015; Schumer et al. 2016). Thus, the potential impact of genome-wide introgression on deeper time scales remains relatively unknown in animals.
One group of organisms in which ancient gene flow may be confounding phylogenetic inference are the darters, a clade of North American freshwater fishes comprised of approximately 250 species (Near et al. 2011). There have been many opportunities for gene flow between darter lineages over their approximately 35 million years (myr) of evolutionary history, with at least 63 documented hybrid combinations (Keck and Near 2009; Near and Keck 2013). Indeed, mtDNA alone identifies 33 cases of cyto-nuclear discordance that is likely the result of introgression between darter lineages (Near et al. 2011). One of the most enigmatic putative introgression events involves the lineage Allohistium, which was originally described as a monotypic subgenus of Etheostoma (Bailey and Gosline 1955). Given the results of our study, hereafter, we will refer to Allohistium as a distinct genus (see Appendix). Allohistium currently contains two species, A. cinereum and A. maydeni (Powers et al. 2012). A third distinct species endemic to the Duck River system is awaiting formal taxonomic description, as A. cinereum is paraphyletic in molecular phylogenies relative to the closely related A. maydeni and exhibits phenotypic divergence (Powers et al. 2004, 2012).
The evolutionary origins of Allohistium have long vexed ichthyologists. (Bailey and Gosline (1955, p. 6) wrote “One species [A. cinereum] that seems to not be intimately allied or properly placed in any of the other groups may best stand alone”. They noted that Allohistium may be most closely related to the Etheostoma subclade Oligocephalus or to another genus, Nothonotus (Bailey and Gosline 1955). Allohistium has been traditionally considered one of the most “primitive” Etheostoma lineages based on the characteristics of its lateralis system and the presence of an LDH isozyme found in species of the darter genus Percina, but not in any species of Etheostoma (Page and Whitt 1973, p. 1977). Unlike most darter lineages, molecular data have failed to resolve the phylogenetic placement of Allohistium. Single locus, concatenated, and species tree analyses resolve Allohistium sister to or slightly nested within the Etheostoma subclade Simoperca (Lang and Mayden 2007; Near et al. 2011; Near and Keck 2013), sister to the genus Nothonotus (Lang and Mayden 2007; Smith et al. 2011, 2014), or sister to the genus Etheostoma (Sloss et al. 2004). Out of nine nuclear gene trees inferred for the same set of 92 species using the same phylogenetic methods, four genes resolve Allohistium sister to the Etheostoma subclade Simoperca, while five genes resolve Allohistium outside of Etheostoma (Near and Keck 2013). Allohistium clearly fits the definition of a phylogenetic “rogue” lineage (Wilkinson 1996). This discordance is ancient, as alternative phylogenetic placements of Allohistium occur deep within the darter tree. For instance, Near and Keck (2013) inferred Allohistium as sister to Simoperca with an estimated divergence time of approximately 22 Ma.
We used several phylogenomic approaches to investigate signatures of ancient introgression in darters. Specifically, we used restriction site associated DNA (RAD) sequencing to examine the genome-wide evolutionary history of Allohistium. By deploying high throughput sequence data in combination with methods that explicitly account for gene flow, we reveal that the evolutionary history of Allohistium involved introgression among stem darter lineages approximately 20–24 Ma. This result demonstrates that introgression presents phylogenetic challenges not only for the most closely related species, but also among the deepest relationships in a clade. Our results provide hope for the phylogenetic resolution of other rogue taxa whose complex evolutionary histories may result from ancient hybridization and introgression.
Materials and Methods
Specimen Selection and RAD Sequencing
We extracted DNA from specimens of 112 darter species sampled from all six genera (Allohistium, Etheostoma, Nothonotus, Percina, Crystallaria, and Ammocrypta) and two outgroup percid species (Perca flavescens and Sander canadensis). Each species was represented by one individual, with the exception of A. maydeni (two individuals) and A. cinereum (two individuals) (Supplementary Table A1 available on Dryad). We strategically selected species to cover most major darter lineages, as identified by previous multilocus phylogenetic efforts (Near et al. 2011; Near and Keck 2013). All extractions were performed using Qiagen DNeasy Blood and Tissue Kits (Qiagen, Valencia, CA, USA). The quality of DNA extractions were checked visually using gel electrophoresis, and DNA concentration was quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Library preparation of the RAD loci was performed by Floragenex Inc. (Portland, OR, USA) using a single digest with the SbfI restriction enzyme (5’ CCTGCA 3’) and sample-specific barcodes. Samples were multiplexed into two 95-sample libraries (including 76 non-darter samples from a different project). Each library was prepared twice in order to minimize the influence of technical errors, resulting in four total libraries. Each library was sequenced twice on an Illumina HiSeq 2000 using single-end sequencing at the University of Oregon GC3F facility (https://gc3f.uoregon.edu/).
We used pyrad v.3.0.61 to assemble the RADseq data set from the raw sequencing reads (Eaton 2014). Individual reads that contained more than four sites with Phred scores 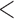 20 were excluded. Reads were clustered using vsearch and an 88% similarity threshold, which allowed approximately 11 base differences between reads within a single cluster. Clusters with a sequencing depth of less than six reads were discarded. Consensus sequences for each cluster were discarded if they contained more than five heterozygous or ambiguous bases. Using an 88% similarity threshold, we clustered consensus sequences across samples. We created several different alignments, requiring loci to be shared by at least 4, 10, 20, or 30 taxa. Hereafter, we refer to these alignments as min4, min10, min20, and min30. Alignments with a higher minimum samples threshold retained fewer loci but contained less missing data.
20 were excluded. Reads were clustered using vsearch and an 88% similarity threshold, which allowed approximately 11 base differences between reads within a single cluster. Clusters with a sequencing depth of less than six reads were discarded. Consensus sequences for each cluster were discarded if they contained more than five heterozygous or ambiguous bases. Using an 88% similarity threshold, we clustered consensus sequences across samples. We created several different alignments, requiring loci to be shared by at least 4, 10, 20, or 30 taxa. Hereafter, we refer to these alignments as min4, min10, min20, and min30. Alignments with a higher minimum samples threshold retained fewer loci but contained less missing data.
Phylogenetic Analyses
We used a concatenated data analysis and several species tree approaches to infer phylogenetic relationships for the 112 sampled species of darters. For each of the four alignments, we concatenated all loci and conducted a maximum likelihood (ML) search plus rapid bootstrapping with RAxML v.8.1.21 (Stamatakis 2014). We applied a GTR+gamma nucleotide substitution model and performed 100 non-parametric bootstrap replicates for each analysis.
To account for ILS, we used two different species tree approaches. The first species tree approach was tetrad v.0.7.19, an implementation of SVDquartets in the software package iPyrad (Chifman and Kubatko 2015, http://github.com/dereneaton/ipyrad). For the tetrad analyses, we used the alignments with the most (min4) and least (min30) amounts of missing data. To avoid issues with linkage, we opted to use only one SNP per locus for a total of 130,095 (min4) and 30,634 SNPs (min30). We inferred all 6,672,876 possible quartets for the 112 sampled taxa. Tetrad joined the individual quartet trees into a supertree using the algorithm implemented by wQMC (Avni et al. 2015). We then constructed a 50% majority-rule consensus tree from the 100 non-parametric bootstrap replicates.
We also employed the summary species tree method ASTRAL v.5.6.2 (Sayyari and Mirarab 2016; Zhang et al. 2018). We inferred gene trees using MrBayes v.3.2.6 (Ronquist et al. 2012) for 30,599 loci in the min30 data set that contained at least one parsimony informative site (PIS). For each gene tree, we used a HKY model of nucleotide substitution and performed two replicate runs of 10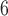 MCMC generations, sampling every 200 generations. If the standard deviation of splits frequencies was greater than 0.05, we continued the MCMC until convergence was achieved. We discarded the first 25% of samples as burn-in and summarized the posterior tree distribution with the “sumt” command. The MrBayes 50% majority-rule consensus gene trees were used as input for ASTRAL. All default settings were used to run ASTRAL. The species trees and concatenated phylogenies were rooted using two outgroup non-darter percid species, S. canadensis and P. flavescens.
MCMC generations, sampling every 200 generations. If the standard deviation of splits frequencies was greater than 0.05, we continued the MCMC until convergence was achieved. We discarded the first 25% of samples as burn-in and summarized the posterior tree distribution with the “sumt” command. The MrBayes 50% majority-rule consensus gene trees were used as input for ASTRAL. All default settings were used to run ASTRAL. The species trees and concatenated phylogenies were rooted using two outgroup non-darter percid species, S. canadensis and P. flavescens.
Time-calibrated Phylogeny
In order to estimate divergence times, we used the species tree method SNAPP v.1.4.1 (Bryant et al. 2012) in BEAST v.2.5.0 (Bouckaert et al. 2014) as described by Stange et al. (2018). We used a reduced data set containing 11 individuals that represented each of the major darter lineages, including two species, E. atripenne and E. pholidotum, that bracket the most recent common ancestor (MRCA) of Simoperca, two species, N. juliae and N. rufilineatus, that bracket the MRCA of Nothonotus, and two species of Allohistium (A. cf. cinereum and A. maydeni). We also selected a single species from Carnipellucida (C. asprella), Percina (P. austropera), Farragoperca (E. artessiae), Goneaperca (E. flabellare), and Gemmaperca (E. vitreum) (Fig. 1b, starred taxa). We constructed a new RAD data subset for these 11 species using the filtering parameters described above, but requiring loci to be shared among all 11 species. We randomly selected one SNP per locus (4,111 total SNPs).
Figure 1.

a) Previous darter phylogenetic hypothesis based on one mtDNA and two nuclear DNA loci, modified from Near et al. (2011). Blue dashed lines indicate alternative phylogenetic placements of Allohistium from previously published studies. b) RAxML phylogeny, minimum 30 specimens sampled per RAD locus. All nodes have 100% bootstrap support unless labeled otherwise. Black nodes indicate clades not present in at least one the species trees. Asterisks indicate species sampled for SNAPP, BUCKy, and phylogenetic network analyses. Allohistium photo by Nate Tessler.
