

Abstract
Oxaliplatin, together with cisplatin, is among the most important drugs used in cancer chemotherapy. Oxaliplatin, which contains a bulky diaminocyclohexane (DACH) moiety, kills cancer cells mainly by producing (DACH)Pt-GpG intrastrand cross-links that impede transcription. The Pt-GpG tolerance by translesion DNA synthesis (TLS) polymerases contributes to the resistance of tumors to platinum-based chemotherapy. In particular, human DNA polymerase η (Polη) readily bypasses Pt-GpG adducts. While many structural studies have addressed how TLS polymerases interact with cisplatin-DNA adducts, a structure of DNA polymerase in complex with oxaliplatin-DNA adducts has not been reported, limiting our understanding of bypass of the bulky (DACH)Pt-GpG lesion by TLS polymerases. Herein, we report the first structure of DNA polymerase bound to oxaliplatinated DNA. We determined a crystal structure of Polη incorporating dCTP opposite the 3’G of the (DACH)Pt-GpG, which provides insights into accurate, efficient bypass of the oxaliplatin-GpG adducts by TLS polymerases. In the catalytic site of Polη, the 3′G of the (DACH)Pt-GpG formed three Watson-Crick hydrogen bonds with incoming dCTP and the primer terminus 3′-OH was optimally positioned for nucleotidyl transfer. To accommodate the bulky (DACH)Pt-GpG lesion, the Val59-Trp64 loop in the finger domain of Polη shifted from the positions observed in the corresponding Polη-cisplatin-GpG and undamaged structures, suggesting that the flexibility of the Val59-Trp64 loop allows the enzyme’s bypass of the (DACH)Pt-GpG adducts. Overall, the Polη-oxaliplatin-GpG structure provides structural basis for TLS-mediated bypass of the major oxaliplatin-DNA adducts and insights into resistance to platinum-based chemotherapy in humans.
INTRODUCTION
The platinum-based “alkylating-like” agents cisplatin [cis-diamminedichloro platinum (II)], carboplatin [cis-diammine(1,1-cyclobutanedicarboxylato) platinum(II)], and oxaliplatin [1,2-diaminocyclohexane oxalatoplatinum(II)] are among the most widely used anticancer drugs (Figure 1)[1–3]. Cisplatin has been shown to effectively treat testicular and ovarian cancer as well as other types of solid tumors [2, 4, 5]. Upon entering tumor cells, the chloride ligands of cisplatin are replaced by water molecules to produce reactive aquated platinum species. The aquated cisplatin attacks the N7 of two nearby guanines to generate DNA intrastrand and interstrand cross-links, which causes cell death by impeding replication and transcription [6–8]. Despite its success, cisplatin’s nephrotoxicity, lack of oral bioavailability, and intrinsic and acquired resistance prompted development of other platinum agents, such as carboplatin and oxaliplatin [6, 9, 10].
Figure 1.
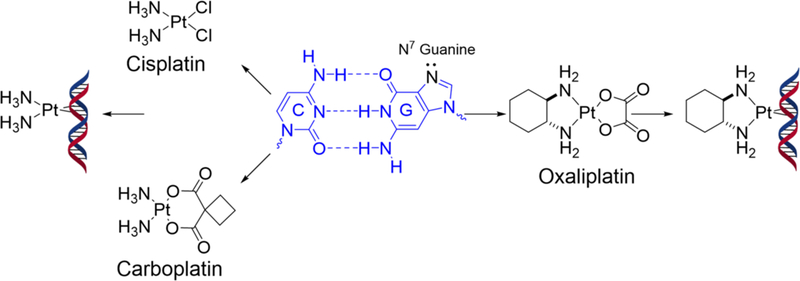
FDA-approved platinum drugs and their DNA intrastrand GpG cross-link adducts. Chloride and dicarboxylate moieties of platinum-based drugs are replaced by water molecules to form reactive aquated platinum species, which preferentially attack the two N7 atoms of adjacent purines. Cisplatin and carboplatin produce the identical (NH3)2Pt-GG adducts, whereas oxaliplatin produces [1,2-cyclohexanediamine]-Pt-GG adducts.
The kinetics of the intrastrand cross-link formation is highly dependent on the nature of the carrier ligand [11, 12]. Cisplatin and carboplatin share the diammine carrier ligand, show similar DNA damage kinetics, give identical Pt-DNA adducts, and are cross-resistant [13, 14] (Figure 1). In contrast, oxaliplatin, which contains a bulky diaminocyclohexane (DACH) ligand, is not cross-resistant with cisplatin. Oxaliplatin shows lower reactivity towards DNA and slower adduct formation, leading to the formation of a significantly smaller number of intrastrand cross-links than cisplatin at equimolar concentrations. Nevertheless, oxaliplatin shows equal or higher cytotoxicity compared to cisplatin at equimolar concentrations, suggesting that the bulkier and more hydrophobic carrier ligand of oxaliplatin is more toxic than that of cisplatin and carboplatin [10, 15–17]. Although oxaliplatin shares the same square-planar geometry with cisplatin, the presence of the bulky DACH ligand likely causes differences in conformational distortions of duplex DNA [10]. In particular, the formation of oxaliplatin-GpG intrastrand cross-links induces 30° DNA-helix bend toward the major groove around the platination site [18]. In stark contrast, cisplatin-GpG intrastrand cross-links causes a greater overall bending of about 80° [19] and a wider minor groove compared to that of oxaliplatin-damaged DNA. The more distorting nature of cisplatin-modified DNA was suggested to be responsible for the more favorable interactions with DNA binding proteins, such as high mobility group proteins [20, 21].
Oxaliplatin is advantageous over cisplatin due to its better safety profile and activity against cisplatin-resistant cancer cells. Many studies focused on understanding the mechanism of cisplatin resistance to gain insight into the molecular basis of oxaliplatin activity [10, 22–25]. Cells can develop resistance to platinum agents in many ways [10], including decreased cellular uptake of drug, increased drug efflux, increased inactivation of the platinum drug with glutathione, increased nucleotide excision repair, and increased bypass of the platinum adduct [10]. Deficiency in mismatch repair (MMR) activity has been shown to be associated with acquired resistance to cisplatin, but not to oxaliplatin [24, 25]. Oxaliplatin is effective at killing colorectal cancer cells that are MMR-deficient and resistant to cisplatin treatment [20, 26–28]. High-mobility group (HMG) proteins have much lower binding affinity to oxaliplatin-DNA adducts than for cisplatin-DNA adducts [15, 29]. Another tolerance mechanism involves translesion synthesis (TLS), whereby Pt-DNA lesions are bypassed by TLS polymerases.
