

Abstract
Nrf2 is essential to antioxidant response element (ARE)-mediated host defense. Sulforaphane (SFN) is a phytochemical antioxidant known to affect multiple cellular targets including Nrf2-ARE pathway in chemoprevention. However, the role of SFN in non-malignant airway disorders remain unclear. To test if pre-activation of Nrf2-ARE signaling protects lungs from oxidant-induced acute injury, wild-type (Nrf2+/+) and Nrf2-deficient (Nrf2−/−) mice were given SFN orally or as standardized broccoli sprout extract diet (SBE) before hyperoxia or air exposure. Hyperoxia-induced pulmonary injury and oxidation indices were significantly reduced by SFN or SBE in Nrf2+/+ mice but not in Nrf2−/− mice. SFN upregulated a large cluster of basal lung genes that are involved in mitochondrial oxidative phosphorylation, energy metabolism, and cardiovascular protection only in Nrf2+/+ mice. Bioinformatic analysis elucidated ARE-like motifs on these genes. Transcript abundance of the mitochondrial machinery genes remained significantly higher after hyperoxia exposure in SFN-treated Nrf2+/+ mice than in SFN-treated Nrf2−/− mice. Nuclear factor-κB was suggested to be a central molecule in transcriptome networks affected by SFN. Minor improvement of hyperoxia-caused lung histopathology and neutrophilia by SFN in Nrf2−/− mice implies Nrf2-independent or alternate effector mechanisms. SFN is suggested to be as a preventive intervention in a preclinical model of acute lung injury by linking mitochondria and Nrf2. Administration of SFN alleviated acute lung injury-like pathogenesis in a Nrf2-dependent manner. Potential AREs in the SFN-inducible transcriptome for mitochondria bioenergetics provided a new insight into the downstream mechanisms of Nrf2-mediated pulmonary protection.
Keywords: lung, broccoli, hyperoxia, microarray, antioxidant response element
INTRODUCTION
NF-E2-related factor 2 (Nrf2) is a transcriptional activator of antioxidant response element (ARE)-bearing defense genes. Acute lung injury (ALI) and its severe form acute respiratory distress syndrome (ARDS) affects over 200,000 annually in the U.S. It is precipitated from various clinical disorders including pneumonia, sepsis, or major trauma, and characterized by increased lung permeability and inflammation accompanying abnormal gas exchange and variable late phase responses (Hendrickson and Matthay, 2018; Matthay and Howard, 2012; Matthay et al., 2012). In laboratory rodents, inhalation of hyperoxia (>85% oxygen) causes ALI-like pulmonary injuries including edema and neutrophilic inflammation and it has been widely used as a model of ALI. We identified Nrf2 as a susceptibility gene in the murine model of ALI (Cho et al., 2015; Cho et al., 2002b) and multiple studies using mice genetically deficient in Nrf2 (Nrf2−/−) determined its protective role in ALI-like phenotypes (Cho and Kleeberger, 2015).
Supplemental antioxidant therapies with N-acetylcysteine (NAC) or various antioxidants have had mild or inconsistent efficacy in clinical settings of oxidative lung disorders. Recent interest has focused on isothiocyanates, bioactive phytochemical antioxidants from cruciferous vegetables, particularly sulforaphane (4-methylsulfinylbutyl isothiocyanate, SFN) present as the precursor form glucoraphanin in broccoli and cauliflower. Glucoraphanin is largely hydrolyzed into SFN by myrosinase in gut microbes or in the plant itself and metabolized through the mercapturic acid pathway (60–90%) starting with glutathione S-transferase (GST)-mediated conjugation followed by sequential cleavage into other dithiocarbamates (i.e., SFN-cysteinyl-glycine, SFN-cysteine, SFN-NAC) or is converted into erucin and its conjugates by S-oxide reduction and dehydration (Budnowski et al., 2015). Metabolites of SFN or isothiocyanate were detected in most rodent tissues, blood, bile, and urine shortly after oral or systemic administration (Budnowski et al., 2015; Yanaka et al., 2009). Relatively higher accumulation of SFN metabolites were found in the lung than in other mouse tissues (Bricker et al., 2014; Clarke et al., 2011). Metabolites were also detected in human blood and urine after single, multiple, or long-term (8–12 weeks) doses (Dinkova-Kostova and Kostov, 2012; Egner et al., 2014; Yanaka et al., 2009).
SFN acts through nuclear factor (NF)-κB inhibition and cell cycle arrest (Kensler et al., 2013). It also binds to inhibit Kelch-like ECH-associated protein 1 (Keap1, a cytoplasmic Nrf2 inhibitor) and thus is a potent inducer of Nrf2-ARE signaling (Kensler et al., 2013). Potential therapeutic spectrum of SFN has expanded from cancer to non-malignant disorders. Dietary SFN decreased gastritis symptoms in Helicobacter pylori (H. pylori) patients (Yanaka et al., 2009). SFN treatments also reduced gluconeogenesis improving glucose control in Type 2 diabetic patients (Axelsson et al., 2017). A series of clinical intervention trials demonstrated that SFN enhanced detoxification of food- and air-borne carcinogens (Egner et al., 2014). SFN-containing juice decreased the number of nasal lavage cells after challenge with diesel exhaust particles in humans (Heber et al., 2014). Further, daily doses of SFN significantly ameliorated bronchoconstriction caused by methacholine challenge in about 60% of asthmatics who participated in a clinical trial (Brown et al., 2015). However, other reports indicate mixed or negative efficacy of sulforaphane as seen in a randomized double-blind study in which oral sulforaphane did neither induce ARE-responsive genes in airway cells nor influence on lung function and inflammatory parameters in a chronic obstructive pulmonary disease cohort (Wise et al., 2016).
The current study tested the hypothesis that SFN prevents oxidant-induced ALI-like disease in a Nrf2-dependent manner. We provided Nrf2-sufficient (Nrf2+/+) and -deficient (Nrf2−/−) mice with SFN orally or as diet rich in SFN precursor glucoraphanin (standardized broccoli sprout extract, SBE) before hyperoxia (>95% O2) exposure to determine its effect on development of lung injury. We also profiled genome-wide transcriptome changes to provide insight into the underlying pulmonary mechanisms of SFN. Results from this study provide novel insights to the role of Nrf2-mediated molecular events on lung gene expression pathways including those for mitochondrial function. Pre-treatment effects of SFN in the hyperoxia-induced model ALI may have important clinical implications.
MATERIALS AND METHODS
Animals, pretreatment, and exposure.
