

Abstract
The classic methodology of inferring a phylogenetic tree from sequence data is composed of two steps. First, a multiple sequence alignment (MSA) is computed. Then, a tree is reconstructed assuming the MSA is correct. Yet, inferred MSAs were shown to be inaccurate and alignment errors reduce tree inference accuracy. It was previously proposed that filtering unreliable alignment regions can increase the accuracy of tree inference. However, it was also demonstrated that the benefit of this filtering is often obscured by the resulting loss of phylogenetic signal. In this work we explore an approach, in which instead of relying on a single MSA, we generate a large set of alternative MSAs and concatenate them into a single SuperMSA. By doing so, we account for phylogenetic signals contained in columns that are not present in the single MSA computed by alignment algorithms. Using simulations, we demonstrate that this approach results, on average, in more accurate trees compared to 1) using an unfiltered MSA and 2) using a single MSA with weights assigned to columns according to their reliability. Next, we explore in which regions of the MSA space our approach is expected to be beneficial. Finally, we provide a simple criterion for deciding whether or not the extra effort of computing a SuperMSA and inferring a tree from it is beneficial. Based on these assessments, we expect our methodology to be useful for many cases in which diverged sequences are analyzed. The option to generate such a SuperMSA is available at http://guidance.tau.ac.il.
Keywords: Alignment reliability, multiple sequence alignment, phylogeny, tree reconstruction
Inference of the phylogenetic relationships among species is one of the most fundamental questions in evolutionary biology. Phylogeny inference from comparative genomics data commonly involves two consecutive steps, the inference of a multiple sequence alignment (MSA) and the inference of the phylogeny assuming the MSA is correct (Holder and Lewis 2003). Therefore, MSAs play a major role in phylogenetic inference. However, accurate MSA reconstruction is a challenging computational task, due to 1) the stochastic nature of the evolutionary process, 2) computational limitations of current bioinformatics methods, and 3) the lack of accurate evolutionary models describing the process of sequence evolution (Do and Katoh 2008; Kemena and Notredame 2009; Loytynoja et al. 2012).
Over the years, various sequence alignment algorithms and methods were developed, showing a steady improvement (Katoh et al. 2002; Loytynoja and Milinkovitch 2003; Edgar 2004; Do et al. 2005; Larkin et al. 2007; Do and Katoh 2008; Katoh and Toh 2008; Loytynoja and Goldman 2008; Sievers et al. 2011; Loytynoja et al. 2012; Katoh and Standley 2013). However, objective large-scale evaluations of alignment methods using both simulated and empirical data sets revealed that alignment methods produce MSAs that are still subject to a considerable number of errors (Blackshields et al. 2006; Nuin et al. 2006; Thompson et al. 2011; Sela et al. 2015). Such alignment errors were shown to impact the accuracy of phylogeny inference (Lake 1991; Morrison and Ellis 1997; Ogden and Rosenberg 2006; Smythe et al. 2006; Talavera and Castresana 2007; Wong et al. 2008; Wang et al. 2011; Md Mukarram Hossain et al. 2015). Previous studies suggested that filtering out unreliable MSA regions prior to phylogeny inference can improve tree reconstruction accuracy (Talavera and Castresana 2007; Dress et al. 2008; Capella-Gutierrez et al. 2009; Kuck et al. 2010; Rajan 2013; Chang et al. 2014). Independently of the employed filtering strategy, the removal of alignment errors is inevitably accompanied by the removal of informative phylogenetic signal. This delicate balance between the amount of signal and the amount of noise removed from the MSA makes it difficult to determine to what extent filtering unreliable alignment regions is beneficial (Gatesy et al. 1993; Lee 2001; Aagesen 2004). For example, highly diverged positions are often harder to align compared to conserved regions. However, diverged regions also contain a substantial amount of the evolutionary signal and therefore filtering them out may impede accurate phylogeny inference. Furthermore, filtering out columns from the MSA requires a score cut-off to determine the low quality columns to be filtered out. Such a cut-off is often data-set specific, resulting in filtering procedures that are both ad hoc and subjective. Recently, it was shown that filtering MSA columns prior to phylogeny inference, using available methods to detect alignment uncertainty, often results in decreased accuracy of the inferred phylogeny (Tan et al. 2015a).
As an alternative to filtering out unreliable alignment columns, several studies advocated minimizing the uncertainty by finding a consensus MSA for a set of possible MSAs or selecting the most consistent MSA (Wallace et al. 2006; Capella-Gutierrez et al. 2009; Collingridge and Kelly 2012). It was previously shown that different alignment methods, or even different choices of parameters of a specific method yield MSAs that substantially differ from one another (Lake 1991; Gatesy et al. 1993; Wheeler 1995; Nelesen et al. 2008; Penn et al. 2010; Blackburne and Whelan 2012; Boyce et al. 2015). This uncertainty in resulting MSAs suggests that MSA consensus methods, similar to the column-filtering methodologies described above, reduce noise at the price of discarding important phylogenetic signal.
Another alternative to filtering out unreliable columns is to weight MSA columns according to their reliability. Within the maximum-parsimony paradigm, early works have shown that applying a different coding scheme to uncertain columns results in more accurate tree reconstruction (Lutzoni et al. 2000; Lee 2001; Geiger 2002). A similar idea of using a coding scheme for unreliable regions was also suggested for the maximum-likelihood (ML) framework (Lucking et al. 2011). More recently, it was shown that when each column in the input MSA is weighted by its reliability score, ML-tree reconstruction is more accurate (Wu et al. 2012; Chang et al. 2014). While such studies show the potential benefit of explicitly accounting for MSA reliability when reconstructing the tree, they still use only one MSA that represents one possible solution to the alignment problem, thus ignoring much of the phylogenetic signal carried by other possible solutions, which are termed hereinafter “alternative MSAs”. Specifically, these methods only weight MSA columns in one particular MSA and implicitly give zero weight to all MSA columns found in alternative MSAs and not in the original MSA, regardless of their frequency in the alternative MSAs.
Within the maximum-parsimony paradigm, it was previously suggested to concatenate alternative MSAs, which were generated by varying the gap penalty parameter. This methodology, termed Elision (Wheeler 1995), uses the concatenated large MSA as input to phylogeny inference. Computationally, this approach is equivalent to weighting MSA columns according to the number of times they appear in the concatenated MSA prior to the tree reconstruction procedure. We have previously developed a method called GUIDANCE, which uses such weights (the number of times a column appears in a set of alternative MSAs) as a measure for the reliability of that column (Landan and Graur 2008; Penn et al. 2010; Sela et al. 2015). We have shown that filtering unreliable positions based on such weighting can improve positive selection inference (Privman et al. 2012). GUIDANCE2 is a further development, which is based on generating alternative MSAs by varying the guide tree used to generate the MSA, the gap-opening penalty and by considering several equally top scoring MSAs (Sela et al. 2015).
In essence, concatenating alternative MSAs is equivalent to generating a unique MSA, in which the weight of each column represents the number of times it appears among the alternative MSAs. This approach is thus similar to the computation of reliability scores used in GUIDANCE. However, while in GUIDANCE, only positions in the best MSA provided by the alignment method (the “base MSA”) were weighted, in the current study, MSA columns that do not appear in the base MSA are weighted as well, that is, all MSAs, original and alternative, are treated equally. This approach can be divided to three steps: 1) generating alternative MSAs; 2) collecting all possible unique alignment columns from the “base MSA” and alternative MSAs. Then, assigning a weight to each such column according to its frequency among the set of alternative MSAs and the base MSA; 3) Using the obtained weighted MSA, termed SuperMSA, and the column weights to reconstruct a phylogenetic tree.
In the original Elision methodology alternative MSAs were generated using the MSA program MALIGN (Wheeler and Gladstein 1994). Alternative MSAs were generated by varying only the gap penalty parameter and trees were inferred only under the maximum parsimony criterion. In this work, we revisit the Elision approach, only this time we 1) generate alternative MSAs by taking into account a wide range of possible gap penalties, guide trees and co-optimal solutions (Landan and Graur 2008; Penn et al. 2010; Sela et al. 2015); 2) generate MSAs by two alignment programs: MAFFT (Katoh and Standley 2013) and PRANK (Loytynoja and Goldman 2008), which were shown to be among the best alignment programs in recent comparative studies (Sievers et al. 2011; Thompson et al. 2011; Sela et al. 2015); 3) infer trees using either an ML or a Bayesian approach. We next analyze real as well as simulated data to test the hypothesis that the accuracy of the inferred phylogeny increases compared to trees inferred by considering only the base MSA, and trees inferred from the base MSA in which each column is weighted according to its reliability score.
