

Abstract
The aim of the present study was to explore the possible mechanisms by which hypertonic saline (HS) effectively ameliorates cerebral oedema via the vascular endothelial growth factor receptor 2 (VEGFR2)-mediated endothelial nitric oxide synthase (eNOS) pathway of endothelial cells in rats. A middle cerebral artery occlusion (MCAO) model in Sprague-Dawley rats and an oxygen-glucose deprivation (OGD) model in cells were used in the present study. Evans blue (EB) staining and a horseradish peroxidase flux assay were performed to evaluate the protective effect of 10% HS on the blood-brain barrier (BBB). The expression levels of vascular endothelial growth factor (VEGF), VEGFR2, zonula occludens 1 (ZO1) and occludin were quantified. The results demonstrated that 10% HS effectively reduced EB extravasation in the peri-ischaemic brain tissue. At 24 h after MCAO, the protein expression levels of VEGF and VEGFR2 in the peri-ischaemic brain tissue were downregulated following treatment with 10% HS. In vitro experiments demonstrated that the permeability of a monolayer endothelial cell barrier was decreased significantly following HS treatment. In addition, VEGF and VEGFR2 protein expression levels were increased in endothelial cells under hypoxic conditions, but that effect was suppressed by HS treatment. Furthermore, HS inhibited the downregulation of ZO1 and occludin effectively, possibly through the VEGFR2/phospholipase C γ1 (PLCγ1)/eNOS signalling pathway. In conclusion, 10% HS may alleviate cerebral oedema through reducing ischaemia-induced BBB permeability, as a consequence of inhibiting VEGFR2/PLCγ1/eNOS-mediated downregulation of ZO1 and occludin.
Keywords: hypertonic saline, blood-brain barrier, vascular endothelial growth factor, endothelial cell, tight junction
Introduction
Ischaemic cerebral stroke is one of the leading causes of cerebral oedema that can lead to malignant brain oedema and brain hernia and can potentially endanger the lives of patients (1). The mechanisms of ischaemic brain oedema are complex, as they involve cytotoxic oedema and vasogenic oedema resulting from disruption of the blood-brain barrier (BBB) (2). A previous study found that BBB hyperpermeability allowed fluid to leak from the intravascular space into the brain parenchyma, which caused vasogenic cerebral oedema and increased interstitial fluid pressure (3).
Osmotherapy is a primary treatment for cerebral oedema. Hypertonic saline (HS), as a common osmotic dehydrating agent, can move free water from the intracellular space into the extracellular space through establishing an osmotic pressure gradient, which lowers peripheral vascular resistance (4). It is well-known that administration of 10% HS is more effective than mannitol for the treatment of cerebral oedema (5,6). However, little is known about the action of HS other than its osmotic effect in the treatment of brain oedema. Zeynalov et al (7) reported that HS attenuated BBB disruption in the presence of perivascular aquaporin 4 (AQP4) in post-ischaemic cerebral oedema. Huang et al (8) also reported that HS downregulated the expression of Na-K-Cl cotransporter 1 in astrocytes. This finding suggested that HS alleviated cerebral oedema not only through an osmotic mechanism but also through regulation of molecular pathways.
Vascular endothelial growth factor (VEGF) is the major regulator of microvascular permeability. It has been reported that BBB permeability was significantly increased and the degree of cerebral oedema decreased abruptly following treatment with VEGF (9-11). It has also been well documented that VEGF expression is increased in various type of cells, including neurons, endothelial cells, astrocytes, pial cells, and microglia/macrophages, following middle cerebral artery occlusion (MCAO) in rats (12,13). Previous studies have focused on astrocyte-derived VEGF (14-16), but the role of VEGF derived from cerebral endothelial cells in the development of brain oedema remains unclear. VEGF receptor 2 (VEGFR2), a receptor of VEGF, serves an important role in regulating VEGF function, and it increases vascular permeability (17,18). Previous studies have demonstrated that VEGF-induced permeability depends on the VEGFR2-mediated endothelial nitric oxide synthase (eNOS) pathway by downregulating the expression of tight junction proteins (19,20). In addition, the tight junction between endothelial cells is the key element in BBB permeability, and disruption of tight junctions leads to BBB breakdown (21). Zonula occludens 1 (ZO1) and occludin are important components of tight junctions, and downregulation of these factors significantly increases the permeability of the BBB (22).
Based on these previous findings, the present study investigated the effect of 10% HS on cerebral oedema induced by VEGF. Additionally, the present study investigated whether HS regulated the expression of ZO1 and occludin via the VEGFR2/phospholipase C γ1 (PLCγ1)/eNOS pathway. The findings are expected to further amplify our understanding of the underlying mechanisms and molecular roles of HS in the clinical management of cerebral oedema.