Male, 6–8-week-old Nrf2+/+ (ICR), Nrf2−/− (ICR.129P2-Nfe2l2tm1Mym), glutathione peroxidase 2-deicifient (Gpx2−/−, B6.129P2-Gpx2tm1Mym), and NAD(P)H:quinone oxidoreductase 1-deficient (Nqo1−/−, B6;129S-Nqo1tm1Akj) mice produced from breeding colonies at the National Institute of Environmental Health Science (NIEHS) were used in the current study. Gender- and age-matched wild-type mice, C57BL/6J (Gpx2+/+) and 129P3/J (Nqo1+/+), were purchased from the Jackson Laboratory (Bar Harbor, ME). Nrf2−/− and Nrf2+/+ mice were provided with modified AIN-76A (AIN) diet; other mice received NIH_31 diet. Food and water were provided ad libitum. A subset of Nrf2+/+ and Nrf2−/− mice were fed either pulverized AIN diet or AIN diet containing lyophilized SBE (Fahey et al., 2012) (as glucoraphanin 5.83 μmol/g diet) using glass jars with mesh inserts for 14 d before inhalation exposure (average consumption 21.35 μmol in 3.66 g diet/animal/day). The SBE preparation used in the current study showed reproducible daily bioavailability of SFN (average 10–18% conversion of glucoraphanin to SFN and metabolites) as determined in urine from healthy volunteers (Fahey et al., 2015; Fahey et al., 2012). Pulverized AIN or SBE diet was provided in glass jars feeders with mesh inserts. Other Nrf2+/+ and Nrf2−/− mice received 9 μmol of pure SFN (R-SFN, LKT Laboratories, Inc., St. Paul, MN) in 100 μl PBS or vehicle (PBS) by oral gavage (100 μl) at 5, 3, and 1 d before inhalation exposure. A subset of Nqo1+/+ mice were fed a diet containing 0.3% dimethyl fumarate (DMF) to induce NQO1 activity for 14 d before inhalation exposure (Begleiter et al., 2004). Mice were continuously exposed to hyperoxia (>95% O2, 48 or 72 h) in a whole-body chamber or to room air as described previously (Cho et al., 2015). Immediately following designated exposure, mice were euthanized by sodium pentobarbital overdose (104 mg/Kg). All animal use was approved by the NIEHS Animal Care and Use Committee. See details in the online data supplement.
Bronchoalveolar lavage (BAL).
Right lungs from each mouse were lavaged in situ with HBSS, and cells and supernatants from BAL returns were analyzed for protein content and cell differentials as described previously (Cho et al., 2015). Colorimetric assay for lactate dehydrogenase (LDH) was performed to detect cell lysis and toxicity by determination of NADH production after addition of NAD+ to aliquots (25 μl) of BAL supernatants (Sigma, St. Louis, MO). Lipid peroxidation was also determined in aliquots of BAL (25 μl) by quantification of TBARS represented by malondialdehyde-TBA using a malondialdehyde equivalence standard curve (Cell Biolabs, Inc., San Diego, CA). Sandwich ELISA was used to detect secreted mucin 5, subtypes A and C (Muc5AC) in BAL (20 μl) using a capture antibody (sc-19603, Santa Cruz Biotechnology, Santa Cruz, CA) and a detection antibody (Clone 45M1, Thermo Scientific, Fremont, CA) following the procedures described previously (Cho et al., 2013).
Lung histopathology.
Formalin-fixed left lungs were processed for H&E and AB/PAS staining. They were immunohistologically stained with voltage-dependent anion-selective channel protein 1 (VDAC1)/porin antibody (Santa Cruz) following routine immunostaining procedures.
cDNA microarray and quantitative reverse transcription-polymerase chain reaction (qRT-PCR).
Total lung RNA (n = 3/group) was applied to mouse 430 2.0 arrays (Affymetrix, Inc., Santa Clara, CA) in the NIEHS Microarray Core Facility as described previously (Cho et al., 2012). Array data were analyzed statistically using GeneSpring (Agilent Technologies, Inc., Santa Clara, CA). Briefly, array raw data were filtered by lower expression percentile (at least 1 sample had values within 20% cut-off rage) and the expression levels were normalized to the mean value of the control (PBS/Air/Nrf2+/+) for each gene by quantile algorithm. Various interpretations were generated for group comparisons by parameters (pretreatment, exposure, genotype). Moderate t-test (PBS vs. SFN in air/Nrf2+/+ or in air/Nrf2−/−, air vs. hyperoxia in PBS/Nrf2+/+ or in PBS/Nrf2−/−) and 2-way ANOVA followed by Student-Newman-Keuls comparison (Nrf2+/+ vs. Nrf2−/− with different pre-treatment/exposure) identified differentially expressed genes (P < 0.01). Ingenuity Pathway Analysis (IPA, Qiagen Inc., Valencia, CA) was used to identify potential molecular mechanisms. Microarray data are deposited in Gene Expression Omnibus (accession number GSE58654) and in the NIEHS Chemical Effects in Biological Systems (CEBS; accession number: 005–00003-0121–000-2). Aliquots of lung RNA were reverse transcribed, and cDNA was subjected to PCR with gene-specific primers and 18s ribosomal RNA primer (an internal control) using an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA).
Protein analysis.
Lung nuclear (7–10 μg) and cytosolic protein (50–100 μg) were analyzed by routine Western blotting using specific antibodies against rodent mitochondrial oxidative phosphorylation (OXPHOS) complex (Abcam, Cambridge, MA), Nrf2 (Santa Cruz), p65 NF-κB (Santa Cruz), VDAC1/porin (Santa Cruz), phopholamban (PPLA, Santa Cruz), lamin B (Santa Cruz), and pan-actin (Santa Cruz). Scanned band intensities of Western blot images (n = 3/antibody) were quantitated by densitometry (ProteinSimple, San Jose, CA). Cytosol fractions were also analyzed for total GSH amount using a commercial kit (Cell Biolabs, Inc.) to detect NADPH/GSH reductase reaction. Briefly, glutathione reductase and NADPH were added to lung cytosol fraction to generate reduced glutathione (GSH) from its oxidized form (GSSG) in the samples, which was followed by incubation with the chromogen reacting with thiol group of GSH to produce a colored compound. Changes of absorbance at 405 nm were immediately measured by 1 min intervals for ~10 min and concentration from the slope of linear portion of each sample kinetic curve was acquired using the net slopes of GSSG standards. Binding activities of nuclear proteins (5 μg) on ARE and NF-κB motifs were determined by gel shift analysis following procedures described previously (Cho et al., 2015). Specific NF-κB binding activity was quantitated by a transcription factor ELISA (Active Motif, Carlsbed, CA). Aliquots of nuclear proteins (2.5 μg) were added to wells in a 96-well plate pre-coated with an NF-κB-binding oligonucleotide. After consecutive incubation with p65 NF-κB antibody and horseradish peroxidase (HRP)-conjugated secondary antibody, binding activity of p65 NF-κB was determined by colorimetric analysis. Cold probes (addition of extra NF-κB binding oligonucleotide) were tested for reaction specificity. Total lung homogenates in radioimmunoprecipitation assay (RIPA) buffer were prepared for Western blotting and detection of oxidized protein by ELISA (Cell Biolabs, Inc.) using an anti-dinitrophenylhydrazine antibody specific to derivatized protein carbonyl groups (Cho et al., 2013).
SFN metabolite detection in urine and sera.
Non-metabolized (unconjugated) SFN and major SFN metabolites, including SFN-GSH, SFN-cysteine, and SFN- NAC, in mouse urine and sera were analyzed by HPLC-mass spectrometry. Briefly, an aliquot (10 μl) of urine was diluted with 90 μl 5% methanol and 0.1% formic acid. The sample was centrifuged at 8° C and 80 μl of supernatant was transferred to an autosampler vial containing a low volume insert. An aliquot (30 μl) of serum was diluted with 270 μl of 0.1% formic acid and loaded onto Oasis HLB solid phase extraction cartridges. Cartridges were then washed with 1 mL of 0.1% formic acid, dried with a stream of nitrogen, and eluted with 2 mL of 9:1 acetonitrile:methanol containing 0.1% formic acid. Serum eluents were dried under vacuum and reconstituted in 50 μl of 5% methanol and 0.1% formic acid solution. The prepared sample (10 μl) was injected onto an ACE Excel 3 CN-ES 2.1 X 100 mm HPLC column with gradient separation at a flow rate of 250 μl/min. Electrospray ionization and selected reaction monitoring mass spectrometry were used for analyte quantitation.