Among TLS DNA polymerases, human DNA polymerase η (hereafter Polη) stands out in its efficiency to bypass lesions as well as its specificity [30–33]. Mutations in Polη that occur in the xeroderma pigmentosum type V (XPV) are responsible for impaired repair of ultraviolet ray-induced damage, which promotes skin cancer development [34, 35]. XPV cells are three times more sensitive to cisplatin treatment than normal fibroblast cells [36], suggesting that Polη plays a role in determining cellular sensitivity to platinum drugs. Polη efficiently bypasses both cisplatin-GpG and oxaliplatin-GpG intrastrand cross-link adducts in vitro [37, 38]. Crystal structures of Polη catalyzing past cisplatin-DNA GpG intrastrand cross-link adducts show that the active site of Polη readily accommodates cisplatin-GpG lesions and accurately incorporates dCTP opposite the (NH3)2Pt-GpG lesion, providing critical insights into TLS polymerasemediated chemoresistance to cisplatin therapy [39–42]. Solution structures of duplex DNA containing oxaliplatin-GG and cisplatin-GpG adducts show that conformations of oxaliplatin-GpG is significantly different from those of cisplatin-GG [43]. Currently, the structure of DNA polymerase bound to oxaliplatin-induced DNA lesions is unknown, thereby limiting our understanding of how DNA polymerases interact with the bulky oxaliplatin-DNA adducts. Herein, we report a crystal structure of Polη incorporating dCTP opposite the 3′G of the Pt(DACH)-GpG adduct. This structure, which represents the first structure of DNA polymerase in complex with oxaliplatinated DNA, provides insight into the efficient bypass of oxaliplatin-DNA adducts by TLS polymerases.
MATERIALS AND METHODS
Human DNA polymerase η expression and purification.
Human Polη (amino acids 1–435) was expressed in E. coli BL21 (DE3) cells. Cultures were grown in LB medium at 37 °C until the OD600 of 0.6, where cells were induced with 0.3 mM of isopropyl β-D-α-thiogalactopyranoside for eighteen hours at 20 °C. Pelleted cells were resuspended in Ni–NTA column binding buffer A (50 mM sodium phosphate, pH 7.8, 500 mM NaCl and 10% glycerol) supplemented with 1 mg/ml lysozyme, 0.25% NP-40, 0.25% Triton X-100, and 0.25 mM phenylmethylsulfonyl fluoride. After sonication for 60 seconds, the lysate was centrifuged at 15000 g at 4 °C for 20 min. The supernatant was then purified using a Ni–NTA column (GE Healthcare). The imidazole-eluted fractions from the Ni column were combined and further purified using Heparin HiTrap column (GE Healthcare) followed by size exclusion chromatography (Superdex75, GE Healthcare). Heparin HiTrap column purification was carried out with a linear gradient of 0.1–1 M NaCl in 50 mM TrisHCl pH 7.4, 5 mM β-ME, 10% glycerol. For experiments performed in this study, the protein was stored at 9 mg/ml in size exclusion buffer (50 mM Tris, pH 7.5, 450 mM KCl, 10% glycerol and 3 mM dithiothreitol), flash frozen in liquid nitrogen and stored at −80 °C.
Preparation of the DNA duplex containing an Pt(DACH) intrastrand cross-link.
To obtain an activated Pt(DACH), 1 mM of dichloro(1,2-diaminocyclohexane) platinum(II) (Sigma-Aldrich, St. Louis, Mo, USA) was incubated with two molar equivalent of silver nitrate in the dark at 37°C for 18 hours. The resulting silver chloride was removed by centrifuging the reaction mixture at 13,000 g for 10 minutes after which the supernatant containing the activated oxaliplatin was recovered. For incorporation of aquated Pt(DACH) on DNA, each oligonucleotide with a single GpG site was mixed with 1.2-fold molar excess of activated Pt(DACH) in 10 mM sodium phosphate buffer (pH 6.8). The mixture was then incubated in the dark at 37°C for 18 hours. Purification of the Pt(DACH) containing oligonucleotide was done by an ion exchange column (Mono Q 5/50 GL, GE Healthcare) using a three-step gradient from 0.1 to 1.0 M NaCl in 10 mM Tris buffer, pH 8.0 (0–15% 1M NaCl buffer in 2 column volumes, 15–50% 1M NaCl buffer in 18 column volumes, and 50–100% 1M NaCl buffer in 0.5 column volume. The site-specifically platinated template DNA was desalted using Sep-Pak C18 cartridges (W aters) and dried with SpeedVac. The modified template was then reconstituted in water and annealed with the complementary upstream primer in 1:1 molar ratio by heating the mixture at 90°C for 10 minutes and slow-cooling over few hours at room temperature.
X-ray crystallographic studies.
For ease of comparison, oligonucleotides used for the crystallographic studies were adopted from the crystal structures of Polη-cisplatin-DNA ternary structure solved by Zhao et al [39]. For co-crystallization of Polη incorporating dCTP opposite the 3′ modified guanine, 12-mer template 5′-ACGGCTCACACT-3′ (GG denotes the modification site) and 9-mer primer 5′-TAGTGTGAG-3′ were used. All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Crystals were grown by the hanging drop vapor-diffusion method in a reservoir buffer containing 0.1M MES (pH 5.5), 5 mM MgCh, and 15 to 20 % PEG 2K-MME. Crystals grew to maximal dimensions over 3–5 days and were cryo-protected in a reservoir buffer supplemented with 25% glycerol. Diffraction data sets for crystals of the Polη-DNA complexes were collected at 100K at the beamline 5.0.3 of the Advanced Light Source (Berkeley, CA), were indexed and scaled using the program HKL2000 [44], and were further processed using CCP4 package programs [45]. The Pt(DACH)-DNA complex structures were solved by molecular replacement with the program Molrep [46] using structures of corresponding Polη-normal DNA complex [Protein Data Bank accession code 4DL4] as the search models [39]. Manual model rebuilding was carried out using the Coot program [47]. Crystallographic figures were prepared using PyMOL [48]. MolProbity was used to make Ramachandran plots. Statistics of data processing, data quality, and the refined models were summarized in Table 1.
Table 1.