Materials and methods
Animals and experimental groups
Male Sprague-Dawley rats (weight, 250-300 g) were housed in groups in a pathogen-free environment (22±2°C, 55±10% humidity and 12-h light/dark cycle) with free access to a standard laboratory diet and water. The animals were randomly divided into the sham group (n=30), cerebral ischaemia-reperfusion group (IR group, n=30), cerebral ischaemia-reperfusion + normal saline group (NS group, n=30) and cerebral ischaemia-reperfusion + 10% HS group (HS group, n=30). Rats in the sham group were subjected to all surgical procedures, except for transient cerebral ischaemia. Rats in the IR group were subjected to cerebral ischemia for 2 h, followed by reperfusion for either 12 or 24 h. Rats in the NS group and HS group were subjected to transient cerebral ischaemia for 2 h, then treatment was administered at the beginning of reperfusion with a continuous intravenous infusion (0.3 ml/h) of normal saline or 10% HS, respectively, via the tail vein, until the end of the experiment. The rats in each group were divided into two groups according to different treatment times: A 12 h subgroup and a 24 h subgroup. All experimental procedures involving the use of animals were approved by the Institutional Animal Care and Use Committee, Guangdong Province, China (approval no. GBREC2012106A). All experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Focal brain ischaemia-reperfusion animal model
The rats were fasted overnight but were allowed free access to water. Focal brain ischaemia was induced by the MCAO method, as previously described (7). First, anaesthesia was injected intra-muscularly with 6 mg/kg diazepam, and then anaesthesia was continued by intraperitoneal injection with 40 mg/kg ketamine. The right common carotid artery (CCA), internal carotid artery, and external carotid artery were then exposed through a midline incision in the neck. A 4-0 head-end spherical nylon suture was inserted into the middle cerebral artery (MCA) via the CCA to occlude the MCA for a total of 2 h. Cerebral reperfusion was restored when the tip of the occluding suture was withdrawn to the carotid bifurcation. The sham group rats were subjected to the same surgical procedures but not MCAO. Neurologic examinations were performed 2 h after the onset of occlusion. Zea-Longa test scores were used to evaluate whether the MCAO was successful (23). The rats with neurologic deficit scores of 1 to 3 were considered to have experienced successful MCAO and were used for subsequent experiments.
Brain microvascular endothelial cell culture and treatment
The brain microvascular endothelial cell (MVEC) line bEnd.3 was obtained from American Type Culture Collection and cultured in DMEM (Thermo Fisher Scientific, Inc.) medium supplemented with 10% foetal bovine serum (FBS; Biological Industries) at 37°C in a humidified incubator with 95% air and 5% CO2. When the culture reached confluence, MVECs were purified by removal of the non-adherent cells and subculturing of only the adherent cells. The purified MVECs were randomly divided into the following groups: Control group, oxygen and glucose deprivation group (OGD group), OGD + 40 mM HS group (HS group) and OGD + 40 mM HS + VEGF-A group (HS+VEGF-A group). The cells in the OGD, HS and HS+VEGF-A groups were all incubated in glucose-free medium (Thermo Fisher Scientific, Inc.) in an airtight hypoxia chamber with 3% O2 and 5% CO2 at 37°C. After 6 h of hypoxic and glucose-free culture, the culture medium of each group was supplemented as follows: The OGD group was cultured with 10% FBS for 24 h; the HS group was cultured with 10% FBS and 40 mM HS for 24 h; and the HS+VEGF-A group medium was supplemented with 40 mM HS and 10 ng/ml VEGF-A simultaneously (PeproTech, Inc.) for 24 h. The cells in the control group were cultured with normal medium containing 10% FBS, and were not subjected to hypoxia.
Evans blue (EB) staining
The permeability of the BBB was assessed by measuring Evans blue extravasation in the ischaemic hemispheric tissue as previously described (24,25). A solution of 2% Evans blue dye in saline (4 ml/kg; Sigma-Aldrich; Merck KGaA) was injected intravenously at the beginning of reperfusion. At 24 h after MCAO and under deep anaesthesia, the rats were subjected to transcardial perfusion with 110 ml saline to remove the intravascular Evans blue dye. After the rats had been sacrificed by decapitation, the hemispheres of the brain were separated along the sagittal suture. Then, both hemispheres were weighed and kept in formamide (1 ml/100 mg) at 60°C for 24 h. The concentration of dye extracted from each brain was determined at 620 nm using spectrophotometry. The quantitative calculation of the dye content in the brain was based on external standards dissolved in the same solvent.
Horseradish peroxidase (HRP) flux assay
The permeability of the MVEC monolayer barrier was detected by the HRP flux assay, as previously described (26). At 3, 6, 12, and 24 h after incubation with 40 mM HS, the culture medium was replaced with DMEM without phenol red or serum. To maintain the levels of culture supernatant, 250 µl of medium containing 500 ng HRP (Sigma-Aldrich; Merck KGaA) was added into the insert, and 1,250 µl of medium was added into each well. After each sample was collected at one of the two time points, 100 µl of peroxidase substrate (Sigma-Aldrich Merck KGaA) containing tetramethyl benzidine and hydrogen peroxide was added to each sample, which was then incubated for 10 min. The reaction was terminated by adding 50 µl of 2 M sulfuric acid. The optical density was measured at 450 nm, and the HRP transmissivity was determined by the standard curve according to the following equation: PHRP %=[(CHRPoxVo/CHRPixVi) ×100%], where CHRPo is the HRP concentration in the well, CHRPi is the HRP concentration in the insert well, Vo is the medium volume in the well, and Vi is the medium volume in the insert.
Immunofluorescence staining
At 24 h after MCAO, frozen sections of coronal brain were cut to 10 µm thickness at the level of the optic chiasma and rinsed in PBS. After fixation with 4% paraformaldehyde at room temperature for 15 min, the samples were permeabilized with methanol, and blocked with 5% BSA in PBS for 20 min. Sections were incubated overnight at 4°C with primary antibodies directed against occludin [rabbit polyclonal (immunoglobulin) Ig G; 1:100; Abcam; cat. no. ab31721), ZO1 (rabbit polyclonal IgG; 1:100; Thermo Fisher Scientific Inc.; cat. no. 61-7300), VEGF (rabbit polyclonal IgG; 1:100; Santa Cruz Biotechnology Inc.; cat. no. sc-152), VEGF receptor 2 (VEGFR2; rabbit polyclonal IgG; 1:100; Santa Cruz Biotechnology Inc.; cat. no. sc-505) and vascular endothelial cell markers von Willebrand factor (VWF; mouse monoclonal IgG; 1:100; Santa Cruz Biotechnology Inc.; cat. no. sc-365712) and CD31 (mouse monoclonal IgG; 1:100; Abcam; cat. no. ab119339). On the following day, the sections were washed with PBS and incubated with the corresponding secondary antibodies as follows: Alexa Fluor® 488-conjugated goat anti-mouse IgG (1:100; Thermo Fisher Scientific Inc.; cat. no. A-28175), Alexa Fluor® 555-conjugated donkey anti-rabbit IgG (1:100; Thermo Fisher Scientific Inc.; cat. no. A-31572), Alexa Fluor® 488-conjugated donkey anti-rabbit IgG (1:100; Thermo Fisher Scientific Inc.; cat. no. A-21206), or Alexa Fluor® 555-conjugated goat anti-mouse IgG (1:100; Thermo Fisher Scientific Inc.; cat. no. A-21424) for 2 h at room temperature. Following rinsing in PBS, samples were mounted with a fluorescent mounting medium containing DAPI (cat. no. DUO82040; Sigma-Aldrich; Merck KGaA). Colocalization was observed by a blinded observer using a fluorescence microscope (Olympus Corporation).