Mitochondrial genome copy number.
Mitochondrial genome copy number was determined using droplet digital PCR (ddPCR) by amplification of a short mitochondria genome fragment at NADH dehydrogenase 6, mitochondrial (mt-Nd6) locus (13731–13831 bp). Briefly, total lung DNA (2 ng) in PCR mixture containing SYBR Green dye and 125 nM of primers (5’-ctcccaaaccatcaagattaattac-3’, 5’-gacttggggatctaactgattaatt-3’) were partitioned into approximately 20,000 individual nanoliter-sized water-in-oil droplets by Bio-Rad QX200 Automated Droplet Generator (Hercules, PA) followed by PCR reaction (C1000 Touch Thermal Cycler, Bio-Rad). The droplets were then read individually in QX200 Droplet Reader (Bio-Rad) and assigned as positive (1) or negative (0). The mitochondrial copy number calculated by the positive droplets was normalized to nuclear DNA copy number determined similarly by ddPCR (primers used for beta-globin locus 54021–54122: 5’-ttctgctcaaccttcctatcag-3’, 5’-ccaaactctagtcaacactcac-3’).
Bioinformatic search of potential ARE.
Potential ARE sequences for Nrf2 binding were determined in selected SFN-responsive genes (n = 193) involved in mitochondrial energy production and metabolism using a position weight matrix (PWM) statistical model (Cho et al., 2012).
Statistics.
Data are expressed as group mean ± standard error (SE). Two-way ANOVA followed by Student-Newman-Keuls test for a posteriori comparisons to evaluate the effect of genotype (Nrf2+/+ vs Nrf2−/−) and treatment (AIN/air vs SFN/Air vs AIN/hyperoxia vs SFN/hyperoxia) on lung injury phenotypes and molecular analyses (P < 0.05). BAL phenotype data from Nqo1−/− and Gpx2−/− mouse studies were also analyzed by two-way ANOVA followed by Fisher LSD Method for a posteriori comparisons (P < 0.05).
RESULTS
Nrf2-dependent acute lung injury prevention by dietary SFN
Consistent with our previous results (Cho et al., 2002a), Nrf2−/− mice developed severe hyperoxic lung injury compared to Nrf2+/+ mice after 48 h of continuous O2 exposure (Figs. 1–3). The SBE preparation used in the current study showed reproducible bioavailability of SFN (average 10–18% conversion of glucoraphanin to SFN and metabolites) in humans (Fahey et al., 2015; Fahey et al., 2012), and has been applied to various clinical studies (Egner et al., 2014; Heber et al., 2014; Kensler et al., 2012). Average glucoraphanin consumption was 21.35 μmol in 3.66g SBE diet/animal/day, which was equivalent to about 6 g of mature broccoli or 0.9 g broccoli sprouts (Fahey et al., 1997). Compared to control (AIN) diet, 14-day SBE diet significantly lowered hyperoxia exposure-induced lung neutrophil number and protein concentration in Nrf2+/+ mice (Fig. 1A) as assessed by BAL analysis. However, no significant diet effects were found on lung phenotypes in Nrf2−/− mice after hyperoxia (Fig.1A, note the differences in y-axis scales for Nrf2+/+ and Nrf2−/−). Lung LDH concentration, a marker of cell lysis, was increased by hyperoxia and SBE significantly reduced this effect Nrf2+/+ mice (Fig. 1B). Compared to AIN, SBE significantly inhibited body weight loss caused by hyperoxia in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 1C). AIN and SBE had no effect on lung histopathology in air-exposed mice (Fig. 2). Hyperoxia-induced key histopathologic changes (e.g., perivascular-peribronchial edema, inflammatory cell infiltration, mucous hypersecretion) in Nrf2+/+ mice were reduced by SBE pre-treatment compared to those with AIN (Fig. 2). Severe histopathologic phenotypes in Nrf2−/− mice by hyperoxia were only marginally improved by SBE compared to AIN (Fig. 2).
Fig. 1. Nrf2-dependent protective effect of dietary glucoraphanin (SBE) on hyperoxia (O2)-induced lung injury.
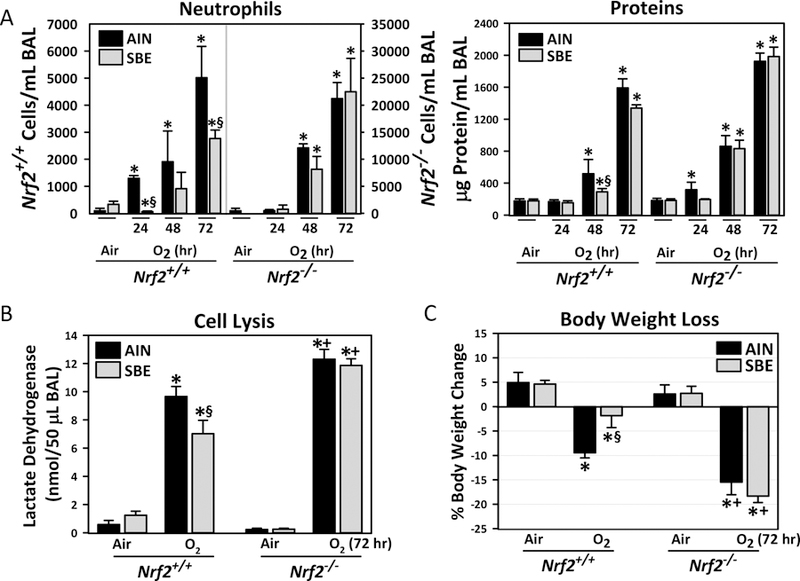
(A) Bronchoalveolar lavage (BAL) fluid analysis determined numbers of neutrophils for inflammation and total protein concentration for vascular permeability (n = 3/group for air and 48 h O2, n = 6/group for 72h O2), and (B) activity of lactate dehydrogenase (LDH) for cytotoxicity (n = 3/group for air, n = 4–5/group for O2). (C) Hyperoxia susceptibility determined by body weight loss was indicated as percent body weight change at the end of 72 h hyperoxia or air exposure (day 18) compared to the onset of the diet (day 1, n = 3/group for air, n = 6/group for O2). Data presented as group mean±SE. Two-way ANOVA used for all statistical analyses. *, P < 0.05 vs. genotype- and diet-matched air controls. +, P < 0.05 vs. diet- and exposure-matched Nrf2+/+ mice. §, P < 0.05 vs genotype- and exposure-matched AIN group.
Fig. 3. Nrf2-dependent protective effect of oral sulforaphane (SFN) on hyperoxia (O2)-induced lung injury.