Materials and Methods
Data Sets
Four data sets were analyzed in this study: PAM100, PAM250, EMPIRICAL, and ENSEMBLsim. The first three data sets were downloaded from http://www.datadryad.org/resource/doi:10.5061/dryad.pc5j0http://www.datadryad.org/resource/doi:10.5061/dryad.pc5j0 (Tan et al. 2015b). PAM100 and PAM250 are simulated data sets. Each of which includes 500 different simulated MSAs. Each of these MSAs was simulated along 30-taxa trees scaled such that the distance from the root to the deepest branch was either 250 point accepted mutation (PAM) units or 100 PAM. Tree topologies and branch lengths were determined using a birth–death process (see Tan et al. 2015b for details).
Our EMPIRICAL data set was derived from the genes analyzed in Tan et al. (2015b). From their empirical data set, we randomly sampled 10% of the genes, generating a total of 1,099 genes, each including six orthologous sequences. The empirical data set covers three taxonomic ranges: fungi, eukaryotes, and bacteria.
The fourth data set examined in this study, ENSEMBLsim, was generated in the following way. We collected human coding genes (GRCh38.p10) and their 1:1 orthologs across vertebrates by querying the ENSEMBL database (Zerbino et al. 2017; version 90, accessed on 04/09/2017) using the Perl scripts described in Levy Karin et al. (2017). We focused only on human coding genes, which had at least 70 orthologs. Out of these, 98 genes were randomly chosen for further processing. If a gene had more than 70 orthologs, we randomly chose 70. We translated each orthologous set and aligned the protein sequences with MAFFT (version 7.123b, Katoh and Standley 2013). We downloaded the ENSEMBL species tree (Herrero et al. 2016; https://github.com/Ensembl/ensembl-compara/blob/release/90/scripts/pipeline/species_tree.ensembl.topology.nw) and pruned it with each coding gene to produce a subtree that contains only taxa from its orthologous set. We then optimized the branch lengths of the pruned tree with respect to its MSA using PhyML (Guindon et al. 2010) with the WAG+I+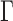 substitution model (Whelan and Goldman 2001). This resulted in 98 pairs of an MSA and its optimized tree. We gave each such pair as input to SpartaABC (Levy Karin et al. 2017) to infer indel parameters. SpartaABC was run in MSA-mode with weights and parameter ranges as described in the “Indel parameters search space” section of Ashkenazy et al. (2017). Next, each tree with its corresponding inferred indel parameters was given to INDELible v1.3 (Fletcher and Yang 2009) to produce a single simulated true MSA. We provide the data set ENSEMBLsim as Supplementary Material available on Dryad at http://dx.doi.org/10.5061/dryad.4j1qt.
substitution model (Whelan and Goldman 2001). This resulted in 98 pairs of an MSA and its optimized tree. We gave each such pair as input to SpartaABC (Levy Karin et al. 2017) to infer indel parameters. SpartaABC was run in MSA-mode with weights and parameter ranges as described in the “Indel parameters search space” section of Ashkenazy et al. (2017). Next, each tree with its corresponding inferred indel parameters was given to INDELible v1.3 (Fletcher and Yang 2009) to produce a single simulated true MSA. We provide the data set ENSEMBLsim as Supplementary Material available on Dryad at http://dx.doi.org/10.5061/dryad.4j1qt.
Alignments
Unaligned sequences (either simulated or empirical data sets) were given as input to GUIDANCE2 (Sela et al. 2015). GUIDANCE2 (version 2.01) then aligned each gene using two alignment programs: 1) PRANK version v. 140603 with the 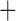 F argument (Loytynoja and Goldman 2008). 2) MAFFT version 7.123b with default parameters (Katoh et al. 2002; Katoh and Standley 2013). Two measures for alignment accuracy were used: 1) column score (CS): a column is scored 1 if it is identically aligned both in the inferred and true MSA and 0 otherwise. 2) sum-of-pairs column (SPC) score: the average of all pair scores in a given column of the inferred alignment, where a pair of residues which is identically aligned both in the inferred and in the true MSA is given a score of 1; all other residue pairs in the inferred alignment are given the score 0 (Thompson et al. 1999).
F argument (Loytynoja and Goldman 2008). 2) MAFFT version 7.123b with default parameters (Katoh et al. 2002; Katoh and Standley 2013). Two measures for alignment accuracy were used: 1) column score (CS): a column is scored 1 if it is identically aligned both in the inferred and true MSA and 0 otherwise. 2) sum-of-pairs column (SPC) score: the average of all pair scores in a given column of the inferred alignment, where a pair of residues which is identically aligned both in the inferred and in the true MSA is given a score of 1; all other residue pairs in the inferred alignment are given the score 0 (Thompson et al. 1999).
Generating a SuperMSA
By default GUIDANCE2 (Sela et al. 2015) provides a set of 400 alternative MSAs for each instance of the sequence data set. The SuperMSA is composed of a randomly selected subset of alternative MSAs concatenated to the base MSA, produced by the alignment program. Throughout this study, unless otherwise stated, the number of alternative MSAs is set to 20. The option to generate such a SuperMSA is implemented as part of the GUIDANCE2 webserver which is available at http://guidance.tau.ac.il.
Phylogenetic Tree Inference
Maximum-likelihood tree inference was performed using RAxML v8.1.3 (Stamatakis 2014). Default parameters were used for tree search. Bayesian tree inference was performed using MrBayes v3.2.2 (Ronquist et al. 2012) with 500,000 generations, and all other parameters set to default. In all cases, the amino acid substitution model was WAG+I+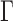 (Whelan and Goldman 2001). Robinson–Foulds (RF) distances (Robinson and Foulds 1981) between trees were computed using the APE package (Paradis et al. 2004) in R. The RF distance was divided by the maximal RF distance 2(
(Whelan and Goldman 2001). Robinson–Foulds (RF) distances (Robinson and Foulds 1981) between trees were computed using the APE package (Paradis et al. 2004) in R. The RF distance was divided by the maximal RF distance 2(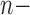 3), where
3), where 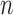 is the number of taxa in the tree, to give the normalized RF distance (normRF) (Kupczok et al. 2010).
is the number of taxa in the tree, to give the normalized RF distance (normRF) (Kupczok et al. 2010).
For Bali-Phy (Redelings and Suchard 2005) analyses, version 2.3.7 was run with default parameters, using the WAG+I+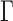 substitution model. Two independent chains were run for each instance of the data set and the results were analyzed using the bp-analyze.pl script, which is part of the Bali-Phy package.
substitution model. Two independent chains were run for each instance of the data set and the results were analyzed using the bp-analyze.pl script, which is part of the Bali-Phy package.
Weighting the Base MSA
The effect of weighting columns of the base MSA on the accuracy of the inferred phylogeny was tested using the column confidence score calculated by three methods: GUIDANCE2 version 2.01 (Sela et al. 2015), ZORRO (Wu et al. 2012), and TCS version 10.00 (Chang et al. 2014). All methods were used with default parameters. For compatibility with the RAxML (Stamatakis 2014) weighting procedure, GUIDANCE2 SPC was multiplied by 10 and rounded to the closest integer. For ZORRO, the CSs were rounded to the closest integer. For TCS, the CSs were used as is. Briefly, the GUIDANCE2 SPC score is a generalization of the definition in “Alignments” section. Here, for each residue pair in the base MSA, the frequency among the alternative MSAs is computed. The GUIDANCE2 SPC score is the average over all pairs in a given column of the base MSA (Sela et al. 2015). ZORRO (Wu et al. 2012) employs a pair-HMM methodology to assign the probability of two residues being aligned together in the pairwise alignments composing the MSA. Such pairwise probabilities are joined to estimate the reliability of columns in the MSA. TCS (Chang et al. 2014) estimates the score of aligning two residues based on a predefined library of pairwise alignments. These pair scores are next joined to estimate the TCS CS for the base MSA.
Results
The Accuracy of MSA Inference Using MAFFT and PRANK on SimulatedData Sets
Throughout this work, we analyzed three simulated protein data sets. Two represent sequences with different divergence level: PAM250 and PAM100 (see Materials and Methods section). Each such data set is comprised of 500 different genes from 30 taxa. The third data set, ENSEMBLsim, represents vertebrate evolution and is comprised of 98 genes from 70 taxa (see Materials and Methods section). We first tested the MSA reconstruction accuracy of both MAFFT and PRANK on each instance of these data sets. For the diverged MSAs (PAM250), the accuracy of both PRANK and MAFFT was relatively low, yielding an average CS (see Materials and Methods section) of 0.19 and 0.17 for MAFFT and PRANK, respectively. In contrast, the accuracy for the relatively conserved MSAs (PAM100) was significantly higher (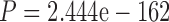 and
and 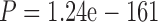 for MAFFT and PRANK, respectively, Mann–Whitney test), with an average CS of 0.83 and 0.84, for MAFFT and PRANK, respectively. The accuracy of MAFFT and PRANK for the ENSEMBLsim data set was in between the accuracies measured for the PAM100 and PAM250 data sets (average CS of 0.28 and 0.31, for MAFFT and PRANK, respectively). These results suggest that aligning instances of the PAM250 data set is much more challenging than instances of the ENSEMBLsim and the PAM100 data sets, and accordingly, we expect that accounting for uncertainty in MSA inference should be most pronounced for the PAM250 data set.
for MAFFT and PRANK, respectively, Mann–Whitney test), with an average CS of 0.83 and 0.84, for MAFFT and PRANK, respectively. The accuracy of MAFFT and PRANK for the ENSEMBLsim data set was in between the accuracies measured for the PAM100 and PAM250 data sets (average CS of 0.28 and 0.31, for MAFFT and PRANK, respectively). These results suggest that aligning instances of the PAM250 data set is much more challenging than instances of the ENSEMBLsim and the PAM100 data sets, and accordingly, we expect that accounting for uncertainty in MSA inference should be most pronounced for the PAM250 data set.