Since gene flow may affect divergence time estimates, we performed an additional time-calibrated species tree analysis after removing loci that may have introgressed between Allohistium and Simoperca. To identify these loci, we examined each gene tree (see Concordance Factor Estimation section). If the gene tree contained a bipartition that grouped Allohistium and Simoperca, we removed the SNP associated with that locus from our species tree analysis. We identified and removed a total of 215 SNPs. Our identification of introgresed loci is likely overly conservative, as not all shared alleles between Allohistium and Simoperca are the result of introgression. We used this reduced data set to infer a second time-calibrated species tree with SNAPP.
The fossil record of darters is limited to the late Pleistocene and does not provide calibrations for relaxed clock analyses (Cavender 1998). We used previously estimated divergence times from Near et al. (2011) that utilized fossil calibrations from the closely related percomorph clade Centrarcidae. We specified a normally distributed calibration prior on the root of darters (mean 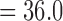 Ma, standard deviation
Ma, standard deviation 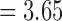 myr). To improve mixing and convergence of the model, we constrained the monophyly of three clades: Etheostoma, Nothonotus, and Allohistium. All three of these clades were consistently resolved as monopyletic with strong support by our other phylogenetic analyses. For each data set, we ran two independent MCMCs for 500,000 generations, discarding 10% of the generations as burn-in. Log and tree traces from the two runs were combined. We estimated the maximum clade credibility tree from the posterior distribution using TreeAnnotator v.2.5.0.
myr). To improve mixing and convergence of the model, we constrained the monophyly of three clades: Etheostoma, Nothonotus, and Allohistium. All three of these clades were consistently resolved as monopyletic with strong support by our other phylogenetic analyses. For each data set, we ran two independent MCMCs for 500,000 generations, discarding 10% of the generations as burn-in. Log and tree traces from the two runs were combined. We estimated the maximum clade credibility tree from the posterior distribution using TreeAnnotator v.2.5.0.
D-statistic Tests for Introgression
In order to detect signatures of introgression within our data set, we applied the D-statistic (ABBA-BABA) test as implemented in pyrad v.3.0.61 (Green et al. 2010; Eaton 2014). The D-statistic test uses a guide species tree in the form of (((P1, P2), P3), O), where O represents an outgroup taxon and P1 and P2 are taxa tested for signatures of introgression with P3. If ILS is the only source of gene tree discordance, then the frequency of derived alleles exclusively present in P1 and P3 should be equal to the frequency of derived alleles exclusively present in P2 and P3 (D  0). However, introgression between P3 and P1 or P2 will result in an excess of shared derived alleles between the taxa that experienced introgression (D
0). However, introgression between P3 and P1 or P2 will result in an excess of shared derived alleles between the taxa that experienced introgression (D 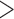 0 or D
0 or D 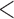 0).
0).
We set up two different species tree scenarios to test for signatures of introgression. For all D-statistic tests, A. pellucida and P. austroperca were combined as the outgroup taxa by pooling SNP frequencies, which increased the number of SNPs available for each D-statistic calculation. For the first set of tests, Allohistium and Nothonotus were specified as sister taxa P1 and P2, and Etheostoma was specified as taxon P3. We performed the D-statistic test using all possible combinations of sampled species following the guide trees outlined above. For each combination of species, we performed 200 bootstrap replicates to estimate the standard deviation in D-statistic scores. We assessed statistical significance by calculating Z-scores from the standard deviations, which were then converted into two-tailed 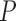 -values. We used alpha = 0.05 as the cutoff for significance after applying a Holm-Bonferroni correction for multiple comparisons using the function p.adjust in R v.3.4.1 (R Core Team 2017).
-values. We used alpha = 0.05 as the cutoff for significance after applying a Holm-Bonferroni correction for multiple comparisons using the function p.adjust in R v.3.4.1 (R Core Team 2017).
The second set of D-statistic tests used a slightly different guide tree that was still consistent with the inferred ML and species tree topologies. Either Nothonotus or Allohistium was specified as taxon P3, while every combination the four major Etheostoma lineages (Simoperca, Gemmaperca, Goneaperca, and Farragoperca) were specified as P1 or P2. This guide tree allowed us to test for the same signals of introgression as our first guide tree (i.e., gene flow between Nothonotus and Etheostoma or between Allohistium and Etheostoma), but without requiring SNPs to be present in both Allohistium and Nothonotus. We performed the D-statistic tests as described above, applying a separate Holm-Bonferroni correction for multiple comparisons.
Concordance Factor Estimation
We assessed the degree of gene tree discordance present in the RAD data by estimating concordance factors (CFs) with BUCKy v.1.4.4 (Ané et al. 2007; Larget et al. 2010). CFs represent the proportion of gene trees that contain a given bipartition (Baum 2007). In order to infer gene trees without any missing data, we used the same subset of RAD data described in the time-calibrated phylogenetic analyses section above. Selecting a subset of 11 taxa allowed us to assemble a complete data matrix and dramatically improved computational efficiency. A reduced number of taxa also allowed us to more easily interpret phylogenetic discordance. We included representatives from each major darter clade, as well as two species each from Simoperca, Nothonotus, and Allohistium to bracket the MRCA of these clades. We used only loci that contained at least one PIS.
First, we inferred a posterior distribution of gene trees using MrBayes v.3.2.6 (Ronquist et al. 2012). For each subsampled RAD locus, we used a HKY model of nucleotide substitution and performed two replicate runs of 10 MCMC generations, sampling every 200 generations. If the standard deviation of splits frequencies was greater than 0.05, we continued the MCMC until convergence was achieved. The posterior distributions of gene trees for each locus was then summarized using the MBSum program in BUCKy, excluding the first 25% of trees as burn-in. This resulted in a posterior distribution of 7500 gene trees for each locus.
MCMC generations, sampling every 200 generations. If the standard deviation of splits frequencies was greater than 0.05, we continued the MCMC until convergence was achieved. The posterior distributions of gene trees for each locus was then summarized using the MBSum program in BUCKy, excluding the first 25% of trees as burn-in. This resulted in a posterior distribution of 7500 gene trees for each locus.
CFs were estimated for three different data subsets: all major darter lineages (11 species), all major darter lineages except Simoperca (nine species), and all major darter lineages except Allohistium (nine species). For each subset, we ran two independent BUCKy analyses for a total of 110 000 MCMC generations, discarding 10 000 MCMC generations as burn-in. We observed no differences in the estimated CF values or topology when using different values for the discordance parameter alpha (0.1, 1, 10, or 100), so we report only the results from analyses with 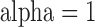 . This choice of discordance parameter resulted in a prior distribution on the number of distinct tree topologies with mean
. This choice of discordance parameter resulted in a prior distribution on the number of distinct tree topologies with mean 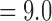 and standard deviation
and standard deviation 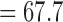 . Our choice of prior corresponds to the assumption that there was a moderate amount of phylogenetic discordance among the individual RAD gene trees. We then reconstructed a consensus tree topology (the primary concordance tree) from the estimated CFs.
. Our choice of prior corresponds to the assumption that there was a moderate amount of phylogenetic discordance among the individual RAD gene trees. We then reconstructed a consensus tree topology (the primary concordance tree) from the estimated CFs.
Phylogenetic Network Analyses
Phylogenetic incongruence can result from either ILS or horizontal gene flow. Therefore, we employed two phylogenetic network approaches in order to distinguish between the two sources of discordance. First, we used the maximum parsimony network analysis (“InferNetwork_MP”) implemented in PhyloNet v.3.6.1 (Than et al. 2008; Yu et al. 2013). The parsimony criterion used to infer the phylogenetic network is a modified version of the minimizing deep coalescences criterion (Maddison 1997). Using the consensus gene trees inferred for the BUCKy analyses, we performed two analyses with the maximum number of reticulations set to either one or two. For each analysis, we performed two independent runs, each with 200 searches. We allowed each search to run until 100 consecutive tree rearrangement proposals failed. All other parameters were left at default settings.
With a moderate number of taxa and loci, full ML or Bayesian inference of phylogenetic networks is computationally prohibitive (Yu et al. 2014). Therefore, as a complementary approach to our maximum parsimony network analyses, we estimated a phylogenetic network using the quartet-based maximum-pseudolikelihood approach implemented in SNaQ v.0.3.0 (Solís-Lemus and Ané 2016). CFs were summarized for each possible quartet using the “bucky.pl” script from the TICR pipeline (https://github.com/nstenz/TICR). We then performed two analyses with the maximum number of reticulations set to either one or two. Each maximum-pseudolikelihood search was started from 30 independent points. To improve search efficiency, the primary concordance tree inferred by BUCKy was used as the starting tree. Optimization parameters were left at default values. Additionally, we performed 100 bootstrap replicates to assess support for the network topology. We calculated 95% credibility intervals (CIs) for the minor hybrid edge inheritance probabilities using the distribution of inheritance probabilities obtained from bootstrap replicates that included that hybrid edge.
In order to test specific cases of gene flow, we performed 22 additional SNaQ analyses with fixed network topologies. For most of these fixed networks, we used the backbone tree as inferred by SNAQ (Allohistium sister to Nothonotus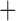 Etheostoma). However, since most other phylogenetic analyses inferred Allohistium as sister to Nothonotus, we also used this arrangement for several of our fixed networks. We specified fixed networks with every possible combination hybrid and parental lineages that could have been detected by our D-statistic tests. We used the “topologyMaxQPseudolik” function to optimize the branch lengths and inheritance probabilities for each network. We then compared the maximum pseudolikelihood scores of each network. Unfortunately, pseudolikelihoods cannot be used in a model fitting framework such as a likelihood ratio test or AIC. Instead, we ranked the networks by their log pseudolikelihood scores. Networks with higher log pseudolikelihood scores were interpreted as a better fit to the data.
