

Abstract
This study aimed to identify DNA methylation markers in oral squamous cell carcinoma (OSCC) and to construct a prognostic prediction model of OSCC. For this purpose, the methylation data of patients with OSCC downloaded from The Cancer Genome Atlas were considered as a training dataset. The methylation profiles of GSE37745 for OSCC samples were downloaded from Gene Expression Omnibus and considered as validation dataset. Differentially methylated genes (DMGs) were screened from the TCGA training dataset, followed by co-methylation analysis using weighted correlation network analysis (WGCNA). Subsequently, the methylation and gene expression levels of DMGs involved in key modules were extracted for correlation analysis. Prognosis-related methylated genes were screened using the univariate Cox regression analysis. Finally, the risk prediction model was constructed and validated through GSE52793. The results revealed that a total of 948 DMGs with CpGs were screened out. Co-methylation gene analysis obtained 2 (brown and turquoise) modules involving 380 DMGs. Correlation analysis revealed that the methylation levels of 132 genes negatively correlated with the gene expression levels. By combining with the clinical survival prognosis of samples, 5 optimized prognostic genes [centromere protein V (CENPV), Tubby bipartite transcription factor (TUB), synaptotagmin like 2 (SYTL2), occludin (OCLN) and CAS1 domain containing 1 (CASD1)] were selected for constructing a risk prediction model. It was consistent in the training dataset and GSE52793 that low-risk samples had a better survival prognosis. On the whole, this study indicates that the constructed risk prediction model based on CENPV, SYTL2, OCLN, CASD1, and TUB may have the potential to be used for predicting the survival prognosis of patients with OSCC.
Keywords: oral squamous cell carcinoma, methylation, gene, prognostic prediction model
Introduction
Oral cancer is a type of head and neck cancer arising in the oral cavity, such as the lips, the anterior two-thirds of the tongue, sublingual region and the hard palate (1,2). There are several histological types of oral cancer, and >90% of oral cancers are squamous cell carcinomas. Oral squamous cell carcinoma (OSCC) is the eighth most common type of cancer worldwide, leading to 144,000 deaths annually (3). Despite many advances being made in the surgical treatment, chemotherapy and radiotherapy used in OSCC, the survival rate of patients with OSCC has not markedly improved (4). Currently, the 5-year survival rate of patients with OSCC is approximately 50% due to the tendency for metastasis (5,6). Therefore, exploring biomarkers with high accuracy, specificity and sensitivity based on the current genome-based approaches underlying OSCC may be helpful for improving its diagnosis, treatment and prognosis.
DNA methylation, mainly referring to the covalent addition of a CH3 on the 5′ position of cytosine, is one of the most important epigenetic modifications involved in the physiological control of genome expression (7). Studies have demonstrated that abnormal methylation mechanisms can result in abnormal gene expression and decreased genomic stability, thereby exerting potent effects on cancer genes (8,9). For OSCC, the silencing of death associated protein kinase 1 (DAPK1), p16INK4a, cadherin 1 (CDH1), O-6-methylguanine-DNA methyltransferase (MGMT) and ABO, alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase (ABO) has been reported to be linked with increased DNA methylation levels of the respective promoters (10-12). Usually, DNA methylation is an earlier event in the process of cell canceration compared with DNA mutation (13). Therefore, it is helpful for diagnostic prediction and therapeutic treatments based on characteristics of DNA methylation patterns across cancers (14). Screening high-risk populations through methylation detection may improve the accuracy of early diagnosis so as to gain more treatment time for cancer patients.
In this study, we aimed to screen some important DNA methylation markers based on the OSCC methylation data in The Cancer Genome Atlas (TCGA). Additionally, a prognostic prediction model was constructed using the screened DNA methylation markers and further validated in another dataset, GSE52793.
Data and methods
Data collection
The methylation data of patients with OSCC, based on the Illumina Infinium Human Methylation 450 BeadChip platform, were downloaded from the TCGA database (https://gdc-portal.nci.nih.gov/). The Cancer Genome Atlas is a public funded project that aims to catalogue and discover major cancer-causing genomic alterations to create a comprehensive 'atlas' of cancer genomic profiles through large-scale genome sequencing (15). According to the definition of oral anatomy, 378 oral samples were retained, including 18 gums, 30 tongue roots, 22 buccal mucosae, 66 mouth floor, 8 dura, 87 oral cavity, and 156 tongue samples. Among these 378 oral samples, 312 samples with survival prognosis information were used as the training dataset. The average age was 61.67±12.89 years, the ratio of males to females was 213:99, the average overall survival (OS) time was 27.61±27.70 months, and the dead to survival ratio was 131:181.
Simultaneously, the GSE52793 (platform: Illumina Infinium Human Methylation 450 BeadChip) was downloaded from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). This dataset contains DNA methylation profiles across approximately 450,000 CpGs in oral rinse samples from a cohort of 82 OSCC patients who had survival prognosis information. This dataset was used as the validation dataset. The average OS time was 41.82±17.45 months, and the dead to survival ratio was 23:59.
