

 We explore the capability of SLIM-based IMS for isomer selectivity in combination with cryogenic, messenger-tagging IR spectroscopy.
We explore the capability of SLIM-based IMS for isomer selectivity in combination with cryogenic, messenger-tagging IR spectroscopy.
Abstract
Double-resonance spectroscopic schemes in combination with cryogenic ion traps are the go-to techniques when isomer-specific high-resolution spectra are required for analysis of molecular ions. Their limitation lies in the requirement for well-resolved, isomer-specific absorption bands as well as in the potentially time-consuming steps to identify each isomer. We present an alternative approach where isomeric species are readily separated using ion mobility spectrometry (IMS) and selected prior to cryogenic spectroscopic analysis. To date, most IMS approaches suffer from relatively low resolution, however, recent technological developments in the field of travelling-wave ion mobility using structures for lossless ion manipulation (SLIM) permit the use of extremely long drift paths, which greatly enhances the resolution. We demonstrate the power of combining this type of ultra-high resolution IMS with cryogenic vibrational spectroscopy by comparing mobility-resolved IR spectra of a disaccharide to those acquired using IR–IR double resonance. This new approach is especially promising for the investigation of larger molecules where spectral congestion interferes with double resonance techniques.
Introduction
The development of atmospheric pressure ionisation (API) sources such as electrospray or MALDI over the last three decades has opened exciting new directions in molecular ion spectroscopy.1–8 Molecules of virtually any size can be transferred into the gas phase, where action-spectroscopic schemes in the IR or UV can reveal details about their structure and intramolecular interactions, aided by quantum-chemical calculations. While vibrational spectra of relatively small molecular ions can be well resolved even at room temperature,9,10 spectral congestion from thermal inhomogeneous broadening and conformational heterogeneity complicates interpretation of spectra for larger molecules. Both effects gain in importance as the size of the molecule, and hence the number of vibrational degrees of freedom, increases. The former can be largely eliminated by cooling molecular ions to their vibrational ground state, which is most commonly achieved using collisions with a buffer gas in cryogenic multipole traps.4,5,8,11–13 When used in combination with either IR-UV photofragment spectroscopy8,12,13 or messenger-tagging spectroscopy,5,14–16 one can generate greatly simplified, linear IR spectra, which facilitates the determination of molecular structure by comparison to calculations. Spectral congestion arising from conformational heterogeneity is more difficult to eliminate, however. Two approaches are commonly employed. By simultaneously requiring molecules to satisfy two or more resonance conditions, one can often separate overlapping spectra. As an example, by fixing a UV probe laser on a conformer-specific electronic transition and then scanning an IR pump laser that arrives prior to the probe, one can induce a “dip” in the UV-induced fragment signal when its wavelength is in resonance with a vibrational transition in the probed conformer.8,12 In this case a UV chromophore and a conformer-resolved electronic spectrum are required, which limits the general applicability of this approach. Alternatively, IR–IR hole-burning on messenger-tagged molecules can achieve similar results while being more general,14 however this still requires the presence of resolved conformer-specific transitions. One could envision higher-order multi-colour schemes in the IR or the UV, but the necessity to identify a conformer-specific transition persists. This can be extremely time consuming and, in many cases, simply impossible.