For the in vitro experiments, after 6 h of hypoxic and glucose-free culture, bEnd.3 endothelial cells were reoxygenated and incubated with the corresponding medium, according to different cell groups as aforementioned, for 24 h. Then the cells were fixed with 4% paraformaldehyde for 20 min at 37°C. Following rinsing with PBS, cells were blocked with 5% goat serum (Jiangxi Haoran Bio-Pharma Co., Ltd.) for 30 min at 37°C and then incubated with antibodies as described above.
For the quantification of protein expression signals, the fluorescence intensity was calculated as follows: Fluorescence intensity=total optical density/total fluorescence area. Quantitative analysis was performed with Image J (v1.8.0; National Institutes of Health).
Western blotting
Total proteins from the peri-ischaemic cerebral cortex and bEnd.3 endothelial cells were extracted using a total protein extraction kit (BestBio Science), according to the manufacturer's instructions. Concentrations of the total proteins were determined by using a bicinchoninic acid protein assay kit (Bioworld Technology, Inc.). Protein samples were separated on SDS-PAGE gels and electroblotted onto nitrocellulose membranes (EMD Millipore). After blocking in TBS with 5% non-fat dry milk for 2 h at 37°C, the membrane was incubated overnight with primary antibodies: Occludin (rabbit polyclonal IgG; 1 µg/ml; Abcam; cat. no. ab31721), ZO1 (rabbit polyclonal IgG; 1:1,000; Thermo Fisher Scientific Inc.; cat. no. 61-7300), VEGF (rabbit polyclonal IgG; 1:500; Santa Cruz Biotechnology. Inc.; cat. no. sc-152), phosphorylated (p-) PLCγ1 (rabbit clonal IgG 1:1,000; Cell Signalling Technology, Inc.; cat. no. 2821), eNOS (rabbit monoclonal IgG; 1:1,000; Cell Signalling Technology, Inc.; cat. no. 32027) and β-actin (mouse monoclonal IgG; 1:1,000; Cell Signalling Technology, Inc.; cat. no. 3700). After washing in TBST three times, the membranes were incubated with HRP-conjugated secondary antibody (1:1,000; Cell Signalling Technology, Inc.; cat. no. 7074 or cat. no. 7076) for 45 min at room temperature. The immunoblots were developed using an enhanced chemiluminescence detection system (ImageQuant LAS 500; GE Healthcare Life Sciences). The band intensity was quantified using ImageJ 1.39u software (National Institutes of Health).
To confirm the regulation of occludin and ZO1 by HS through the VEGFR2/PLCγ1/eNOS pathway, bEnd.3 cells were first stimulated with OGD for 6 h as aforementioned. Then, cells were transferred to DMEM medium supplemented with 10% FBS and SU5416 (VEGFR2 antagonist; 10 ng/ml; Sigma-Aldrich; Merck KGaA; cat. no. S8442), U73122 (PLCγ1 inhibitor; 1 µM; Sigma-Aldrich; Merck KGaA; cat. no. U6756), L-NAME (eNOS inhibitor; 100 µM; Sigma-Aldrich; Merck KGaA; cat. no. N5751) or HS (40 mM) for 24 h at 37°C in a humidified incubator with 95% air and 5% CO2. bEnd.3 cells, that did not undergo OGD, and were cultured in DMEM/10% FBS, with or without 1 mM DMSO, were used as controls.
Statistical analysis
SPSS 19.0 (IBM Corp.) was used to analyse the data. The results are expressed as means ± standard deviation. Differences among multiple groups were statistically analysed using one-way ANOVA and post hoc comparisons (Bonferroni test). P<0.05 was considered to indicate a statistically significant difference.
Results
Determination of BBB permeability
Fig. 1A shows a schematic diagram of EB staining. The extravasation of EB was significantly increased in samples from the IR group, NS group and HS group at 12 and 24 h compared with those in the corresponding sham groups (P<0.05; Fig. 1B). However, following HS treatment, the concentration of EB in the HS group was significantly decreased compared with that in the IR group (P<0.05; Fig. 1B). At 24 h after MCAO, the EB staining area was obviously increased in brain sections from the IR and NS groups, but was noticeably reduced following 10% HS treatment in the HS group (Fig. 1C).
Figure 1.

Assessment of Evans blue dye extravasation. (A) Schematic diagram of the experimental procedure. (B) Quantitative analysis of the extravasated Evans blue dye at 12 and 24 h in the different experimental groups. §P<0.05 vs. IR at 12 h; &P<0.05 vs. IR at 24 h; *P<0.05 vs. sham at 12 h; and #P<0.05 vs. sham at 24 h. (C) Evans blue extravasation in each group at 24 h after MCAO. (D) Evans blue staining (red) in tissues sections was observed in different groups at 24 h after MCAO by fluorescence microscopy (scale bar, 20 µm). The white arrows in the sham group indicate that no visible red Evans blue extravasation was observed around the cerebral vessels. The white arrows in the IR, NS and HS groups indicate the red Evans blue extravasation around the cerebral vessels. IR, ischaemia-reperfusion; MCAO, middle cerebral artery occlusion; NS, normal saline; HS, hypertonic saline; i.v., intravenous; EB, Evans blue; ns, non-significant.