Screening of differentially methylated genes (DMGs)
The samples in the TCGA training dataset were divided into the bad prognostic group (survival time of <12 months and died) and the good prognostic group (survival time of >36 months and still survived) according to the OS time recorded in the clinical information. Subsequently, the detected methylation sites were annotated according to the platform annotation information and the meth-ylation sites located in the CpGs regions of the genes were selected for differential methylation analysis using the limma package (16) (version 3.34.7, https://bioconductor.org/packages/release/bioc/html/limma.html) in R3.4.1. Limma package is a R-based open-source software development project in statistical genomics that provides enhanced possibilities for biological interpretation of both differential expression and differential splicing analyses of RNA sequencing data (16). False discovery rate (FDR) <0.05 and |log fold change (FC)|>0.2 were used as the thresholds.
Co-methylation gene analysis
In order to evaluate the internal correlations between CpG islands, we performed co-methylation analysis of all CpG island methylation data using weighted correlation network analysis (WGCNA) package (17) (version 1.63, https://cran.r-project.org/web/packages/WGCNA/index.html) in R3.4.1. The WGCNA package is a comprehensive collection of R functions for performing various aspects of weighted correlation network analysis that includes functions for network construction, module detection, gene selection, calculations of topological properties, data simulation, visualization, and interfacing with external software (17). In this method, the module was defined as a group of genes that had similar methylation levels. Based on this algorithm, we screened the modules that were significantly related to methylation levels. The CpGs in the same module highly correlated with the meth-ylation level.
Subsequently, the screened CpGs were mapped to each WGCNA module to calculate the fold enrichment ratio and the P-value of target CpGs in each module significantly correlated with the methylation level using the hypergeometric algorithm. The formula is f (k, N, M, N) = C (k, M) x C (n-k, n-m)/C (N, N) (17), where 'N' indicates all methylation sites involved in the WGCNA algorithm analysis; 'M' indicates the number of genes in each module obtained from the WGCNA algorithm; and 'n' indicates the number of significant DMGs. The thresholds for module screening were P<0.05 and fold enrichment >1. Finally, the genes in the screened modules were subjected to biology process enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8, https://david.ncifcrf.gov/) (18,19). DAVID is an online available bioinformatics resources that consists of an integrated biological knowledgebase and analytic tools aimed at systematically extracting biological meaning from large gene/protein lists (18,19).
Correlation analysis between the methylation level and expression level
From the TCGA training dataset, the OSCC mRNA-seq samples matching methylation profiles were selected. The methylation levels and gene expression levels of DMGs involved in key modules were extracted as well. The overall Pearsons' correlation coefficients (PCC) (20) between the methylation levels and gene expression levels were calculated using cor.test (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/cor.test.html) in R3.4.1. The cor.test is a program of R functions that can be used for exploring the correlations between variables by Pearsons' correlation coefficient (20). The correlation between the methylation level and gene expression level of each gene was then calculated and the DMGs whose methylation levels negatively correlated with the expression levels were selected for further analysis (P<0.05).
Screening of prognosis-related methylated genes
Based on the expression levels of the screened DMGs in TCGA samples, as well as the clinical survival prognosis information, the prognosis-related methylated genes were screened using the univariate Cox regression analysis implemented in survival package (21) (version 2.41-1, http://bioconductor.org/packages/survivalr/) of R3.4.1. The log-rank tested P-value <0.05 was considered as the significance threshold.
Establishment of prognostic prediction model based on methylated genes
According to the methylation levels of the prognosis-related methylated genes, the optimized prognostic genes were screened by the Cox-Proportional Hazards (Cox-PH) model (22) based on L1 penalized regularized regression algorithm (23) (version 0.9-50, http://bioconductor.org/packages/penalized/) of R3.4.1. The Cox-PH model is a predictive model of overall and disease-free survival, based on demographic and clinical covariates (24).
Subsequently, the risk prediction model was constructed based on the prognosis coefficients of optimized prognostic genes. The risk score of each sample was calculated as follows: Risk score=∑coefgene × Methylationgene, where coefgene represents the regression coefficient obtained in the previous step, and Methylationgene represents the methylation level of the corresponding gene.
Finally, the samples in TCGA training dataset were divided into the high- and low-risk groups according to the median of the risk score. The association between risk model and prognosis was assessed by the Kaplan-Meier (KM) survival curve (25) in survival package (version 2.41-1). The KM survival curve is the probability of surviving in a given length of time while considering time in many small intervals (25). Moreover, the discriminant efficacy of the prediction model on survival prognosis was evaluated through the validation dataset GSE52793.
Results
Screening of significant DMGs
A total of 15,654 gene methylation sites containing CpGs were obtained through the annotation information on the Illumina 450 K methylation platform. Subsequently, in the TCGA training dataset, 54 samples were divided into the bad prognostic group and 46 were divided into the good prognostic group. Finally, a total of 948 DMGs with CpGs were screened out and the volcano plot is presented in Fig. 1A. The log2 Kernel density curve revealed that 64.56% (612/948) of the DMGs were significantly hypomethylated in the good prognostic group, and 35.44% (336/948) were significantly hypermethylated (Fig. 1B). The bidirectional hierarchical cluster heatmap revealed that the samples were clearly divided into 2 groups based on the screened DMGs (Fig. 1C).
Figure 1.

The significant differentially methylated genes. (A) Volcano plot of significant differentially methylated genes. The red dots represent differentially methylated genes; the black dots represent non-differentially methylated genes; the green horizontal dotted line represents the false discovery rate (FDR) <0.05; the two green vertical dotted lines represent the |log2 fold change (FC)|>0.2. A total of 948 DMGs with CpGs were screened out. (B) Log2 Kernel density curve based on differentially methylated genes. The proportion of hypomethylated genes in the good prognostic group was 64.56% (612/948), and 35.44% (336/948) were significantly hypermethylated. (C) The hierarchical clustering heatmaps of significant differentially methylated genes. The red and green bars represent the samples in good and bad prognostic groups, respectively. The samples were clearly divided into 2 groups based on the screened differentially methylated genes.