Data collection and refinement statistics.
| Pt(DACH)- 3′G・dCTP (6MXO) |
|
|---|---|
| Data Collection | |
| Space Group | P 61 |
| Cell constants | |
| a (Å) | 98.560 |
| b | 98.560 |
| c | 81.713 |
| α (°) | 90.00 |
| β | 90.00 |
| γ | 120.0 |
| Resolution (Å)a | 37.86–2.04 |
| Rmerge b (%) | (2.08–2.04) 0.133 |
| <I/σ> | (0.727) 19.54 (3.00) |
| Completeness (%) | 99.2 (98.3) |
| Redundancy | 11.3 (9.7) |
| Refinement | |
| Rwork c/Rfree d (%) | 17.5/23 |
| Unique reflections | 28856 |
| Mean B factor (Å2) | |
| Protein | 24.41 |
| Ligand | 25.61 |
| Solvent | 21.57 |
| Ramachandran plot | |
| Most favored (%) | 97.4 |
| Additional allowed (%) | 2.6 |
| RMSD | |
| Bond lengths (Å) | 0.018 |
| Bond angles (°) | 1.86 |
Values in parentheses are for the highest resolution shell.
Rmerge = Σ|I-<I>|/ ΣI where I is the integrated intensity of a given reflection.
Rwork = Σ|F(obs)-F(calc)|/ΣF(obs).
Rfree = Σ|F(obs)-F(calc)|/ΣF(obs), calculated using 5% of the data.
RESULTS AND DISCUSSION
Structure of Polη incorporating dCTP opposite the 3′G of Pt(DACH)-GpG cross-link
To understand how Polη accurately and efficiently bypasses oxaliplatin-GpG intrastrand cross-link adducts, we determined a crystal structure of Polη incorporating dCTP opposite the 3′G of the Pt(DACH)-GpG adduct, which represents the first insertion stage. To allow a direct comparison of our work on oxaliplatin with the previous work on cisplatin, we used the same duplex DNA that was used for the structural study of the Polη-cisplatin-GpG-dCTP* complex [39]. The purity of Pt(DACH)-modified template DNA was assessed by using matrix-assisted laser desorption/ionization (MALDI-TOF) mass spectrometry (Figure 2). In this structure, Polη was crystalized in the presence of a template-primer duplex and an incoming nonhydrolyzable dCTP analog (dCTP* hereafter). Duplex DNA was composed of a dodecamer template (5′-ACGGCTCACACT-3′) and a nonamer primer (5′-TAGTGTGAG-3′), where GG denotes the platination site for the Pt(DACH)-GpG cross-link.
Figure 2.
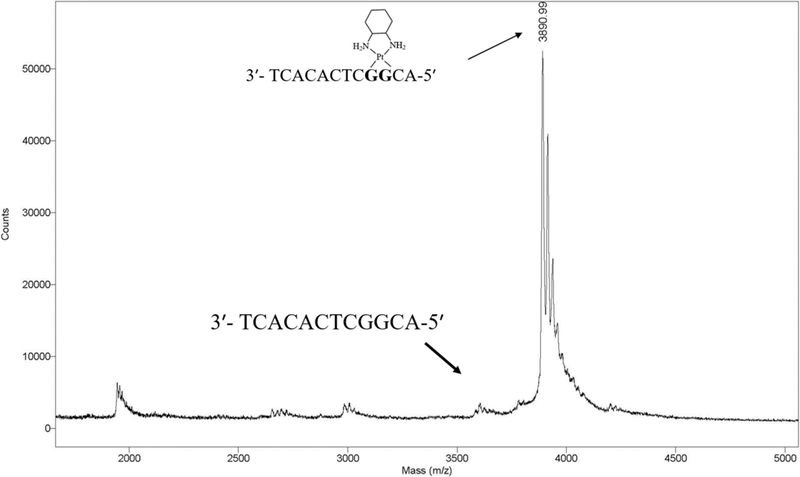
MALDI-TOF mass spectrometry analysis of oxaliplatinated DNA that was purified by urea PAGE.
The Polη-Pt(DACH)-GpG:dCTP* complex (Figure 3A), which was refined to 2.0 Å resolution, clearly showed the presence of the platinum atom, which was found at the same position seen in the corresponding cisplatin-Polη structure (PDB ID 4DL4 [39]) (Figure 3C). Raising sigma level to 30 still showed electron density for the platinum atom. Electron density was also apparent for 1,2-carbons and exocyclic amines of the cyclohexane moiety (Figure 3C). Pt(DACH) was positioned in the major groove without any interaction with the protein (Figure 3A). The catalytic and nucleotide-binding Mg2+ ions were coordinated with the catalytic carboxylates and the triphosphate of dCTP* (Figure 3D). In addition, the incoming dCTP* formed a Watson-Crick base pairing with the 3′-G of the Pt(DACH)-GpG cross-link. The 3′-OH of the primer terminus was coordinated to the catalytic metal ion (2.2 Å) and was aligned with the α-phosphate at an optimum distance (3.3 Å) for catalysis (Figure 3D). The templating cytosine base upstream of the Pt(DACH)-GpG maintained its stacking interaction with the 3′-G of the Pt(DACH)-GpG as was seen in the undamaged GpG DNA.
Figure 3. Structure of Polη incorporating dCTP opposite the 3′G of the Pt(DACH)GpG adducts.
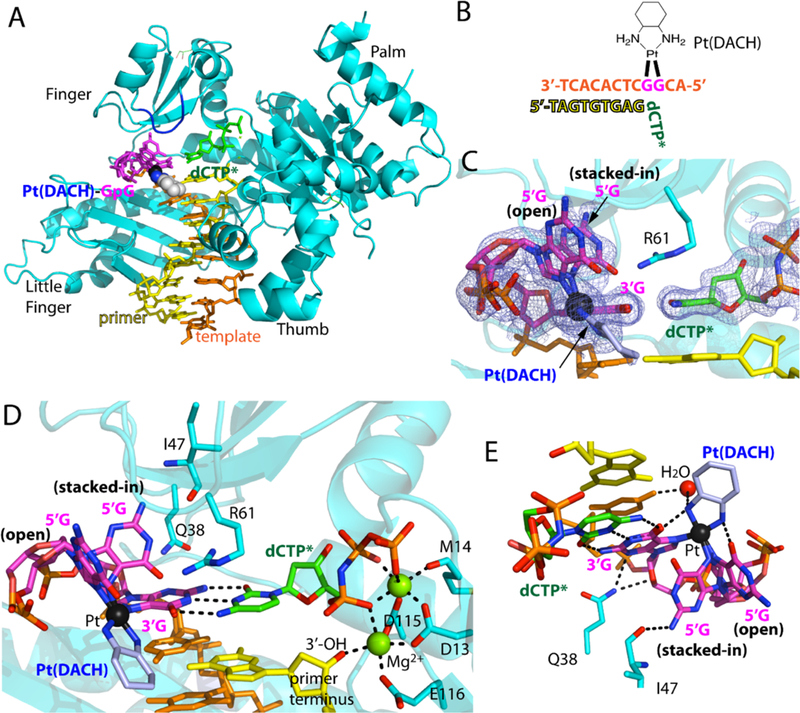
(A) Overall structure of Polη incorporating dCTP* opposite Pt(DACH)−3′G. The protein chain is colored in cyan, the template DNA that contains the two modified guanines (magenta) is colored in orange, the DNA primer in yellow, and the incoming nucleotide in green. The Pt(DACH) moiety is shown in spheres. The Val59-Trp64 loop near the Pt(DACH)GpG is shown in blue. (B) DNA sequences used for the crystallographic studies. (C) A 2Fo-Fc electron density map contoured at 1σ around the Pt(DACH):dCTP* base pair. Pt is shown in black sphere. The 5′G of the Pt(DACH)GpG exists as a mixture of an “open” and “stacked-in” conformations. (D) A close-up view of the active site of the Pt(DACH)−3′G-Polη complex. The catalytic triad (Asp13, Asp115, and Glu116) and two Mg2+ metal ions (binding and catalytic metals) are indicated. The incoming nonhydrolyzable dCTP* forms Watson-Crick base pair with the Pt(DACH)−3′G. The primer terminus dG is shown in yellow. Hydrogen bonds are indicated as dashed lines (E) Interaction of the Pt(DACH) moiety with the surrounding environment.