The Accuracy of Alternative Alignments
The approach suggested here relies on integrating phylogenetic signals from alternative MSAs as part of the tree inference procedure. However, it may be that alternative MSAs are on average less accurate than the base MSA. If this is the case, accounting for alternative MSAs may introduce noise, which could obscure the phylogenetic signal. To test this hypothesis, we compared the average CS of the base MSA against 400 alternative MSAs generated using GUIDANCE2 (Sela et al. 2015) (see Materials and Methods section). This comparison was repeated for each of the different genes in each data set. When MSAs are computed with either MAFFT or PRANK, often alternative MSAs are more accurate than the base MSA (Fig. 1). For the PAM250 data set, when using MAFFT and PRANK as the alignment method, 28.8% and 62.39% of the alternative MSAs, respectively, had a higher CS compared to the base MSA (Fig. 1a, d). For the PAM100 data set, 26.04% and 41.14% of the alternative alignments had a higher CS compared to the base MSA, for MAFFT and PRANK, respectively (Fig. 1b, e). The results for the ENSEMBLsim data set show a similar pattern (Fig. 1c, f). The distributions of the CS scores (Fig. 1) demonstrate the potential of sampling and concatenating alternative MSAs for improving phylogeny inference.
Figure 1.

The difference in average CS between alternative MSAs and the base MSA when using MAFFT (a–c) and PRANK (d–f) as the alignment method, for the PAM250 (a, d), PAM100 (b, e), and ENSEMBLsim (c, f) data sets. A positive value indicates that an alternative MSA is more accurate than the base MSA.
As part of its procedure to generate alternative MSAs, GUIDANCE2 varies the gap-opening penalty score, the guide tree provided to the alignment program, and samples from different co-optimal solutions. The above results suggest that, the default values of the alignment program may not fit some of the analyzed data sets. To test this hypothesis, we ran the alignment program with various input parameters and studied the effect on the MSA accuracy. Our results show that there is no single gap-opening parameter that fits all data sets. That is, there is a high variability in the gap-opening parameter that best fits each data set (Supplementary Fig. S1 available on Dryad). The same trend is observed when studying the effect of the guide tree. For many data sets, the default guide tree produced by the MSA program is not the guide tree that maximized the MSA accuracy (Supplementary Fig. S2 available on Dryad). These results suggest that accounting for uncertainty in these input parameters may be beneficial for phylogeny inference.
The Accuracy of ML Trees Given the Correct MSA Versus the Base MSA
Our aim is to develop a more accurate phylogeny inference procedure, accounting for alignment uncertainty. Our first effort was to compute the level of tree inference error in a procedure which does not account for alignment uncertainty. For the PAM250 data set, when the true MSA was provided as input to the tree reconstruction program, the average normRF distance (see Materials and Methods section) was 0.103 (Table 1). This value can be regarded as a baseline for comparison when using an inferred MSA as input, rather than the true one. Indeed, when inferred MSAs were given as input, the accuracy substantially decreased, resulting in average normRF distances of 0.187 for MAFFT-based MSAs and 0.169 for PRANK-based MSAs. For instances of the PAM100 data set, only a modest decrease in accuracy was observed (less than 0.001 difference in the average normRF distance; Table 2). These results and those obtained for the ENSEMBLsim data set (Table 3) support two previous observations: 1) For the PAM250 data set, ML trees reconstructed from PRANK-based MSAs are significantly more accurate than MAFFT-based MSAs (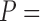 4.235e
4.235e 07 paired Wilcoxon test; see also Loytynoja and Goldman (2008)); 2) Alignment errors in diverged data sets (such as those in the PAM250 data set compared to the PAM100 data set) substantially reduce the accuracy of phylogenetic inference (
07 paired Wilcoxon test; see also Loytynoja and Goldman (2008)); 2) Alignment errors in diverged data sets (such as those in the PAM250 data set compared to the PAM100 data set) substantially reduce the accuracy of phylogenetic inference (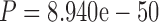 and
and 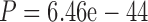 for MAFFT and PRANK MSAs, respectively, paired Wilcoxon test; see also Chang et al. 2014).
for MAFFT and PRANK MSAs, respectively, paired Wilcoxon test; see also Chang et al. 2014).
Table 1.
The effect of MSA weighting (GUIDANCE2, ZORRO, TCS) and averaging on tree inference accuracy for the PAM250 data set
| No weighting | GUIDANCE2 | ZORRO | TCS | SuperMSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| Alignment method | Average normalized RF distance | Average normalized RF distance |
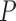 -value -value |
Average normalized RF distance |
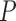 -value -value |
Average normalized RF distance |
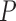 -value -value |
Average normalized RF distance |
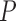 -value -value |
| True MSA | 0.103 (0.07) | ||||||||
| MAFFT | 0.187 (0.125) | 0.184 (0.123) | 0.022 | 0.187 (0.129) | 0.295 | 0.188 (0.127) | 0.661 | 0.149 (0.099) | 1.596e–24 |
| PRANK | 0.169 (0.108) | 0.168 (0.109) | 0.286 | 0.174 (0.111) | 0.999 | 0.176 (0.114) | 0.999 | 0.162 (0.105) | 0.00011 |
Note: All 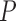 -values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
Table 2.
The effect of MSA weighting (GUIDANCE2, ZORRO, TCS) and averaging on tree inference accuracy for the PAM100 data set
| No weighting | GUIDANCE2 | ZORRO | TCS | SuperMSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| Alignment method | Average normalized RF distance | Average normalized RF distance |
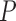 -value -value |
Average normalized RF distance |
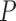 -value -value |
Average normalized RF distance |
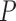 -value -value |
Average normalized RF distance |
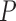 -value -value |
| True MSA | 0.085 (0.066) | ||||||||
| MAFFT | 0.086 (0.066) | 0.086 (0.067) | 0.347 | 0.086 (0.066) | 0.414 | 0.085 (0.066) | 0.029 | 0.084 (0.066) | 0.039 |
| PRANK | 0.086 (0.067) | 0.086 (0.067) | 0.242 | 0.087 (0.068) | 0.703 | 0.087 (0.069) | 0.613 | 0.085 (0.067) | 0.146 |
Note: All 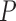 -values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
Table 3.
The effect of MSA weighting (GUIDANCE2, ZORRO, TCS) and averaging on tree inference accuracy for the ENSEMBLsim data set
| No weighting | GUIDANCE2 | ZORRO | TCS | SuperMSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| Alignment method | Average normalized RF distance | Average normalized RF distance |
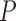 -value -value |
Average normalized RF distance |
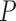 -value -value |
Average normalized RF distance |
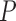 -value -value |
Average normalized RF distance |
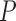 -value -value |
| True MSA | 0.285 (0.186) | ||||||||
| MAFFT | 0.301 (0.192) | 0.303 (0.191) | 0.624 | 0.306 (0.192) | 0.836 | 0.307 (0.269) | 0.856 | 0.285 (0.186) | 0.0003 |
| PRANK | 0.283 (0.182) | 0.281 (0.184) | 0.378 | 0.286 (0.187) | 0.948 | 0.286 (0.187) | 0.901 | 0.281 (0.186) | 0.447 |
Note: All 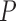 -values were computed using a onesided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a onesided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
The Accuracy of ML Trees When Weighting the Base MSA Columns According to Their Reliability Score
We next evaluated a method that accounts for MSA uncertainty by weighting columns of the base MSA according to their reliability score. The effect of columns weighting was tested using three methods: ZORRO (Wu et al. 2012), TCS (Chang et al. 2014) and GUIDANCE2 (Sela et al. 2015) on the three simulated data sets (PAM100, PAM250, and ENSEMBLsim) aligned using two methods (MAFFT and PRANK) as described in Materials and Methods section. In comparison to the trees reconstructed using the base MSA, the column weighting showed statistically significant increase in the accuracy only for PAM250 MAFFT MSAs weighted by GUIDANCE2 scores and for PAM100 MAFFT MSAs weighted by TCS scores (Tables 1–3). Importantly, for instances of the PRANK-PAM250 data set, weighting columns by TCS or ZORRO resulted in inferred phylogenies, which are significantly less accurate than those obtained using the base MSA (Table 1; 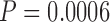 and
and 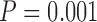 for TCS and ZORRO, respectively, paired Wilcoxon test).
for TCS and ZORRO, respectively, paired Wilcoxon test).