Etheostoma). However, since most other phylogenetic analyses inferred Allohistium as sister to Nothonotus, we also used this arrangement for several of our fixed networks. We specified fixed networks with every possible combination hybrid and parental lineages that could have been detected by our D-statistic tests. We used the “topologyMaxQPseudolik” function to optimize the branch lengths and inheritance probabilities for each network. We then compared the maximum pseudolikelihood scores of each network. Unfortunately, pseudolikelihoods cannot be used in a model fitting framework such as a likelihood ratio test or AIC. Instead, we ranked the networks by their log pseudolikelihood scores. Networks with higher log pseudolikelihood scores were interpreted as a better fit to the data.
Results
RAD-Seq Data
We collected RAD data for 112 species that represent all major darter lineages. Our Illumina sequencing produced an average of 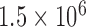 reads per sample after quality filtering. These reads were reduced to an average of 72 000 clusters per sample, with an average sequencing depth of 19 reads per locus. After removing clusters with less than 6X coverage and calling consensus sequences, we were left with an average of 46,000 loci per sample. We created four different alignments, requiring loci to be shared by at least 4, 10, 20, or 30 samples. These data sets ranged from 82% missing data to 44% missing data. Loci counts, number of informative sites, and percent missing data are reported in Supplementary Table A1 available on Dryad.
reads per sample after quality filtering. These reads were reduced to an average of 72 000 clusters per sample, with an average sequencing depth of 19 reads per locus. After removing clusters with less than 6X coverage and calling consensus sequences, we were left with an average of 46,000 loci per sample. We created four different alignments, requiring loci to be shared by at least 4, 10, 20, or 30 samples. These data sets ranged from 82% missing data to 44% missing data. Loci counts, number of informative sites, and percent missing data are reported in Supplementary Table A1 available on Dryad.
Phylogenetic Trees
The maximum likelihood (ML) phylogenies for each of the four concatenated data sets were congruent despite variation in the proportion of missing data. There were only two topological differences and eight nodes that differed in their bootstrap support values among the four concatenated phylogenies (Supplementary Table A2 available on Dryad). Nearly all nodes in the concatenated phylogenies inferred from each alignment were supported by 100% of the bootstrap replicates. Since there was almost no variation between the different concatenated phylogenies, we hereafter only refer to the min30 ML phylogeny. Interestingly, all of the concatenated ML phylogenies resolved, with strong support, an undiscovered clade of darters that contains the Etheostoma subclades Oligocephalus, Microperca, and Hololepis, plus several species that are not part of any Etheostoma subclades outlined in Near et al.’s (2011) phylogenetic classification of darters (Fig. 1b). We refer to this unnamed clade as Farragoperca (see Appendix).
The concatenated ML tree differed from the species trees in the resolution of 13 different clades (Fig. 1). Nine of these conflicts occurred within one of the major clades of darters (Fig. 1). The remaining four topological disagreements involved relationships between the major darter lineages (Fig. 2). None of the species trees perfectly matched the ML concatenated tree, but there were also topological differences among all of the species tree methods. The ASTRAL tree topology was a very close match to the concatenated ML tree, differing at only five nodes. Two of these differences were a result of phylogenetic placement of Gemmaperca (Fig. 2, Supplementary Fig. A2 available on Dryad). Local posterior probabilities were high across the ASTRAL species tree, with only three nodes with posterior probabilities less than 1.0.
Figure 2.

Summary of topological differences involving the major lineages of darters between the concatenated ML tree and the species trees. Dashed lines indicate disagreement between the concatenated tree and the species tree. All nodes are supported with a Bayesian posterior of 1.0 or a bootstrap support value of 100 unless otherwise indicated.
The tetrad species trees had much lower bootstrap support values across many nodes, with five nodes receiving less than 50% bootstrap support in both the min4 and min30 data sets (Supplementary Fig. A1 available on Dryad). There were four topological differences between the min4 tetrad species tree and the min30 tetrad species tree, as well as additional topological conflicts resulting from low bootstrap support values (Supplementary Fig. A1 available on Dryad). There were a number of differences between the tetrad species tree topologies and the concatenated ML tree topology, particularly concerning relationships near the MRCA of Etheostoma. Between both tetrad species trees and the concatenated ML tree, there were a total of 10 topological conflicts (Fig. 1b). These conflicts were found at both deep nodes subtended by major darter lineages and at shallow nodes subtended by only a few species. Nine of the 10 conflicts were resolved with low bootstrap support (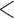 90%) in either the tetrad species trees or the concatenated ML tree. However, one of these conflicting topologies was well supported in both the species trees and the concatenated ML tree. In the concatenated ML tree, Simoperca formed a clade with Gemmaperca with 100% bootstrap support, whereas in the min30 species tree, Gemmaperca formed a clade with all other Etheostoma lineages excluding Simoperca with 100% bootstrap support. Allohistium and Nothonotus were resolved as a clade with 100% bootstrap support in the ML tree, but with only 67% bootstrap support in the min4 species tree. In the min30 species tree, Nothonotus and all lineages of Etheostoma were resolved as a clade, but with only 67% bootstrap support.
90%) in either the tetrad species trees or the concatenated ML tree. However, one of these conflicting topologies was well supported in both the species trees and the concatenated ML tree. In the concatenated ML tree, Simoperca formed a clade with Gemmaperca with 100% bootstrap support, whereas in the min30 species tree, Gemmaperca formed a clade with all other Etheostoma lineages excluding Simoperca with 100% bootstrap support. Allohistium and Nothonotus were resolved as a clade with 100% bootstrap support in the ML tree, but with only 67% bootstrap support in the min4 species tree. In the min30 species tree, Nothonotus and all lineages of Etheostoma were resolved as a clade, but with only 67% bootstrap support.
Time-Calibrated Phylogeny
We inferred a time-calibrated phylogeny using the species tree method SNAPP for two different SNP data sets. The first data set contained 4111 unlinked SNPs shared by 11 taxa that represented each of the major darter lineages. In the second data set, SNPs that have potentially introgressed between Allohistium and Simoperca were removed, leaving 3896 SNPs. Both data sets produced identical tree topologies with very similar age estimates (Fig. 3, Supplementary Fig. A5 available on Dryad). For instance, with all SNPs included, the crown age of Simoperca was estimated as 20.1 Ma (95% HPD 15.9–24.1 Ma), but removing the introgressed SNPs did not change the age estimate for this clade. The only notable difference between the two phylogenies was that when potentially introgressed SNPs were removed, the posterior probability for Goneaperca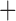 Gemmaperca sister to Simoperca increased from 0.48 to 0.65. All other nodes were supported with Bayesian posterior probability values of 1.0.
Gemmaperca sister to Simoperca increased from 0.48 to 0.65. All other nodes were supported with Bayesian posterior probability values of 1.0.
Figure 3.

Time-calibrated maximum clade credibility species tree inferred using SNAPP, with potentially introgressed SNPs discarded. Grey bars on the nodes indicate 95% highest posterior density for the node heights. Bayesian posterior support for all nodes is 1.0 unless otherwise indicated. Arrows indicate the potential window of time for introgression between the stem lineages of Allohistium and Simoperca.
D-statistic Tests for Introgression
The D-statistic test provides a measure of introgression between taxa given a guide tree topology. We performed two sets of D-statistic tests with different guide tree topologies that were consistent with the results from our phylogenetic analyses. In the first set of 3120 tests, Nothonotus and Allohistium were designated as P1 and P2, while Etheostoma was designated as P3. This allowed us to test for signals of introgression between any Etheostoma species and any species of Allohistium or Nothonotus. Given this guide tree, we found no significant signals of introgression involving Gemmaperca species or most Goneaperca species (Fig. 4). However, 54.2% of all D-statistic tests involving E. boschungi, E. tuscumbia, and E. palididrosum indicated introgression with Nothonotus (Fig. 4, Supplementary Table A4 available on Dryad). This result was not limited to any particular Nothonotus species. Every Nothonotus species except N. acuticeps had some proportion of significant D-statistic tests, ranging from 25% (for all tests involving N. sanguifluus) to 83% (for all tests involving N. juliae) (Supplementary Table A5 available on Dryad). There was also a significant signal of introgression with Nothonotus in 85% of the tests involving Farragoperca species (except for E. trisella). This signal was largely consistent across all species of Nothonotus (Supplementary Table A5 available on Dryad) and across all species of Farragoperca except for E. trisella (Supplementary Table A6 available on Dryad). The strongest signal of introgression from the first set of D-statistic tests was between all species of Allohistium and all species of Simoperca, with 87% of the tests indicating significant gene flow. Again, this signal was consistent across all species of Simoperca (Supplementary Table A6 available on Dryad) and all species of Allohistium (Supplementary Table A7 available on Dryad).
Figure 4.

First set of D-statistic test results. a) Concatenated ML RADseq phylogeny. All nodes are supported with 100% bootstrap support unless otherwise indicated. b) Boxplots show distribution of D-statistic values for each combination of Nothonotus, Allohistium, and the adjacent Etheostoma taxon (40 combinations of Nothonotus and Allohistium per Etheostoma species). D-statistic values less than zero indicate gene flow between Allohistium and the Etheostoma species (more ABBA site patterns compared with BABA site patterns). D-statistic values greater than zero indicate gene flow between Nothonotus and the Etheostoma species (more ABBA site patterns compared with BABA site patterns). c) Guide trees used to generate D-statistic test comparisons (3120 total species combinations).