Using fluorescence microscopy, the contours of the microvessels were observed to be well-delineated in the sham group. However, in the IR and NS groups at 24 h, the vascular contours were less evident, and increased extravasated EB was observed in the tissue parenchyma. Following treatment with 10% HS, the extravasation of EB was noticeably decreased (Fig. 1D).
VEGF protein expression in peri-ischaemic brain tissue
Western blot analysis revealed moderate expression of VEGF in the sham group (Fig. 2A). At 12 and 24 h after MCAO, VEGF protein expression levels in the IR and NS groups were significantly increased compared with those in the sham group (Fig. 2A). However, after treatment with 10% HS, VEGF expression levels were significantly decreased when compared with those in the corresponding ischaemic rats (Fig. 2A). Accordingly, quantification of the western blot signals demonstrated that the optical density of the VEGF bands in the peri-ischaemic brain tissue increased significantly at 12 and 24 h for the IR and NS groups compared with those in the sham group (P<0.05; Fig. 2B). By contrast, VEGF protein expression levels decreased significantly at 12 and 24 h in the HS group compared with those in the IR and NS groups (P<0.05; Fig. 2B).
Figure 2.


VEGF protein expression in peri-ischaemic brain tissue. (A) VEGF protein expression levels in the different experimental groups at 12 and 24 h, as detected by western blotting. (B) Quantification of VEGF levels. §P<0.05 vs. IR at 12 h; &P<0.05 vs. IR at 24 h; *P<0.05 vs. sham at 12 h; and #P<0.05 vs. sham at 24 h. (C) Fluorescence intensity changes of VEGF protein in longitudinal and cross sections of the vessels from each group. §P<0.05 vs. IR; and *P<0.05 vs. sham. (D) Representative images. Scale bar, 5 µm for longitudinal sections and 10 µm for the cross sections. VEGF, vascular endothelial growth factor; IR, ischaemia-reperfusion; NS, normal saline; HS, hypertonic saline.
Immunofluorescence staining analysis revealed that VEGF was expressed specifically in the MVECs, as confirmed by double labelling with VWF (Fig. 2D). The staining was evident in the cross sections and in longitudinal sections of the blood vessels (Fig. 2D). Intense VEGF immunofluorescence was detected at 24 h in samples from the IR and NS groups compared with the sham group (P<0.05; Fig. 2C and D). Treatment with 10% HS attenuated VEGF immunofluorescence in samples from the HS group compared with those from the IR and NS groups (P<0.05; Fig. 2C and D).
VEGFR2 protein expression in the peri-ischaemic brain tissue
Immunofluorescence staining analysis revealed that VEGFR2 expression was specifically detected in MVECs, as confirmed by double labelling with CD31 (Fig. 3A). At 24 h after MCAO, very intense VEGFR2 immunoreactivity was detected in the IR and NS groups. In the HS group, the immunoreactivity was obviously decreased compared with that of the IR and NS groups (Fig. 3A). Quantification of the fluorescence intensity demonstrated that this was significantly increased in the IR and NS groups at 24 h when compared with that in the sham group (P<0.05; Fig. 3B). However, in the HS group, VEGFR2 immunofluorescence was significantly decreased compared with that in the IR and NS groups (P<0.05; Fig. 3B).
Figure 3.

VEGFR2 protein expression of cerebral microvessels in the peri-ischaemic brain tissue. (A) Immunofluorescence staining of VEGFR2 (red) protein on the cerebrovascular endothelial cells (as identified by the green staining for the endothelial cell-specific marker CD31) at 24 h after MCAO in each experimental group. Scale bar, 20 µm. The arrows indicate the cerebral vessels around the ischemic lesion. (B) Fluorescence intensity changes of VEGFR2 protein in cerebral microvessels in each group. §P<0.05 vs. IR; and *P<0.05 vs. sham. VEGFR2, vascular endothelial growth factor receptor 2; MCAO, MCAO, middle cerebral artery occlusion; IR, ischaemia-reperfusion; NS, normal saline; HS, hypertonic saline.
Expression of occludin and ZO1 in the peri-ischaemic brain tissue
Western blotting analysis demonstrated that the expression levels of occludin and ZO1 were markedly lower in the IR and NS groups compared with that in the sham group at 12 and 24 h after MCAO. However, after 10% HS treatment, the expression levels of occludin and ZO1 were markedly enhanced compared with that of the IR and the NS groups at the corresponding time points (Fig. 4A). Accordingly, quantification of the optical densities of occludin and ZO1 revealed that their levels were significantly decreased in the IR and NS groups compared with the sham group at 12 and 24 h post-reperfusion (P<0.05; Fig. 4B). Remarkably, treatment with 10% HS markedly increased the optical density of occludin and ZO1 compared with the IR and NS groups (P<0.05; Fig. 4B).
Figure 4.

Occludin and ZO1 protein expression in the peri-ischaemic brain tissue. (A) Representative images from western blot analysis for the proteins occludin (63 kDa), ZO1 (220 kDa) and β-actin (43 kDa). (B) Quantification of western blotting results in each experimental group at 12 and 24 h. §P<0.05 vs. IR at 12 h; &P<0.05 vs. IR at 24 h; *P<0.05 vs. sham at 12 h; and #P<0.05 vs. sham at 24 h. (C) Fluorescence intensity changes of ZO1 and occludin proteins in each group 24 h after treatment. §P<0.05 vs. IR; and *P<0.05 vs. sham. (D) Representative images (scale bar, 5 µm). ZO1, zonula occludens 1; IR, ischaemia-reperfusion; NS, normal saline; HS, hypertonic saline; VWF, von Willebrand factor.