The Accuracy of ML Trees Based on a SuperMSA Approach
We next tested the hypothesis that accounting for MSA columns absent from the base MSA increases tree reconstruction accuracy. To this end, we computed trees based on a SuperMSA – a concatenation of 20 alternative MSAs (see Materials and Methods section). Using a SuperMSA significantly improved the accuracy in all cases, except for PRANK-PAM100 and PRANK-ENSEMBLsim (Tables 1–3). Notably, for the PAM250 data set, when accounting for alternative MSAs, the accuracy of the inferred trees was significantly higher than those inferred based on weighting MSA columns from the base alignment using either GUIDANCE2, ZORRO, or TCS (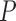 < 2.2e
< 2.2e 16 and
16 and 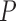 < 0.0003, for MAFFT and PRANK, respectively, paired Wilcoxon test). The accuracy of the SuperMSA approach was also significantly higher than the other weighting methods for the ENSEMBLsim data set when aligned using MAFFT (
< 0.0003, for MAFFT and PRANK, respectively, paired Wilcoxon test). The accuracy of the SuperMSA approach was also significantly higher than the other weighting methods for the ENSEMBLsim data set when aligned using MAFFT (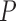 < 0.0003). As the most significant increase in accuracy was obtained for PAM250 and ENSEMBLsim (which includes MSAs that are more divergent than PAM100), our results suggest that accounting for alternative MSAs increases phylogenetic accuracy mostly when analyzing diverged sequences. For less diverged sequences, this procedure results in only a modest increase in tree accuracy. We stress that the average trends are similar across all data sets and alignment methods, however, only when MSA inaccuracy is substantial (i.e., a large difference in normRF distance between the tree inferred given the true MSA and the tree inferred assuming the base MSA, Tables 1–3), there is enough room for improvement and the increased accuracy is statistically significant.
< 0.0003). As the most significant increase in accuracy was obtained for PAM250 and ENSEMBLsim (which includes MSAs that are more divergent than PAM100), our results suggest that accounting for alternative MSAs increases phylogenetic accuracy mostly when analyzing diverged sequences. For less diverged sequences, this procedure results in only a modest increase in tree accuracy. We stress that the average trends are similar across all data sets and alignment methods, however, only when MSA inaccuracy is substantial (i.e., a large difference in normRF distance between the tree inferred given the true MSA and the tree inferred assuming the base MSA, Tables 1–3), there is enough room for improvement and the increased accuracy is statistically significant.
It may be claimed that the improvement in tree accuracy obtained by averaging over alternative MSAs is a result of a few outlier instances of the data set, while for the rest of the instances, accounting for alternative MSAs may even reduce the inference accuracy. To test this hypothesis, we compared the number of instances for which accounting for uncertainty improved tree accuracy to that for which it reduced tree accuracy. For most cases considered (combination of data set and alignment method), the number of instances in which accounting for MSA uncertainty by the SuperMSA approach increased inference accuracy was significantly greater than the number of cases in which the accuracy was decreased, as determined by a Binomial test (Supplementary Table S1 available on Dryad). Taken together, accounting for alternative MSAs increases tree inference accuracy, both when considering the average normRF distance between the inferred and the true tree, as well as when considering the number of cases in which the true tree was inferred.
Components and Mechanism of the SuperMSA Approach.—
Next, we quantified the relevant contribution of the GUIDANCE2 components to the observed increase in tree accuracy. To this end, we compared the average normRF distance between the inferred tree and the true tree, when alternative MSAs were generated 1) considering only co-optimal solutions, that is, using the Head or Tails (Landan and Graur 2008) algorithm; 2) considering only alternative guide trees, that is, using the original GUIDANCE algorithm (Penn et al. 2010); 3) considering only alternative MSAs generated by varying the parameter of gap-opening penalty; 4) combining all these components, as implemented in GUDIANCE2 (Sela et al. 2015). These analyses were performed for each instance of the PAM250 data set with either MAFFT or PRANK as the alignment method (Supplementary Table S2 available on Dryad). Based on MAFFT alignments, in all cases except for the HoT method, using alternative MSAs to produce a SuperMSA resulted in a significant increase in tree inference accuracy compared to using the base MSA (paired Wilcoxon test, all 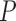 < 2.2e
< 2.2e 16). Furthermore, we found that SuperMSAs constructed by GUIDANCE2-generated alternative MSAs significantly improved the accuracy of tree inference compared to using SuperMSAs based only on a single GUIDANCE2 component (paired Wilcoxon test
16). Furthermore, we found that SuperMSAs constructed by GUIDANCE2-generated alternative MSAs significantly improved the accuracy of tree inference compared to using SuperMSAs based only on a single GUIDANCE2 component (paired Wilcoxon test 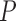
 2.071e
2.071e 08 and
08 and  0.001, compared to GUIDANCE and to varying the gap-opening penalty, respectively). When using PRANK as the alignment method, SuperMSAs produced based on alternative MSAs computed by any of the methods resulted in a significant increase in tree inference accuracy compared to the base MSA (paired Wilcoxon test, all
0.001, compared to GUIDANCE and to varying the gap-opening penalty, respectively). When using PRANK as the alignment method, SuperMSAs produced based on alternative MSAs computed by any of the methods resulted in a significant increase in tree inference accuracy compared to the base MSA (paired Wilcoxon test, all 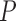 < 0.002). The highest effect was achieved when using MSAs generated by varying the guide-tree component (GUIDANCE). When using PRANK, we found that the accuracy of phylogenetic trees inferred based on SuperMSAs comprised of alternative MSAs based on a single component were not significantly different from phylogenetic trees inferred based on SuperMSAs comprised of GUIDANCE2-generated alternative MSAs.
< 0.002). The highest effect was achieved when using MSAs generated by varying the guide-tree component (GUIDANCE). When using PRANK, we found that the accuracy of phylogenetic trees inferred based on SuperMSAs comprised of alternative MSAs based on a single component were not significantly different from phylogenetic trees inferred based on SuperMSAs comprised of GUIDANCE2-generated alternative MSAs.
Finally, we aimed at characterizing the mechanism behind the ability of the SuperMSA approach to improve tree reconstruction. For each protein of the PAM250 data set and MAFFT as the alignment method, we divided the 20 alternative MSAs comprising the SuperMSA into two groups. Alternative MSAs with a better SPC score than the base MAFFT MSA are termed “bMSAs,” and those with a worse score—“wMSAs.” We inferred a phylogenetic tree based on each single MSA in the bMSA (wMSA) group. We computed the normRF distance between each bMSA (wMSA) tree and the true tree. We next recorded the median distance within the bMSA (wMSA) group. We thus obtained 249 median bMSA (wMSA) values. We found that, as expected, trees inferred based on bMSAs are significantly closer to the true tree than trees inferred based on wMSAs (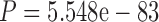 , paired Wilcoxon test, Fig. 2a). Next, we compared the level of agreement between pairs of trees inferred based on bMSAs to the level of agreement between pairs of trees inferred from wMSAs. To this end, we measured the median normRF distance between each pair of trees within the bMSA (wMSA) group, with a lower median normRF distance indicating a higher level of agreement within the group. Overall, we found that trees induced by wMSAs are in stronger disagreement with each other compared to the disagreement between trees induced by bMSAs (in 165 cases out of 249 (66.27%), the bMSAs group had better agreement,
, paired Wilcoxon test, Fig. 2a). Next, we compared the level of agreement between pairs of trees inferred based on bMSAs to the level of agreement between pairs of trees inferred from wMSAs. To this end, we measured the median normRF distance between each pair of trees within the bMSA (wMSA) group, with a lower median normRF distance indicating a higher level of agreement within the group. Overall, we found that trees induced by wMSAs are in stronger disagreement with each other compared to the disagreement between trees induced by bMSAs (in 165 cases out of 249 (66.27%), the bMSAs group had better agreement, 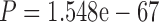 , paired Wilcoxon test, Fig. 2b). Put together, these results suggest that bMSAs increase the support of the true tree, while the total phylogenetic signal carried by wMSAs does not point at a specific wrong tree.
, paired Wilcoxon test, Fig. 2b). Put together, these results suggest that bMSAs increase the support of the true tree, while the total phylogenetic signal carried by wMSAs does not point at a specific wrong tree.
Figure 2.

Inaccurate alternative MSAs do not point at a single wrong topology. For each of the 249 MAFFT-PAM250 cases, the SuperMSA’s alternative MSAs are divided into two sets: bMSAs (more accurate than the base MSA) and wMSAs (less accurate than the base MSA). We inferred a phylogenetic tree based on each single MSA in the bMSA (wMSA) group. Each point represents a single case, for which two median normRF distances were computed: one for the bMSA (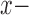 axis) and one for the wMSA (
axis) and one for the wMSA (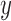 -axis). Trees inferred based on bMSAs are generally closer to the true tree than trees inferred based on wMSAs (a). The level of agreement among trees of each set is measured by the median normRF distance of all pairs in the group, with a lower median distance indicating a higher level of agreement within the set. Sets of bMSA trees have higher agreement than sets of wMSA trees (b).