The second set of D-statistic tests used a slightly different guide tree, with either Allohistium or Nothonotus as P3 and every combination of Simoperca, Gemmaperca, Goneaperca, and Farragoperca as P1 or P2. The second guide tree should have allowed us to detect most of the same signals of introgression as the first guide tree, but while treating Allohistium and Nothonotus independently. For instance, the first guide tree could not have detected gene flow between Simoperca and both Nothonotus and Allohistium. Regardless of the other Etheostoma lineage included in the test, nearly 100% of the D-statistic tests involving Allohistium and Simoperca detected significant gene flow, even after correcting for 26,243 total comparisons (Fig. 5, Supplementary Table A8 available on Dryad). We did not consistently observe signatures of introgression between any other taxa in this second set of D-statistic tests. For instance, while 25% of the D-statistic tests indicated significant gene flow between Farragoperca and Nothonotus with Goneaperca as the P2 lineage, that result disappeared when either Simoperca or Gemmaperca was the P2 lineage (Fig. 5). Thus, the only consistent signature of gene flow detected by all of our D-statistic tests occcured between all species of Simoperca and Allohistium.
Figure 5.

Second set of D-statistic test results. Each density plot shows the distribution of D-statistic values for a particular combination of P1, P2, and P3 lineages. Rows are the different P1 lineages, columns are the different P2 lineages. Plots below the diagonal surrounded by the green line had Nothonotus as the P3 lineage, whereas plots above the diagonal surrounded by the blue line had Allohistium as the P3 lineage. Vertical black dashed line indicates the average D-statistic value for tests involving that group of lineages. Number of D-statistic tests for each combination of lineages is noted on the x-axis of each plot. D-statistic values less than zero indicate gene flow between P1 and P3, whereas D-statistic values greater than zero indicate gene flow between P2 and P3. Red numbers indicate the percent of D-statistic tests for a particular combination of lineages that were statistically significant after correcting for multiple comparisons (alpha 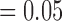 ).
).
Concordance Factors
We investigated genealogical discordance using concordance factors (CFs), which represent the proportion of gene trees that contain a given phylogenetic bipartition. We estimated CFs using a reduced data set that included 11 individuals representing all major darter lineages. We recovered 3516 loci with at least one parsimony informative site (PIS) among the 11 sampled species representing all major darter lineages. The CF consensus tree topology was congruent with the SNAPP species tree topology, depending on how the CF tree is rooted (Figs. 3 and 6). Similar to the concatenated ML tree and most of the species trees, Allohistium and Nothonotus were resolved as sister lineages in the CF consensus tree. CF estimation allows us to examine gene tree support for alternate clades that are not present in the primary concordance tree. We defined conflicting biparitions as clades with CF 95% credible intervals overlapping that of a clade present in the primary concordance tree. We considered that clade a conflicting bipartition. We observed three conflicting bipartitions (Fig. 6a–c), two of which reflect uncertainty regarding the relationships between the subclades of Etheostoma (Fig. 6a and b). The third conflicting bipartition reflects uncertainty in the phylogenetic position of Allohistium and Simoperca (Fig. 6c).
Figure 6.

BUCKy primary concordance tree. Species sampled for each clade are indicated below the clade names. Nodes are labeled with concordance factors, 95% credible interval (CI) in parentheses. Dotted lines indicate conflicting topologies (a-c) with concordance factor 95% CIs that overlap (a-c) with the concordance factor 95% CI of a bipartition in the primary concordance tree.
Using the approach outlined above, we created two data sets that differed in taxon sampling. In the first taxon subsample, we excluded the two species of Allohistium, resulting in 3722 loci with at least one PIS. The primary concordance tree topology and the conflicting bipartition in this analysis matched the topology and one of the conflicting bipartitions from the full data set, though CF values are higher across the tree (Supplementary Fig. A2 available on Dryad). For the second taxon subsample, we excluded the two species of Simoperca but retained the two species of Allohistium, resulting in a data set of 4010 loci with at least one PIS. Again, the primary concordance tree topology did not change, but with Simoperca excluded, we observed no conflicting bipartitions (Supplementary Fig. A3 available on Dryad). Therefore, discordance relating to the phylogenetic position of Allohistium appears linked to Simoperca, but not to any other subclade within Etheostoma.
Phylogenetic Networks
In order to simultaneously account for ILS and horizontal gene flow, we reconstructed phylogenetic networks using both a parsimony criterion and a maximum pseudolikelihood framework (Yu et al. 2013; Solís-Lemus and Ané 2016) (Fig. 7). The network topologies differed from each other in the placement of Nothonotus, which was sister to Allohistium in the parsimony network (Fig. 7b) but sister to Etheostoma in the maximum pseudolikelihood network (Fig. 7a). The networks also differed in the placement of Simoperca, resolved as sister to Gemmaperca in the parsimony network (Fig. 7b) but sister to the clade containing Gemmaperca and Goneaperca in the maximum pseudolikelihood network (Fig. 7a).
Figure 7.

a) Maximum pseudolikelihood network, nodes labeled with bootstrap support values. Gamma is the inheritance probability for the minor hybrid edge, 95% confidence interval in parentheses. b) Maximum parsimony network. Gamma is the inheritance probability for the minor hybrid edge.
In both phylogenetic networks, the stem lineage of Simoperca was connected by a minor hybrid edge to the stem lineage of Allohistium (Fig. 7). This connection was observed in 90% of the bootstrap replicates in the maximum-pseudolikelihood analysis with a maximum of one allowed reticulation. Even when we allowed for a maximum of two reticulations in the pseudolikelihood network analyses, the connection between Allohistium and Simoperca was the only strongly supported reticulation. With two reticulations, 87% of bootstrap replicates contained a minor hybrid edge between Allohistium and Simoperca, while the next best supported hybrid connection (between Gemmaperca and the stem of Etheostoma) was observed in only 25% of the bootstrap replicates. The inferred inheritance probabilities for the minor hybrid edge differed substantially between the pseudolikelihood and parsimony networks (Fig. 7). The minor inheritance probability for the parsimony network was not within the 95% CI for the maximum-pseudolikelihood network minor inheritance probability. Although it is difficult to determine the exact amount of horizontally inherited genetic material, both networks had similar topologies with Simoperca and Allohistium linked by a minor hybrid edge.
We compared the pseudolikelihood scores of several fixed networks to test different gene flow scenarios. Unfortunately, pseudolikelihood scores cannot be used in a statistical model comparison framework. Instead we simply ranked networks based on their pseudolikelihood scores. The pseudolikelihood scores provided similar support for Nothonotus as the sister lineage of Allohistium versus Nothonotus 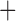 Etheostoma as the sister lineage of Allohistium (Table 1). The network inferred by our unconstrained analysis had the highest pseudolikelihood score (Fig. 7, Table 1). Several other networks with a hybrid edge connecting the stem lineage of Allohistium and the stem lineage of Simoperca were among the highest scoring networks. These included networks where the direction of introgression was reversed (i.e., gene flow from Simoperca to Allohistium rather than from Allohistium to Simoperca) and networks with Allohistium sister to Nothonotus rather than sister to all of Etheostoma. Thus, while Simoperca was identified as the hybrid lineage in our unconstrained maximum pseudolikelihood network analysis (Fig. 7a), the inference of Allohistium as a hybrid lineage was also well supported. Therefore, the optimal network reticulation connects the stem lineage of Allohistium and the stem lineage of Simoperca rather than any particular species or subclades.
Etheostoma as the sister lineage of Allohistium (Table 1). The network inferred by our unconstrained analysis had the highest pseudolikelihood score (Fig. 7, Table 1). Several other networks with a hybrid edge connecting the stem lineage of Allohistium and the stem lineage of Simoperca were among the highest scoring networks. These included networks where the direction of introgression was reversed (i.e., gene flow from Simoperca to Allohistium rather than from Allohistium to Simoperca) and networks with Allohistium sister to Nothonotus rather than sister to all of Etheostoma. Thus, while Simoperca was identified as the hybrid lineage in our unconstrained maximum pseudolikelihood network analysis (Fig. 7a), the inference of Allohistium as a hybrid lineage was also well supported. Therefore, the optimal network reticulation connects the stem lineage of Allohistium and the stem lineage of Simoperca rather than any particular species or subclades.
Table 1.
Results of optimizing branch lengths and inheritance probabilities on fixed networks using SNAQ
| Hybrid lineage | Parental lineage | Allohistium sister group | inheritance probability | score |
|---|---|---|---|---|
| No reticulation | No reticulation | Nothonotus + Etheostoma | N/A |
 459.26 459.26 |
| No reticulation | No reticulation | Nothonotus | N/A |
 460.84 460.84 |
| Simoperca | Allohistium | Nothonotus + Etheostoma | 0.178 | –202.41 |
| Allohistium | Simoperca | Nothonotus | 0.174 | –210.65 |
| Allohistium | E. pholidotum (Simopercaa) | Nothonotus + Etheostoma | 0.173 | –253.62 |
| Allohistium | E. atripinne (Simopercaa) | Nothonotus + Etheostoma | 0.179 | –254.57 |
| Allohistium | Simoperca | Nothonotus + Etheostoma | 0.175 | –254.88 |
| Simoperca | Allohistium | Nothonotus | 0.161 | –275.79 |
| Nothonotus | Farragoperca | Nothonotus + Etheostoma | 0.10 |
 377.29 377.29 |
| Farragoperca | Nothonotus | Nothonotus + Etheostoma | 0.12 |
 381.53 381.53 |
| E. atripinne (Simopercaa) | Allohistium | Nothonotus + Etheostoma | 0.0069 | –398.18 |
| E. pholidotum (Simopercaa) | Allohistium | Nothonotus + Etheostoma | 0.0023 | –398.19 |
| Simoperca | Nothonotus | Nothonotus + Etheostoma | 0.10 |
 437.24 437.24 |
| Allohistium | Gemmaperca | Nothonotus + Etheostoma | 0 |
 458.26 458.26 |
| Gemmaperca | Allohistium | Nothonotus + Etheostoma | 0 |
 458.30 458.30 |
| Goneaperca | Allohistium | Nothonotus + Etheostoma | 0 |
 458.30 458.30 |
| Goneaperca | Nothonotus | Nothonotus + Etheostoma | 0 |
 458.45 458.45 |
| Allohistium | Goneaperca | Nothonotus + Etheostoma | 0 |
 458.52 458.52 |
| Allohistium | Farragoperca | Nothonotus + Etheostoma | 0 |
 458.55 458.55 |
| Nothonotus | Gemmaperca | Nothonotus + Etheostoma | 0 |
 458.63 458.63 |
| Allohistium | Nothonotus | Nothonotus + Etheostoma | 0 |
 458.70 458.70 |
| Farragoperca | Allohistium | Nothonotus + Etheostoma | 0 |
 458.74 458.74 |
| Nothonotus | Allohistium | Nothonotus + Etheostoma | 0 |
 458.81 458.81 |
| Gemmaperca | Nothonotus | Nothonotus + Etheostoma | 0 |
 460.71 460.71 |
| Nothonotus | Simoperca | Nothonotus + Etheostoma | 0.0799 |
 462.92 462.92 |
| Nothonotus | Goneaperca | Nothonotus + Etheostoma | 0.10 |
 1117.4 1117.4 |
Note: Networks involving gene flow between Allohistium and Simoperca are in bold.
