

ABSTRACT
Wilson disease is an autosomal recessive disorder of abnormal copper metabolism that is prevalent in the younger population, rarely presenting in patients older than 40 years. Clinical presentation may be variable, and diagnosis is often aided by clinical and biochemical tests. We report the case of a 72-year-old woman who presented with acute liver failure initially of unclear etiology. Our patient was initially managed for presumed drug-induced liver injury but ultimately diagnosed with Wilson disease on the basis of clinical presentation, laboratory testing, liver biopsy, quantitative hepatic copper, and abnormal genetic testing.
INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder of abnormal copper metabolism, with the prevalence of approximately 1 in 30,000 individuals. First described by Kinnier Wilson as “always developing in young people,” WD is more prevalent in a younger population, with a mean age of 15–20 years and age of presentation, and seldom older than 40 years.1–5 Although there has been a mention of WD in patients as old as 72 years, such cases are extremely rare.6 Current practice guidelines issued by the American Association for the Study of Liver Diseases on diagnosis and management of WD specify that advanced age alone is not a basis for eliminating WD from the differential diagnosis.
WD has a wide spectrum of presentation. Patients may present with primary hepatic or neurologic symptoms; a minority are diagnosed through family history screening.7 Diagnosis remains laboratory based; although there is an emerging role for genetic testing, it must be underscored that there is high alleleic heterogeneity among patients.8 In cases of equivocal clinical presentation, laboratory values and pathologic findings prove to be particularly useful in aiding the diagnosis of WD.
CASE REPORT
A 72-year-old woman presented to our institution for acute liver failure. Her medical history was significant for rheumatoid arthritis and lupus, which had been managed on methotrexate for 1 year. She denied history of alcohol use, recreational drug use, or nonprescription health supplement use. There was no family history of liver disease. Our patient initially presented to an outside hospital with acute pancreatitis. Admission liver chemistries were total bilirubin 0.7 mg/dL, direct bilirubin 0.3 mg/dL, aspartate aminotransferase 125 U/L, alanine aminotransferase 94 U/L, alkaline phosphatase 468 U/L, and international normalized ratio 1.3. Etiology of the pancreatitis was unknown despite extensive workup.
During this hospitalization, the patient's liver function progressively deteriorated. Initial concern for possible drug-induced liver injury prompted initiation of intravenous methylprednisone and N-acetylcysteine. Despite this intervention, her synthetic function continued to deteriorate. She underwent transjugular liver biopsy, which revealed steatohepatitis, minimal fibrosis 1a/4, cholestasis, and anisonucleosis consistent with methotrexate-induced hepatotoxicity vs WD. Liver biopsy was sent for quantitative copper determination.
The patient was transferred to our center with progressive hepatocellular injury. Initial management was conservative with working diagnosis of presumed methotrexate-induced hepatotoxicity. Over the course of the hospitalization, complications included hemolytic anemia, acute renal failure, depressed mental status, and worsening jaundice. Increasing suspicion for WD prompted evaluation by ophthalmology for assessment of Kayser-Fleischer rings, ultimately with equivocal findings. She demonstrated reluctance in undergoing brain magnetic resonance imaging. Notable laboratory values included the following: serum copper 79 μg/dL, 24-hour urinary copper excretion 31 μg/24 hr (with a glomerular filtration rate of 34 mL/min/1.73 m2 at the time of collection), and ceruloplasmin 14 mg/dL. Nonceruloplasmin bound copper was 37 μg/dL. Her quantitative hepatic copper returned elevated at 298 μg Cu/g. Other laboratory testing for acute liver failure workup was negative.