In this structure, the 5′-G of Pt(DACH)-GpG displayed two conformations. One, referred to as “stacked-in” conformation, was with an acute roll angle between the 3′-and 5′-modified G and another — “open” conformation —with an obtuse roll angle between the 3′-and 5′-modified G (Figures 3C and 3D). Similar conformations were observed for the corresponding cisplatin-modified DNA [39]. In the “stacked-in” conformation, the 5′-G fitted well in the active site without interfering with the incoming nucleotide. In this conformation, the 5′-G was stabilized by a hydrogen bond between the N2 of the 5′-G and the backbone carbonyl oxygen of Ile47 (2.9 Å, Figure 3E). In addition, the N3 and O4 of the 3′-G engaged in hydrogen bond interactions with Gln38 (Figure 3E). According to the occupancy analysis with PHENIX suite software, the Polη-Pt(DACH)-DNA complex structure showed a higher occurrence of the “open” conformation, which was in contrast with the published Polη-cisplatin-DNA complex structure that showed a greater population for the “stacked-in” conformation [39]. In the “open” conformation, interaction between Pt(DACH) and duplex DNA was observed (Figure 3E). In particular, the two exocyclic N atoms of the DACH moiety were hydrogen bonded to the O6 atoms of 3′-G (2.8 Å) and 5′-G (2.7 Å). In addition, the N atom of the DACH moiety that is proximal to the 3′-G engaged in a hydrogen bond with an ordered water molecule (3.3 Å), which, in turn, interacted with the N4 of the adjacent cytosine (3.0 Å) (Figure 3E).
Notable conformational differences among the Polη-oxaliplatin, -cisplatin, and -undamaged DNA structures were found in Arg61, which has been shown to contribute to the catalytic properties of Polη [49]. Arg61 engaged in stacking interaction with the 3′-G:dCTP base pair through its guanidium moiety (Figure 3D). In the Polη-Pt(DACH)-GpG structure, the guanidinium moiety of Arg61 pointed toward the 5′-G of the lesioned bases and was proximal to the “stacked-in” 5′-G conformation (Figure 3D). This was in contrast with the published Polη-dG:dCTP ternary complex structure, where the side chain of Arg61 was projected toward the incoming dCTP, forming a hydrogen bond with a phosphate of dCTP (Figure 4C). This was also in contrast with the corresponding Polη-Pt(NH3)2-GpG structure showing a mixture of the two orientations for Arg61 (Figure 4B).
Figure 4. Comparison of the Val59-Trp64 loop in structures of Polη incorporating dCTP opposite the 3′G, Pt(NH3)2−3′G, and Pt(DACH)−3′G.
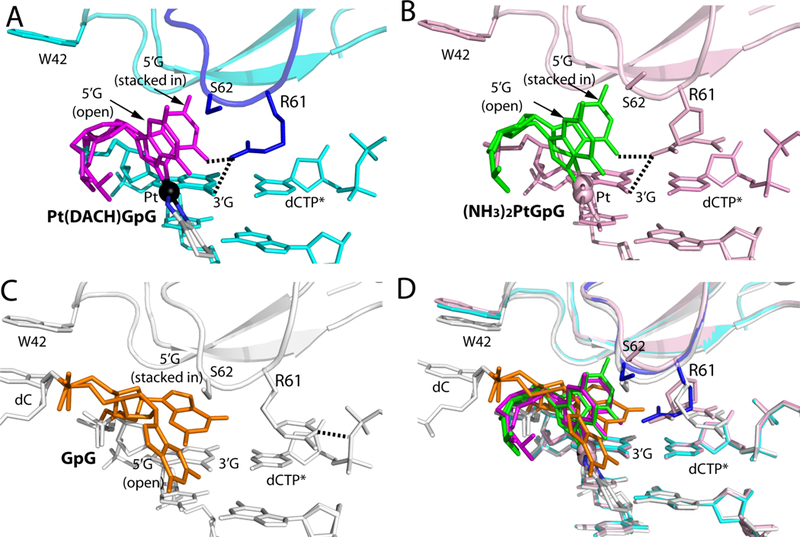
(A) A close-up view showing the Val59-Trp64 loop in the structure of the Polη:Pt(DACH)−3′G:dCTP complex. The 5′G is colored in magenta and its stacked-in and open conformations are indicated. The Val59-Trp64 loop is colored in blue and Pt is shown in a black sphere. Arg61, Ser62, and Trp42 are shown in sticks. Hydrogen bonds between Arg61 and Pt(DACH)GpG are indicated as dotted lines. (B) Structure of Polη incorporating dCTP* opposite Pt(NH3)2-3′G (PDB ID: 4DL4 [39]). Polη and DNA are colored in pink and the 5′G in green. (C) Structure of Polη incorporating dCTP* opposite undamaged 3′G (PDB ID: 4DL2 [39]). The 5′G is colored in orange, whereas protein and DNA are shown in white. Arg61 in the Val59-Trp64 loop engages in a hydrogen bond with a phosphate of dCTP. Note that a stacked-in 5′G is in a syn conformation, whereas an open 5′G is in an anti conformation. (D) Superposition of the Pt(DACH)−3′G, Pt(NH3)2-3′G, and undamaged 3′G structures.