Another approach is to physically separate molecules of different conformations or isomeric forms prior to spectroscopic investigation using, for example, ion mobility spectrometry (IMS).17 The technique makes use of the difference in the mobility of isomers or conformers when they drift through an inert buffer gas under the influence of an electric field. In addition to reducing the conformational complexity of an ensemble of ions, IMS can determine an orientationally-averaged collision cross section (CCS), which can then serve as a filter for calculated structures, greatly simplifying the conformational search.18–21 Ion mobility spectrometry has gained popularity in analytical applications since IMS-MS instruments became commercially available about a decade ago.22 In different IMS variants, ions of a particular mobility can either be continuously filtered using, for example, differential IMS or FAIMS,23–27 or they can be selected in a pulsed fashion after separation in time as occurs, for example, in drift tube (DT) IMS,28–30 where a constant electric field propels the ions through the drift region. Recent reports using IMS-MS as a conformational pre-filter for spectroscopic investigation have applied this approach to study small drug molecules,10 carbohydrates,31–33 peptides and proteins,15,34–37 and even weakly bound peptide aggregates or clusters.38,39
How does the combination of IMS-MS with spectroscopic methods compare to double-resonance spectroscopic schemes in terms of isomer selectivity? While spectroscopic approaches can have extremely high sensitivity to the slightest structural differences, they rely on one’s ability to find and resolve transitions that are characteristic of a particular conformer. For an IMS technique to be similarly sensitive to different isomers of the same molecule, its resolving power would need to be sufficiently high to distinguish minute differences in CCS. While the resolving power of linear IMS instruments is limited by constraints in the use of high voltages and instrument size, new technological developments in travelling wave (TW) IMS using so-called structures for lossless ion manipulation (SLIM) have emerged as a promising alternative.40,41 In its most recent implementation, ions can drift in a nearly lossless manner inside travelling-wave electric potentials produced by mirrored pairs of electrodes on planar printed circuit boards (PCBs), separated by a gap of a few millimetres. Using SLIM, ions can be directed around corners, enabling the entire two-dimensional space of the SLIM device to be used for IM separation. This poses a tremendous advantage over previous IM techniques in terms of resolving power, since it is approximately proportional to the square root of the drift-path length.42 Moreover, the drift length can be further extended to almost arbitrary length by cycling ions repeatedly through the same SLIM structure until the desired resolving power is reached.43
Here we explore the capability of SLIM-based IMS for isomer selectivity in combination with cryogenic, messenger-tagging IR spectroscopy. To demonstrate the promise of this approach, we directly compare it with IR–IR double resonance spectroscopy on the same disaccharide molecules. We expect the rapidly evolving techniques of ultra-high resolution IMS to soon be able to separate molecules with the most minuscule differences in molecular structure, which can then be probed by cryogenic messenger-tagging spectroscopy to provide their fingerprints for identification and structural analysis.
Experimental methods
Sample preparation
The disaccharide Galβ(1-4)GlcNAc was purchased from Dextra UK and used without further purification. Solvents were purchased from Sigma-Aldrich. For (nano) electrospray ionisation, samples were diluted with water/methanol v/v 50/50 to yield a concentration of 100 μM. To enhance the formation of singly sodiated disaccharides, 1.5 equivalents of NaCl were added.
IR–IR double resonance messenger-tagging spectroscopy
Experiments were performed on a home-built tandem quadrupole instrument equipped with a cryogenically cooled octupole ion trap to allow for messenger-tagging spectroscopy. A detailed description of the instrument can be found elsewhere.44 Briefly, ions are electrosprayed into the instrument using commercially available nano-electrospray emitters (Thermo Fisher) and transferred into high vacuum. Here, they are m/z selected by a quadrupole mass filter before being trapped and cooled by collisions with helium buffer gas inside an octupole ion trap, which is maintained at a temperature below 5 K using a closed-cycle helium cryostat. Helium atoms condense onto the molecular ions due to their low internal temperature. The contents of the ion trap can be analysed using a second quadrupole mass filter and a channeltron ion detector. In order to acquire an IR vibrational spectrum, the helium-tagged ions are irradiated with light from a pulsed OPO laser system. When the light is resonant with a molecular vibration, the absorption of a single photon by the molecule provides enough energy to dissociate the helium-ion complex, which results in a change in mass. We then generate the IR spectrum by monitoring the wavelength-dependent ion signal of the singly helium-tagged ions. To achieve conformer selectivity, we first use an IR pulse from a second OPO at a fixed wavelength corresponding to a conformer-specific IR transition in the molecule to deplete the helium-tagged ions corresponding to this particular conformation from the ensemble. Scanning the wavelength of the second laser pulse then results in an IR spectrum of the remaining conformations. This so-called IR–IR hole-burning spectroscopic scheme14 is applied to various absorption bands to sample all the isomers present.