Given our patient's failure to improve with supportive measures, low ceruloplasmin, histological findings of steatohepatitis and anisonucelosis, equivocal findings of Kayser-Fleischer rings, and elevated quantitative hepatic copper, suspicion for WD increased. She was urgently evaluated for liver transplant. Unfortunately, the course was complicated by anuric renal failure and hypoxic respiratory failure, and she eventually died in the intensive care unit. At death, the patient's laboratory values trended to total bilirubin 34.7 mg/dL, direct bilirubin 30.8 mg/dL, aspartate aminotransferase 124 U/L, alanine aminotransferase 38 U/L, and alkaline phosphatase 475 U/L. International normalized ratio during hospitalization peaked at 2.58 despite administration of vitamin K. Genetic testing with whole-gene sequencing revealed heterozygosity for a single copy of a point mutation in the ATP7B gene (A to G substitution at position 226).
DISCUSSION
WD is diagnosed on the basis of several clinical and biochemical tests. Classic presentation has been characterized by age at onset between 5 and 40 years, detectable Kayser-Fleischer rings, and decreased serum ceruloplasmin.8 However, half of patients presenting with liver disease do not meet two of these three criteria, leading to diagnostic uncertainty.9 Recently, diagnostic advances have allowed for a more systematic approach to diagnosis. American Association for the Study of Liver Diseases guidelines provide a systematic approach in assisting with the diagnosis of WD (Figure 1).
Figure 1.

Approach to diagnosis of Wilson disease in a patient with unexplained liver disease from the “AASLD Practice Guidelines—Diagnosis and Treatment of Wilson Disease: An Update.” (Printed with permission from the the copyright holder, the American Association for the Study of Liver Diseases (AASLD). Reproduced from Roberts EA and Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008:47;2089-2111. doi:10.1002/hep.22261. All permission requests for this image should be made to the copyright holder.) CPN, ceruloplasmin; KF, Kayser-Fleischer.
In our patient, symptoms consistent with WD included cirrhosis, neuropsychiatric depression, hemolytic anemia, and acute renal failure. Clinical suspicion prompted further testing notable for equivocal Kaiser-Fleischer rings, low ceruloplasmin 14 mg/dL, and 24-hour urine copper 31 μg, although the diminished glomerular filtration rate at the time of collection suggested an inadequate urine sample. Notably, urinary copper has been shown to be <100 μg in 16%–23% of WD cases.10 Per guidelines by the American Association for the Study of Liver Diseases, liver biopsy was obtained; findings included anisonucleosis and hepatitis on pathology and an elevated quantitative hepatic copper of 298 μg Cu/g, thus confirming our suspicion.
Our patient's genetic testing revealed a variant of unclear significance in the ATP7B gene. Recent literature has cited over 500 disease-causing mutations of WD in the ATP7B gene.11 Importantly, as many as 1 in 4 cases of WD may have only a single copy of an identifiable point mutation, supporting the importance of this or other possible mechanisms for phenotypic WD in the absence of homozygosity or compound heterozygosity for known pathogenic point mutations.12 As a result of alleleic heterogeneity and sequence variations, genetic testing may prove helpful in diagnosing patients with biallelic pathogenic mutations, but its role is still evolving as a first-line diagnostic test. In situations of genetic equivocality, other laboratory results must be considered.
Finally, pathologic findings of anisonucleosis may be helpful in uncovering WD. Histologically, “the constellation of microvesicular steatosis, ballooning change, glycogenated nuclei, anisonucleosis, and binucleation is very suggestive of WD.”13 Further studies correlate these findings—in 34 cases of WD in 1980, all cirrhotic livers showed anisocytosis and anisonucleosis.14 Similar histologic findings can be found in our patient (Figure 2).
Figure 2.