Among the CpGs of the 948 DMGs, 15 were in the transcription start site (TSS) area, 432 in the gene body area, 104 in the 5′UTR region, 17 in the 3′UTR region, and 380 in the promoter region. The top 20 DMGs with CpGs sorted according to FDR are presented in Table I.
Table I.
The top 20 significant differentially methylated genes (DMGs) with CpGs between bad prognostic group and good prognostic group sorted according to false discovery rate (FDR).
| Methylation loci | Chr. | Position | Genes | Location | β-bad | β-good | Effect | Pnominal | FDR |
|---|---|---|---|---|---|---|---|---|---|
| cg03904042 | chr20 | 31719152 | NECAB3 | Body | 0.3413 | 0.2296 | −0.5719 | 8.05×10−7 | 2.21×10−5 |
| cg04488521 | chr5 | 178420322 | ZNF354C | Promoter | 0.3382 | 0.2272 | −0.5737 | 3.42×10−6 | 9.37×10−5 |
| cg09595479 | chr12 | 47975479 | PRPH | 1stExon | 0.4229 | 0.3151 | −0.4244 | 5.31×10−6 | 1.45×10−4 |
| cg00054702 | chr5 | 135556442 | LOC389332 | Body | 0.1967 | 0.1149 | −0.7755 | 8.34×10−6 | 2.29×10−4 |
| cg07040405 | chr2 | 174537121 | SP3 | Promoter | 0.0306 | 0.0383 | 0.3253 | 9.43×10−6 | 2.59×10−4 |
| cg13146839 | chr3 | 192063116 | LOC647309 | 1stExon | 0.4032 | 0.5159 | 0.3556 | 1.12×10−5 | 3.08×10−4 |
| cg00741900 | chr14 | 101097550 | DIO3 | 5′UTR | 0.3288 | 0.2510 | −0.3892 | 1.15×10−5 | 3.14×10−4 |
| cg27229100 | chr20 | 47330667 | C20orf199 | Body | 0.4259 | 0.5299 | 0.3153 | 1.44×10−5 | 3.94×10−4 |
| cg13316171 | chr2 | 187059280 | ZC3H15 | 1stExon | 0.0212 | 0.0273 | 0.3649 | 1.56×10−5 | 4.28×10−4 |
| cg01494348 | chr8 | 144731538 | NAPRT1 | Promoter | 0.1567 | 0.0991 | −0.6612 | 1.64×10−5 | 4.50×10−4 |
| cg24960763 | chr12 | 132217389 | ZNF10 | Promoter | 0.0867 | 0.0578 | −0.5848 | 1.90×10−5 | 5.22×10−4 |
| cg18113994 | chr19 | 62817679 | ZNF134 | 1stExon | 0.0784 | 0.1796 | 1.1953 | 2.04×10−5 | 5.60×10−4 |
| cg06815419 | chr15 | 81526866 | BTBD1 | Promoter | 0.0215 | 0.0265 | 0.3018 | 2.05×10−5 | 5.63×10−4 |
| cg25019777 | chr4 | 53220160 | USP46 | Promoter | 0.0432 | 0.0641 | 0.5716 | 2.08×10−5 | 5.70×10−4 |
| cg00004421 | chr17 | 7415803 | SENP3 | TSS1500 | 0.4066 | 0.4947 | 0.2828 | 2.35×10−5 | 6.44×10−4 |
| cg01331992 | chr9 | 19369118 | RPS6 | Body | 0.2316 | 0.3035 | 0.3899 | 2.95×10−5 | 8.09×10−4 |
| cg00767496 | chr15 | 43458571 | GATM | Body | 0.5105 | 0.4140 | −0.3020 | 3.00×10−5 | 8.22×10−4 |
| cg21858255 | chr12 | 103133739 | TXNRD1 | Promoter | 0.1570 | 0.0696 | −1.1737 | 3.15×10−5 | 8.64×10−4 |
| cg02605461 | chr20 | 32047137 | RALY | 5′UTR | 0.3931 | 0.4871 | 0.3095 | 3.37×10−5 | 9.25×10−4 |
| cg26658728 | chr16 | 65195711 | CMTM3 | 5′UTR | 0.1783 | 0.1032 | −0.7891 | 3.38×10−5 | 9.26×10−4 |
The first column indicates the cg number of the methylation site; the second column indicates the chromosome number of detected methylation site; the third column indicates the chromosome position of the detected methylation site; the fourth column indicates the gene name of methylation area; the fifth column represents the CpGs location in methylation regional; the sixth and seventh columns represent the average methylation level β of CpGs in bad and good prognostic groups; the eighth column represents log2FC (good/bad); the ninth column represents the P-value; and the tenth column represents the corrected FDR value.