-axis). Trees inferred based on bMSAs are generally closer to the true tree than trees inferred based on wMSAs (a). The level of agreement among trees of each set is measured by the median normRF distance of all pairs in the group, with a lower median distance indicating a higher level of agreement within the set. Sets of bMSA trees have higher agreement than sets of wMSA trees (b).
Improvement as a Function of the Base MSA Accuracy.
Next we asked when the SuperMSA approach is expected to be most beneficial for phylogeny reconstruction. We hypothesized that the proportion of alternative MSAs that are more accurate than the base MSA decreases as base accuracy increases.
This is indeed the case for the PAM250 and PAM100 data sets with either MAFFT or PRANK as the alignment method (Fig. 3 and Supplementary Fig. S3 available on Dryad). Despite the large variance observed, this suggests a potential increased benefit in considering alternative alignments when the base MSA accuracy is low. Notably, this effect was not observed for the ENSEMBLsim data set (Fig. fig3 and Supplementary Fig. S3 available on Dryad). Further support for this observation was obtained when we repeated the tree accuracy analyses, this time, aligning the sequences using the high accuracy mode LINSI of the MAFFT algorithm. MAFFT-LINSI alignments were, on average, more accurate than those of default MAFFT, and as a result, the increase in accuracy, albeit statistically significant, was smaller when compared to the default MAFFT (MAFFT-LINSI results are given in Supplementary Table S3 available on Dryad).
Figure 3.

The fraction of alternative MSAs that are more accurate than the base MSA as a function of the base MSA accuracy (measured by the SPC score). Each data set is plotted separately: MAFFT-PAM250 (a), MAFFT-PAM100 (b), MAFFT-ENSEMBLsim (c), PRANK-PAM250 (d), PRANK-100 (e), and PRANK-ENSEMBLsim (f). The linear regression line, R-squared, and 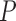 -value are indicated for each data set.
-value are indicated for each data set.
In practice, however, the base MSA accuracy is unknown. We have previously shown that GUIDANCE2 scores are highly predictive of MSA accuracy (Sela et al. 2015) and we show this trend again on the data analyzed in this work. To this end, we computed the correlation between the MSA accuracy and the GUIDANCE2 SPC scores for the base MSA. We found that the Spearman correlations are high for all examined data sets (Supplementary Fig. S4 available on Dryad). Taken together, these results led us to hypothesize that the GUIDANCE2 score of the base MSA can be informative regarding the probability that a more accurate tree is reconstructed when the SuperMSA approach is used.
We next tested the relationship between the GUIDANCE2 SPC score of the base MSA and the ability to improve accuracy of the inferred phylogeny by considering alternative MSAs. For all data sets the trends are clear: the lower the GUIDANCE2 SPC score of the base MSA is, the higher the probability to improve the inferred phylogeny by considering alternative MSAs (Fig. 4 and Supplementary Figs. S5 and S6 available on Dryad). Notably, for lower GUIDANCE2 SPC score, the number of trees that are less accurate does not substantially increase (Supplementary Fig. S7 available on Dryad). Inspection of these results reveals a clear association between the alignment reliability score, the number of better alternative MSAs and the probability to improve the inferred tree. We note that this association is more prominent for MAFFT. We observe that a GUIDANCE2 SPC score for the base MSA lower than 0.6 is associated with a fraction of better MSAs that is greater than 50% and this is when it is most beneficial to use the SuperMSA approach.
Figure 4.

The fraction of improved trees when considering alternative MSAs as a function of the base MSA accuracy scored by GUIDANCE2. Base MSAs were divided into bins according to their GUIDANCE2 SPC score, where lower scores represent less accurate MSAs. The number of improved trees and the total number of trees are indicated inside the bar for each bin. Each data set is plotted separately: MAFFT-PAM250 (a), MAFFT-PAM100 (b), MAFFT-ENSEMBLsim (c), PRANK-PAM250 (d), PRANK-100 (e), and PRANK-ENSEMBLsim (f).
Running Time Analysis.
While the SuperMSA approach has the potential to improve tree reconstruction accuracy it is expected to increase the total computation time. Using the SuperMSA approach requires two computational steps: 1) generating alternative MSAs to produce the SuperMSA and 2) performing tree inference based on the SuperMSA. The increase in runtime due to the first step is linear in the number of alternative MSAs produced by GUIDANCE2. For the PAM250 data set, the first step took, on average, 16 min and 32 h when aligning using MAFFT and PRANK, respectively (Supplementary Table S4 available on Dryad). Similar results were obtained for the PAM100 data set (Supplementary Table S4 available on Dryad). The run time of the second step is expected to increase linearly in the number of alternative MSAs included in the SuperMSA. Moreover, it is possible that considering too many alternative alignments will introduce additional noise that will eventually decrease the accuracy of the inferred phylogeny. For each instance of the PAM250 data set aligned with MAFFT we tested the trade-off between the improvement in tree reconstruction accuracy and running time as a function of the number of alternative alignments included in the SuperMSA (Fig. 5). As expected, the running time of the tree inference step increases linearly with the number of alternative alignments considered. The accuracy of the inferred tree increases in a nonlinear fashion as more alternative alignments are used. Considering 20 alternative MSAs seems to provide a good balance between the running time and the improvement in tree accuracy.
Figure 5.

The accuracy of the inferred tree and running time as a function the number of alternative MSAs in the SuperMSA calculated for the MAFFT-PAM250 data set.
Improving Bayesian Tree Inference Using Alternative Alignments
In the following section, we demonstrate that the SuperMSA approach improves Bayesian tree inference. In this section, we focused on the PAM100 and PAM250 data sets as they represent the least and most diverged data sets, respectively. Considering alternative MSAs improved the accuracy of Bayesian tree inference as implemented in MrBayes (Ronquist et al.2012), similar to the improvement observed when trees were reconstructed using maximum-likelihood (Table 4). Specifically, for both the MAFFT-PAM250 and the PRANK-PAM250 MSAs, the improvement in tree accuracy was statistically significant (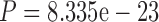 and
and 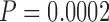 for MAFFT and PRANK, respectively, paired Wilcoxon test). For the PAM100 analysis, a modest improvement in tree accuracy was observed, although, it was only statistically significant for PRANK (
for MAFFT and PRANK, respectively, paired Wilcoxon test). For the PAM100 analysis, a modest improvement in tree accuracy was observed, although, it was only statistically significant for PRANK (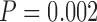 ).
).
Table 4.
The effect of MSA averaging on Bayesian tree inference accuracy
| Base MSA | SuperMSA | |||
|---|---|---|---|---|
| Data set | Alignment method | Average normalized RF distance | Average normalized RF distance |
 -value -value |
| PAM250 | True MSA | 0.098 (0.07) | ||
| MAFFT | 0.18 (0.119) | 0.148 (0.098) | 8.335e–23 | |
| PRANK | 0.168 (0.109) | 0.161 (0.105) | 0.0002 | |
| PAM100 | True MSA | 0.085 (0.068) | ||
| MAFFT | 0.086 (0.068) | 0.084 (0.067) | 0.112 | |
| PRANK | 0.087 (0.072) | 0.083 (0.064) | 0.002 | |
Note: In parenthesis are standard deviations. Statistically significant values are in bold.
Comparison to Bayesian Coestimation of the MSA and Tree.
In the analyses presented above, two methods for tree reconstruction were employed: the ML method RAxML (Stamatakis 2014), and the Bayesian method MrBayes (Ronquist et al. 2012). Notably, both these methods rely on a single MSA as input. Here, we compare the performance of the SuperMSA approach to Bali-Phy (Redelings and Suchard 2005)—a full Bayesian framework, in which both the tree and the MSA are inferred. Bali-Phy allows sampling the joint MSA and tree space, and thus, it also accounts for alignment uncertainty. We aimed to compare the accuracy of the Bali-Phy maximum a posteriori (MAP) tree to the accuracy of MrBayes MAP tree, obtained by using either the base MSA or the SuperMSA. Due to the computational high demand of running Bali-Phy, we limited the comparison to 50 instances randomly sampled from the PAM250 data set. On this subset, the average normRF distance between the MrBayes MAP tree and the true tree was 0.174 (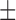 0.137) and 0.177 (
0.137) and 0.177 (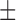 0.131), when using MAFFT and PRANK base MSA, respectively. When the base MSA was replaced with the SuperMSA, the average normRF distance between the MrBayes MAP tree and the true tree was reduced to 0.15 (
0.131), when using MAFFT and PRANK base MSA, respectively. When the base MSA was replaced with the SuperMSA, the average normRF distance between the MrBayes MAP tree and the true tree was reduced to 0.15 (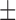 0.112) and 0.175 (
0.112) and 0.175 (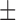 0.126) for MAFFT and PRANK respectively, in accordance with the results presented for the larger data set (Table 4). The average normRF distance of the MAP tree computed using Bali-Phy was substantially smaller (0.091
0.126) for MAFFT and PRANK respectively, in accordance with the results presented for the larger data set (Table 4). The average normRF distance of the MAP tree computed using Bali-Phy was substantially smaller (0.091 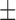 0.069) than that obtained using the SuperMSA approach. As can be expected, for this subset, the average normRF distances obtained by Bali-Phy were similar to those obtained when providing MyBayes with the “true” alignment (0.091
0.069) than that obtained using the SuperMSA approach. As can be expected, for this subset, the average normRF distances obtained by Bali-Phy were similar to those obtained when providing MyBayes with the “true” alignment (0.091  0.064). The computational run times of Bali-Phy and MrBayes (including the time to produce the alternative MSAs) for an MSA were on average 15.39 days and 4.31 days, respectively.