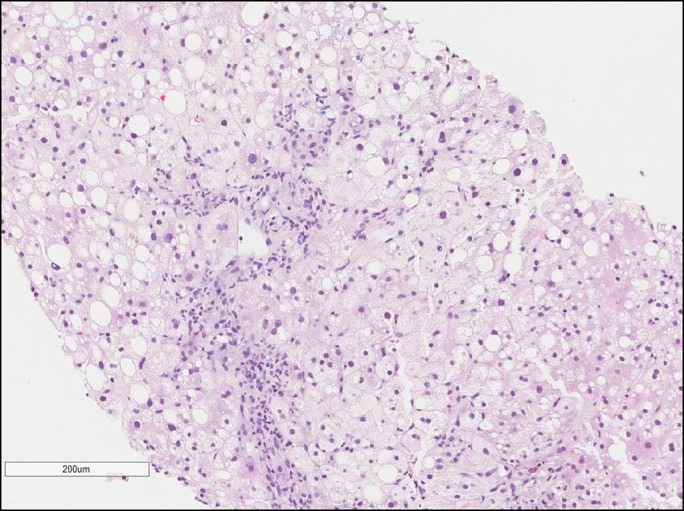
Liver biopsy revealing steatohepatitis with moderate activity (6/8), minimal increase in fibrosis (1a/4), cholestasis, and anisonucleosis.
Ultimately, diagnosis of WD was made by a combination of presentation, laboratory results, equivocal genetic testing, and suggestive liver pathology. As per AASLD guidelines, diagnosis of WD would be established. Our case provides a unique learning opportunity—the atypical presentation of WD in an elderly 72-year-old patient. This case highlights the importance of maintaining clinical suspicion for WD even in older patients and the uncertainty that can be involved in the interpretation of genetic testing for WD.
DISCLOSURES
Author contributions: All authors were directly involved in conception, data collection, interpretation of data, and drafting of the manuscript. JM Civan is the article guarantor.
Financial disclosure: None to report.
Informed consent was obtained for this case report.
REFERENCES
- 1.Wilson SAK. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver. Brain 1912;34:295–509. [DOI] [PubMed] [Google Scholar]
- 2.Scheinberg IH, Sternleib I. Wilson's Disease. WB Saunders: Philadelphia, 1984. [Google Scholar]
- 3.Taly AB, Meenakshi-Sundaram S, Sinha S, et al. Wilson disease: Description of 282 patients evaluated over 3 decades. Medicine (Baltimore) 2007;86:112. [DOI] [PubMed] [Google Scholar]
- 4.Machado A, Chien HF, Deguti MM, et al. Neurological manifestations in Wilson's disease: Report of 119 cases. Mov Disord 2006;21:2192. [DOI] [PubMed] [Google Scholar]
- 5.Walshe JM, Yealland M. Wilson's disease: The problem of delayed diagnosis. J Neurol Neurosurg Psychiatry 1992;55:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ala A, Borjigin J, Rochwarger A, Schilsky M. Wilson disease in septagenarian siblings: Raising the bar for diagnosis. Hepatology 2005;41:668–70. [DOI] [PubMed] [Google Scholar]
- 7.Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long‐term outcome of Wilson's disease: A cohort study. Gut 2007;56:115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sternlieb I. Perspectives on Wilson's disease. Hepatology 1990;12:1234–9. [DOI] [PubMed] [Google Scholar]
- 9.Steindl P, Ferenci P, Dienes HP, et al. Wilson's disease in patients presenting with liver disease: A diagnostic challenge. Gastroenterology 1997;113:212–8. [DOI] [PubMed] [Google Scholar]
- 10.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008;47:2089–111. [DOI] [PubMed] [Google Scholar]
- 11.Hedera P. Update on the clinical management of Wilson's disease. Appl Clin Genet 2017;10:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferenci P. Phenotype-genotype correlations in patients with Wilson's disease. Ann N Y Acad Sci 2014;1315:1–5. [DOI] [PubMed] [Google Scholar]
- 13.Cheng L, Bostwick DG. Essentials of Anatomic Pathology. 3rd edn Springer: New York, 2011. [Google Scholar]
- 14.Stromeyer FW, Ishak KG. Histology of the liver in Wilson's disease: A study of 34 cases. Am J Clin Pathol 1980;73:12–24. [DOI] [PubMed] [Google Scholar]