Overall, the accommodation of the Pt(DACH)-GpG in the replicating site of Polη, the formation of an ideal Watson-Crick base pair between dCTP* and the 3′-G of Pt(DACH)-GpG, the coordination of two metal ions in the catalytic site, and an optimal geometry for the primer terminus 3′-OH toward the nucleophilic attack on the Pα of dCTP are consistent with the previously observed efficient, accurate bypass of oxaliplatin-DNA adducts by Polη [37, 38].
The role of the Val59-Trp64 loop in accommodating bulky DNA lesions in the replicating base pair site.
Comparison of ternary complex structures of Polη incorporating dCTP opposite the 3′G of the Pt(DACH)GpG, Pt(NH3)2-GpG, and undamaged GpG provides into the mechanism by which Polη counteracts the effect of oxaliplatin-based chemotherapy. The overall structure of the Polη-Pt(DACH) structure was very similar to those of the corresponding Polη-normal DNA and Polη-Pt(NH3)2-DNA complexes (Figure S2) [39]. The root-mean-square deviation (RMSD) between the Polη-Pt(DACH) structure and the corresponding controls (Polη-normal DNA: 4DL2, 4DL4 [39]) ranged from 0.2 to 0.4, indicating that Polη accommodates various DNA lesions with a minimal conformational reorganization. Nevertheless, noticeable deviations were observed in the flexible Val59-Trp64 loop of the finger domain (shown in a box in Figure S2), which interacts with the replicating base pair (Figure 4).
The flexibility of the Val59-Trp64 loop appears to be important for the accommodation of various lesions in the replicating site of Polη. In the presence of Pt(DACH)GpG lesion, the Val59-Trp64 loop containing Arg61 and Ser62 takes on a conformation that is suitable for stabilizing the major “open” 5′G conformation (Figure 4A). Comparison of this structure with the undamaged Polη-dG:dCTP ternary complex structure showed that the Cα of Ser62 shifted toward the major groove by 0.8 Å from the position observed in the control structure (Figure 5B), suggesting that the catalytic site of the enzyme readily accommodated the platinum lesion. In the case of the cisplatin-Polη complex structure, the Cα of Ser62 moved to a greater extent (1.8 Å) than that observed in the oxaliplatin-Polη complex structure [39] (Figure 5A). The conformational difference of the Val59-Trp64 loops in the cisplatin-Polη and oxaliplatin-Polη structures appears to be caused by the conformational difference of the stacked-in 5′G. More specifically, the stacked-in 5′G of the Pt(NH3)2GpG structure shifted ~0.8 Å toward the Val59-Trp64 loop from the position of the corresponding 5′G of the Pt(DACH)GpG structure (Figure 5A), which would result in steric clash if the Val59-Trp64 loop stays at the position observed in the Pt(DACH)GpG structure. Overall, conformational differences in the Val59-Trp64 loop of Polη structures suggest that the flexibility of the Val59-Trp64 loop in the finger domain plays a key role in the accommodation of various DNA lesions by the enzyme.
Figure 5. Comparison of the conformation of Ser62 in structures of Polη bypassing Pt(DACH)GpG, Pt(NH3)2GpG, and undamaged 3′G.
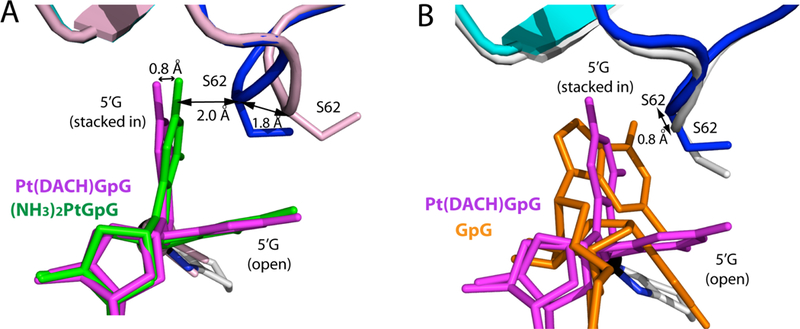
(A) Superposition of the Pt(DACH)GpG and Pt(NH3)2GpG structures. Ser62 in the Pt(NH3)2GpG and Pt(DACH)GpG structures is colored pink and blue, respectively. The distance (1.8 Å) between Ser62 Cα atoms in the Pt(NH3)2GpG and Pt(DACH)GpG structures is shown. (B) Superposition of the Pt(DACH)GpG (PDB ID: 4DL4 [39]) and undamaged GpG (PDB ID: 4DL2 [39]) structures. The distance between the two Ser62 Cα atoms is indicated.
Polη-mediated bypass of oxaliplatin-induced DNA lesions.
Polη is the only DNA polymerase, the defect of which is unambiguously linked to cancer development [30]. Polη protects cells from the damage incurred by ultraviolet radiation. Irradiation of two neighboring pyrimidines by ultraviolet rays gives rise to the formation of cyclobutane pyrimidine dimers, which block DNA replication activity of most DNA polymerases but not that of Polη. In addition to bypassing cyclobutane pyrimidine dimers, Polη bypasses 8-oxoguanine, cisplatin, oxaliplatin, and helix-distorting lesions. The enzyme shares a spacious active site with Y-family polymerases but executes a unique mechanism that guarantees efficiency and enhanced specificity.
A back-side view of the Pt(DACH)−3′GdCTP* complex illustrates how Polη works as a “molecular splint” (Figure 6). Amino acids 317 to 324 of the β-sheet in the little finger domain trace the template containing the cross-linked guanines by forming multiple hydrogen bonds to the phosphate backbone. These extensive interactions facilitate a fine control of the conformation of the lesioned bases. The Asp13-Leu18 fragment of the finger domain aligns with the incoming nucleotide and interact with it directly or through the intermediary of a Mg2+ ion, ensuring good stacking with the primer terminus nucleotide and Watson-crick base pairing with the lesioned-templating base. Gln38 interacts specifically with the replicating base (Pt(DACH)−3′G). Gln38, the mutation of which decreases polymerization fidelity opposite thymine-thymine dimers [50], forms hydrogen bonds with the O4′ of the Pt(DACH)−3′G deoxyribose sugar ring and the N3 of the guanine base in the Pt(DACH) structure (Figures 3D and 6). Other amino acids, such as Asp116, Tyr118, Ser257, Leu262, Arg382, and Cys384 interact with the phosphate backbone of the primer strand.