To confirm the effect of focal ischaemia-reperfusion on occludin and ZO1, immunofluorescence staining analysis was also performed. As shown in Fig. 4D, occludin and ZO1 expression in brain tissue was colocalized with VWF. Very weak occludin and ZO1 immunoreactivity was detected in the IR and NS groups compared with that in the sham group. However, after 10% HS treatment, occludin and ZO1 immunoreactivity was markedly enhanced. Quantification of the fluorescent signals revealed a significant decrease in the mean fluorescence intensity values for occludin and ZO1 in the IR and NS groups compared with the sham group (P<0.05; Fig. 4C). Of note, a significance increase in fluorescence intensity was observed in the HS group for both ZO1 and occludin, compared with the IR and NS groups (P<0.05; Fig. 4C).
Determination of endothelial cell monolayer permeability
To determine the optimal concentration of HS on bEnd.3 cells, first the effect of HS on bEnd.3 cell viability was evaluated. The viability of the OGD-induced bEnd.3 cells increased significantly at 24 h after treatment with 40 mM HS compared with other concentrations of HS (10, 20, 80 and 160 mM; Fig. 5B). Therefore, the concentration of 40 mM HS was selected for the subsequent experiments in vitro. Fig. 5A shows a schematic diagram of the experiments conducted in vitro.
Figure 5.

Assessment of HRP permeability. (A) Schematic diagram of the experimental procedure in vitro. (B) Cell viability analysis of OGD-induced bEnd.3 ECs following treatment with different concentrations of HS. (C) HRP permeability of the cerebral microvascular EC monolayer barrier. #P<0.05 vs. OGD; and *P<0.05 vs. control. HRP, horseradish peroxidase; OGD, oxygen-glucose deprivation; HS, hypertonic saline; Con, control; EC, endothelial cell; ZO1, zonula occludens 1.
The permeability of the bEnd.3 cell monolayer was assessed by measuring the permeability coefficient of HRP. As shown in Fig. 5C, compared with the control group, the HRP permeability increased significantly at 3, 6, 12, and 24 h after 6 h of OGD (P<0.05). However, when compared with the OGD group, the HRP permeability decreased significantly at 12 h and 24 h after treatment with 40 mM HS (P<0.05; Fig. 5C).
HS upregulates VEGF, VEGFR2, pPLCγ1, and eNOS protein expression in bEnd.3 endothelial cells
Immunofluorescence staining analysis demonstrated that VEGF was expressed in the bEnd.3 MVECs, as confirmed by double staining with CD31 (Fig. 6A). When compared with the control group, VEGF immunofluorescence intensity in the bEnd.3 endothelial cells was enhanced at 24 h after OGD (P<0.05; Fig. 6A and C), but was significantly decreased after treatment with 40 mM HS for 24 h compared with the OGD group (P<0.05; Fig. 6A and C). Similar changes were observed for VEGFR2 immunofluorescence expression in the bEnd.3 endothelial cells (Fig. 6B and D).
Figure 6.


VEGF, VEGFR2, p-PLCγ1, PLCγ1 and eNOS protein expression in bEnd.3 endothelial cells. (A) Immunofluorescence staining of VEGF (green) in bEnd.3 endothelial cells. Co-localization with the endothelial cell marker CD31 (red) was observed. (B) Immunofluorescence staining of VEGFR2 in bEnd.3 endothelial cells. Scale bar, 20 µm. (C) Fluorescence intensity changes of VEGF protein and (D) VEGFR2 protein in each experimental group. §P<0.05 vs. OGD; and *P<0.05 vs. control. (E) Representative images from western blot analysis for the proteins VEGF (21 and 42 kDa), VEGFR2 (154 kDa), p-PLCγ1 (155 kDa), PLCγ1 (155 kDa), eNOS (140 kDa) and β-actin (43 kDa). (F) Quantification of western blotting results in each group. #P<0.05 vs. OGD; and *P<0.05 vs. control. VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; p-, phosphorylated; PLCγ1, phospholipase C γ1; eNOS, endothelial nitric oxide synthase; OGD, oxygen-glucose deprivation; HS, hypertonic saline; Con, control.
To confirm whether the upregulated expression of VEGF and VEGFR2 occurs through the VEGF/VEGFR2/pPLCγ1/eNOS signalling pathway, the expression of each of these proteins was investigated by western blotting. As shown in Fig. 6E, the expression levels of VEGF, VEGFR2, p-PLCγ1 and eNOS were significantly increased 24 h following OGD compared with the control (Fig. 6E and 6F). After treatment with 40 mM HS, however, VEGF, VEGFR2, p-PLCγ1 and eNOS protein expression levels in the HS group were significantly decreased compared with those in the OGD group (Fig. 6E and F).
HS regulates occludin and ZO1 expression through VEGFR2/PLCg1/eNOS in bEnd.3 endothelial cells
In the OGD group, occludin and ZO1 protein expression levels were decreased in the bEnd.3 endothelial cells compared with the sham group (P<0.05; Fig. 7A and B). Twenty-four hours after treatment with 40 mM HS, however, the protein expression levels of occludin and ZO1 in the HS group were higher than in the OGD group (P<0.05; Fig. 7A and B). Following a combined treatment of 10 ng/ml VEGF-A and 40 mM HS (HS+VEGF-A group), the expression levels of both proteins were significantly decreased at 24 h when compared with those in the HS group (P<0.05; Fig. 7A and B).
Figure 7.