Co-methylation gene analysis
In the WGCNA algorithm, we selected the power value (power=6) when the square value of the correlation coefficient reached up to 0.9 for the first time. The topological overlap matrix was then calculated to evaluate the correlation between the methylation levels of two genes, subsequently constructing the system clustering tree. With parameters of minSize=100 and cutHeight=0.99, 15 modules were obtained (Fig. 2A and Table II). The CpGs gene expression levels in the modules were significantly associated with the methylation levels, apart from the grey module (P<0.05). The average of significant correlation coefficients was 0.547, ranging from 0.138 to 0.777. The number of DMGs in each module and the fold enrichment ratio of each module are shown in Fig. 2B and C, respectively. According to the hyper-geometric enrichment algorithm, 2 (brown and turquoise) modules were obtained. There were 208 and 172 DMGs in the brown and turquoise modules, respectively. Functional enrichment analysis revealed these 380 DMGs were significantly related to 24 biological processes, such as vasculogenesis and response to hypoxia (Fig. 3).
Figure 2.

Screening of modules related to gene CpG methylation by weighted correlation network analysis (WGCNA). (A) The modules related to gene CpGs methylation. A total of 15 modules were identified, and they were presented by 15 different colors (black, blue, brown, cyan, green, green-yellow, grey, magenta, pink, purple, red, salmon, tan, turquoise and yellow). (B) The number of differentially methylated genes in the brown, blue, black, yellow, turquoise, tan, red, purple, pink, magenta, green-yellow, green, cyan models. (C) Fold enrichment column graph of the brown, blue, black, yellow, turquoise, tan, red, purple, pink, magenta, green-yellow, green, cyan models. The green horizontal dotted line represents a fold enrichment ratio of 1.
Table II.
Information of the 15 modules identified by weighted correlation network analysis.
| Color | #CpGs | Correlation | Pcorr | DE CpGs | Enrichment fold (95% CI) | Phyper |
|---|---|---|---|---|---|---|
| Black | 290 | 0.7553 | 5.3×10−29 | 10 | 0.2477 | 1.439×10−7 |
| Blue | 468 | 0.6377 | 3.33×10−3 | 29 | 0.4452 | 5.483×10−6 |
| Brown | 430 | 0.7665 | 8.58×10−8 | 208 | 3.4742 | 2.200×10−16 |
| Cyan | 110 | 0.7766 | 3.93×10−9 | 2 | 0.1306 | 1.104×10−4 |
| Green | 334 | 0.5926 | 7.60×10−15 | 16 | 0.3442 | 2.647×10−6 |
| Green-yellow | 157 | 0.6143 | 4.24×10−20 | 12 | 0.5491 | 4.249×10−2 |
| Grey | 2,667 | 0.2203 | 7.42×10−2 | 433 | 1.1662 | 1.408×10−2 |
| Magenta | 261 | 0.6383 | 1.38×10−25 | 9 | 0.2477 | 7.327×10−7 |
| Pink | 276 | 0.6787 | 1.27×10−13 | 15 | 0.3904 | 9.668×10−5 |
| Purple | 222 | 0.6008 | 4.42×10−3 | 17 | 0.5501 | 1.520×10−2 |
| Red | 333 | 0.6209 | 6.26×10−15 | 2 | 0.0432 | 5.343×10−16 |
| Salmon | 120 | 0.2454 | 4.37×10−11 | - | - | - |
| Tan | 147 | 0.1381 | 5.75×10−10 | 21 | 1.0262 | 9.052×10−1 |
| Turquoise | 585 | 0.5115 | 1.02×10−15 | 172 | 2.112 | 3.342×10−14 |
| Yellow | 410 | 0.4044 | 4.85×10−10 | 2 | 0.03503 | 2.200×10−16 |
The first column represents the module color; the second column represents the number of CpGs genes in module; the third column represents the correlation coefficient between CpGs gene and methylation level; the fourth column represents the P-value; the fifth column represents the number of differentially methylated CpGs gene mapping to each module; the sixth and seventh columns represent Enrichment fold and P-values obtained based on hypergeometric enrichment algorithm. CI, confidence interval.
Figure 3.

The significantly enriched biological processes for the differentially methylated genes in brown and turquoise modules. The horizontal axis represents the number of genes involved in the biological process; the vertical axis represents the name of the biological process; the height of the column represents the number of genes involved in biological process; and the color of the column represents P-value. These 380 differentially methylated genes were significantly related to 24 biological processes.
Correlation analysis between the methylation level and expression level of key DMGs
The overall correlation analysis between the methylation levels and expression levels of the 380 DMGs mentioned above revealed that there was a significant negative correlation between the gene methylation and expression level (Cor.=−0.3379, P=2.978e-07; Fig. 4). After searching the published studies on line about the correlation analysis between the methylation levels and expression levels, some correlation coefficients of revealed significant correlations were below 0.5. For example, in the study by Győrffy et al, the significant correlations between the methylation of gene regions and expression levels among prognostic genes in the internal cohort of breast cancer patients revealed that most of the absolute correlation coefficients were <0.5 (26). Therefore, the results may be not lower than expected. The analysis of the correlation between the methylation and expression levels for each gene revealed that the methylation levels of 132 genes negatively correlated with the expression levels (data not shown).
Figure 4.