Figure 6. Interactions between Polη and Pt(DACH)-DNA adducts.
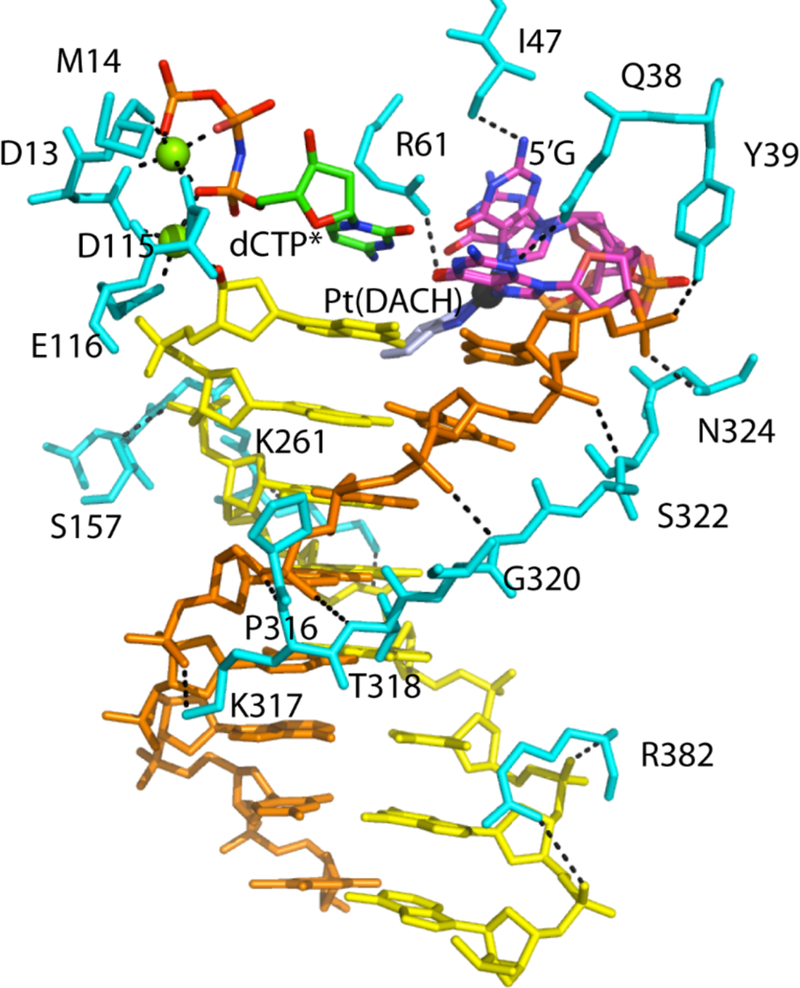
Amino acid residues interacting with DNA, an incoming nucleotide, and the Pt(DACH)GpG moiety are shown in cyan. Cross-linked DNA template is shown in orange and magenta. Primer DNA is colored in yellow with incoming dCTP*. Magnesium ions are shown in green spheres. Black dashed lines represent polar interactions.
Comparison of DNA conformations in the Polη Pt(DACH)−3′G dCTP*, Poln:Pt(NH3)2-3’GdCTP*, and Poln:3′GdCTP* structures shows that conformational differences are mainly confined to the 5′G and its downstream (Figure 7). The 5′G in the Polη structures exists as a mixture of stacked-in and open conformations. The 5′G in the Polη Pt(DACH)−3′G dCTP* complex favors an open conformation, whereas the 5′G in the Polη:Pt(NH3)2-3′GdCTP* complex favors a stacked-in conformation. In the Poln:3′GdCTP* structure, the stacked-in 5′G is in a syn conformation, whereas the open 5′G adopts an anti conformation. The cytosine on the 5′-side of the 5′G in the Poln:3′GdCTP complex engages in π-stacking interaction with Trp42 (Figure 4C). By contrast, the corresponding cytosines in the Pt(DACH)−3′GdCTP* and Polη:Pt(NH3)2-3′GdCTP* complexes are disordered.
Figure 7. Conformations of DNA in Polη structures.
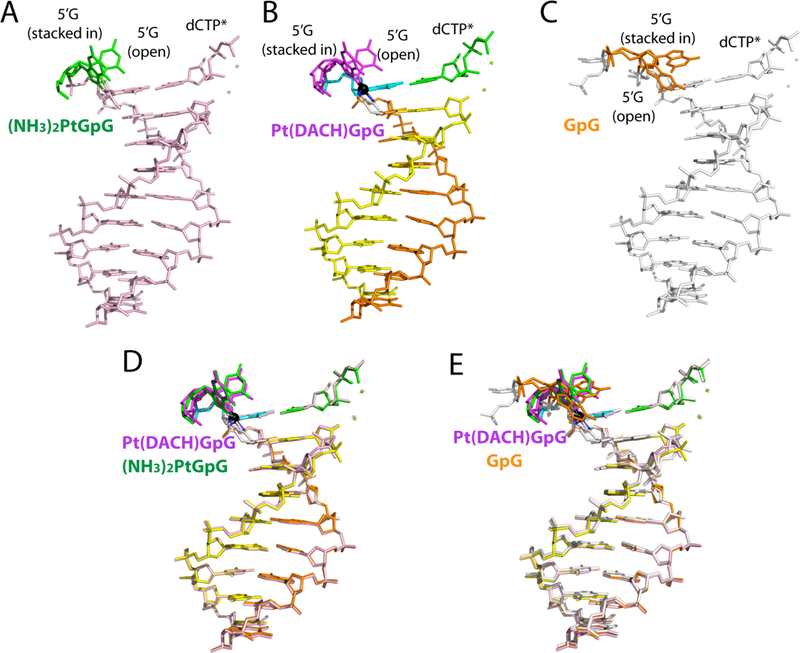
Comparison of DNA conformations in (A) the Pt(NH3)2–3′G・dCTP* (PDB ID: 4DL4 [39]); (B) the Pt(DACH)−3′G・dCTP*; and (C) the normal-3′G・dCTP* (PDB ID: 4DL2 [39]) structures. Superposition of (D) the Pt(DACH)−3′G・dCTP* and Pt(NH3)2–3′G・dCTP* structures and (E) the Pt(DACH)−3′G・dCTP* and normal-3′G・dCTP* structures.