Occludin and ZO1 protein expression in bEnd.3 endothelial cells. (A) Representative images from western blot analysis for the proteins ZO1 (220 kDa), occludin (63 kDa) and β-actin (43 kDa). (B) Quantification of western blotting results in each experimental group. (C) Immunofluorescence staining of ZO1 and occludin in bEnd.3 endothelial cells at 24 h after treatment. Scale bar, 20 µm. (D) Fluorescence intensity changes of ZO1 and occludin proteins in each group. *P<0.05 vs. control; #P<0.05 vs. OGD; and &P<0.05 vs. HS. ZO1, zonula occludens 1; OGD, oxygen-glucose deprivation; Con, control; HS, hypertonic saline; VEGF-A, vascular endothelial growth factor A.
By immunofluorescence staining analysis of the bEnd.3 cell monolayers, weak occludin and ZO1 immunoreactivity was detected in the OGD group at 24 h compared with that in the control group (P<0.05; Fig. 7C and D). However, after treatment with 40 mM HS, occludin and ZO1 immunoreactivity was significantly increased compared with that in the OGD group (P<0.05; Fig. 7C and D). Following the combined treatment of 10 ng/ml VEGF-A and 40 mM HS, expression of both proteins was decreased compared with that in the HS group (P<0.05; Fig. 7C and D).
To further confirm the regulation of occludin and ZO1 by HS through the VEGFR2/PLCγ1/eNOS pathway, bEnd.3 cell were incubated with SU5416 (inhibitor of VEGFR2), U73122 (inhibitor of PLCγ1), and L-NAME (inhibitor of eNOS). As shown in Fig. 8A and B, the protein expression levels of occludin and ZO1 were decreased after OGD compared with the control (P<0.05), but significantly increased following treatment with SU5416, U73122, L-NAME and 40 mM HS (all P<0.05).
Figure 8.

VEGF regulated occludin and ZO1 expression via the VEGFR2/PLCγ1/eNOS signalling pathway in bEnd.3 endothelial cells. (A) Representative images from western blot analysis for the proteins occludin (63 kDa), ZO1 (220 kDa) and β-actin (43 kDa), following different treatments, including OGD, 40 mM HS, SU5416 (VEGFR2 inhibitor), U73122 (PLCγ1 inhibitor) and L-NAME (eNOS inhibitor). (B) Quantification of western blotting results in each experimental group. *P<0.05 vs. control; and #P<0.05 vs. OGD. VEGF, vascular endothelial growth factor; ZO1, zonula occludens 1; VEGFR2, vascular endothelial growth factor receptor 2; PLCγ1, phospholipase C γ1; eNOS, endothelial nitric oxide synthase; OGD, oxygen-glucose deprivation; HS, hypertonic saline; Con, control.
Discussion
The BBB is a selective semipermeable membrane and the regulated interface between the peripheral circulation and the central nervous system (CNS) (27). The anatomical substrate of the BBB is the cerebral microvascular endothelium, which, together with astrocytes, pericytes, neurons, and the extracellular matrix, constitutes a 'neurovascular unit' that is essential for the health and function of the CNS (27). The BBB separates the brain parenchyma from the peripheral circulation and stabilizes the microenvironment of neurons, protecting them from deleterious effects of certain substances in the blood (28). The disruption of the BBB leads to extravasation of intravascular substances into the extracellular spaces of the brain. The present study demonstrated that EB dye extravasation in the ischaemic hemisphere gradually increased up to 24 h. These results are in accord with a previous report that ischaemia-reperfusion enhanced BBB permeability and breakdown (29). HS is a common osmotic dehydrating agent that exerts anti-oedema effects on an intact BBB, and the anti-oedema effect in the brain may be associated with alleviating the disruption of the BBB (5). As expected, the present results demonstrated that treatment with 10% HS resulted in a significant decrease in EB extravasation. This was further confirmed in vitro; HRP extravasation was significantly decreased, compared to that in the OGD group, in a bEnd.3 cell-derived cerebral MVEC monolayer barrier following incubation with 40 mM HS for 24 h. Thus, the present study demonstrated that 10% HS ameliorated cerebral oedema through reduction of BBB permeability and protection of BBB integrity.
VEGF is known to be an important vascular permeability factor that can increase cerebral vascular permeability and induce vasogenic brain oedema in multiple diseases, such as cerebral ischaemic infarction (30), brain tumour (11), and subarachnoid haemorrhage (9). It has been reported that increased VEGF induces BBB leakage (31). VEGFR2, a major receptor of VEGF, is also an important mediator of vascular permeability. It is overexpressed in endothelial cells, astrocytes, neuronal stomata and tissues adjacent to areas of damage by brain injury (32). A previous report suggested that the effects of VEGF on the BBB depended on VEGFR2, and inhibition of VEGF/VEGFR2 was beneficial for ameliorating oedema (33). In view of this, the present study measured the expression of VEGF and VEGFR2. The results demonstrated that the protein expression levels of VEGF and VEGFR2 were significantly upregulated after MCAO. Notably, when compared with the IR group, the HS group exhibited lower VEGF and VEGFR2 expression levels. These changes correlated with the reduced BBB permeability after treatment with 10% HS. These findings suggested that HS may alleviate BBB permeability through inhibiting VEGF and VEGFR2 expression.
It is known that endothelial cells have a significant role in maintaining BBB permeability and integrity (34,35), and it has been reported that VEGF expression is also detected in vascular endothelial cells and neurons (36). To detect whether VEGF and VEGFR2 exhibit the same expression pattern in endothelial cells, an in vitro hypoxia model with endothelial cells was generated. The results were in agreement with the in vivo experiments. Western blot analysis revealed that the protein expression levels of VEGF and VEGFR2 were significantly increased upon OGD, but they were markedly decreased after treatment with 40 mM HS. Immunofluorescence assays confirmed this finding, demonstrating that HS directly suppressed the expression of VEGF and VEGFR2 in endothelial cells.