Ion-mobility pre-selection for isomer-selective messenger tagging spectroscopy
Mobility-selective IR spectroscopic experiments were performed on a second home-built instrument that combines a newly developed travelling wave IMS technique with messenger-tagging IR spectroscopy. A detailed description of the instrument will be published elsewhere, however, the part of the instrument used for spectroscopic investigation was described recently.15,45 A schematic overview of the modified setup can be found in Fig. 1(a). Briefly, ions are electrosprayed and transferred into the instrument where they are accumulated inside the hour-glass section of a dual ion funnel trap.46 From there, packets of ions are injected into the IMS region, where they are separated based on their size and shape. Subsequently, m/z selection occurs in a quadrupole mass filter. So-called arrival time distributions (ATDs) can be acquired by monitoring the drift time of m/z selected ions on a channeltron detector. Alternatively, size- and m/z-selected species can be investigated spectroscopically using a messenger-tagging spectroscopic scheme.47 For this, ions are injected into a planar ion trap15 held at a temperature of 40 K using a helium cryostat, where they are trapped and cooled by collisions with a He/N2 (90/10) buffer gas mixture. Nitrogen molecules condense on the internally cold ions, which can be observed as a shift in mass of +28 u in a subsequent time-of-flight (TOF) analysis. In the case of the here-investigated disaccharide ions, mainly singly tagged ions are observed. To obtain IR spectra, we irradiate the nitrogen-tagged ions with an OPO laser pulse (5–10 mJ) and monitor the wavelength dependent loss of N2 tag molecules as a shift in the TOF mass spectrum.
Fig. 1. (a) Schematic overview of the instrument combining ultra-high resolution SLIM IMS techniques with messenger-tagging IR spectroscopy. (b) IMS resolution of the new instrument as a function of SLIM cycles (IMS path length) determined by monitoring the peak widths and positions of two isomeric MS standard peptides GRGDS and SDGRG in their doubly protonated forms.

High-resolution ion mobility separation is achieved using so-called structures for lossless ion manipulation (SLIM),40 which employs electrodes on printed circuit boards (PCBs) to generate travelling wave (TW) electric potentials that propel the ions forward. We implemented this technique using a 15 cm × 15 cm SLIM IMS device, featuring a single-pass pathlength L of 1.8 m, following a previously published electrode design.40 To increase IM resolution, ions can by cycled through the same SLIM structure multiple times. Typical IMS parameters were: RF frequency 800 kHz; TW amplitude 27 V; TW speed 600 m s–1; He buffer gas pressure 3 mbar.
Results and discussion
Ion-mobility selective IR spectroscopy
To date, ion mobility spectrometry (IMS) has been considered a low-resolution technique, and with commercial instruments featuring IMS resolutions typically well below 100, isomers need to differ in their size by a few percent for them to be distinguishable. Cyclic IMS instruments in which ions pass the same drift region multiple times are a promising approach to increase the resolving power while maintaining a reasonable size of instrument. The recently developed structures for lossless ion manipulation (SLIM) allow for the straightforward implementation of cyclic IMS.43 Ions that complete a cycle on the serpentine drift path of length L of the SLIM ion mobility region of our new instrument can be either sent further downstream towards the cryogenic ion trap or they can be routed back to the beginning of the drift section to complete another IM separation cycle until the desired resolving power is reached. Fig. 1(b) shows the typical IM resolution measured for the [M+2H]2+ ions of the ion mobility standard peptides GRGDS and SDGRG as a function of the number of cycles n. Here, the expected 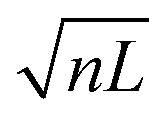 dependency of the resolution42 is confirmed by the good fit of the data points to a function of the form
dependency of the resolution42 is confirmed by the good fit of the data points to a function of the form 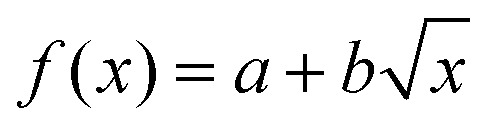 . The instrument reaches a resolution of ∼280 at n = 10, although this is by no means the limit.
. The instrument reaches a resolution of ∼280 at n = 10, although this is by no means the limit.