0.064). The computational run times of Bali-Phy and MrBayes (including the time to produce the alternative MSAs) for an MSA were on average 15.39 days and 4.31 days, respectively.
We next examined a larger data set of 200 sequences simulated using INDELible (Fletcher and Yang 2009). While the SuperMSA approach on this data set resulted in significantly more accurate ML trees compared to using a single base MSA (see Supplementary Text available on Dryad for a full description of the simulation scheme and the detailed results), running Bali-Phy on this large input failed at an early step on every run. This is in accordance with previous reports of failed Bali-Phy runs for data sets of > 200 species (Nute and Warnow 2016) and limiting running times (weeks) for data sets with 70–100 species (Gaya et al. 2011; McKenzie et al. 2014). Taken together, these results strengthen the observation that accounting for MSA uncertainty, either using Bali-Phy or using the SuperMSA approach, is highly beneficial. While Bali-Phy reduces the normRF distance more than the SuperMSA approach, using Bali-Phy entails a heavy computational cost and hence, its applicability might be limited when hundreds of sequences and MSAs are analyzed.
Improving Branch Length Estimates Using Alternative Alignments
Accounting for alternative MSAs may also improve the accuracy of branch lengths estimation. To demonstrate this, we used RAxML to estimate branch lengths constraining on the true topology, using either the base MSA or the SuperMSA. The estimated branch lengths were compared to the true ones, by computing the Euclidean distances between the two branch lengths vectors. For MAFFT-PAM250 when using the base MSA, the mean Euclidean distance was 1.029 substitutions per site. This mean distance was reduced to 0.804, when replacing the base MSA with the SuperMSA (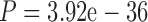 , paired Wilcoxon test). A similar significant reduction was observed when PRANK was used to align the same sequences (Table 5). Of note, the improvement in branch length accuracy when averaging over alternative MSAs was greater compared to the improvement obtained by weighting columns according to their reliability, regardless of the method used to quantify the column reliability (GUIDANCE2, ZORRO, or TCS) (Table 5). Consistent with the results presented above, when less divergent sequences were considered (i.e., the PAM100 data set), no significant improvement was observed (Table 6). We additionally tested whether averaging over alternative MSAs results in branch lengths overestimation or underestimation. Our results (Supplementary Table S5 and S6 available on Dryad) show that averaging over alternative MSAs does not significantly reduce error compared to the base MSA (P
, paired Wilcoxon test). A similar significant reduction was observed when PRANK was used to align the same sequences (Table 5). Of note, the improvement in branch length accuracy when averaging over alternative MSAs was greater compared to the improvement obtained by weighting columns according to their reliability, regardless of the method used to quantify the column reliability (GUIDANCE2, ZORRO, or TCS) (Table 5). Consistent with the results presented above, when less divergent sequences were considered (i.e., the PAM100 data set), no significant improvement was observed (Table 6). We additionally tested whether averaging over alternative MSAs results in branch lengths overestimation or underestimation. Our results (Supplementary Table S5 and S6 available on Dryad) show that averaging over alternative MSAs does not significantly reduce error compared to the base MSA (P  1, paired Wilcoxon test), but rather, results in a slight (less than 5%) overestimation of the total branch lengths. We could not detect any significant difference between internal and external branches (Supplementary Table S6 available on Dryad). We conclude that the major effect of the SuperMSA approach is on the tree topology rather than on branch length estimation.
1, paired Wilcoxon test), but rather, results in a slight (less than 5%) overestimation of the total branch lengths. We could not detect any significant difference between internal and external branches (Supplementary Table S6 available on Dryad). We conclude that the major effect of the SuperMSA approach is on the tree topology rather than on branch length estimation.
Table 5.
The effect of MSA weighting (GUIDANCE2, ZORRO, TCS) and averaging on tree branch length accuracy for the PAM250 data set
| No weighting | GUIDANCE2 | ZORRO | TCS | SuperMSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| Alignment method | Average BL distance | Average BL distance |
 -value -value |
Average BL distance |
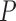 -value -value |
Average BL distance |
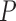 -value -value |
Average BL distance |
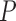 -value -value |
| True MSA | 0.598 (0.269) | ||||||||
| MAFFT | 1.029 (0.664) | 0.987 (0.631) | 1.257e–24 | 1.018 (0.673) | 0.0002 | 1.028 (0.689) | 0.0242 | 0.804 (0.403) | 3.92e–36 |
| PRANK | 0.968 (0.483) | 0.965 (0.483) | 0.0515 | 1.002 (0.609) | 0.9902 | 1.069 (1.882) | 0.9822 | 0.953 (0.59) |
2.862e 05 05
|
Notes: Euclidean branch length (BL) distance was calculated compared to the true tree used in simulation. All 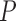 -values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
Table 6.
The effect of MSA weighting (GUIDANCE2, ZORRO, TCS) and averaging on tree branch length accuracy for the PAM100 data set
| No weighting | GUIDANCE2 | ZORRO | TCS | SuperMSA | |||||
|---|---|---|---|---|---|---|---|---|---|
| Alignment method | Average BL distance | Average BL distance |
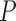 -value -value |
Average BL distance |
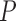 -value -value |
Average BL distance |
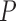 -value -value |
Average BL distance |
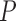 -value -value |
| True MSA | 0.261 (0.11) | ||||||||
| MAFFT | 0.264 (0.111) | 0.263 (0.111) |
1.09e 06 06
|
0.263 (0.111) | 0.0001 | 0.263 (0.112) | 0.0036 | 0.264 (0.113) | 0.1966 |
| PRANK | 0.262 (0.111) | 0.262 (0.111) | 0.0269 | 0.262 (0.112) | 0.4273 | 0.262 (0.112) | 0.9259 | 0.262 (0.111) | 0.6645 |
Notes: Euclidean branch length (BL) distance was calculated compared to the true tree used in simulation. All 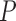 -values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
Empirical Data Sets
We tested whether alternative alignments can improve the phylogeny using empirical rather than simulated sequences. For that purpose, we used sets of sequences from Fungi, Bacteria and Eukaryota for which the phylogeny is widely accepted (Tan et al. 2015a). Our analysis indicates that this data set mostly includes relatively reliable alignments, as reflected by their high GUIDANCE2 scores: an average SPC of 0.89 for all data sets (Table 7). Consequently, the accuracy of the inferred tree using the base MSA was relatively high and the effect of considering alternative MSAs was modest and statistically significant for the Fungi and Bacteria data sets (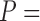 1.46e
1.46e 4 and
4 and 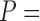 0.023, respectively, paired Wilcoxon test). These results are consistent with the results obtained for the simulated data sets: as the GUIDANCE2 score is relatively high, only limited improvement is expected when incorporating alternative MSAs.
0.023, respectively, paired Wilcoxon test). These results are consistent with the results obtained for the simulated data sets: as the GUIDANCE2 score is relatively high, only limited improvement is expected when incorporating alternative MSAs.
Table 7.
The effect of MSA weighting and averaging on tree accuracy for empirical data
| Base MSA (MAFFT) | GUIDANCE2 weighting | SuperMSA | ||||
|---|---|---|---|---|---|---|
| Average normalized RF distance | Average normalized RF distance |
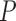 -value -value |
Average normalized RF distance |
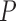 -value -value |
GUIDANCE2 SPC score for base MSA | |
| Bacteria | 0.571 (0.34) | 0.565 (0.338) | 0.1154 | 0.56 (0.333) | 0.023 | 0.89 (0.09) |
| Fungi | 0.158 (0.241) | 0.141 (0.23) | 0.0116 | 0.128 (0.218) | 0.000146 | 0.89 (0.1) |
| Eukaryota | 0.087 (0.185) | 0.087 (0.181) | 0.5125 | 0.087 (0.172) | 0.504 | 0.89 (0.12) |
Notes: All  -values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
-values were computed using a one-sided Wilcoxon test. In parenthesis are standard deviations. Statistically significant values are in bold.