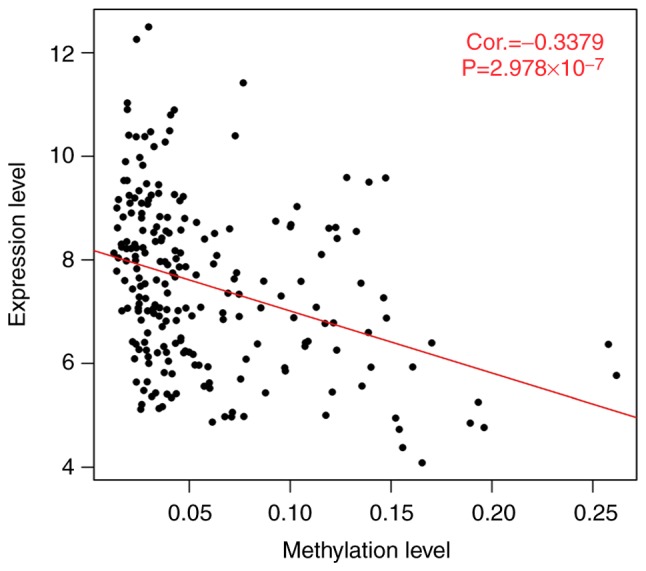
The overall correlation analysis between methylation levels and expression levels of the 380 differentially methylated genes in brown and turquoise modules. The red line is the trend line of point distribution; Cor. represents the Pearson's correlation coefficient between the methylation levels and expression levels; 'P' represents the significance of the correlation. There was a significant negative correlation between the gene methylation and expression level.
Screening of prognosis-related methylated genes
Based on the methylation levels of CpGs in these 132 genes in the TCGA samples and the clinical survival prognosis of samples, we identified 27 methylated genes that were significantly associated with prognosis. Subsequently, 5 optimized prognostic genes [centromere protein V (CENPV), Tubby bipartite transcription factor (TUB), synaptotagmin like 2 (SYTL2), occludin (OCLN) and CAS1 domain containing 1 (CASD1)] were selected through the Cox-PH model (Fig. 5A). The prognosis coefficients of these 5 genes are shown in Fig. 5B. Subsequently, according to the median of the methylated signal value, the samples were divided into the hypomethylation and hypermethylation groups. The KM curves revealed that the samples with hypomethylation levels had a better OS (P<0.05; Fig. 6). Additionally, according to the median of the gene expression level, the samples were divided into the high expression and low expression groups. The KM curves revealed that the samples with high expression levels had a better OS (P<0.05; Fig. 6).
Figure 5.

Identification of optimized prognostic genes through the Cox-Proportional Hazards (Cox-PH) model. (A) The lambda parameter curve selected by cross-validation likelihood. The horizontal and vertical axes respectively represent different values of lambda and cross-validation likelihood. (B) The prognosis coefficients of the 5 optimized prognostic genes (CENPV, TUB, SYTL2, OCLN and CASD1) selected through the Cox-PH model. CENPV, centromere protein V; TUB, Tubby bipartite transcription factor; SYTL2, synaptotagmin like 2; OCLN, occluding; CASD1, CAS1 domain containing 1.
Figure 6.

The Kaplan-Meier curves for patients with different methylation or expression levels of CASD1, OCLN, SYTL2, TUB and CENPV. According to the median of the methylated signal value, the samples were divided into the hypomethylation and hypermethylation groups (upper panels). The KM curves also revealed that the samples with high expression levels had a better overall survival prognosis (lower panels). CENPV, centromere protein V; TUB, Tubby bipartite transcription factor; SYTL2, synaptotagmin like 2; OCLN, occluding; CASD1, CAS1 domain containing 1.
Prognosis prediction model based on optimized methylated genes
Based on the Cox-PH prognostic coefficients of the 5 genes, the gene-based risk prediction model was constructed as follows: Risk score=(0.2666) × Methylationcg08214689 + (0.2937) × Methylationcg09498146 + (1.5954) × Methylationcg02716252 + (0.6491) × Methylationcg00562641 + (0.3989) × Methylationcg11341528.
The risk score of each sample was calculated using the above-mentioned formula. In order to evaluate the discriminant efficacy of the prediction model on the survival prognosis, we divided the samples in the TCGA training dataset into the high- and low-risk groups based on the median of the risk score. The KM curves of the TCGA dataset revealed that the low-risk samples had a better survival prognosis (P=4.698e-03; Fig. 7A). Additionally, the discriminant efficacy of the prediction model was also evaluated in the validation dataset GSE52793, and the results were consistent with those of the TCGA training dataset (P=1.466e-02; Fig. 7B).
Figure 7.

The Kaplan-Meier curves for patients based on the risk score prediction model. (A) Kaplan-Meier curves of patients in TCGA training dataset revealed that the low-risk samples had a better survival prognosis. (B) Kaplan-Meier curves of patients in the validation dataset GSE52793 were consistent with those of the TCGA training dataset.
Discussion
In the present study, we analyzed the methylation data of patients with OSCC downloaded from the TCGA database, and selected 5 optimized genes (CENPV, TUB, SYTL2, OCLN and CASD1) associated with methylation through a series of bioinformatics methods. Based on these 5 genes, a prognostic prediction model was constructed and its discriminant efficacy on survival prognosis was confirmed by the validation dataset GSE52793.