Conclusion:
The first structure of a TLS polymerase bypassing the major oxaliplatin-DNA adducts provides insights into the mechanism by which resistance to oxaliplatin chemotherapy occurs. In the replicating base pair site of Polη, the 3′G of the oxaliplatin-GpG and dCTP form Watson-Crick base pair, the geometry of which is indistinguishable from those observed in the 3′G:dCTP and cisplatin-3′G:dCTP base pairs. In the catalytic site of Polη, the 5′G of the oxaliplatin-GpG favors an open conformation, which is in contrast to the corresponding 5′G of the cisplatin-GpG that favors a “stacked-in” conformation. Polη readily accommodates and efficiently bypass the oxaliplatin-GpG and cisplatin-GpG adducts in the replicating base pair site by locally changing the conformation of the Val59-Trp64 loop, suggesting that the plasticity of the Val59-Trp64 loop contribute to chemoresistance to platinum-based anticancer treatment. Overall, these results suggest that Polη does not significantly contribute to differences in chemoresistance to oxaliplatin and cisplatin, which is consistent with the published kinetic studies that show Polη efficiently bypasses both cisplatin-GpG and oxaliplatin-GpG adducts [37, 38].
Supplementary Material
Acknowledgments
Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The Berkeley Center for Structural Biology is supported in part by the National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Computational resources are provided from the Swedish National Infrastructure for Computing (SNIC) and the National Energy Research Scientific Computing Center (NERSC).
Funding
This work was supported in part by grants from the National Institutes of Health Grant ES26676 and the Robert Welch Foundation (F-1741).
Footnotes
Accession Numbers
The atomic coordinate of Polη-DNA complexes has been deposited in the Protein Data Bank with the following accession code: 6MXO (structure of Pt(DACH)−3′G:dCTP*).
Competing Interests
The authors declare that there are no competing interests.
References
- 1.Johnstone TC, Park GY and Lippard SJ (2014) Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer research. 34, 471–476 PMID: [PMC free article] [PubMed] [Google Scholar]
- 2.Dasari S and Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. European journal of pharmacology. 740, 364–378 doi: 10.1016/j.ejphar.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wheate NJ, Walker S, Craig GE and Oun R (2010) The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton transactions. 39, 8113–8127 doi: 10.1039/c0dt00292e [DOI] [PubMed] [Google Scholar]
- 4.Helm CW (2009) Enhancing the efficacy of cisplatin in ovarian cancer treatment-could arsenic have a role. Journal of ovarian research. 2, 2 doi: 10.1186/1757-2215-2-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung Y and Lippard SJ (2007) Direct cellular responses to platinum-induced DNA damage. Chemical reviews. 107, 1387–1407 doi: 10.1002/chin.200731270 [DOI] [PubMed] [Google Scholar]
- 6.Kelland L (2007) The resurgence of platinum-based cancer chemotherapy. Nature Reviews Cancer. 7, 573 doi: 10.1038/nrc2167 [DOI] [PubMed] [Google Scholar]
- 7.Loeb LA and Monnat RJ Jr (2008) DNA polymerases and human disease. Nature Reviews Genetics. 9, 594 doi: 10.1038/nrg2345 [DOI] [PubMed] [Google Scholar]
- 8.Florea A-M and Busselberg D (2011) Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 3, 1351–1371 doi: 10.3390/cancers3011351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M and Kroemer G (2012) Molecular mechanisms of cisplatin resistance. Oncogene. 31, 1869 doi: 10.1038/onc.2011.384 [DOI] [PubMed] [Google Scholar]
- 10.Di Francesco A, Ruggiero A and Riccardi R (2002) Cellular and molecular aspects of drugs of the future: oxaliplatin. Cellular and Molecular Life Sciences CMLS. 59, 1914–1927 doi: 10.1007/pl00012514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasparkova J, Farrell N and Brabec V (2000) Sequence specificity, conformation, and recognition by HMG1 protein of major DNA interstrand cross-links of antitumor dinuclear platinum complexes. Journal of Biological Chemistry. 275, 15789–15798 doi: 10.1074/jbc.m000777200 [DOI] [PubMed] [Google Scholar]
- 12.Aris SM and Farrell NP (2009) Towards Antitumor Active trans-Platinum Compounds. European journal of inorganic chemistry. 2009, 1293–1302 doi: 10.1002/ejic.200801118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gore M, Fryatt I, Wiltshaw E, Dawson T, Robinson B and Calvert A (1989) Cisplatin/carboplatin cross-resistance in ovarian cancer. British journal of cancer. 60, 767 doi: 10.1038/bjc.1989.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, Paull K and Fojo T (1996) Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute’s Anticancer Drug Screen panel. Biochemical pharmacology. 52, 1855–1865 doi: 10.1016/s0006-2952(97)81490-6 [DOI] [PubMed] [Google Scholar]
- 15.Rabik CA and Dolan ME (2007) Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer treatment reviews. 33, 9–23 doi: 10.1016/j.ctrv.2006.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu G (1994) Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. Journal of Biological Chemistry. 269, 787–790 PMID [PubMed] [Google Scholar]
- 17.Mehmood RK (2014) Review of cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncology reviews. 8doi: 10.4081/oncol.2014.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Pradhan P, Havener J, Boysen G, Swenberg JA, Campbell SL and Chaney SG (2004) NMR solution structure of an oxaliplatin 1, 2-d (GG) intrastrand cross-link in a DNA dodecamer duplex. Journal of molecular biology. 341, 1251–1269 doi: 10.2210/pdb1pgc/pdb [DOI] [PubMed] [Google Scholar]
- 19.Gelasco A and Lippard SJ (1998) NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum (II) d (GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry. 37, 9230–9239 doi: 10.1021/bi973176v [DOI] [PubMed] [Google Scholar]
- 20.Zdraveski ZZ, Mello JA, Farinelli CK, Essigmann JM and Marinus MG (2002) MutS preferentially recognizes cisplatin-over oxaliplatin-modified DNA. Journal of Biological Chemistry. 277, 1255–1260 doi: 10.1074/jbc.m105382200 [DOI] [PubMed] [Google Scholar]
- 21.Ohndorf U-M, Rould MA, He Q, Pabo CO and Lippard SJ (1999) Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 399, 708 doi: 10.1038/21460 [DOI] [PubMed] [Google Scholar]
- 22.Hector S, Bolanowska-Higdon W, Zdanowicz J, Hitt S and Pendyala L (2001) In vitro studies on the mechanisms of oxaliplatin resistance. Cancer chemotherapy and pharmacology. 48, 398–406 doi: 10.1007/s002800100363 [DOI] [PubMed] [Google Scholar]