Discussion
Analyses of RADseq data result in a unique phylogenetic hypothesis for darters despite discordance among inference methods (Figs. 1b, 2, and 3, Supplementary Figs. A1 and A2 available on Dryad). There are 13 topological differences between the species trees and the concatenated ML phylogeny (Figs. 1b and 2). One of the most notable differences is the phylogenetic placement of Allohistium. Our traditional phylogenetic analyses utilizing tens of thousands of RAD loci are not able to resolve the placement of Allohistium with greater confidence than analyses of three Sanger sequenced loci (Near et al. 2011). However, additional analyses reveal that the elusive evolutionary history of Allohistium is the product of ancient gene flow that occurred approximately 20–24 Ma.
Darter Evolutionary Relationships
Phylogenies inferred from thousands of RAD loci offer important insights into the evolutionary relationships of darters. Previous phylogenies inferred using DNA sequences provided poor support for the relationships among the major darter lineages or resolved Percina as the sister lineage of all other darters (Near et al. 2011; Near and Keck 2013). The concatenated ML RADseq phylogeny and the ASTRAL species tree provide high support for the deepest nodes in the darter tree and resolve the earliest bifurcation between a clade containing Carnipellucida and Percina as the sister lineage of a clade containing Etheostoma, Nothonotus, and Allohistium (Fig. 2). However, our other species tree approaches (SNAPP and tetrad) resolve Carnipellucida as the sister lineage of the rest of darters, although this inference is poorly supported in the tetrad species trees (Fig. 2). There is much less phylogenetic resolution among the major lineages of darters in some of the species trees, indicating a complex history of lineage sorting deep in the darter phylogeny (Supplementary Fig. A1 available on Dryad). Within Etheostoma, all of the species trees and the concatenated ML RADseq phylogeny resolve a new and inclusive clade that we name Farragoperca (see Appendix), which includes the subclades Oligocephalus, Fuscatellum, Microperca, Hololepis, and several species that were not classified in any subclade due to lack of resolution in previous molecular phylogenies (Near et al. 2011).
While providing new insights into the relationships of darters, the RADseq phylogenies include relationships that were initially discovered in the first comprehensive molecular analyses of the clade, including the monophyly of both Goneaperca and Gemmaperca, the relationships within Simoperca, and a clade containing E. baileyi and E. histrio (Fig. 1b, Supplementary Figs. A1 and A2 available on Dryad; Near et al. 2011; Near and Keck 2013). There are still important areas of darter phylogenetics requiring additional research as evidenced by the differences between the concatenated ML phylogeny and the species tree inferences. For example, the clade Catonotus, as traditionally delimited, is paraphyletic in the ML tree (Fig. 1b) and the ASTRAL species tree (Supplementary Fig. A2 available on Dryad), but is resolved as monophyletic with moderate node support in the min4 tetrad species tree (Supplementary Fig. A1 available on Dryad). A similar result is observed in analyses of 13 Sanger sequenced nuclear genes, where the concatenated Bayesian analysis resolves Catonous as paraphyletic, but a species tree analysis supports monophyly of the group (Harrington and Near 2015). The ML and species tree analyses demonstrate the potential for RADseq data to provide a phylogenomic perspective on the evolutionary history of darter diversification.
Substantial Gene Tree Discordance
In order to determine gene tree support for the deepest nodes in the darter phylogeny, we estimated concordance factors (CFs) for a subset of 11 taxa. CFs represent the proportion of gene trees that contain a given phylogenetic bipartition. The CF consensus tree topology did not match the backbone topology of the ML tree or any of the species trees except for the SNAPP tree, depending on how the CF consensus tree is rooted. A closer examination reveals three alternative topologies with statistically significant gene tree support that are not present in the consensus tree (Fig. 6a–c). Two of the three alternative topologies reflect uncertainty in the relationships between the major lineages of Etheostoma (Fig. 6a and b). One of these conflicting topologies is a clade containing Farragoperca, Goneaperca, and Gemmaperca (Fig. 6a), which is also resolved in the ASTRAL species tree and the tetrad min30 tree (Supplementary Figs. A1 and A2 available on Dryad). The other conflicting topology is a clade containing Gemmaperca and Simoperca, which was inferred by our concatenated ML analyses. Gene tree discordance among the lineages that subtend the MRCA of Etheostoma could be the result of ILS, as each conflicting bipartition is only separated by two nodes in the phylogeny (Fig. 6a and b).
The third alternative topology in our CF analyses involves three divergent lineages: Allohistium, Nothonotus, and Simoperca. In the consensus tree (Fig. 6), Allohistium and Nothonotus form a clade (CF 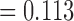 ) and Simpoerca is the sister lineage of a clade containing Gemmaperca and Goneaperca (CF
) and Simpoerca is the sister lineage of a clade containing Gemmaperca and Goneaperca (CF 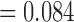 ). However, there is also substantial gene tree support for monophyly of Allohistium and Simoperca (CF
). However, there is also substantial gene tree support for monophyly of Allohistium and Simoperca (CF 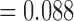 ). This discordance cannot be accounted for by ILS. For unlinked loci, sorting of ancestral variation into descendant lineages should proceed randomly (Tajima 1983; Hudson 2002). Therefore, ILS could produce shared ancestral variation between Allohistium and Simoperca, but it should also produce equal amounts of shared ancestral variation between Allohistium and the other major lineages of Etheostoma (i.e., Farragoperca, Goneaperca, and Gemmaperca). However, gene trees indicate that Allohistium is the sister lineage of either Nothonotus or Simoperca, but not any other lineage of Etheostoma. Therefore, a mechanism other than ILS is necessary to explain the gene tree discordance involving Allohistium, Simoperca, and Nothonotus.
). This discordance cannot be accounted for by ILS. For unlinked loci, sorting of ancestral variation into descendant lineages should proceed randomly (Tajima 1983; Hudson 2002). Therefore, ILS could produce shared ancestral variation between Allohistium and Simoperca, but it should also produce equal amounts of shared ancestral variation between Allohistium and the other major lineages of Etheostoma (i.e., Farragoperca, Goneaperca, and Gemmaperca). However, gene trees indicate that Allohistium is the sister lineage of either Nothonotus or Simoperca, but not any other lineage of Etheostoma. Therefore, a mechanism other than ILS is necessary to explain the gene tree discordance involving Allohistium, Simoperca, and Nothonotus.
Evidence of Ancient Gene Flow
One potential reason for phylogenetic discordance involving Allohistium and Simoperca is gene flow via introgression. Our D-statistic tests indicate a strong signal of introgression between all species of Allohistium and Simoperca. This result was largely insensitive to the specified guide tree topology. By comparison, all other signals of introgression detected in our D-statistic tests (e.g., Farragoperca-Nothonotus gene flow) were only significant for a particular set of guide trees. It remains unclear why other signals of gene flow were not consistently uncovered across all D-statistic test. D-statistic tests can be misled by various factors such as ancestral population structure (Eriksson and Manica 2012) or low effective population sizes (Martin et al. 2015). Even with the shortcomings of the D-statistic, there is a robust signal of gene flow across all tests between Allohistium and Simoperca.
To supplement our D-statistic tests, we used two phylogenetic network approaches to determine whether simultaneously accounting for ILS and gene flow can explain genealogical discordance involving Allohistium. Though the inferred networks differ slightly in topology, none of them identify reticulation among the sampled Etheostoma subclades, an area of the phylogeny with two conflicting bipartitions in the CF analyses (Fig. 6a and b). Therefore, uncertainty in the relationships among the major Etheostoma subclades is best explained by ILS. However, both phylogenetic network approaches consistently uncover the same minor hybrid edge connecting the stem lineage of Allohistium to the stem lineage of Simoperca, an inference that was insensitive to the number of reticulations allowed in the network (Fig. 7).
The directionality of gene flow inferred from the network analysis indicates that Simoperca was the recipient of genetic material from Allohisitum. However, we found that networks with Allohistium as the hybrid lineage have very similar pseudolikelihood scores. Unfortunately, the models used in both network approaches do not allow for bidirectional gene flow. SNaQ in particular assumes that the true network is a level-1 network, meaning no two edges can be part of the same cycle (Solís-Lemus and Ané 2016). This assumption precludes the possibility of inferring gene flow from Allohistium to Simoperca and from Simoperca to Allohistium in the same analysis. In addition, some network topologies produce identical gene tree distributions and are thus indistinguishable (Pardi and Scornavacca 2015). These limitations make it challenging to interpret the biological significance of the inferred direction of introgression. However, we suspect that Allohistium is more likely to be the recipient of gene flow than Simoperca. In all prior molecular phylogenetic studies of darters, Allohistium exhibits “rouge” lineage behavior, whereas Simoperca is consistently nested within Etheostoma (Song et al. 1998; Smith et al. 2011, 2014; Near et al. 2011; Near and Keck 2013).