Our focus here lies on the preparation of conformer-pure ion ensembles for spectroscopic investigation. We test this multi-dimensional IMS-MS-IR spectroscopic approach on carbohydrates – a class of biomolecules that poses a particular challenge to established analytical methods because of their immense structural complexity resulting from various types of isomerism.48 An IMS arrival time distribution (ATD) of the singly sodiated disaccharide Galβ(1-4)GlcNAc is depicted in Fig. 2 (top). After a 13 m drift path (i.e., seven SLIM cycles), we separate two distinct conformations of this molecule. Since we start with an isomerically pure sample, these two species cannot result from different linkage positions or isomerisation of the glycosidic bond. However, different drift times can be a result of multiple stable conformations of the monosaccharide ring structures, different positions of the sodium ion on the molecule, or the difference that results from the C1–OH of the reducing end being in an axial or equatorial configuration with respect to the cyclic structure of the first monosaccharide subunit. These so-called α and β anomers at the reducing end interconvert in solution through mutarotation of the C1–OH in a ring-opening event and have recently been suggested to be responsible for multiple IMS features of a range of glycans studied with an ultra-high resolution SLIM IMS instrument.49 The separation of the two observed species in our disaccharide sample required seven IMS cycles, at which our instrument features a resolving power of approximately 230 (see Fig. 1). This translates into a difference in collisional cross section (CCS) of ∼0.5% between the almost baseline separated ions. The difference in orientation of a single OH group that distinguishes two anomers would result in a change in CCS consistent with what we measure. After IM separation, ions of a particular mobility can be selected by electrostatically deflecting the unwanted ions as shown in the middle and bottom traces of Fig. 2. We then transfer either selected ion packet into the cryogenic ion trap, where its vibrational spectrum is measured.
Fig. 2. (Top) ion mobility arrival time distribution (ATD) of the sodiated disaccharide Galβ(1-4)GlcNAc after seven cycles of SLIM mobility separation. The portions selected by electrostatic deflection of unwanted ions for further spectroscopic analysis are displayed in red (species 1) and blue (species 2), respectively.

The large number of OH and NH oscillators in carbohydrate molecules provide excellent structural probes. We thus focus on the wavenumber range between 3300 cm–1 and 3700 cm–1, where the corresponding stretching bands are expected, as shown in Fig. 3. For both species 1 and 2 (red and blue, respectively), we find five of the eight expected absorption bands in the wavenumber region of 3580 cm–1 and above, in which free or weakly hydrogen-bonded OH groups are expected.50 Vibrational bands in the lower wavenumber range are considered to originate from strongly hydrogen-bonded OH and NH oscillators.50 It is noteworthy that both 1 and 2 feature unique bands that cannot be found in the spectrum of the other, and hence are characteristic for their respective isomeric structures. This observation alone illustrates the power of ion-mobility isomer pre-selection for spectroscopic studies. When no ion-mobility separation is applied prior to spectroscopic analysis of the disaccharide, the spectrum in the upper panel (grey) is obtained. From this spectrum alone, it is not evident that two distinct species are present. To emphasize this, a linear combination of spectra 1 and 2 at a ratio of 4/1 as found for species 1 and 2 in Fig. 2, respectively, is displayed (purple) below the spectrum of the non-IMS selected species in Fig. 3. This synthetic mixture is virtually identical with the experimentally obtained spectrum, which is evident by visual comparison as well as by the almost featureless difference spectrum in Fig. 3 (in grey) obtained by subtracting the synthetic spectrum from the isomer-unresolved spectrum.
Fig. 3. IR spectra of the nitrogen-tagged disaccharide Galβ(1-4)GlcNAc at 40 K when no ion-mobility separation is applied (top, grey) and after ion-mobility separation and selection of the first (red) and second (blue) drift peak (compare Fig. 2). Adding the spectra of species 1 and 2 in a 4 : 1 ratio yields the synthetic mixture displayed in pink (2nd from top), and subtraction from the conformer unresolved spectrum yields the difference spectrum displayed in grey for comparison.

IR–IR double resonance spectroscopy
The conformer-selective messenger-tagging IR–IR double resonance technique was first developed for cold neutral molecules in supersonic jet experiments51 but quickly found application for ionised species.14 It presents a more generally applicable alternative to IR-UV spectroscopic schemes, which require a UV chromophore to be present in the molecule. Here we apply an IR–IR hole burning double resonance spectroscopic scheme,14 using a previously described instrument that allows us to perform helium-tagging spectroscopic experiments on m/z-selected ions at temperatures just below 5 K.
The resulting IR spectra of two different conformers of the sodiated disaccharide Galβ(1-4)GlcNAc are depicted in Fig. 4 as downward-pointing depletion signals of the singly tagged ions. The pump laser was fixed at 3536 cm–1 to acquire the upper spectrum (red) and at 3498 cm–1 for the lower spectrum (blue). Also here, the spectra exhibit distinct features that are characteristic for the probed isomers, and the number of observed absorption bands is consistent with the total of eight OH and NH oscillators present in the molecule. Despite having fixed the pump wavenumber on every resolved absorption signal (data not shown), we only find the two presented spectra that differ from one another. These are likely a result of the two anomeric forms of the disaccharide that are expected to coexist in solution and in the gas phase.