Among the 5 genes, SYTL2, OCLN and CASD1 have been reported to be associated with tumorigenesis. SYTL2 possesses a C-terminal tandem C2 domain, N-terminal Slp homology domain and Rab-binding region (27), which is primarily characterized as an effector of Rab27, a Ras-related small GTPase (28). SYTL2 can regulate tubulogenesis and cell signaling by promoting the trafficking of the signaling molecule, podocalyxin, to the apical surface through a Rab27-dependent manner (29). Recently, SYTL2 has been reported to be implicated in cancer pathogenesis. Ho et al (30) analyzed 223 Rab effector genes in bladder cancer and found that the Rab27 gene cluster was deregulated in the carcinoma in situ pathway and Ta pathway. Specifically, SYTL2 was downregulated in the two above-mentioned pathways and was associated with differentiation. More recently, SYTL2 was demonstrated to serve as an epigenetically regulated pro-metastatic factor and to be associated with a poorer survival in patients with ovarian cancer (31). These findings suggest that SYTL2 plays critical roles in human cancers, although its role in OSCC has not been reported previously, at least to the best of our knowledge.
OCLN encodes an integral membrane protein which plays an important role in cytokine-induced regulation of the tight junction (32). The disruption of the structure of tight junction or occludin is related to cancer invasion and metastasis (33). A previous study reported that OCLN downregulation is a common feature of epithelial-mesenchymal-transition in tumors derived from simple epithelial cells (34). To date, the downregulation of OCLN has been reported in breast cancer, liver tumors, endometrial carcinoma and lung cancer (33,35-37). Rachow et al (32) observed the loss of OCLN in cutaneous squamous cell carcinoma and suggested that OCLN loss was related to cell adhesion, apoptosis and proliferation (32). Importantly, OCLN has been demonstrated to be methylated in the promoter and endogenous region of a breast cancer cell line (38). Therefore, we speculated that OCLN might serve as an important prognostic maker in OSCC.
A recent study demonstrated that CASD1 is a sialate O-acetyltransferase (SOAT), a key enzyme in the biosyn-thesis of 9-O-acetylated sialoglycans (39). Evidence has also indicated an enhanced SOAT activity in the lymphoblasts of children with acute lymphoblastic leukemia and SOAT is thus considered a possible biomarker for monitoring acute lymphoblastic leukemia (40). Currently, its role in other solid cancers, particularly OSCC has not been reported, at least to the best of our knowledge. According to the results of this study, we hypothesized that the methylation of CASD1 may be a key predictor for the prognosis of patients with OSCC. Taken together, in consideration of the reports associated with the functions of SYTL2, OCLN and CASD1 in human cancers, it can be concluded that the constructed risk prediction model may have the potential to be used in predicting the survival of patients with OSCC.
CENPV encodes a centrosome-associated protein. Presently, there are very few specific studies available on the role of CENPV in human cancers (41). Notably, there are a number of studies available on the other centromere proteins in cancers. It has been reported that the enhanced expression of centromere protein A is implicated in the poor survival of patients with human cancers (42,43). Centromere protein A has been considered as a prognostic and predictive biomarker in human cancers (44). Additionally, the elevated expression of centromere protein F can predict clinical progression and prognosis in patients with prostate cancer (45). Importantly, human centromere protein H has been suggested to be closely linked to the increased or abnormal cell proliferation in human OSCC (46). These findings mentioned above suggest the critical roles of centromere protein in predicting human cancers. CENPV may thus serve as a potential prognostic factor in human OSCC.
For the marker gene TUB, it encodes a member of the Tubby family of bipartite transcription factors. The Tubby proteins are a unique protein family sharing a highly conserved C-terminal domain, playing a role in obesity and sensorineural degradation (47). However, the function of TUB in cancers has not yet been reported, at least to the best of our best knowledge. In view of the reliability of the risk prediction models in the present study, we considered that methylated TUB may be a prognostic factor in OSCC.
In spite of these findings mentioned above, there were some limitations to our study. Although RNA sequencing is able to identify and quantify transcripts, isoforms, novel transcripts, gene fusions, and non-coding RNAs among a wide range of samples rapidly and accurately, the methylation level and expression level of the 5 predicted genes should be further detected using laboratory experiments, such as PCR. Additionally, the constructed risk prediction model should also be applied to other cohorts of patients with OSCC to further confirm the discriminant efficacy of the prediction model on survival prognosis, even though this prediction model has been verified in a validation dataset GSE52793.
In conclusion, the present study identified 5 key methylated genes (CENPV, SYTL2, OCLN, CASD1 and TUB) for OSCC based on the methylation data in the TCGA database. Moreover, the constructed risk prediction model was validated in GSE52793 suggesting that this prediction model may be helpful for predicting the survival prognosis of patients OSCC.
Acknowledgments
Not applicable.