Discussion
A common practice in bioinformatics employs only a single base MSA for downstream analyses (e.g., phylogeny reconstruction, ancestral sequence reconstruction, selective force inference). In order to reduce the effect of MSA uncertainty, two main approaches were suggested: 1) filtering or masking unreliable alignment columns (Talavera and Castresana 2007; Dress et al. 2008; Capella-Gutierrez et al. 2009; Kuck et al. 2010; Jordan and Goldman 2012; Privman et al. 2012; Rajan 2013) or 2) weighting alignment columns according to their estimated reliability (Wu et al. 2012; Chang et al. 2014). Specifically, for the tree inference problem, it was shown that these methods may sometimes be inefficient and even reduce the inferred tree accuracy (Tan et al. 2015a). Here, we demonstrated that tree inference using a set of alternative alignments as input is superior over weighting columns and can significantly improve the inferred tree accuracy (topology and branch lengths). Producing alternative alignments using GUIDANCE2 is also modular and can be easily incorporated in bioinformatics pipelines, which take an MSA as input, avoiding the complexity and computational burden associated with the full statistical framework discussed below.
One possible conceptual criticism regarding the work presented in this study is that concatenating alternative MSA columns contradicts one of the philosophical assumptions at the base of phylogenetic studies, which is that alignment columns represent homologous characters. When considering concatenated alignments, the same character in a specific sequence can appear homologous to two different characters in a second sequence, allegedly violating this basic assumption. In this work, we follow the statistical framework (Lunter et al. 2005b; Redelings and Suchard 2005; Novak et al. 2008; Herman et al. 2014) that considers the MSA as a parameter (hidden state) of the phylogenetic inference, and advocate that for this parameter too, similar to all other model parameters, tree, and branch lengths, uncertainty should be accounted for. Furthermore, we claim that our methodology does not violate the assumption of site-specific homology in the same sense as accounting for tree uncertainty does not violate the assumption of sequence evolution along a tree. Rather, as site-specific homology is unknown, uncertainty in it should be accounted for both when reconstructing the tree, and for downstream phylogenetic inference.
Our procedure to generate alternative alignments is an ad hoc methodology. Notably, the statistical alignment framework (Holmes and Bruno 2001; Lunter et al. 2005a; Metzler et al. 2005) can be used to rigorously sample MSAs from the posterior distribution. Furthermore, it can offer a joint estimation of both the MSA and the phylogeny (Lunter et al. 2005b; Redelings and Suchard 2005; Novak et al. 2008; Herman et al. 2014). However, statistical alignment methods are still computationally very intensive and are therefore limited in practice to the analysis of a small number of sequences (Lunter et al. 2005b; Herman et al. 2014). In addition, advanced evolutionary models were recently developed, which provide a more realistic representation of the evolutionary dynamic in terms of substitutions and how they vary across sequence sites and tree branches (e.g., Galtier 2001; Quang et al. 2008; Rubinstein et al. 2011; Zaheri et al. 2014). All these models assume that the MSA is given (i.e., fixed) and thus uncertainty in the MSA is not accounted for. Unfortunately, these advanced evolutionary models are currently not implemented within any statistical alignment software. The SuperMSA approach suggested here allows a practical solution for using these advanced models while also accounting for alignment uncertainty.
An interesting research direction is related to estimating posterior probabilities of alignments given a specific indel and substitution model. It is still an open question how well an ad hoc sampling approach, such as the one implemented in GUIDANCE2, approximates a sample from the posterior MSA space. Answers to this question would potentially lead to better sampling strategies and thus, to even more accurate tree reconstructions.
In this work, the possible benefit of accounting for MSA uncertainty in tree reconstruction was studied. As both the tree and the alignment are usually unknown, the two types of uncertainties (that of the tree and that of the alignment) are inherent to most downstream molecular evolution inference methodologies. Notably, while accounting for tree uncertainty within a Bayesian framework is now integrated in various molecular evolution applications, such as the inference of ancestral character states (Pagel et al. 2004), the reconciliation of gene trees and species trees (Arvestad et al. 2003), and the inference of site-specific evolutionary conservation (Mayrose et al. 2005), accounting for MSA uncertainty is remarkably less common. We hope that the fast heuristics presented here for generating alternative MSAs will help diminish the above difference between the way these two types of uncertainty are accounted for.
Acknowledgments
We thank the Associate Editor and two anonymous reviewers for helpful comments and suggestions.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.4j1qt.
Funding
This work was supported by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University to H.A. and E.L.K.; ISF [Grant No. 802/16 to T.P.]; European Research Council to G.L. [Grant No. 281357 to Tal Dagan].
References
- Aagesen L. 2004. The information content of an ambiguously alignable region, a case study of the trnL intron from the Rhamnaceae. Organ. Divers. Evol. 4:35–49. [Google Scholar]
- Arvestad L., Berglund A.C., Lagergren J., Sennblad B.. 2003. Bayesian gene/species tree reconciliation and orthology analysis using MCMC. Bioinformatics 19(Suppl 1):i7–15. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H., Levy Karin E., Mertens Z., Cartwright R.A., Pupko T.. 2017. SpartaABC: a web server to simulate sequences with indel parameters inferred using an approximate Bayesian computation algorithm. Nucleic Acids Res. 45:W453–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburne B.P., Whelan S.. 2012. Measuring the distance between multiple sequence alignments. Bioinformatics 28:495–502. [DOI] [PubMed] [Google Scholar]
- Blackshields G., Wallace I.M., Larkin M., Higgins D.G.. 2006. Analysis and comparison of benchmarks for multiple sequence alignment. In Silico Biol. 6:321–339. [PubMed] [Google Scholar]
- Boyce K., Sievers F., Higgins D.G.. 2015. Instability in progressive multiple sequence alignment algorithms. Algorithms Mol. Biol. 10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.M., Di Tommaso P., Notredame C.. 2014. TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol. Biol. Evol. 31:1625–1637. [DOI] [PubMed] [Google Scholar]
- Collingridge P.W., Kelly S.. 2012. MergeAlign: improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. BMC Bioinformatics 13:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do C.B., Katoh K.. 2008. Protein multiple sequence alignment. Methods Mol. Biol. 484:379–413. [DOI] [PubMed] [Google Scholar]
- Do C.B., Mahabhashyam M.S., Brudno M., Batzoglou S.. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res., 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dress A.W., Flamm C., Fritzsch G., Grunewald S., Kruspe M., Prohaska S.J., Stadler P.F.. 2008. Noisy: identification of problematic columns in multiple sequence alignments. Algorithms Mol. Biol. 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher W., Yang Z.. 2009. INDELible: a flexible simulator of biological sequence evolution. Mol. Biol. Evol. 26:1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N. 2001. Maximum-likelihood phylogenetic analysis under a covarion-like model. Mol. Biol. Evol., 18:866–873. [DOI] [PubMed] [Google Scholar]
- Gatesy J., DeSalle R., Wheeler W.. 1993. Alignment-ambiguous nucleotide sites and the exclusion of systematic data. Mol. Phylogenet. Evol. 2:152–157. [DOI] [PubMed] [Google Scholar]
- Gaya E., Redelings B.D., Navarro-Rosines P., Llimona X., De Caceres M., Lutzoni F.. 2011. Align or not to align? Resolving species complexes within the Caloplaca saxicola group as a case study. Mycologia, 103:361–378. [DOI] [PubMed] [Google Scholar]
- Geiger D.L. 2002. Stretch coding and block coding: two new strategies to represent questionably aligned DNA sequences. J. Mol. Evol. 54:191–199. [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321. [DOI] [PubMed] [Google Scholar]
- Herman J.L., Challis C.J., Novak A., Hein J., Schmidler S.C.. 2014. Simultaneous Bayesian estimation of alignment and phylogeny under a joint model of protein sequence and structure. Mol. Biol. Evol. 31:2251–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero J.,, Muffato M.,, Beal K.,, Fitzgerald S.,, Gordon L.,, Pignatelli M.,, Vilella A.J.,, Searle S.M.,, Amode R.,, Brent S.,, Spooner W.,, Kulesha E.,, Yates A.,, Flicek P. 2016. Ensembl comparative genomics resources. Database 2016. 10.1093/database/bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder M., Lewis P.O.. 2003. Phylogeny estimation: traditional and Bayesian approaches. Nat. Rev. Genet. 4:275–284. [DOI] [PubMed] [Google Scholar]
- Holmes I., Bruno W.J.. 2001. Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics 17:803–820. [DOI] [PubMed] [Google Scholar]
- Jordan G., Goldman N.. 2012. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol. Biol. Evol. 29:1125–1139. [DOI] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Toh H.. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9:286–298. [DOI] [PubMed] [Google Scholar]