ZO1 and occludin are important tight junction proteins, and their downregulation significantly increases the permeability of the BBB (22). A previous study demonstrated that VEGF could downregulate ZO1 and occludin expression, leading to the disruption of the BBB (37). The present study confirmed through western blot and immunofluorescence assays that HS could also significantly inhibit the downregulation of ZO1 and occludin. Therefore, HS may ameliorate cerebral oedema via inhibition of VEGF-mediated downregulation of ZO1 and occludin, which may then protect the integrity of the BBB. It was reported that VEGF-induced permeability depends on the VEGFR2-mediated eNOS pathway (19,20), via inhibiting the expression of tight junction proteins. Another study also reported that the expression levels of occludin and claudin-5 were downregulated in microvascular endothelial cells when treated with VEGF-A; however, they were upregulated after treatment with an anti-VEGFR2 blocking antibody, a PLCγ1 inhibitor or eNOS inhibitor (15). In the present study, the results demonstrated that ZO1 and occludin protein expression levels were increased significantly after cells were incubated with SU5416, U73122 or L-NAME. These findings suggested that the VEGFR2/PLCγ1/eNOS signalling pathway may be involved in this process, but further studies will be needed to fully elucidate this mechanism.
In conclusion, upregulated VEGF and VEGFR2 expression in endothelial cells induced by ischaemia-reperfusion resulted in subsequent downregulation of ZO1 and occludin. This downregulation of tight junction proteins may contribute to BBB dysfunction by increasing its permeability. Finally, HS treatment effectively alleviated this damage to the BBB, at least partly via the VEGFR2/PLCγ1/eNOS signalling pathway (Fig. 9).
Figure 9.
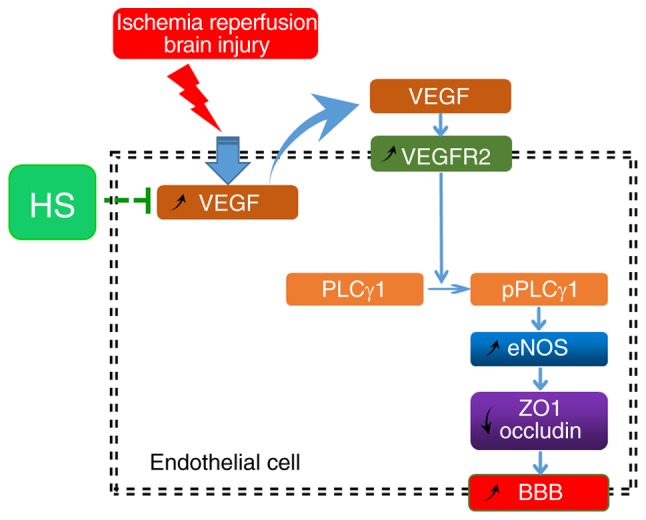
Schematic of the molecular mechanism by which HS may reduce BBB permeability induced by IR brain injury via the VEGFR2-mediated eNOS pathway in endothelial cells. HS, hypertonic saline; BBB, blood-brain barrier; IR, ischaemia-reperfusion; VEGFR2, vascular endothelial growth factor receptor 2; eNOS, endothelial nitric oxide synthase; VEGF, vascular endothelial growth factor; PLCγ1, phospholipase C γ1; ZO1, zonula occludens 1.
Acknowledgments
The authors would like to thank Mr Jiening Zhu and Qiuxiong Lin for technical assistance.
Funding
This study was supported by the Natural Science Foundation of Guangdong province (grant nos. 2016A030311043, 2017A030313691 and 2015A030313004), the Natural Science Foundation of Hunan province (grant no. 2017JJ2229), the National Natural Science Foundation for Young Scientists of China (grant nos. 81701939 and 81701875) and the Science and Technology Project of Guangdong (grant no. 2016A020215209).
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
HZ, QW, YD and WZ conceived the study. YH, HD, SC and BL provided materials and samples. LH, MW, YH, WJ and YD participated in data collection and analysis. All authors read and approved the final manuscript and consented to publish this manuscript.
Ethics approval and consent to participate
All experimental procedures involving the use of animals were approved by the Institutional Animal Care and Use Committee, Guangdong Province, China (approval no. GBREC2012106A). All experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing interests.