The network analyses estimate that 17–47% of the genome is shared between Allohistium and Simoperca due to reticulation rather than vertical inheritance. Based on both the network and D-statistic analyses, the admixture of Allohistium is not derived from a specific Simoperca species or subclade, but rather from the stem lineage of Simoperca. There are two potential explanations for the reticulation involving Allohistium. First, species currently represented by the stem lineage of Allohistium could have hybridized with those that comprise the stem lineage of Simoperca. Introgression between these stem lineages would have left all extant Allohistium species with an appreciable fraction of their genomes derived from the stem lineage of Simoperca. Another potential explanation is that Allohistium is the result of ancient homoploid hybrid speciation (HHS) between the stem lineage of Simoperca and another darter lineage. In several of our phylogenetic analyses, Allohistium is sister to Nothonotus, suggesting that Nothonotus could be the other parental darter lineage.
Genomic mosaicism is often used as evidence of HHS, but genomic mosaicism alone cannot show that hybridization was critical to development of reproductive isolation (Schumer et al. 2014). Gross and Rieseberg (2005) suggest that ecological selection is an important factor in the formation of hybrid species. Hybrids may possess intermediate or extreme phenotypes that allow them to exploit niche space that is unoccupied by either parental lineage, leading to reduced levels of gene flow by habitat segregation (Gross and Rieseberg 2005; Chapman and Burke 2007; Nolte and Tautz 2010). In addition to genomic mosaicism, Allohistium also exhibits morphological and ecological divergence relative to its putative parental lineages, Simoperca and Nothonotus. Among darters, Allohistium has a unique body shape, with the longest snout-to-body ratio of any Etheostoma or Nothonotus species and papillose lips not found in any other darter species (Kuehne and Barbour 1983, p. 1983). The elongated snout and papillose lips might serve as adaptations for feeding on burrowing invertebrates (Shepard and Burr 1984). Allohistium also occupies a different habitat than Simoperca or Nothonotus. All 20 species of Nothonotus and 32 out of 35 species of Simoperca occupy riffles, shallow areas of streams and rivers characterized by rocky substrate and substantial current (Kuehne and Barbour 1983, p. 1983). Many species of Simoperca species utilize deeper pool habitats in rivers, but most of those species are also associated with nearby riffle habitat (Kuehne and Barbour 1983, p. 1983). Allohistium, on the other hand, is found only in deeper pools or eddies (0.5–1.75 m) with sluggish current that are often heavily vegetated (Page 1981; Kuehne and Barbour 1983; Shepard and Burr 1984). Thus, it appears that the unique morphology of Allohistium may have allowed it to exploit habitat unoccupied by either of its putative parental lineages. Additionally, Allohistium and Simoperca share similar reproductive strategies by attaching eggs to rocks or vegetation without parental care, whereas species of Nothonotus bury eggs without parental care or clump eggs in nests cavities that are tended by a male (Kelly et al. 2012). Though it is difficult to prove whether these unique traits arose via hybridization, phenotypic and ecological divergence suggests that HHS is a possible explanation for the genomic mosaicism of Allohistium.
Biogeography potentially holds some clues to the historical gene flow between Simoperca and Allohistium. Currently, Allohistium is sporadically distributed in the Tennessee, Duck, and Cumberland River systems. It appears to be rare over most of its range, with many local extirpations in the last few decades (Powers et al. 2012). Given the current patchiness of its distribution, it seems plausible that Allohistium was once widespread throughout the Cumberland, Duck, and Tennessee Rivers prior to modern impoundments. Only 15 of the 35 species of Simoperca occupy the same river drainages as Allohistium. However, our D-statistic tests indicate strong signals of introgression between Allohistium and all sampled species of Simoperca, including many that do not co-occur (e.g., E. bellator and E. rupestre, both Mobile River drainage endemics). Gene flow between the stem lineages of Allohistium and Simoperca would have required overlap between the ancestral ranges of these lineages, but species that arose and subsequently dispersed after hybridization would still retain signatures of ancestral gene flow. While there has been no attempt to reconstruct ancestral areas for darter lineages, the presence of some extant Simoperca species in the same river drainages as Allohistium suggests that their ancestral ranges may have overlapped, allowing for gene flow. Alternatively, if Allohistium was the product of hybrid speciation between Nothonotus and Simoperca, there must have been range overlap between these parental lineages. Currently, 11 of the 20 Nothonotus species are found in the Cumberland, Duck, and/or Tennessee River systems, suggesting that the ancestral range of Nothonotus overlapped with the ancestral ranges of Allohistium and Simoperca.
Timing of Reticulate Evolution
Regardless of whether Allohistium is a homoploid hybrid lineage, it represents the oldest known example of genome-wide introgression in animals. Our D-statistic tests indicate strong signals of gene flow between all extant Allohistium and Simoperca species, and our phylogenetic networks infer a minor hybrid edge connecting the stem lineage of Allohistium to the stem lineage of Simoperca. These results are consistent with a scenario of ancient gene flow deep within the darter phylogeny. Our RADseq time-calibrated tree estimates that the stem lineage of Simoperca existed from approximately 20.1 Ma to 24.3 Ma, while the stem lineage of Nothonotus existed from approximately 16.8 Ma to 24.9 Ma (Fig. 3), consistent with previous age estimates for these clades (Near et al. 2011; Near and Keck 2013). Since the genome of Allohistium appears to be derived from the stem lineages of Simoperca and Nothonotus, the reticulation event must have occurred between approximately 20.1 Ma and 24.3 Ma. Ancient hybridization is supported by mitochondrial-nuclear gene tree incongruence (Near et al. 2011), discordance between a handful of nuclear gene trees (Near and Keck 2013), and genealogical conflict we uncovered in thousands of genome-wide RAD loci (Fig. 6).
Our results demonstrate that genomic analyses can address difficult phylogenetic questions, but not always with a simple answer. We reveal that Allohistium, long recognized by ichthyologists for its unusual morphology and taxonomy, has an equally unusual evolutionary history. Its complicated evolutionary history reconstructed from thousands of genomic loci cannot be explained by ILS alone. Rather, the genome of Allohistium appears to be the product of ancient gene flow approximately 20–24 Ma between the stem lineages of two major darter clades. Thorough investigation of phylogenomic discordance in other clades using models that account for gene flow may begin to uncover many examples of ancient non-treelike evolutionary history.
Acknowledgments
The authors thank D.A.R. Eaton for guidance with collecting RADseq data and pyrad assembly/analysis. A. Dornburg and M.A. Correa assisted with extraction preparation and quantification. C. Ané and N. Stenz provided help with bioinformatics for the SNaQ network analysis. G.J. Watkins-Colwell assisted with museum collections. Colleagues and members of the Near and Donoghue lab provided valuable discussion and feedback, including E. Benavides, P.F. Cowman, A. Ghezelayagh, J.R. Glass, R.H. Harrington, B.P. Keck, D. Kim, C.E. Parker, R.G. Simpson, S. Federman, M.J. Landis, B. Park, M. Sinnott-Armstrong, and E.L. Spriggs. DJM thanks his dissertation committee members M.J. Donoghue, F.T. Burbrink, and D.M. Post for their support. Two anonymous reviewers and our associate editor provided thoughtful comments that helped improve an earlier version of this manuscript.
Appendix
Phylogenetic Definitions of Darter Clade Names Introduced in this Study
Allohistium Bailey and Gosline 1955:6 [D.J. MacGuigan & T.J. Near], converted clade name. Previously recognized as a subgenus (Bailey and Gosline 1955). Though we define Allohistium as a rank-free clade, we apply the name Allohistium as a genus in our binomial nomeclature to ensure that the genus Etheostoma represents a monophyletic group. Definition (node-based). The least inclusive clade containing Allohistium cinereum (Storer 1845), Allohistium maydeni (Powers & Kuhajda 2012: 52–53) in Powers et al. (2012), and Allohistium cf. cinerium, an undescribed species endemic to the Duck and Bufflalo River systems (Powers et al. 2012). Refernce phylogeny. Figure 1b. Composition. Includes the species designated in the definition. Synapomorphies. Species of Allohistium are distinguished from all other Etheostomatinae by the presence of unique allele of Ldh-B and papillose lips (Shepard and Burr 1984, Dimmick and Page 1992). Type species. Allohistium cinereum (Storer 1845:49).
Farragoperca D.J. MacGuigan & T.J. Near, new clade name. Definition (node-based). The least inclusive clade containing Etheostoma trisella, E. parvipinne, E. fusiforme, E. mariae, E. microperca, E. proliare, E. edwini, E. hopkinsi, E. spectabile, E. radiosum, E. caeruleum, and E. lepidum. Etymology. From the Latin farrago meaning a mixed fodder or mash and the Greek word 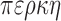 meaning perch. Refernce phylogeny. Figure 1b. Composition. Includes the species designated in the definition and the previosly defined clades Oligocephalus, Fuscatellum, Microperca, and Hololepis (Near et al. 2011). In addition, Farragoperca includes E. collis, E. saludae, E. fricksium, E. binotatum, E. okaloosae, E. gracile and, E. zonifer, which were not classified in any subclae of Etheostoma (Near et al. 2011: Fig. 4). Type species: Etheostoma trisellaBailey & Richards 1963:14.
meaning perch. Refernce phylogeny. Figure 1b. Composition. Includes the species designated in the definition and the previosly defined clades Oligocephalus, Fuscatellum, Microperca, and Hololepis (Near et al. 2011). In addition, Farragoperca includes E. collis, E. saludae, E. fricksium, E. binotatum, E. okaloosae, E. gracile and, E. zonifer, which were not classified in any subclae of Etheostoma (Near et al. 2011: Fig. 4). Type species: Etheostoma trisellaBailey & Richards 1963:14.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.1c9g5mm. Raw sequence data is available for download from the NCBI Sequence Read Archive (SRA) (BioProject PRJNA505339).