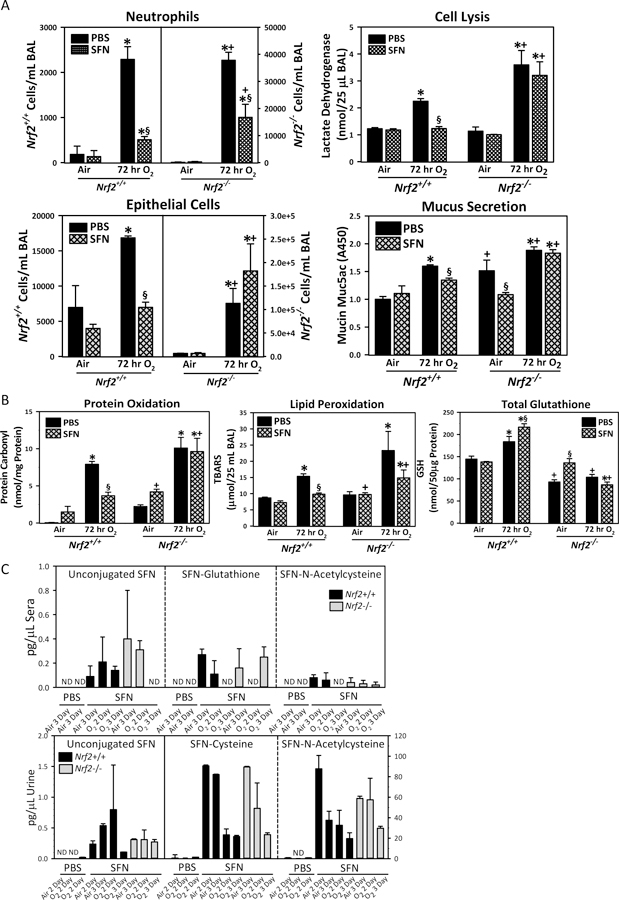
(A) Bronchoalveolar lavage (BAL) fluids were analyzed for the numbers of lung epithelial cells and neutrophils (n = 3/group for air, n = 6/group for O2), activity of lactate dehydrogenase (n = 3/group for air, n = 4/group for O2), and the amount of secreted mucin proteins (Muc5AC, n = 3/group for air, n = 5/group for O2). (B) Lung oxidative stress indicated as oxidized protein (protein carbonyl), lipid peroxidation (thiobarbituric acid reactive substances, TBARS) in BAL, and total glutathione (GSH) levels in lung homogenates. Data presented as mean ± SE (n = 3/group). Two-way ANOVA used for all statistical analyses. *, P < 0.05 vs. genotype- and pretreatment-matched air controls. +, P < 0.05 vs. pretreatment- and exposure-matched Nrf2+/+ mice. §, P < 0.05 vs. genotype- and exposure-matched PBS. (C) Major SFN metabolites including SFN-glutathione (GSH), SFN-cysteine, and SFN-N-acetylcysteine (NAC) as well as non-metabolized (unconjugated) SFN were determined in aliquots of diluted urine or deproteinated sera at 2 or 3 days after air or O2 exposure following oral dose of PBS or SFN by HPLC-mass spectrometry. Data presented as mean ± SE (n=3/group). ND=not detected.
Fig. 2. Nrf2-dependent mitigation of hyperoxia (O2)-induced pulmonary histopathology by dietary glucoraphanin (SBE).
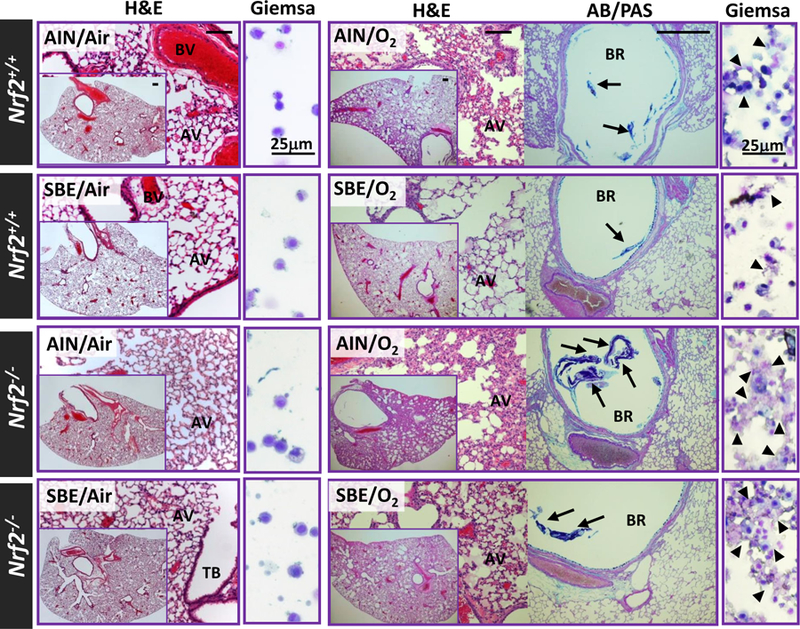
Representative light photomicrographs of lung tissue sections stained with H&E and AB/PAS or cytocentrifuged BAL cells stained with Giemsa after 14 days of normal (AIN) or standardized broccoli extract-containing diet (SBE) followed by 72 h exposure to normoxia (room air) or O2. AV, alveoli; BR, bronchi; BV, blood vessel; TB, terminal bronchiole. Arrows=secreted mucus stained. Arrow heads=lysed cells. Bars (unlabeled) = 100 μm.
Nrf2-dependent acute lung injury prevention by oral SFN
Oral administration of pure SFN (9 μmol/animal at 5-, 3-, and 1-day before start of inhalation exposure) significantly reduced BAL neutrophil and epithelial cell numbers as well as mucus hypersecretion (determined by measurement of Muc5AC) in hyperoxia-exposed Nrf2+/+ mice (Fig. 3A). The SFN dose was determined based on bioavailability of SFN metabolites in murine lungs reported in previous studies (Clarke et al., 2011). Hyperoxia-increased BAL neutrophils in Nrf2−/− mice were significantly lowered by SFN pretreatment, but no SFN effect was found in hyperoxia-induced lung cell death and mucus hypersecretion in these mice (Fig. 3A). Relative to vehicle (PBS), SFN significantly lowered the magnitude of hyperoxia-increased lung protein oxidation (determined by protein carbonyl) and lipid peroxidation (determined by thiobarbituric acid reactive substance, TBARS) in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 3B). Total lung glutathione level (reduced GSH form) was also significantly elevated by SFN in Nrf2+/+ mice after hyperoxia (Fig. 3B). Although SFN increased basal GSH level in Nrf2−/− lungs, the overall GSH pool was significantly lower after air or hyperoxia in Nrf2−/− mice compared to Nrf2+/+ mice (Fig. 3B).
SFN and metabolites were detected in mouse sera and urine until 3–4 days after the last oral SFN administration (Fig. 3C). In urine, primary metabolites were SFN-cysteine and SFN-NAC while the unconjugated SFN and SFN-GSH levels were negligible. Less than 1 pg/μl of unconjugated SFN, SFN-GSH, and SFN-NAC was detected but not SFN-cysteine in sera. No genotype or exposure effects were found on metabolite levels or composition.