- 23.Reardon JT, Vaisman A, Chaney SG and Sancar A (1999) Efficient Nucleotide Excision Repair of Cisplatin, Oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum (IV)(JM216) PlatinumIntrastrand DNA Diadducts. Cancer research. 59, 3968–3971 PMID: [PubMed] [Google Scholar]
- 24.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehmé A, Christen RD and Howell SB (1996) The role of DNA mismatch repair in platinum drug resistance. Cancer research. 56, 4881–4886 PMID: [PubMed] [Google Scholar]
- 25.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin T-P, Nehmé A, Christen RD, Haas M and MacLeod CL (1997) In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer research. 57, 1841–1845 PMID: [PubMed] [Google Scholar]
- 26.Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W and Woodgate R (2004) Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. Journal of Biological Chemistry. 279, 32932–32940 doi: 10.1074/jbc.m405249200 [DOI] [PubMed] [Google Scholar]
- 27.Rothenberg M (2000) Efficacy of oxaliplatin in the treatment of colorectal cancer. Oncology-Williston Park. 14, 9–14 PMID: [PubMed] [Google Scholar]
- 28.Comella P, Casaretti R, Sandomenico C, Avallone A and Franco L (2009) Role of oxaliplatin in the treatment of colorectal cancer. Therapeutics and clinical risk management. 5, 229 doi: 10.2147/tcrm.s3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaisman A, Lim SE, Patrick SM, Copeland WC, Hinkle DC, Turchi JJ and Chaney SG (1999) Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum–DNA adducts. Biochemistry. 38, 11026–11039 doi: 10.1021/bi9909187 [DOI] [PubMed] [Google Scholar]
- 30.Biertümpfel C, Zhao Y, Kondo Y, Ramón-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F and Yang W (2010) Structure and mechanism of human DNA polymerase η. Nature. 465, 1044 doi: 10.1038/nature09196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masutani C, Kusumoto R, Iwai S and Hanaoka F (2000) Mechanisms of accurate translesion synthesis by human DNA polymerase η. The EMBO journal. 19, 3100–3109 doi: 10.1093/emboj/19.12.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RE, Prakash S and Prakash L (1999) Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science. 283, 1001–1004 doi: 10.1126/science.283.5404.1001 [DOI] [PubMed] [Google Scholar]
- 33.Haracska L, Yu S-L, Johnson RE, Prakash L and Prakash S (2000) Efficient and accurate replication in the presence of 7, 8-dihydro-8-oxoguanine by DNA polymerase η. Nature genetics. 25, 458 doi: 10.1038/78169 [DOI] [PubMed] [Google Scholar]
- 34.Johnson RE, Kondratick CM, Prakash S and Prakash L (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 285, 263–265 doi: 10.1126/science.285.5425.263 [DOI] [PubMed] [Google Scholar]
- 35.Auclair Y, Rouget R, Belisle JM, Costantino S and Drobetsky EA (2010) Requirement for functional DNA polymerase eta in genome-wide repair of UV-induced DNA damage during S phase. DNA repair. 9, 754–764 doi: 10.1016/j.dnarep.2010.03.013 [DOI] [PubMed] [Google Scholar]
- 36.Chen Y. w., Cleaver JE, Hanaoka F, Chang C. f. and Chou K. m. (2006) A novel role of DNA polymerase η in modulating cellular sensitivity to chemotherapeutic agents. Molecular Cancer Research. 4, 257–265 doi: 10.1158/1541-7786.mcr-05-0118 [DOI] [PubMed] [Google Scholar]
- 37.Vaisman A, Masutani C, Hanaoka F and Chaney SG (2000) Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase η. Biochemistry. 39, 4575–4580 doi : 10.1021/bi000130k [DOI] [PubMed] [Google Scholar]
- 38.Chaney SG, Campbell SL, Bassett E and Wu Y (2005) Recognition and processing of cisplatin-and oxaliplatin-DNA adducts. Critical reviews in oncology/hematology. 53, 3–11 doi: 10.1016/j.critrevonc.2004.08.008 [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y, Biertümpfel C, Gregory MT, Hua Y-J, Hanaoka F and Yang W (2012) Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proceedings of the National Academy of Sciences, 201202681 doi: 10.1073/pnas.1202681109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner K-P and Carell T (2007) Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase η. Science. 318, 967–970 doi: 10.1126/science.1148242 [DOI] [PubMed] [Google Scholar]
- 41.Ummat A, Rechkoblit O, Jain R, Choudhury JR, Johnson RE, Silverstein TD, Buku A, Lone S, Prakash L and Prakash S (2012) Structural basis for cisplatin DNA damage tolerance by human polymerase η during cancer chemotherapy. Nature structural & molecular biology. 19, 628 doi: 10.1038/nsmb.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L and Aggarwal AK (2001) Structure of the catalytic core of S. cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Molecular cell. 8, 417–426 doi: 10.1016/s1097-2765(01)00306-9 [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Bhattacharyya D, King CL, Baskerville-Abraham I, Huh S-H, Boysen G, Swenberg JA, Temple B, Campbell SL and Chaney SG (2007) Solution structures of a DNA dodecamer duplex with and without a cisplatin 1, 2-d (GG) intrastrand cross-link: comparison with the same DNA duplex containing an oxaliplatin 1, 2-d (GG) intrastrand cross-link. Biochemistry. 46, 6477–6487 doi: 10.1021/bi062291f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otwinowski Z and Minor W (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. In Methods in enzymology. pp. 307–326 doi: 10.1016/s0076-6879(97)76066-x [DOI] [PubMed] [Google Scholar]
- 45.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG and McCoy A (2011) Overview of the CCP4 suite and current developments. Acta Crystallographica Section D. 67, 235–242 doi: 10.1107/s0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vagin A and Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallographica Section D: Biological Crystallography. 66, 22–25 doi: 10.1107/s0021889897006766 [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D. 53, 240–255 doi: 10.1107/s0907444996012255 [DOI] [PubMed] [Google Scholar]
- 48.DeLano W The PyMOL Molecular Graphics System, Version 1.5. 0.4, Schrödinger, LLC, 2002. Schrödinger PyMOL.org [Google Scholar]
- 49.Su Y, Patra A, Harp JM, Egli M and Guengerich FP (2015) Roles of Residues Arg-61 and Gln-38 of Human DNA Polymerase Eta in Bypass of Deoxyguanosine and 7, 8-Dihydro-8-oxo-2’-deoxyguanosine. Journal of Biological Chemistry, jbc. M115. 653691 doi: 10.1074/jbc.m115.653691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suarez SC, Beardslee RA, Toffton SM and McCulloch SD (2013) Biochemical analysis of active site mutations of human polymerase nη. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 745, 46–54 doi: 10.1093/protein/6.supplement.41-b [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.