Funding
This work was supported by the Bingham Oceanographic Fund administered by the Peabody Museum of Natural History, Yale University. D.J.M. was supported by the Yale Training Program in Genetics (National Institute of Health grant number T32 GM007499).
References
- Ané C., Larget B., Baum D.A., Smith S.D., Rokas A.. 2007. Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 24:412–426. [DOI] [PubMed] [Google Scholar]
- Arcila D., Orti G., Vari R., Armbruster J.W., Stiassny M.L.J., Ko K.D., Sabaj M.H., Lundberg J., Revell L.J., Betancur-R R.. 2017. Genome-wide interrogation advances resolution of recalcitrant groups in the tree of life. Nat. Ecol. Evol. 1. [DOI] [PubMed] [Google Scholar]
- Avni E., Cohen R., Snir S.. 2015. Weighted quartets phylogenetics. Syst. Biol. 64:233–242. [DOI] [PubMed] [Google Scholar]
- Bailey R.M., Gosline W.A.. 1955. Variation and systematic significance of vertebral counts in the American fishes of the family Percidae. Misc. Publ. Mus. Zool. Univ. Mich. 93:1–44. [Google Scholar]
- Baum D.A. 2007. Concordance trees, concordance factors, and the exploration of reticulate genealogy. Taxon. 56:417–426. [Google Scholar]
- Blaimer B.B., Brady S.G., Schultz T.R., Lloyd M.W., Fisher B.L., Ward P.S.. 2015. Phylogenomic methods outperform traditional multi-locus approaches in resolving deep evolutionary history: a case study of formicine ants. BMC Evol. Biol. 15:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R.R., Heled J., Kuehnert D., Vaughan T.G., Wu C.-H., Xie D., Suchard M.A., Rambaut A., Drummond A.J.. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10.e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D., Bouckaert R., Felsenstein J., Rosenberg N.A., RoyChoudhury A.. 2012. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29:1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavender T.M. 1998. Development of the North American Tertiary freshwater fish fauna. Ital. J. Zool. 65S:S149–S161. [Google Scholar]
- Chapman M.A., Burke J.M.. 2007. Genetic divergence and hybrid speciation. Evolution. 61:1773–1780. [DOI] [PubMed] [Google Scholar]
- Chifman J., Kubatko L.. 2015. Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes. J. Theor. Biol. 374:35–47. [DOI] [PubMed] [Google Scholar]
- Cui R., Schumer M., Kruesi K., Walter R., Andolfatto P., Rosenthal G.G.. 2013. Phylogenomics reveals extensive reticulate evolution in Xiphophorus fishes. Evolution. 67:2166–2179. [DOI] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2006. Discordance of species trees with their most likely gene trees. PLoS Genet. 2:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Dornburg A., Townsend J.P., Brooks W., Spriggs E., Eytan R.I., Moore J.A., Wainwright P.C., Lemmon A., Moriarty E., Near T.J.. 2017. New insights on the sister lineage of percomorph fishes with an anchored hybrid enrichment dataset. Mol. Phylogenet. Evol. 110:27–38. [DOI] [PubMed] [Google Scholar]
- Dunn C.W., Hejnol A., Matus D.Q., Pang K., Browne W.E., Smith S.A., Seaver E., Rouse G.W., Obst M., Edgecombe G.D., Sørensen M.V., Haddock S.H.D., Schmidt-Rhaesa A., Okusu A., Kristensen R.M., Wheeler W.C., Martindale M.Q., Giribet G.. 2008. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 452:745–749. [DOI] [PubMed] [Google Scholar]
- Eaton D.A.R. 2014. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics. 30:1844–1849. [DOI] [PubMed] [Google Scholar]
- Eaton D.A.R., Hipp A.L., Gonzalez-Rodriguez A., Cavender-Bares J.. 2015. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution. 69:2587–2601. [DOI] [PubMed] [Google Scholar]
- Edwards S.V. 2009. Is a new and general theory of molecular systematics emerging? Evolution. 63:1–19. [DOI] [PubMed] [Google Scholar]
- Eriksson A., Manica A.. 2012. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proc. Natl. Acad. Sci. USA. 109:13956–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth B.C., Sorenson L., Santini F., Alfaro M.E.. 2013. A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs). PLoS One. 8.e65923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z., Zhao G., Li P., Osada N., Xing J., Yi Y., Du L., Silva P., Wang H., Sakate R., Zhang X., Xu H., Yue B., Li J.. 2014. Whole-genome sequencing of Tibetan macaque (Macaca thibetana) provides new insight into the macaque evolutionary history. Mol. Biol. Evol. 31:1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J., Pons J., Liu L., Ericson P.G.P., Couloux A., Pasquet E.. 2013. A multi-locus phylogeny suggests an ancient hybridization event between Campephilus and melanerpine woodpeckers (Aves: Picidae). Mol. Phylogenet. Evol. 67:578–588. [DOI] [PubMed] [Google Scholar]
- Gante H.F., Matschiner M., Malmstrom M., Jakobsen K.S., Jentoft S., Salzburger W.. 2016. Genomics of speciation and introgression in Princess cichlid fishes from Lake Tanganyika. Mol. Ecol. 25:6143–6161. [DOI] [PubMed] [Google Scholar]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.-Y., Hansen N.F., Durand E.Y., Malaspinas A.-S., Jensen J.D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L.F., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S.. 2010. A draft sequence of the neandertal genome. Science. 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross B.L., Rieseberg L.H.. 2005. The ecological genetics of homoploid hybrid speciation. J. Hered. 96:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.W., Nakhleh L.. 2015. Irrational exuberance for resolved species trees. Evolution. 70:7–17. [DOI] [PubMed] [Google Scholar]
- Harrington R.C., Near T.J.. 2015. Phylogenetic relationships of Goneaperca and the evolution of parental care in darters (Teleostei: Percidae). Mol. Phylogenet. Evol. 34:158–165. [DOI] [PubMed] [Google Scholar]
- Hudson R.R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 18:337–338. [DOI] [PubMed] [Google Scholar]
- Hughes L.C., Ortí G., Huang Y., Sun Y., Baldwin C.C., Thompson A.W., Arcila D., Betancur-R R., Li C., Becker L., Bellora N., Zhao X., Li X., Wang M., Fang C., Xie B., Zhou Z., Huang H., Chen S., Venkatesh B., Shi Q.. 2018. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. USA. 115:6249–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H., Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. [DOI] [PubMed] [Google Scholar]
- Jarvis E.D., Mirarab S., Aberer A.J., Li B., Houde P., Li C., Ho S.Y.W., Faircloth B.C., Nabholz B., Howard J.T., Suh A., Weber C.C., da Fonseca R.R., Li J., Zhang F., Li H., Zhou L., Narula N., Liu L., Ganapathy G., Boussau B., Bayzid M.S., Zavidovych V., Subramanian S., Gabaldón T., Capella-Gutiérrez S., Huerta-Cepas J., Rekepalli B., Munch K., Schierup M., Lindow B., Warren W.C., Ray D., Green R.E., Bruford M.W., Zhan X., Dixon A., Li S., Li N., Huang Y., Derryberry E.P., Bertelsen M.F., Sheldon F.H., Brumfield R.T., Mello C.V., Lovell P.V., Wirthlin M., Schneider M.P.C., Prosdocimi F., Samaniego J.A., Velazquez A.M.V., Alfaro-Núñez A., Campos P.F., Petersen B., Sicheritz-Ponten T., Pas A., Bailey T., Scofield P., Bunce M., Lambert D.M., Zhou Q., Perelman P., Driskell A.C., Shapiro B., Xiong Z., Zeng Y., Liu S., Li Z., Liu B., Wu K., Xiao J., Yinqi X., Zheng Q., Zhang Y., Yang H., Wang J., Smeds L., Rheindt F.E., Braun M., Fjeldsa J., Orlando L., Barker F.K., Jønsson K.A., Johnson W., Koepfli K.-P., O’Brien S., Haussler D., Ryder O.A., Rahbek C., Willerslev E., Graves G.R., Glenn T.C., McCormack J., Burt D., Ellegren H., Alström P., Edwards S.V., Stamatakis A., Mindell D.P., Cracraft J., Braun E.L., Warnow T., Jun W., Gilbert M.T.P., Zhang G.. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 346:1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson H., Schubert M., Seguin-orlando A., Ginolhac A., Petersen L., Fumagalli M., Albrechtsen A., Petersen B., Korneliussen T.S., Vilstrup J.T., Lear T., Myka J.L., Lundquist J., Miller D.C., Alfarhan A.H., Alquraishi S.A., Al-Rasheid K.A.S., Stagegaard J., Strauss G., Bertelsen M.F., Sicheritz-Ponten T., Antczak D.F., Bailey E., Nielsen R., Willerslev E., Oralndo L.. 2014. Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc. Natl. Acad. Sci. USA. 111:18655–18660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck B.P., Near T.J.. 2009. Patterns of natural hybridization in darters (Percidae: Etheostomatinae). Copeia. 2009:758–773. [Google Scholar]
- Kelly N.B., Near T.J., Alonzo S.H.. 2012. Diversification of egg-deposition behaviours and the evolution of male parental care in darters (Teleostei: Percidae: Etheostomatinae). J. Evol. Biol. 25:836–846. [DOI] [PubMed] [Google Scholar]
- Kuehne R.A., Barbour R.W.. 1983. The American Darters. Lexington, KY: The University Press of Kentucky. [Google Scholar]
- Kutschera V.E., Bidon T., Hailer F., Rodi J.L., Fain S.R., Janke A.. 2014. Bears in a forest of gene trees: phylogenetic inference is complicated by incomplete lineage sorting and gene flow. Mol. Biol. Evol. 31:2004–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang N.J., Mayden R.L.. 2007. Systematics of the subgenus Oligocephalus (Teleostei: Percidae: Etheostoma) with complete subgeneric sampling of the genus Etheostoma. Mol. Phylogenet. Evol. 43:605–615. [DOI] [PubMed] [Google Scholar]