Nrf2-dependent lung transcriptomics
1. SFN effects on air control mice
Supplemental SFN significantly altered the basal lung transcriptome in air-exposed Nrf2+/+ mice (n = 1,187, P < 0.01). Hierarchical clustering indicated these genes responded to SFN in Nrf2+/+ but not in Nrf2−/− mice (Fig. 4A). Transcripts were highly enriched for mitochondrial functions, OXPHOS, TCA cycle, substrate carriers and transporters, and biogenesis (Figs. 4B–C, Tables 1 and E1). Genes including NADH dehydrogenases (e.g., Ndufs1), cytochrome c oxidases (e.g., Cox7a1), isocitrate dehydrogenase 3 (NAD+) alpha (Idh3a), ATP synthases (e.g., Atp5g1), peroxisome proliferator activated receptor, gamma, coactivators (e.g., Ppargc1a), aconitase 2, mitochondrial (Aco2), and mitochondrial symporters (e.g., Mpc1) were largely upregulated (≤ 4-fold). In addition to nuclear-encoded mitochondria genes, SFN induced (≤ 32-fold) genes related to mitochondrial energy metabolism (Figs. 4B and 4D, Tables 1 and E1) including lipid-carbohydrate-amino acid catabolism (e.g., Adipoq, Fabp4, Acadl, Slc2a4) and cardiovascular-muscular contractility and calcium homeostasis including natriuretic peptide type A (Nppa), phopholamban (Pln), corin (Corin), and myosins (e.g., Myl3). SFN also significantly increased many antioxidant (e.g., Ephx2, Car3) and immunity genes (e.g., Ltf, Muc5b) in Nrf2+/+ mice (Tables 1 and E1). In contrast, SFN significantly decreased genes encoding tissue injury (e.g., Tnf receptor-associated factor 1, Traf1), oxidant production (e.g., NADPH oxidase 4, Nox4) and downstream targets of vitamin D3 receptor/retinoid X receptor (RXR) signaling in Nrf2+/+ lungs, and multiple mitogen-activated protein kinase (MAPK) cascade enzymes (e.g., Map3k8) and NF-κB were predicted to play central roles in their networks (Table E1, Fig. 4E).
Fig. 4. Nrf2-dependent changes of basal lung transcriptome by sulforaphane (SFN) pretreatment.
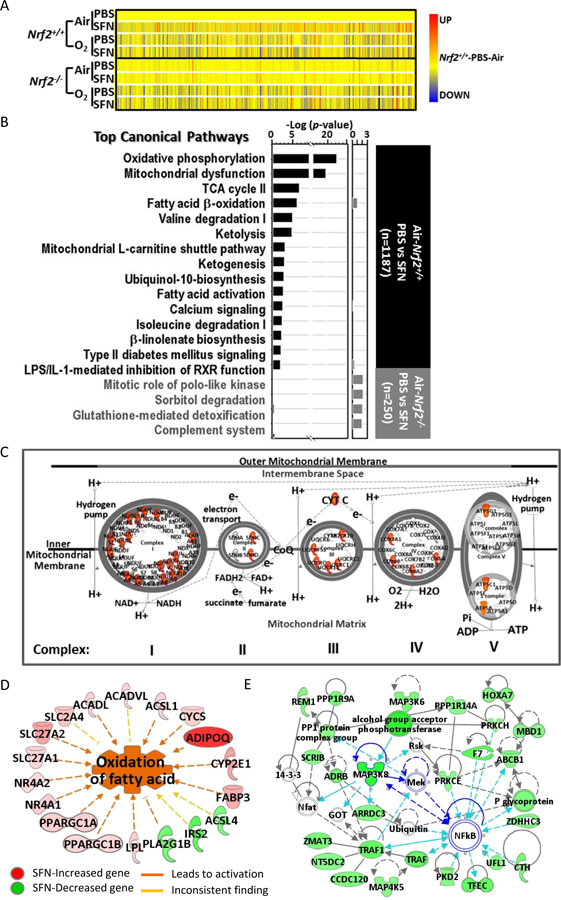
(A) Heat map from hierarchical clustering analysis depicts expression profiles of SFN-responded genes in Nrf2+/+ after 72 h hyperoxia (O2) exposure (n = 1,187, P < 0.01 with moderated t-test). Heat map for the same genes in Nrf2−/− mice were shown for comparison. Color bar indicates average expression intensity (n = 3/group) normalized to Nrf2+/+-PBS-Air group. (B) Top canonical pathways of lung genes significantly altered by SFN in air-exposed Nrf2+/+ (black bars) and in air-exposed Nrf2−/− mice (grey bars). (C) Mitochondrial oxidative phosphorylation complex is illustrated with genes (in red) that were induced by SFN treatment in Nrf2+/+ mice exposed to normoxia (room air). (D) Top diseases and bio-functions of SFN-responsive lung genes in Nrf2+/+ mice included energy metabolism such as fatty acid beta-oxidation. (E) Lung genes significantly reduced by SFN in Nrf2+/+ mice were involved in the network of organismal injury and abnormality (scores 32–41), in which key molecules such as TNF receptor associated factor 1 (Traf1) and multiple mitogen-activated protein kinase (MAPK) cascade enzymes (e.g., Map3k8) were predicted to play central roles with NF-κB. Analysis was done by Ingenuity Pathway Analysis software.
Table 1.
Representative lung genes induced by sulforaphane and potential antioxidant response elements (AREs).