Funding
The study was supported by Scientific Research Projects of Shanghai Municipal Commission of Health and Family Planning (no. 201840040).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
QZ and GT performed data analyses and wrote the manuscript. JG conceived and designed the study. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All patient data were derived from online datasets; thus, no ethics approval was required.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Werning JW. Thieme. 2007. Oral cancer: Diagnosis, management, and rehabilitation. [Google Scholar]
- 2.World Health Organization (WHO) In: International classification of diseases for oncology (ICD-O)-3rd edition, 1st revision. Fritz A, Percy C, Jack A, Shanmugaratnam K, Sobin L, Parkin DM, Whelan S, editors. WHO; Geneva: 2013. [Google Scholar]
- 3.Wu Y, Zhang L, Zhang L, Wang Y, Li H, Ren X, Wei F, Yu W, Liu T, Wang X, et al. Long non-coding RNA HOTAIR promotes tumor cell invasion and metastasis by recruiting EZH2 and repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol. 2015;46:2586–2594. doi: 10.3892/ijo.2015.2976. [DOI] [PubMed] [Google Scholar]
- 4.Wikner J, Gröbe A, Pantel K, Riethdorf S. Squamous cell carcinoma of the oral cavity and circulating tumour cells. World J Clin Oncol. 2014;5:114–124. doi: 10.5306/wjco.v5.i2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya A, Roy R, Snijders AM, Hamilton G, Paquette J, Tokuyasu T, Bengtsson H, Jordan RC, Olshen AB, Pinkel D, et al. Two distinct routes to oral cancer differing in genome instability and risk for cervical node metastasis. Clin Cancer Res. 2011;17:7024–7034. doi: 10.1158/1078-0432.CCR-11-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choo KB. Epigenetics in disease and cancer. Malays J Pathol. 2011;33:61–70. [PubMed] [Google Scholar]
- 8.Deng J, Liang H, Zhang R, Ying G, Xie X, Yu J, Fan D, Hao X. Methylated CpG site count of dapper homolog 1 (DACT1) promoter prediction the poor survival of gastric cancer. Am J Cancer Res. 2014;4:518–527. [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura J, Tanaka T, Kitajima Y, Noshiro H, Miyazaki K. Methylation-mediated gene silencing as biomarkers of gastric cancer: A review. World J Gastroenterol. 2014;20:11991–12006. doi: 10.3748/wjg.v20.i34.11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakayama S, Sasaki A, Mese H, Alcalde R, Tsuji T, Matsumura T. The E-cadherin gene is silenced by CpG methylation in human oral squamous cell carcinomas. Int J Cancer. 2001;93:667–673. doi: 10.1002/ijc.1386. [DOI] [PubMed] [Google Scholar]
- 11.Rosas SL, Koch W, da Costa Carvalho MG, Wu L, Califano J, Westra W, Jen J, Sidransky D. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001;61:939–942. [PubMed] [Google Scholar]
- 12.Gao S, Worm J, Guldberg P, Eiberg H, Krogdahl A, Liu CJ, Reibel J, Dabelsteen E. Genetic and epigenetic alterations of the blood group ABO gene in oral squamous cell carcinoma. Int J Cancer. 2004;109:230–237. doi: 10.1002/ijc.11592. [DOI] [PubMed] [Google Scholar]
- 13.Delpu Y, Cordelier P, Cho W, Torrisani J. DNA methylation and cancer diagnosis. Int J Mol Sci. 2013;14:15029–15058. doi: 10.3390/ijms140715029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang C, Zhao H, Li J, Liu H, Wang F, Wei Y, Su J, Zhang D, Liu T, Zhang Y. The identification of specific methylation patterns across different cancers. PLoS One. 2015;10:e0120361. doi: 10.1371/journal.pone.0120361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68–A77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritchie M, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman B, Lempicki R. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang da W, Sherman B, Lempicki R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 20.Zou K, Tuncali K, Silverman SG. Correlation and simple linear regression. Radiology. 2003;227:617–622. doi: 10.1148/radiol.2273011499. [DOI] [PubMed] [Google Scholar]
- 21.Wang P, Wang Y, Hang B, Zou X, Mao J. A novel gene expression-based prognostic scoring system to predict survival in gastric cancer. Oncotarget. 2016;7:55343–55351. doi: 10.18632/oncotarget.10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16:385–395. doi: 10.1002/(SICI)1097-0258(19970228)16:4<385::AID-SIM380>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Goeman JJ. L1 penalized estimation in the Cox proportional hazards model. Biom J. 2010;52:70–84. doi: 10.1002/bimj.200900028. [DOI] [PubMed] [Google Scholar]
- 24.Shahraki HR, Salehi A, Zare N. Survival prognostic factors of male breast cancer in southern Iran: A LASSO-Cox regression approach. Asian Pac J Cancer Prev. 2015;16:6773–6777. doi: 10.7314/APJCP.2015.16.15.6773. [DOI] [PubMed] [Google Scholar]