- Kemena C., Notredame C.. 2009. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25:2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuck P., Meusemann K., Dambach J., Thormann B., von Reumont B.M., Wagele J.W., Misof B.. 2010. Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front. Zool. 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupczok A., Schmidt H.A., von Haeseler A.. 2010. Accuracy of phylogeny reconstruction methods combining overlapping gene data sets. Algorithms Mol. Biol. 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake J.A. 1991. The order of sequence alignment can bias the selection of tree topology. Mol. Biol. Evol. 8:378–385. [DOI] [PubMed] [Google Scholar]
- Landan G., Graur D.. 2008. Local reliability measures from sets of co-optimal multiple sequence alignments. Pac. Symp. Biocomput. 13:15–24. [PubMed] [Google Scholar]
- Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., Thompson J.D., Gibson T.J., Higgins D.G.. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lee M.S.Y. 2001. Unalignable sequences and molecular evolution. Trends Ecol. Evol. 16:681–685. [Google Scholar]
- Levy Karin E., Shkedy D., Ashkenazy H., Cartwright R.A., Pupko T.. 2017. Inferring rates and length-distributions of indels using approximate Bayesian computation. Genome Biol. Evol. 9:1280–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A. 2012. Alignment methods: strategies, challenges, benchmarking, and comparative overview. Methods Mol. Biol. 855:203–235. [DOI] [PubMed] [Google Scholar]
- Loytynoja A., Goldman N.. 2008. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320:1632–1635. [DOI] [PubMed] [Google Scholar]
- Loytynoja A., Milinkovitch M.C.. 2003. A hidden Markov model for progressive multiple alignment. Bioinformatics 19:1505–1513. [DOI] [PubMed] [Google Scholar]
- Loytynoja A., Vilella A.J., Goldman N.. 2012. Accurate extension of multiple sequence alignments using a phylogeny-aware graph algorithm. Bioinformatics 28:1684–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking R., Hodkinson B.P., Stamatakis A., Cartwright R.A.. 2011. PICS-Ord: unlimited coding of ambiguous regions by pairwise identity and cost scores ordination. BMC Bioinformatics 12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G., Drummond A.J., Miklós I., Hein J.. 2005a. Statistical alignment: recent progress, new applications, and challenges. In: Nielsen R., editor. Statistical methods in molecular evolution. New York: Springer; p. 375–405. [Google Scholar]
- Lunter G., Miklos I., Drummond A., Jensen J.L., Hein J.. 2005b. Bayesian coestimation of phylogeny and sequence alignment. BMC Bioinformatics 6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzoni F., Wagner P., Reeb V., Zoller S.. 2000. Integrating ambiguously aligned regions of DNA sequences in phylogenetic analyses without violating positional homology. Syst. Biol. 49:628–651. [DOI] [PubMed] [Google Scholar]
- Mayrose I., Mitchell A., Pupko T.. 2005. Site-specific evolutionary rate inference: taking phylogenetic uncertainty into account. J. Mol. Evol. 60:345–353. [DOI] [PubMed] [Google Scholar]
- McKenzie S.K., Oxley P.R., Kronauer D.J.. 2014. Comparative genomics and transcriptomics in ants provide new insights into the evolution and function of odorant binding and chemosensory proteins. BMC Genomics 15:718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Md Mukarram Hossain A.S., Blackburne B.P., Shah A., Whelan S.. 2015. Evidence of statistical inconsistency of phylogenetic methods in the presence of multiple sequence alignment uncertainty. Genome Biol. Evol. 7:2102–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler D., Fleißner R., Wakolbinger A., von Haeseler A.. 2005. Stochastic insertion-deletion processes and statistical sequence alignment. In: Deuschel JD,, Greven A., editors. Interacting stochastic systems. Berlin, Heidelberg: Springer; p. 247–267. [Google Scholar]
- Morrison D.A., Ellis J.T.. 1997. Effects of nucleotide sequence alignment on phylogeny estimation: a case study of 18S rDNAs of apicomplexa. Mol. Biol. Evol. 14:428–441. [DOI] [PubMed] [Google Scholar]
- Nelesen S., Liu K., Zhao D., Linder C.R., Warnow T.. 2008. The effect of the guide tree on multiple sequence alignments and subsequent phylogenetic analyses. Pac. Symp. Biocomput. 13:25–36. [DOI] [PubMed] [Google Scholar]
- Novak A., Miklos I., Lyngso R., Hein J.. 2008. StatAlign: an extendable software package for joint Bayesian estimation of alignments and evolutionary trees. Bioinformatics 24:2403–2404. [DOI] [PubMed] [Google Scholar]
- Nuin P.A., Wang Z., Tillier E.R.. 2006. The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinformatics 7:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nute M., Warnow T.. 2016. Scaling statistical multiple sequence alignment to large datasets. BMC Genomics 17:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden T.H., Rosenberg M.S.. 2006. Multiple sequence alignment accuracy and phylogenetic inference. Syst. Biol., 55:314–328. [DOI] [PubMed] [Google Scholar]
- Pagel M., Meade A., Barker D.. 2004. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 53:673–684. [DOI] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Penn O., Privman E., Landan G., Graur D., Pupko T.. 2010. An alignment confidence score capturing robustness to guide tree uncertainty. Mol. Biol. Evol. 27:1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privman E., Penn O., Pupko T.. 2012. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol. Biol. Evol. 29:1–5. [DOI] [PubMed] [Google Scholar]
- Quang le S., Gascuel O., Lartillot N.. 2008. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 24:2317–2323. [DOI] [PubMed] [Google Scholar]
- Rajan V. 2013. A method of alignment masking for refining the phylogenetic signal of multiple sequence alignments. Mol. Biol. Evol. 30:689–712. [DOI] [PubMed] [Google Scholar]
- Redelings B.D., Suchard M.A.. 2005. Joint Bayesian estimation of alignment and phylogeny. Syst. Biol. 54:401–418. [DOI] [PubMed] [Google Scholar]
- Robinson D.F., Foulds L.R.. 1981. Comparison of phylogenetic trees. Math. Biosci. 53:131–147. [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Hohna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P.. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein N.D., Doron-Faigenboim A., Mayrose I., Pupko T.. 2011. Evolutionary models accounting for layers of selection in protein-coding genes and their impact on the inference of positive selection. Mol. Biol. Evol. 28:3297–3308. [DOI] [PubMed] [Google Scholar]
- Sela I., Ashkenazy H., Katoh K., Pupko T.. 2015. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43:W7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J., Thompson J.D., Higgins D.G.. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe A.B., Sanderson M.J., Nadler S.A.. 2006. Nematode small subunit phylogeny correlates with alignment parameters. Syst. Biol. 55:972–992. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G., Castresana J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56:564–577. [DOI] [PubMed] [Google Scholar]
- Tan G., Muffato M., Ledergerber C., Herrero J., Goldman N., Gil M., Dessimoz C.. 2015a. Current methods for automated filtering of multiple sequence alignments frequently worsen single-gene phylogenetic inference. Syst. Biol. 64:778–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G., Muffato M., Ledergerber C., Herrero J., Goldman N., Gil M., Dessimoz C.. 2015b. Data from: current methods for automated filtering of multiple sequence alignments frequently worsen single-gene phylogenetic inference. Dryad Data Repository. 10.5061/dryad.pc5j0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Linard B., Lecompte O., Poch O.. 2011. A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives. PLoS One 6:e18093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Plewniak F., Poch O.. 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27:2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace I.M., O’Sullivan O., Higgins D.G., Notredame C.. 2006. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34:1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.S., Leebens-Mack J., Kerr Wall P., Beckmann K., dePamphilis C.W., Warnow T.. 2011. The impact of multiple protein sequence alignment on phylogenetic estimation. IEEE/ACM Trans. Comput. Biol. Bioinform. 8:1108–1119. [DOI] [PubMed] [Google Scholar]
- Wheeler W.C., Gladstein D.. 1994. MALIGN: a multiple sequence alignment program. J. Heredity 85:417–418. [Google Scholar]
- Wheeler W.C. 1995. Sequence alignment, parameter sensitivity, and the phylogenetic analysis of molecular data. Syst. Biol. 44:321–331. [Google Scholar]
- Wheeler W.C., Gatesy J., DeSalle R.. 1995. Elision: a method for accommodating multiple molecular sequence alignments with alignment-ambiguous sites. Mol. Phylogenet. Evol. 4:1–9. [DOI] [PubMed] [Google Scholar]
- Whelan S., Goldman N.. 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18:691–699. [DOI] [PubMed] [Google Scholar]
- Wong K.M., Suchard M.A., Huelsenbeck J.P.. 2008. Alignment uncertainty and genomic analysis. Science 319:473–476. [DOI] [PubMed] [Google Scholar]
- Wu M., Chatterji S., Eisen J.A.. 2012. Accounting for alignment uncertainty in phylogenomics. PLoS One 7:e30288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheri M., Dib L., Salamin N.. 2014. A generalized mechanistic codon model. Mol. Biol. Evol. 31:2528–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O.G., Janacek S.H., Juettemann T., To J.K., Laird M.R., Lavidas I., Liu Z., Loveland J.E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D.N., Newman V., Nuhn M., Ogeh D., Ong C.K., Parker A., Patricio M., Riat H.S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S.E., Kostadima M., Langridge N., Martin F.J., Muffato M., Perry E., Ruffier M., Staines D.M., Trevanion S.J., Aken B.L., Cunningham F., Yates A., Flicek P.. 2017. Ensembl 2018. Nucleic Acids Res. 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]