| Category | RefSeq ID | †FI | Gene Symbol | Gene Title | Functions |
‡Potential AREs No: PWM |
|---|---|---|---|---|---|---|
| Mitochondrial Function & Energy Metabolism | NM_009605 | 8.72 | Adipoq* | adiponectin, C1Q and collagen domain containing | fat metabolism, glucose uptake and insulin sensitivity | 10: 6.9–10.6 |
| NM_024406 | 4.89 | Fabp4 | fatty acid binding protein 4, adipocyte | long chain fatty acid/ retinoic acid delivery | 10: 6.4–15.6 | |
| NM_146050 | 4.01 | Oit1 | oncoprotein induced transcript 1 | negative regulation of insulin secretion | 19: 6.4–13.5 | |
| NM_011978 | 3.50 | Slc27a2 | solute carrier family 27 (fatty acid transporter), member 2 | very long-chain fatty acid transporter | 14: 6.5–11.5 | |
| NM_009204 | 2.18 | Slc2a4 | solute carrier family 2 (facilitated glucose transporter), member 4 | glucose transmembrane transporter (uptake), GLUT4 | 4: 7.9–9.5 | |
| NM_007381 | 1.75 | Acadl* | acyl-Coenzyme A dehydrogenase, long-chain | fatty acid β-oxidation | 15: 6.8–14.2 | |
| NM_009949 | 1.52 | Cpt2* | carnitine palmitoyltransferase 2 | fatty acid β-oxidation | 9: 6.8–15.0 | |
| NM_146108 | 1.44 | Hibadh | 3-hydroxyisobutyryl-coenzyme A hydrolase | amino acid catabolic process | 6: 6.6–14.7 | |
| NM_011099 | 1.23 | Pkm2* | pyruvate kinase, muscle | glycolysis | 10: 6.8–15.1 | |
| NM_009944 | 4.05 | Cox7a1 | cytochrome c oxidase, subunit VIIa 1 | mt OXPHOS | 11: 6.9–14.2 | |
| NM_001161419 | 1.83 | Atp5g1* | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c1 (subunit 9) | mt OXPHOS | 9: 7.0–14.1 | |
| NM_008904 | 1.76 | Ppargc1a | peroxisome proliferative activated receptor, gamma, coactivator 1 alpha | mt biogenesis | 8: 6.6–9.5 | |
| NM_025567 | 1.68 | Cyc1 | cytochrome c-1 | mt OXPHOS | 7: 7.1–12.0 | |
| NM_001160038 | 1.63 | Ndufs1* | NADH dehydrogenase (ubiquinone) Fe-S protein 1 | mt OXPHOS | 11: 6.4–11.0 | |
| NM_026452 | 1.62 | Coq9* | coenzyme Q9 homolog (yeast) | mt OXPHOS | 10: 6.6–17.4 | |
| NM_023374 | 1.58 | Sdhb* | succinate dehydrogenase complex, subunit B, iron sulfur | mt OXPHOS | 12: 7.2–10.6 | |
| NM_025710 | 1.57 | Uqcrfs1 | ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | mt OXPHOS | 6: 6.7–12.8 | |
| NM_029573 | 1.55 | Idh3a* | isocitrate dehydrogenase 3 (NAD+) alpha | mt TCA cycle | 12: 6.5–14.9 | |
| NM_133201 | 1.41 | Mfn2 | mitofusin 2 | mt fusion | 4: 7.8–11.2 | |
| NM_018819 | 1.41 | Mpc1* | mitochondrial pyruvate carrier 1 | mt pyruvate transporter | 11: 6.4–16.5 | |
| Cardio-vascular Tone & Muscular Function | NM_008725 | 31.74 | Nppa | natriuretic peptide type A | blood pressure | 14: 6.4–11.8 |
| NM_001141927 | 10.12 | Pln | phospholamban | Ca2+ homeostasis | 9: 6.6–14.8 | |
| NM_001122756 | 7.76 | Corin* | corin | blood pressure | 12: 6.6–17.2 | |
| NM_009406 | 6.94 | Tnni3 | troponin I, cardiac 3 | Ca2+ homeostasis | 9: 7.1–12.0 | |
| NM_010859 | 5.90 | Myl3 | myosin, light polypeptide 3 | cardiac muscle contraction | 10: 6.8–14.1 | |
| Antioxidant & Defense | NM_008522 | 13.07 | Ltf | lactotransferrin | anti-microbial | 7: 7.0–11.8 |
| NM_028801 | 5.74 | Muc5b | mucin 5, subtype B, tracheobronchial | mucosal defense | 9: 6.5–14.6 | |
| NM_029803 | 5.06 | Ifi27l2a | interferon, alpha-inducible protein 27 like 2A | adipocytokine, mt biogenesis | 7: 7.2–11.4 | |
| NM_016771 | 4.76 | Sult1d1 | sulfotransferase family 1D, member 1 | metabolic process | 7: 8.1–10.4 | |
| NM_007940 | 4.36 | Ephx2 | epoxide hydrolase 2, cytoplasmic | lipid phosphatase | 10: 6.9–12.6 | |
| NM_007606 | 4.08 | Car3 | carbonic anhydrase 3 | antioxidant, muscle | 14: 6.7–13.1 | |
| NM_026183 | 4.02 | Slc47a1 | solute carrier family 47, member 1 | multidrug and toxic compound transporters | 9: 6.5–13.3 | |
| NM_010362 | 2.21 | Gsto1* | glutathione S-transferase omega 1 | L-ascorbic acid biosynthesis | 9: 7.8–13.7 | |
| NM_007452 | 1.34 | Prdx3* | peroxiredoxin 3 | antioxidant, mt | 11: 6.6–10.9 |
mt, mitochondria. OXPHOS, oxidative phosphorylation. PWM, position weight matrix. ARE, antioxidant response element.
Genes with ChIP-Seq data published by the Encyclopedia of Mouse DNA Elements Consortium (Mouse et al., 2012; Yue et al., 2014).
Fold increase in SFN/air vs. PBS/air in Nrf2+/+ mice.
Number of potential AREs and their PWM range. Expanded data for full gene list and ARE search in Supplementary Tables E1 and E3.
In Nrf2−/− lungs, SFN-modulated genes (n = 250, P < 0.01) were involved in inflammation and organismal injury pathways (Table E2). Chemokine (C-C motif) ligand 11 (Ccl11), Ccl20, and sorbitol dehydrogenase (Sord) are examples of genes affected by SFN only in Nrf2−/− mice. A limited number (n = 17) of genes (e.g., Myoz2, Gsto1) were modulated commonly by SFN in both genotypes, which indicates that SFN differentially influenced some pulmonary transcriptomics independent of Nrf2.
Selected SFN-induced genes related to mitochondria, energy metabolism, and cardiovascular function (n = 193) have multiple ARE-like motifs (PWM score of ≥ 6.4) for Nrf2 binding (Tables 1, E3). Some potential AREs have been functionally verified by chromatin immunoprecipitation followed by DNA sequencing (Mouse et al., 2012; Yue et al., 2014), supporting roles as Nrf2 downstream effectors.
2. Hyperoxia effects
As we previously reported (Cho et al., 2005), hyperoxia significantly altered lung transcriptomics in Nrf2+/+ mice (n = 7,162, P < 0.01). Pathway analyses suggested that p53, together with PDGF, coagulation factor 2 (F2), TNF, and Nrf2 were key upstream regulators for transcriptome changes by O2 (Fig. 5A, Table E4). A role for p53 in this ALI model is explained by its functions in anti-oxidant induction, mitochondrial energy metabolism, and co-regulation with the Keap1-Nrf2 pathway and DNA damage response (Bensaad and Vousden, 2007). During the development of hyperoxic lung injury, genes involved in extracellular matrix degradation [e.g., matrix metalloproteinase 3 (Mmp3), heparin-binding EGF-like growth factor (Hbegf)] and smooth muscle cell migration in response to vascular injury (as suggested by increased gene transcripts such as plasminogen activator, urokinase receptor, Plaur) may be activated by MAPKs, IL-1, or PDGF. Nrf2-mediated oxidative responses by well-known ARE-bearing antioxidants as well as Nrf2-dimerizing transcription factors (e.g., Mafk), protein ubiquitination and proteasomal degradation (e.g., ubiquitin-conjugating enzyme E2K, Ube2k), and phase 3 transporters (e.g., Apcc1) were the most affected pathways during hyperoxia exposure (Table E4), further supporting Nrf2 as an important susceptibility gene in this model. Hyperoxia also upregulated multiple amino acyl tRNA synthetases (e.g., seryl-tRNA synthetase, Sars) for tRNA charging and integrin linked kinase (ILK) signaling in Nrf2+/+ mice (Table E4). Coordinated transcriptional induction of sequestrome 1 (Sqstrm1) with vacuolar proton pumps (e.g., ATPase, H+ transporting, lysosomal such as Atp6v0a1), cathepsins (e.g., Ctsh), and vacuolar protein sorting proteins (e.g., Vps8) suggested lysosomal degradative function and autophagy was also triggered by hyperoxia in Nrf2+/+ mice (Perera et al., 2015).
Fig. 5. Nrf2-dependent lung transcriptome changes by hyperoxia (O2).