Fig. 4. IR spectra of two conformers of Galβ(1-4)GlcNAc acquired with the IR–IR double resonance method (downward pointing) with the pump laser fixed at 3536 cm–1 (red) and at 3498 cm–1 (blue), respectively. The spectra of the ion-mobility separated species 1 and 2 (upward pointing) are displayed for comparison in red and blue, respectively.

In Fig. 4 the spectra obtained from the double resonance experiments are directly compared to those obtained after ion-mobility separation. Upon visual inspection one finds many similarities between the spectra obtained through the two different methods. Most strikingly, the bands observed above 3600 cm–1, corresponding to reasonably free OH oscillators,50 are conserved in their peak positions and intensities. For lower wavenumbers, where hydrogen-bonded oscillators are probed, absorption bands of the mobility-selected ions can be slightly shifted or broadened with respect to those measured using IR–IR double resonance. This is to be attributed to the higher trap temperature of 40 K in the IMS method versus a temperature of ∼5 K in the double-resonance experiments and the different tag molecules used in the experiments. Nitrogen, which was used as a tag molecule for the mobility-separated species, has a polarizability that exceeds that of helium by a factor of about nine52 and will therefore have a stronger charge-induced dipole interaction with the molecule than the helium tag. As a consequence, nitrogen tags will have a more prominent structure-disturbing effect than helium tags as previously observed for small molecules and clusters.53,54 As the molecules under investigation get larger, the effect of a single tag molecule on the analyte structure will be less pronounced, and the overall good agreement of the spectra acquired with the two different methods justifies the use of nitrogen tagging at higher temperature.
The key difference between ion-mobility isomer pre-selection and double-resonance spectroscopic simplification is in the generality and ease of implementation of the former. For the latter to be effective, an IR spectrum with well-resolved isomer-specific transitions is required. For larger molecules this can be challenging, since the vibrational degrees of freedom quickly increase with the molecular size. On the other hand, mobility pre-selection can only help to simplify an ensemble of conformers if the potentially minuscule size difference between two species can be resolved in the experiment. With the advent of cyclic travelling-wave ion-mobility methods such as SLIM, the resolution-defining drift length can be extended into the kilometre range.43 Considering the experimental parameters used here, a one-kilometre drift path would result in a resolving power of about 2000, enough to separate two ions that differ in their collision cross section by only 0.05%. We are in the process of constructing an instrument with such capabilities. Finally, one must consider the time that is needed for analysis of all components in the ion ensemble; the IR–IR method requires probing each absorption line for potential isomer specificity, which is extremely time consuming. In contrast, ions can be readily separated with IMS after typically tens to hundreds of milliseconds, thereby facilitating single-laser, isomer-pure IR spectroscopic experiments.
Conclusions
The combination of ultra-high resolution ion mobility spectrometry with high-resolution (cryogenic) IR spectroscopy presents an alternative to IR–IR double resonance techniques for isomer or conformer-selective vibrational spectroscopic experiments. We use structures for lossless ion manipulation (SLIM) to achieve high IMS resolution on a cyclic IMS device in which ions cycle the same serpentine drift path multiple times. Different species with small structural differences can thereby readily be separated in time followed by spectroscopic analysis. The approach is particularly promising for larger molecules that possess inherently congested vibrational spectra due to conformational heterogeneity. In this case, sampling of the entire conformational ion ensemble can be cumbersome using double resonance techniques since the required knowledge about the conformer specificity of the vibrational bands is not a priori given. A direct comparison between the two techniques using data on a sodiated disaccharide acquired on two different platforms shows an excellent agreement in both the conformer selectivity of the techniques and quality of the spectra. The latter is particularly interesting as it shows that the experimentally more accessible nitrogen-tagging method represents a good alternative to helium tagging for probing molecular structures the size of disaccharides and larger. The fact that we can separate the same isomeric species of the analyte ions in both experiments represents a success for the SLIM IMS method that we employ with moderately sized PCBs (15 cm × 15 cm). The two separated disaccharide species differ in their size by less than 1% and can possibly be attributed to the α and β anomers of the reducing-end C1–OH group. Anticipated increases in the resolving power of TW IMS will make this approach even more attractive.