References
- 1.Wijdicks EF, Sheth KN, Carter BS, Greer DM, Kasner SE, Kimberly WT, Schwab S, Smith EE, Tamargo RJ, Wintermark M, et al. Recommendations for the management of cerebral and cerebellar infarction with swelling: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:1222–1238. doi: 10.1161/01.str.0000441965.15164.d6. [DOI] [PubMed] [Google Scholar]
- 2.Gasche Y, Copin JC. Blood-brain barrier pathophysiology and ischaemic brain oedema. Ann Fr Anesth Reanim. 2003;22:312–319. doi: 10.1016/S0750-7658(03)00069-8. In French. [DOI] [PubMed] [Google Scholar]
- 3.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Nout YS, Mihai G, Tovar CA, Schmalbrock P, Bresnahan JC, Beattie MS. Hypertonic saline attenuates cord swelling and edema in experimental spinal cord injury: A study utilizing magnetic resonance imaging. Crit Care Med. 2009;37:2160–2166. doi: 10.1097/CCM.0b013e3181a05d41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng HK, Wang QS, Deng YY, Jiang WQ, Fang M, Chen CB, Jiang X. A comparative study on the efficacy of 10% hypertonic saline and equal volume of 20% mannitol in the treatment of experimentally induced cerebral edema in adult rats. BMC Neurosci. 2010;11:153. doi: 10.1186/1471-2202-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarz S, Georgiadis D, Aschoff A, Schwab S. Effects of hypertonic (10%) saline in patients with raised intracranial pressure after stroke. Stroke. 2002;33:136–140. doi: 10.1161/hs0102.100877. [DOI] [PubMed] [Google Scholar]
- 7.Zeynalov E, Chen CH, Froehner SC, Adams ME, Ottersen OP, Amiry-Moghaddam M, Bhardwaj A. The perivascular pool of aquaporin-4 mediates the effect of osmotherapy in postischemic cerebral edema. Crit Care Med. 2008;36:2634–2640. doi: 10.1097/CCM.0b013e3181847853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang LQ, Zhu GF, Deng YY, Jiang WQ, Fang M, Chen CB, Cao W, Wen MY, Han YL, Zeng HK. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-alpha and IL-1beta-induced Na-K-Cl Cotransporter up-regulation. J Neuroinflammation. 2014;11:102. doi: 10.1186/1742-2094-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu L, Fujimoto M, Kawakita F, Ichikawa N, Suzuki H. Vascular endothelial growth factor in brain edema formation after subarachnoid hemorrhage. Acta Neurochir Suppl. 2016;121:173–177. doi: 10.1007/978-3-319-18497-5_31. [DOI] [PubMed] [Google Scholar]
- 10.Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metal-loproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30:837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerstner ER, Duda DG, di Tomaso E, Ryg PA, Loeffler JS, Sorensen AG, Ivy P, Jain RK, Batchelor TT. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229–236. doi: 10.1038/nrclinonc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.STR.28.10.2039. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg DA, Jin K. Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci. 2013;70:1753–1761. doi: 10.1007/s00018-013-1282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, Loo H, Laitman BM, Mariani JN, Straus Farber R, et al. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain. 2015;138:1548–1567. doi: 10.1093/brain/awv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobbs CS, Chen J, Greenberg DA, Graham SH. Vascular endothelial growth factor expression in transient focal cerebral ischemia in the rat. Neurosci Lett. 1998;249:79–82. doi: 10.1016/S0304-3940(98)00377-2. [DOI] [PubMed] [Google Scholar]
- 17.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling-in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 18.Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–349. doi: 10.1161/STROKEAHA.109.564229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2002;39:225–237. doi: 10.1016/S1537-1891(03)00011-9. [DOI] [PubMed] [Google Scholar]
- 20.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44:130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 23.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- 24.Jiang S, Xia R, Jiang Y, Wang L, Gao F. Vascular endothelial growth factors enhance the permeability of the mouse blood-brain barrier. PLoS One. 2014;9:e86407. doi: 10.1371/journal.pone.0086407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- 26.Wang LF, Li X, Gao YB, Wang SM, Zhao L, Dong J, Yao BW, Xu XP, Chang GM, Zhou HM, et al. Activation of VEGF/Flk-1-ERK pathway induced blood-brain barrier injury after microwave exposure. Mol Neurobiol. 2015;52:478–491. doi: 10.1007/s12035-014-8848-9. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 28.Anfuso CD, Lupo G, Romeo L, Giurdanella G, Motta C, Pascale A, Tirolo C, Marchetti B, Alberghina M. Endothelial cell-pericyte cocultures induce PLA-2 protein expression through activation of PKCalpha and the MAPK/ERK cascade. J Lipid Res. 2007;48:782–793. doi: 10.1194/jlr.M600489-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Cui HS, Shin SK, Kim JM, Kim SY, Lee JE, Koo BN. Effect of propofol post-treatment on blood-brain barrier integrity and cerebral edema after transient cerebral ischemia in rats. Neurochem Res. 2013;38:2276–2286. doi: 10.1007/s11064-013-1136-7. [DOI] [PubMed] [Google Scholar]
- 30.Chi OZ, Hunter C, Liu X, Weiss HR. Effects of anti-VEGF antibody on blood-brain barrier disruption in focal cerebral ischemia. Exp Neurol. 2007;204:283–287. doi: 10.1016/j.expneurol.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen Nv, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafuente JV, Argandona EG, Mitre B. VEGFR-2 expression in brain injury: Its distribution related to brain-blood barrier markers. J Neural Transm (Vienna) 2006;113:487–496. doi: 10.1007/s00702-005-0407-0. [DOI] [PubMed] [Google Scholar]
- 33.Vohra PK, Hoeppner LH, Sagar G, Dutta SK, Misra S, Hubmayr RD, Mukhopadhyay D. Dopamine inhibits pulmonary edema through the VEGF-VEGFR2 axis in a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302:L185–L192. doi: 10.1152/ajplung.00274.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roe K, Orillo B, Verma S. West Nile virus-induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood-brain barrier model. PLoS One. 2014;9:e102598. doi: 10.1371/journal.pone.0102598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma SC, Li Q, Peng JY, Zhouwen JL, Diao JF, Niu JX, Wang X, Guan XD, Jia W, Jiang WG. Claudin-5 regulates blood-brain barrier permeability by modifying brain microvascular endothelial cell proliferation, migration, and adhesion to prevent lung cancer metastasis. CNS Neurosci Ther. 2017;23:947–960. doi: 10.1111/cns.12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang L, Cao W, Deng Y, Zhu G, Han Y, Zeng H. Hypertonic saline alleviates experimentally induced cerebral oedema through suppression of vascular endothelial growth factor and its receptor VEGFR2 expression in astrocytes. BMC Neurosci. 2016;17:64. doi: 10.1186/s12868-016-0299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L, Ye Z, Pan Y, Li X, Fu X, Zhang B, Li Y, Lin W, Li X, Gao Q. Vascular endothelial growth factor aggravates cerebral ischemia and reperfusion-induced blood-brain-barrier disruption through regulating LOC102640519/HOXC13/ZO-1 signaling. Exp Cell Res. 2018;369:275–283. doi: 10.1016/j.yexcr.2018.05.029. [DOI] [PubMed] [Google Scholar]