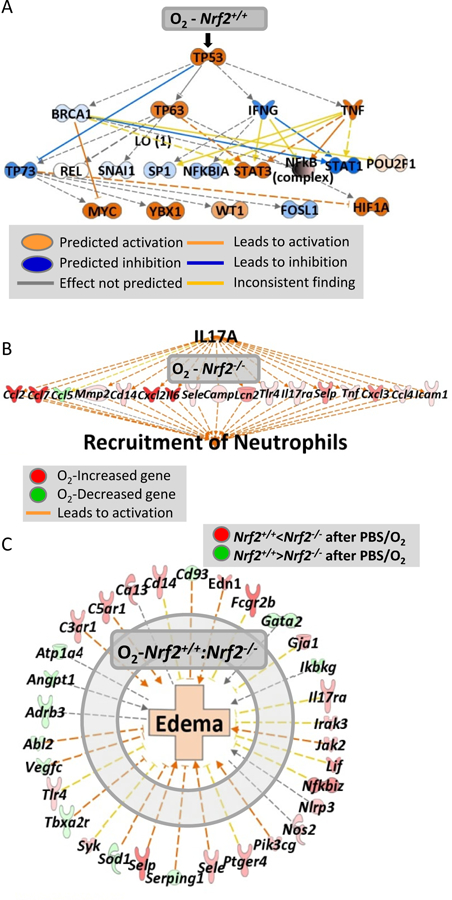
(A) Pathway analysis for hyperoxia-responsive genes in PBS-received Nrf2+/+ mice (n= 7162 genes, P < 0.01, Moderated t-test) demonstrated p53 as a key upstream regulator for the hyperoxia-altered lung genes, which may sequentially modulate other signal transducers. (B) In Nrf2−/− mice that received PBS, O2 altered genes (n = 4,799, P < 0.01) involved predominantly in IL-17A signaling pathway, which may lead to severe neutrophil infiltration. (C) Nrf2-dependently modulated genes during hyperoxia (n = 816, P < 0.01) such as Selp and Fcgr2b may contribute to the differential lung edema between Nrf2+/+ and Nrf2−/− mice given PBS.
In Nrf2−/− lungs, hyperoxia-induced transcriptome changes (n = 4,799, P < 0.01) were associated with acute and acquired immune responses and immune cell trafficking [e.g., serum amyloid A 3 (Saa3), Il6, Ccl7, Mmp3], and IL-17α, TNF, IL-1, NF-κB, and toll like receptors (TLRs) were suggested as upstream regulators of the hyperoxia-response genes in Nrf2−/− lungs (Fig. 5B, Table E5). Many of these hyperoxia-responsive genes in Nrf2−/− lungs were not affected in Nrf2+/+ lungs. Similar pathways were demonstrated by Nrf2-dependently altered genes during hyperoxia (n = 816, P < 0.01, Table E6). Overall analyses indicated that immune and inflammatory signaling through IL-17α and pattern recognition receptor-mediated signaling on downstream genes including selectin, platelet (Selp), CD14 antigen (Cd14), Ccl7, and Fc receptor, IgG, l low affinity IIb (Fcgr2b) correlate with more severe lung injury including protein edema in Nrf2−/− mice relative to Nrf2+/+ mice (Figs. 5C and 6A-top panel, Table E6).
Fig. 6. Nrf2-dependent lung transcriptome changes by sulforaphane (SFN) pretreatment during hyperoxia (O2) exposure.
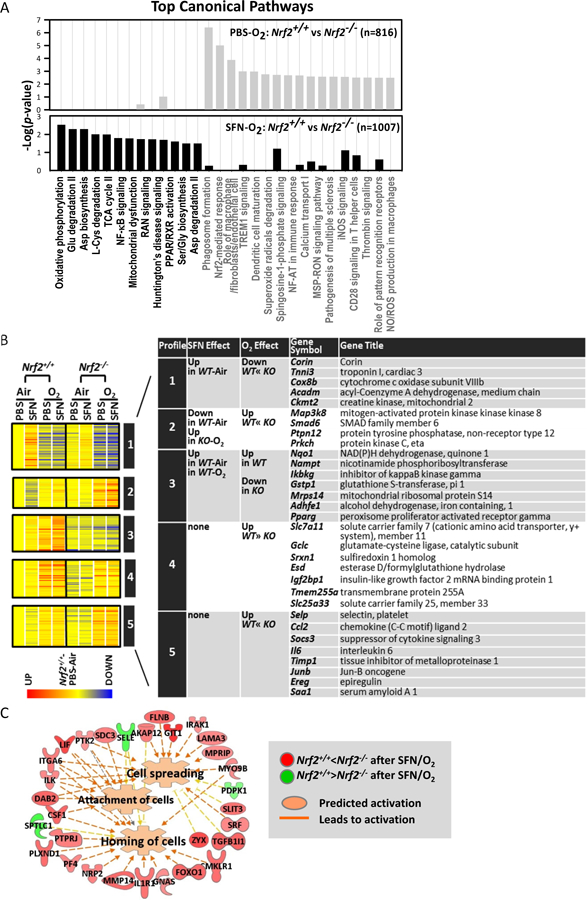
(A) Top canonical pathways of Nrf2-dependently changed gene transcripts with PBS (top, gray bars) or SFN (bottom, black bars) pretreatment in response to hyperoxia. (B) Profile analysis classified Nrf2-dependently regulated genes by similar expression patterns. (C) Pathway analysis for SFN-responsive genes in hyperoxia-exposed Nrf2−/− mice (n= 533, P < 0.01) depicted that genes altered by SFN only in these mice (e.g., Sele, Itga5, Lif, Flnb) may stimulate cellular movement and interaction by activating cell spreading, attachment, and homing, through which SFN may exert Nrf2-independent responses against hyperoxia in Nrf2−/− mice. Analyses were done using Ingenuity Pathway Analysis and GeneSpring software.
3. SFN and hyperoxia effects
In Nrf2+/+ mice, SFN pretreatment significantly altered 264 lung genes (P < 0.01, 2-Way ANOVA, Table E7) during ALI-like phenotype development by hyperoxia. IPA indicated that they were involved in protein post-translational modification (e.g., Ube2d3), vascular-endothelial development (e.g., Ddah1, Cdc16), and phospholipid signaling/metabolism (e.g., S1pr1), suggesting that SFN predisposed to regulatory protein degradation, cell membrane signaling, and endothelial integrity to counter pulmonary hyperoxia toxicity.