This study opens potential applications in the field of glycan analysis, where one’s ability to separate and identify different isomeric species in a timely manner is of the utmost importance. The combination of short isomer-separation times with isomer-characteristic fingerprint IR spectra could result in new analytical workflows that are orders of magnitude faster and more accurate than established methods.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
The authors thank the Swiss National Science Foundation (200020_165908), the European Research Council (788697-GLYCANAL), and the EPFL for their generous support of this work. We thank Valeriu Scutelnic for fruitful discussions during all phases of the experiments. We would also like to acknowledge R. D. Smith and his group for helpful discussions on the implementation of the SLIM technology.
References
- Gas-Phase IR Spectroscopy and Structure of Biological Molecules, A. M. Rijs and J. Oomens, Springer International Publishing, 2015. [Google Scholar]
- Oomens J., Sartakov B. G., Meijer G., von Helden G. Int. J. Mass Spectrom. 2006;254:1–19. [Google Scholar]
- Polfer N. C., Oomens J. Mass Spectrom. Rev. 2009;28:468–494. doi: 10.1002/mas.20215. [DOI] [PubMed] [Google Scholar]
- Heine N., Asmis K. R. Int. Rev. Phys. Chem. 2014;34:1–34. [Google Scholar]
- Wolk A. B., Leavitt C. M., Garand E., Johnson M. A. Acc. Chem. Res. 2014;47:202–210. doi: 10.1021/ar400125a. [DOI] [PubMed] [Google Scholar]
- Zabuga A. V., Kamrath M. Z., Boyarkin O. V., Rizzo T. R. J. Chem. Phys. 2014;141:154309. doi: 10.1063/1.4897158. [DOI] [PubMed] [Google Scholar]
- Manz C., Pagel K. Curr. Opin. Chem. Biol. 2018;42:16–24. doi: 10.1016/j.cbpa.2017.10.021. [DOI] [PubMed] [Google Scholar]
- Redwine J. G., Davis Z. A., Burke N. L., Oglesbee R. A., McLuckey S. A., Zwier T. S. Int. J. Mass Spectrom. 2013;348:9–14. [Google Scholar]
- Kamariotis A., Boyarkin O. V., Mercier S. R., Beck R. D., Bush M. F., Williams E. R., Rizzo T. R. J. Am. Chem. Soc. 2006;128:905–916. doi: 10.1021/ja056079v. [DOI] [PubMed] [Google Scholar]
- Warnke S., Seo J., Boschmans J., Sobott F., Scrivens J. H., Bleiholder C., Bowers M. T., Gewinner S., Schollkopf W., Pagel K., von Helden G. J. Am. Chem. Soc. 2015;137:4236–4242. doi: 10.1021/jacs.5b01338. [DOI] [PubMed] [Google Scholar]
- Jašík J., žabka J., Roithová J., Gerlich D. Int. J. Mass Spectrom. 2013;354–355:204–210. [Google Scholar]
- Stearns J. A., Seaiby C., Boyarkin O. V., Rizzo T. R. Phys. Chem. Chem. Phys. 2009;11:125–132. doi: 10.1039/b814143f. [DOI] [PubMed] [Google Scholar]
- Stearns J. A., Mercier S., Seaiby C., Guidi M., Boyarkin O. V., Rizzo T. R. J. Am. Chem. Soc. 2007;129:11814–11820. doi: 10.1021/ja0736010. [DOI] [PubMed] [Google Scholar]
- Leavitt C. M., Wolk A. B., Fournier J. A., Kamrath M. Z., Garand E., Van Stipdonk M. J., Johnson M. A. J. Phys. Chem. Lett. 2012;3:1099–1105. doi: 10.1021/jz3003074. [DOI] [PubMed] [Google Scholar]
- Masson A., Kamrath M. Z., Perez M. A., Glover M. S., Rothlisberger U., Clemmer D. E., Rizzo T. R. J. Am. Soc. Mass Spectrom. 2015;26:1444–1454. doi: 10.1007/s13361-015-1172-4. [DOI] [PubMed] [Google Scholar]
- Goebbert D. J., Wende T., Bergmann R., Meijer G., Asmis K. R. J. Phys. Chem. A. 2009;113:5874–5880. doi: 10.1021/jp809390x. [DOI] [PubMed] [Google Scholar]