- Larget B.R., Kotha S.K., Dewey C.N., Ané C.. 2010. BUCKy: gene tree reconciliation with concordance. Bioinformatics. 26:2910–2911. [DOI] [PubMed] [Google Scholar]
- Lavretsky P., Engilis A., Eadie J.M., Peters J.L.. 2015. Genetic admixture supports an ancient hybrid origin of the endangered Hawaiian duck. J. Evol. Biol. 28:1005–1015. [DOI] [PubMed] [Google Scholar]
- Lemmon E.M., Lemmon A.R.. 2013. High-throughput genomic data in systematics and phylogenetics. Annu. Rev. Ecol. Evol. Syst. 44:99–121. [Google Scholar]
- Li G., Davis B.W., Eizirik E., Murphy W.J.. 2016. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 26:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison W. 1997. Gene trees in species trees. Syst. Biol. 46:523–536. [Google Scholar]
- Martin S.H., Davey J.W., Jiggins C.D.. 2015. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol. Biol. Evol. 31:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack J.E., Faircloth B.C.. 2013. Next-generation phylogenetics takes root. Mol. Ecol. 22:19–21. [DOI] [PubMed] [Google Scholar]
- McCormack J.E., Hird S.M., Zellmer A.J., Carstens B.C., Brumfield R.T.. 2013. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 66:526–538. [DOI] [PubMed] [Google Scholar]
- Near T.J., Bossu C.M., Bradburd G.S., Carlson R.L., Harrington R.C., Hollingsworth P.R., Keck B.P., Etnier D.A.. 2011. Phylogeny and temporal diversification of darters (Percidae: Etheostomatinae). Syst. Biol. 60:565–595. [DOI] [PubMed] [Google Scholar]
- Near T.J., Keck B.P.. 2013. Free from mitochondrial DNA: nuclear genes and the inference of species trees among closely related darter lineages (Teleostei: Percidae: Etheostomatinae). Mol. Phylogenet. Evol. 66:868–876. [DOI] [PubMed] [Google Scholar]
- Near T.J., MacGuigan D.J., Parker E., Struthers C.D., Jones C.D., Dornburg A.. 2018. Phylogenetic analysis of Antarctic notothenioids illuminates the utility of RADseq for resolving Cenozoic adaptive radiations. Mol. Phylogenet. Evol. 129:268–279. [DOI] [PubMed] [Google Scholar]
- Nolte A.W., Tautz D.. 2010. Understanding the onset of hybrid speciation. Trends Genet. 26:54–58. [DOI] [PubMed] [Google Scholar]
- Page L.M. 1977. The lateralis system of darters (Etheostomatini). Copeia. 1977:472–475. [Google Scholar]
- Page L.M. 1981. The genera and subgenera of darters (Percidae, Etheostomatini). Occas. Pap. Mus. Nat. Hist. Univ. Kansas 90:1–69. [Google Scholar]
- Page L.M. 1983. Handbook of Darters. Neptune City, NJ: T.F.H Publications. [Google Scholar]
- Page L.M., Whitt G.S.. 1973. Lactate dehydrogenase isozymes of darters and the inclusiveness of the genus Percina. Ill. Nat. Hist. Sur. Biol. Notes. 82:1–7. [Google Scholar]
- Pardi F., Scornavacca C.. 2015. Reconstructible phylogenetic networks: do not distinguish the indistinguishable. PLoS Comput. Biol. 11:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard D.A., Iyer V.N., Moses A.M., Eisen M.B.. 2006. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2:e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers S.L., Kuhajda B.R., Ahlbrand S.E.. 2012. Systematics of the Etheostoma cinereum (Teleostei: Percidae) species complex (subgenus Allohistium). Zootaxa. 3277:43–55. [Google Scholar]
- Powers S.L., Mayden R.L., Etnier D.A.. 2004. Conservation genetics of the ashy darter, Etheostoma cinereum (Percidae: Subgenus Allohistium), in the Cumberland and Tennessee rivers of the southeastern United States. Copeia. 2004:632–637. [Google Scholar]
- Prum R.O., Berv J.S., Dornburg A., Field D.J., Townsend J.P., Lemmon E.M., Lemmon A.R.. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 526:569–573. [DOI] [PubMed] [Google Scholar]
- R Core Team 2017. R: a language and environment for statistical computing [Internet]. Vienna (Austria): R Foundation for Statistical Computing. Available from: https://www.R-project.org/. [Google Scholar]
- Rokas A., Williams B.L., King N., Carroll S.B.. 2003. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 425:798–804. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Höhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P.. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salichos L., Rokas A.. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 497:327–331. [DOI] [PubMed] [Google Scholar]
- Sayyari E., Mirarab S.. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Molecular Biology and Evolution. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M., Cui R., Pwell D.L., Rosenthal F.G., Andolfatto P.. 2016. Ancient hybridization and genomic stabilization in a swordtail fish. Mol. Ecol. 25:2661–2679. [DOI] [PubMed] [Google Scholar]
- Schumer M., Rosenthal G.G., Andolfatto P.. 2014. How common is homoploid hybrid speciation? Evolution. 68:1553–1560. [DOI] [PubMed] [Google Scholar]
- Shen X.-X., Hittinger C.T., Rokas A.. 2017. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard T.E., Burr B.M.. 1984. Systematics, status, and life history aspects of the ashy darter, Etheostoma cinereum (Pisces: Percidae). Proc. Biol. Soc. 97:693–715. [Google Scholar]
- Sloss B.L., Billington N., Burr B.M.. 2004. A molecular phylogeny of the Percidae (Teleostei, Perciformes) based on mitochondrial DNA sequence. Mol. Phylogenet. Evol. 32:545–562. [DOI] [PubMed] [Google Scholar]
- Smith T.A., Ciccotto P.J., Mendelson T.C., Page L.M.. 2014. Dense taxon sampling using AFLPs leads to greater accuracy in phylogeny estimation and classification of darters (Percidae: Etheostomatinae). Copeia. 2014:257–268. [Google Scholar]
- Smith T.A., Mendelson T.C., Page L.M.. 2011. AFLPs support deep relationships among darters (Percidae: Etheostomatinae) consistent with morphological hypotheses. Heredity. 107:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís-Lemus C., Ané C.. 2016. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.B., Near T.J., Page L.M.. 1998. Phylogenetic relations among percid fishes as inferred from mitochondrial cytochrome b DNA sequence data. Mol. Phylogenet. Evol. 10:343–353. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange M., Sánchez-Villagra M.R., Salzburger W., Matschiner M.. 2018. Bayesian divergence-time estimation with genome-wide single-nucleotide polymorphism data of sea catfishes (Ariidae) supports miocene closure of the Panamanian isthmus. Systematic Biology. 67:681–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics. 105:437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C., Ruths D., Nakhleh L.. 2008. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics. 9:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Yu Y., Hahn M.W., Nakhleh L.. 2016. Reticulate evolutionary history and extensive introgression in mosquito species revealed by phylogenetic network analysis. Mol. Ecol. 25:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M. 1996. Majority-rule reduced consensus trees and their use in bootstrapping. Mol. Biol. Evol. 13:437–444. [DOI] [PubMed] [Google Scholar]
- Wolf Y.I., Rogozin I.B., Grishin N.V., Koonin E.V.. 2002. Genome trees and the tree of life. Trends Genet. 18:472–479. [DOI] [PubMed] [Google Scholar]
- Yu Y., Barnett R.M., Nakhleh L.. 2013. Parsimonious inference of hybridization in the presence of incomplete lineage sorting. Syst. Biol. 62:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Dong J., Liu K.J., Nakhleh L.. 2014. Maximum likelihood inference of reticulate evolutionary histories. Proc. Natl. Acad. Sci. USA. 111:16448–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarza E., Faircloth B.C., Tsai W.L.E., Bryson R.W. Jr, Klicka J., McCormack J.E. 2016. Hidden histories of gene flow in highland birds revealed with genomic markers. Mol. Ecol. 25:5144–5157. [DOI] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S.. 2018. ASTRAL-III: oolynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Dasmahapatra K.K., Mallet J., Moreira G.R.P., Kronforst M.R.. 2016. Genome-wide introgression among distantly related Heliconius butterfly species. Genome Biol. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- Bailey R.M., Gosline W.A.. 1955. Variation and systematic significance of vertebral counts in the American fishes of the family percidae. Misc. Publ. Museum Zool. Univ. Michigan. 93:1–44. [Google Scholar]
- Bailey R.M., Richards W.. 1963. Status of Poecilichthys hopkinsi Fowler and Etheostoma trisella, a new species, percid fishes from Alabama, Georgia, and South Carolina. Occas. Pap. Mus. Zool. Univ. Mich. 630:1–21. [Google Scholar]
- Dimmick W.W., Page L.M.. 1992. Systematic significance of lactate dehydrogenase variation at the generic level in percids. Copeia. 1992:535–537. [Google Scholar]
- Near T.J., Bossu C.M., Bradburd G.S., Carlson R.L., Harrington R.C., Hollingsworth P.R., Keck B.P., Etnier D.A.. 2011. Phylogeny and Temporal Diversification of Darters (Percidae: Etheostomatinae). Syst. Biol. 60:565–595. [DOI] [PubMed] [Google Scholar]
- Powers S.L., Kuhajda B.R., Ahlbrand S.E.. 2012. Systematics of the Etheostoma cinereum (Teleostei: Percidae) species complex (subgenus Allohistium). Zootaxa. 3277:43–55. [Google Scholar]
- Storer, D.H. 1845. Proceedings of the Boston Society of Natural History 2:47–50. [Google Scholar]