- 25.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Győrffy B, Bottai G, Fleischer T, Munkácsy G, Budczies J, Paladini L, Børresen-Dale AL, Kristensen VN, Santarpia L. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int J Cancer. 2016;138:87–97. doi: 10.1002/ijc.29684. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda M, Saegusa C, Mikoshiba K. Novel splicing isoforms of synaptotagmin-like proteins 2 and 3: Identification of the Slp homology domain. Biochem Biophys Res Commun. 2001;283:513–519. doi: 10.1006/bbrc.2001.4803. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda M. Rab27 and its effectors in secretory granule exocytosis: A novel docking machinery composed of a Rab27.effector complex. Biochem Soc Trans. 2006;34:691–695. doi: 10.1042/BST0340691. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda T, Saegusa C, Kamakura S, Sumimoto H, Fukuda M. Rab27 effector Slp2-a transports the apical signaling molecule podocalyxin to the apical surface of MDCK II cells and regulates claudin-2 expression. Mol Biol Cell. 2012;23:3229–3239. doi: 10.1091/mbc.e12-02-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho JR, Chapeaublanc E, Kirkwood L, Nicolle R, Benhamou S, Lebret T, Allory Y, Southgate J, Radvanyi F, Goud B. Deregulation of Rab and Rab effector genes in bladder cancer. PLoS One. 2012;7:e39469. doi: 10.1371/journal.pone.0039469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sung HY, Han J, Ju W, Ahn JH. Synaptotagmin-like protein 2 gene promotes the metastatic potential in ovarian cancer. Oncol Rep. 2016;36:535–541. doi: 10.3892/or.2016.4835. [DOI] [PubMed] [Google Scholar]
- 32.Rachow S, Zorn-Kruppa M, Ohnemus U, Kirschner N, Vidal-y-Sy S, von den Driesch P, Börnchen C, Eberle J, Mildner M, Vettorazzi E, et al. Occludin is involved in adhesion, apoptosis, differentiation and Ca2+-homeostasis of human keratinocytes: Implications for tumorigenesis. PLoS One. 2013;8:e55116. doi: 10.1371/journal.pone.0055116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tobioka H, Isomura H, Kokai Y, Tokunaga Y, Yamaguchi J, Sawada N. Occludin expression decreases with the progression of human endometrial carcinoma. Hum Pathol. 2004;35:159–164. doi: 10.1016/j.humpath.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Sehrawat A, Singh SV. Benzyl isothiocyanate inhibits epithelial-mesenchymal transition in cultured and xenografted human breast cancer cells. Cancer Prev Res (Phila) 2011;4:1107–1117. doi: 10.1158/1940-6207.CAPR-10-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orbán E, Szabó E, Lotz G, Kupcsulik P, Páska C, Schaff Z, Kiss A. Different expression of occludin and ZO-1 in primary and metastatic liver tumors. Pathol Oncol Res. 2008;14:299–306. doi: 10.1007/s12253-008-9031-2. [DOI] [PubMed] [Google Scholar]
- 36.Martin TA, Mansel RE, Jiang WG. Loss of occludin leads to the progression of human breast cancer. Int J Mol Med. 2010;26:723–734. doi: 10.3892/ijmm_00000519. [DOI] [PubMed] [Google Scholar]
- 37.Tobioka H, Tokunaga Y, Isomura H, Kokai Y, Yamaguchi J, Sawada N. Expression of occludin, a tight-junction-associated protein, in human lung carcinomas. Virchows Arch. 2004;445:472–476. doi: 10.1007/s00428-004-1054-9. [DOI] [PubMed] [Google Scholar]
- 38.Osanai M, Murata M, Nishikiori N, Chiba H, Kojima T, Sawada N. Epigenetic silencing of occludin promotes tumori-genic and metastatic properties of cancer cells via modulations of unique sets of apoptosis-associated genes. Cancer Res. 2006;66:9125–9133. doi: 10.1158/0008-5472.CAN-06-1864. [DOI] [PubMed] [Google Scholar]
- 39.Baumann AM, Bakkers MJ, Buettner FF, Hartmann M, Grove M, Langereis MA, de Groot RJ, Mühlenhoff M. 9-O-Acetylation of sialic acids is catalysed by CASD1 via a covalent acetyl-enzyme intermediate. Nat Commun. 2015;6:7673. doi: 10.1038/ncomms8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandal C, Mandal C, Chandra S, Schauer R, Mandal C. Regulation of O-acetylation of sialic acids by sialate-O-acetyltransferase and sialate-O-acetylesterase activities in childhood acute lymphoblastic leukemia. Glycobiology. 2012;22:70–83. doi: 10.1093/glycob/cwr106. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Y, Zhou J, Tong Y. Gene signatures of drug resistance predict patient survival in colorectal cancer. Pharmacogenomics J. 2015;15:135–143. doi: 10.1038/tpj.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Han Y, Huang H, Min L, Qu L, Shou C. Integrated analysis of expression profiling data identifies three genes in correlation with poor prognosis of triple-negative breast cancer. Int J Oncol. 2014;44:2025–2033. doi: 10.3892/ijo.2014.2352. [DOI] [PubMed] [Google Scholar]
- 43.Qiu JJ, Guo JJ, Lv TJ, Jin HY, Ding JX, Feng WW, Zhang Y, Hua KQ. Prognostic value of centromere protein-A expression in patients with epithelial ovarian cancer. Tumor Biol. 2013;34:2971–2975. doi: 10.1007/s13277-013-0860-6. [DOI] [PubMed] [Google Scholar]
- 44.Sun X, Clermont PL, Jiao W, Helgason CD, Gout PW, Wang Y, Qu S. Elevated expression of the centromere protein-A (CENP-A)-encoding gene as a prognostic and predictive biomarker in human cancers. Int J Cancer. 2016;139:899–907. doi: 10.1002/ijc.30133. [DOI] [PubMed] [Google Scholar]
- 45.Zhuo YJ, Xi M, Wan YP, Hua W, Liu YL, Wan S, Zhou YL, Luo HW, Wu SL, Zhong WD, Wu CL. Enhanced expression of centromere protein F predicts clinical progression and prognosis in patients with prostate cancer. Int J Mol Med. 2015;35:966–972. doi: 10.3892/ijmm.2015.2086. [DOI] [PubMed] [Google Scholar]
- 46.Shigeishi H, Higashikawa K, Ono S, Mizuta K, Ninomiya Y, Yoneda S, Taki M, Kamata N. Increased expression of CENP-H gene in human oral squamous cell carcinomas harboring high-proliferative activity. Oncol Rep. 2006;16:1071–1075. [PubMed] [Google Scholar]
- 47.Carroll K, Gomez C, Shapiro L. Tubby proteins: The plot thickens. Nat Rev Mol Cell Biol. 2004;5:55–63. doi: 10.1038/nrm1278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.