- Wyttenbach T., Pierson N. A., Clemmer D. E., Bowers M. T. Annu. Rev. Phys. Chem. 2014;65:175–196. doi: 10.1146/annurev-physchem-040513-103644. [DOI] [PubMed] [Google Scholar]
- Lapthorn C., Pullen F. S., Chowdhry B. Z., Wright P., Perkins G. L., Heredia Y. Analyst. 2015;140:6814–6823. doi: 10.1039/c5an00411j. [DOI] [PubMed] [Google Scholar]
- Schubert F., Rossi M., Baldauf C., Pagel K., Warnke S., von Helden G., Filsinger F., Kupser P., Meijer G., Salwiczek M., Koksch B., Scheffler M., Blum V. Phys. Chem. Chem. Phys. 2015;17:7373–7385. doi: 10.1039/c4cp05541a. [DOI] [PubMed] [Google Scholar]
- Schubert F., Pagel K., Rossi M., Warnke S., Salwiczek M., Koksch B., von Helden G., Blum V., Baldauf C., Scheffler M. Phys. Chem. Chem. Phys. 2015;17:5376–5385. doi: 10.1039/c4cp05216a. [DOI] [PubMed] [Google Scholar]
- Voronina L., Masson A., Kamrath M., Schubert F., Clemmer D., Baldauf C., Rizzo T. J. Am. Chem. Soc. 2016;138:9224–9233. doi: 10.1021/jacs.6b04550. [DOI] [PubMed] [Google Scholar]
- Pringle S. D., Giles K., Wildgoose J. L., Williams J. P., Slade S. E., Thalassinos K., Bateman R. H., Bowers M. T., Scrivens J. H. Int. J. Mass Spectrom. 2007;261:1–12. [Google Scholar]
- Buryakov I. A., Krylov E. V., Nazarov E. G., Rasulev U. K. Int. J. Mass Spectrom. Ion Processes. 1993;128:143–148. [Google Scholar]
- de la Mora J. F., Ude S., Thomson B. A. Biotechnol. J. 2006;1:988–997. doi: 10.1002/biot.200600070. [DOI] [PubMed] [Google Scholar]
- Guevremont R. J. Chromatogr. A. 2004;1058:3–19. [PubMed] [Google Scholar]
- Hogan Jr C. J., Ruotolo B. T., Robinson C. V., Fernandez de la Mora J. J. Phys. Chem. B. 2011;115:3614–3621. doi: 10.1021/jp109172k. [DOI] [PubMed] [Google Scholar]
- Kolakowski B. M., Mester Z. Analyst. 2007;132:842–864. doi: 10.1039/b706039d. [DOI] [PubMed] [Google Scholar]
- Kemper P. R., Dupuis N. F., Bowers M. T. Int. J. Mass Spectrom. 2009;287:46–57. [Google Scholar]
- Tang K., Shvartsburg A. A., Lee H. N., Prior D. C., Buschbach M. A., Li F., Tolmachev A. V., Anderson G. A., Smith R. D. Anal. Chem. 2005;77:3330–3339. doi: 10.1021/ac048315a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrer B. C., Merenbloom S. I., Koeniger S. L., Hilderbrand A. E., Clemmer D. E. Annu. Rev. Anal. Chem. 2008;1:293–327. doi: 10.1146/annurev.anchem.1.031207.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal N., Masellis C., Kamrath M. Z., Clemmer D. E., Rizzo T. R. Analyst. 2018;143:1846–1852. doi: 10.1039/c8an00230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masellis C., Khanal N., Kamrath M. Z., Clemmer D. E., Rizzo T. R. J. Am. Soc. Mass Spectrom. 2017;28:2217–2222. doi: 10.1007/s13361-017-1728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez O., Isenberg S., Steinmetz V., Glish G. L., Maitre P. J. Phys. Chem. A. 2015;119:6057–6064. doi: 10.1021/jp511975f. [DOI] [PubMed] [Google Scholar]
- Papadopoulos G., Svendsen A., Boyarkin O. V., Rizzo T. R. J. Am. Soc. Mass Spectrom. 2012;23:1173–1181. doi: 10.1007/s13361-012-0384-0. [DOI] [PubMed] [Google Scholar]
- Seo J., Hoffmann W., Warnke S., Bowers M. T., Pagel K., von Helden G. Angew. Chem., Int. Ed. Engl. 2016;55:14173–14176. doi: 10.1002/anie.201606029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W., Marianski M., Warnke S., Seo J., Baldauf C., von Helden G., Pagel K. Phys. Chem. Chem. Phys. 2016;18:19950–19954. doi: 10.1039/c6cp03827a. [DOI] [PubMed] [Google Scholar]
- Papadopoulos G., Svendsen A., Boyarkin O. V., Rizzo T. R. Faraday Discuss. 2011;150:243. doi: 10.1039/c0fd00004c. [DOI] [PubMed] [Google Scholar]
- Seo J., Warnke S., Pagel K., Bowers M. T., von Helden G. Nat. Chem. 2017;9:1263–1268. doi: 10.1038/nchem.2821. [DOI] [PubMed] [Google Scholar]
- Seo J., Hoffmann W., Warnke S., Huang X., Gewinner S., Schollkopf W., Bowers M. T., von Helden G., Pagel K. Nat. Chem. 2017;9:39–44. doi: 10.1038/nchem.2615. [DOI] [PubMed] [Google Scholar]
- Hamid A. M., Ibrahim Y. M., Garimella S. V., Webb I. K., Deng L., Chen T. C., Anderson G. A., Prost S. A., Norheim R. V., Tolmachev A. V., Smith R. D. Anal. Chem. 2015;87:11301–11308. doi: 10.1021/acs.analchem.5b02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L., Ibrahim Y. M., Hamid A. M., Garimella S. V., Webb I. K., Zheng X., Prost S. A., Sandoval J. A., Norheim R. V., Anderson G. A., Tolmachev A. V., Baker E. S., Smith R. D. Anal. Chem. 2016;88:8957–8964. doi: 10.1021/acs.analchem.6b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A., Smith R. D. Anal. Chem. 2008;80:9689–9699. doi: 10.1021/ac8016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L., Webb I. K., Garimella S. V. B., Hamid A. M., Zheng X., Norheim R. V., Prost S. A., Anderson G. A., Sandoval J. A., Baker E. S., Ibrahim Y. M., Smith R. D. Anal. Chem. 2017;89:4628–4634. doi: 10.1021/acs.analchem.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen A., Lorenz U. J., Boyarkin O. V., Rizzo T. R. Rev. Sci. Instrum. 2010;81:073107. doi: 10.1063/1.3458014. [DOI] [PubMed] [Google Scholar]
- Kamrath M. Z., Rizzo T. R. Acc. Chem. Res. 2018;51:1487–1495. doi: 10.1021/acs.accounts.8b00133. [DOI] [PubMed] [Google Scholar]
- Chen T. C., Fillmore T. L., Prost S. A., Moore R. J., Ibrahim Y. M., Smith R. D. Anal. Chem. 2015;87:7326–7331. doi: 10.1021/acs.analchem.5b01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamrath M. Z., Garand E., Jordan P. A., Leavitt C. M., Wolk A. B., Van Stipdonk M. J., Miller S. J., Johnson M. A. J. Am. Chem. Soc. 2011;133:6440–6448. doi: 10.1021/ja200849g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, 3rd edn, 2017. [PubMed] [Google Scholar]
- Nagy G., Attah I. K., Garimella S. V. B., Tang K., Ibrahim Y. M., Baker E. S., Smith R. D. Chem. Commun. 2018;54:11701–11704. doi: 10.1039/c8cc06966b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss J. M., Kregel S. J., Fischer K. C., Garand E. J. Am. Soc. Mass Spectrom. 2018;29:42–50. doi: 10.1007/s13361-017-1813-x. [DOI] [PubMed] [Google Scholar]
- Elliott B. M., Relph R. A., Roscioli J. R., Bopp J. C., Gardenier G. H., Guasco T. L., Johnson M. A. J. Chem. Phys. 2008;129:094303. doi: 10.1063/1.2966002. [DOI] [PubMed] [Google Scholar]
- Haynes W. M. and Lide D. R., CRC handbook of chemistry and physics, CRC Press, Boca Raton, Fla, 92nd edn, 2011. [Google Scholar]
- Johnson C. J., Wolk A. B., Fournier J. A., Sullivan E. N., Weddle G. H., Johnson M. A. J. Chem. Phys. 2014;140:221101. doi: 10.1063/1.4880475. [DOI] [PubMed] [Google Scholar]
- Masson A., Williams E. R., Rizzo T. R. J. Chem. Phys. 2015;143:104313. doi: 10.1063/1.4930196. [DOI] [PubMed] [Google Scholar]