Comparative analysis between Nrf2+/+ and Nrf2−/− mice identified Nrf2-dependently changed genes by SFN during hyperoxia (n = 1,007, P < 0.01). Most of the mitochondrial function and energy metabolism genes (e.g., Ndufs1, Atp5b, Idh2, Acadl, Slc25a17, Dnaja3) remained consistently higher in SFN-treated Nrf2+/+ than in SFN-treated Nrf2−/− in response to hyperoxia (Table E8, Fig. 6A). This suggests their roles in SFN-Nrf2-mediated lung protection against hyperoxia. Genes encoding NF-κB (e.g., Traf3, Casp8) and PPAR/RXR (e.g., Il1r1, Nr2f1) signal transducers were also Nrf2-dependently expressed in SFN-treated lungs during hyperoxia (Table E8, Fig. 6A-bottom graph), which indicated that SFN inhibited lung inflammatory response in a Nrf2-dependent manner for lung protection. In addition, SFN heightened the expression of cancer (e.g., Fgfr1, Kit, Pdgfrb) and immunity (e.g., Prg4, Itga9) genes but suppressed metabolic antioxidant genes (e.g., Ces1g, Got1, Akr1b7) in Nrf2−/− mice relative to Nrf2+/+ mice after hyperoxia. Pearson and Spearman metric analyses (r > 0.95) clustered Nrf2-dependently modulated genes by similar expression profiles (Fig. 6B). Corin-like genes that were SFN-inducible in air-Nrf2+/+ lungs and suppressed by hyperoxia more significantly in Nrf2−/− mice than in Nrf2+/+ mice (Profile 1) were involved in cardiovascular tone and energy metabolism. Signal transducers including Map3k8 (Profile 2) were decreased by SFN in air-exposed Nrf2+/+ mice but induced by SFN in hyperoxia-Nrf2−/− mice. Nqo1-like genes that were upregulated by SFN and hyperoxia only in Nrf2+/+ mice (Profile 3) encoded antioxidants and mitochondrial/lipid metabolism proteins. Profiles 4 and 5 included solute carrier family 7 member 11 (Slc7a11)- or Selp-like genes modulated Nrf2-dependently by hyperoxia, but not by SFN.
In Nrf2−/− mice, hyperoxia- and SFN-responsive genes (n = 533, P < 0.01) were dissociated from those in Nrf2+/+ mice. This indicated that SFN triggered different pulmonary response mechanisms in the absence of Nrf2. Pathway analysis demonstrated that these Nrf2−/− unique genes (e.g., Sele, Itga5, Lif, Flnb) may stimulate cellular movement and interaction in hyperoxia-injured extracellular matrix and connective tissues by activating cell spreading, attachment, and homing (Fig. 6C), through which SFN may exert Nrf2-independent, mild protection against hyperoxia.
4. Validation of microarray data
Effect of SFN on mitochondrial proteins
Lung mitochondrial genome copy number determined by PCR method (Furda et al., 2014) was not significantly affected by genotype, exposure, or pretreatment (Table 2). Immunohistochemical localization of VDAC1 for mitochondrial membrane potential, a marker of mitochondrial function or biogenesis, was dense in alveolar macrophages, type 2 cells, and bronchial epithelium and smooth muscle as well as endothelium and vascular myocytes of pulmonary arteries in normal mice (PBS/Air), and little variation was found between two genotypes (Fig. 7A). In air-exposed control Nrf2+/+ mice, SFN increased VDAC1-positive cells in pulmonary arteries and terminal bronchioles, but not in Nrf2−/− mice. This observation was consistent with SFN-induced transcriptional induction of vascular-muscular genes (e.g., Nppa, Corin, Pln, Myl3) and muscle-related mitochondrial genes (e.g., Fabp3, Cox7a1) in air-Nrf2+/+ mice (see Fig. 4B). Hyperoxia reduced VADC1 staining in alveoli, arterial endothelium, and myocytes in both genotypes, indicating mitochondrial dysfunction or mitophagy in these cells. In contrast, hyperoxia increased VDAC1 proteins in injured small vessels (perivascular edema, hypertrophy) and hyperplastic bronchial/terminal bronchial epithelium in Nrf2+/+ mice. SFN pretreatment rescued hyperoxia-induced loss of alveolar VDAC1 and enhanced VDAC-positive cells in pulmonary artery and distal airway epithelium of Nrf2+/+ mice. Hyperoxia and SFN effect on VDAC1 expression was relatively marginal in Nrf2−/− mice.
Table 2.
Mitochondrial genome copy numbers determined by droplet digital PCR (ddPCR).
| Nrf2+/+ | Nrf2−/− | |||
|---|---|---|---|---|
| Pre-treatment | PBS | SFN | PBS | SFN |
| Air | 148.81 ± 10.53 | 255.31 ± 44.73 | 175.38 ± 17.39 | 190.78 ± 12.04 |
| Hyperoxia (72 h) | 168.48 ± 39.99 | 223.43 ± 27.10 | 141.54 ± 24.95 | 172.36 ± 51.76 |
Mitochondria copy number determined by comparing the abundance of mitochondrial DNA to nuclear DNA in 1 ng total DNA. Mean ± SE presented (n=3/group). SFN=sulforaphane.
Fig. 7. Validation of lung gene expression array analysis.
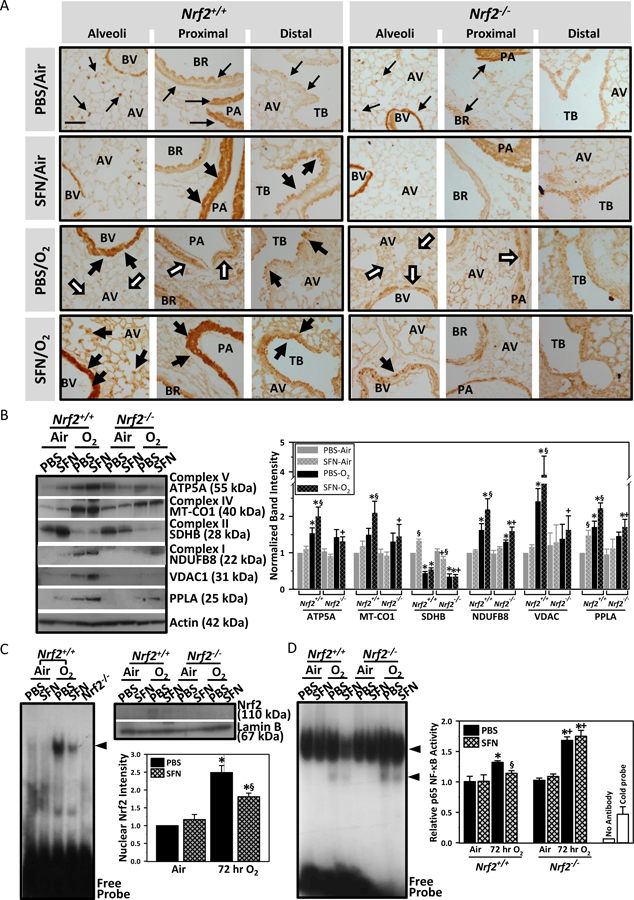
(A) Immunohistochemical localization of voltage-dependent anion-selective channel 1 (VDAC1)/porin, a mitochondrial membrane potential marker, in lung tissue sections. Brown dots indicate VDAC1-positive cells. Representative light photomicrographs are shown (n = 3–4/group). VDAC1 localization at baseline lung (PBS/Air) is indicated by small arrows. Thick black arrows indicate areas with increased VDAC1 expression compared to genotype-matched PBS/Air. White arrows indicate areas with decreased VDAC1 compared to genotype-matched PBS/Air. SFN = sulforaphane. O2 = Hyperoxia. AV, alveoli; BR, bronchi; BV, blood vessel; PA, pulmonary artery; TB, terminal bronchiole. Bar = 100 μm. (B) Aliquots of lung cytosolic proteins were subjected for Western blotting using specific antibodies. Representative images from multiple analyses of pooled proteins (n = 3/antibody) presented. ATP5A = ATP synthase subunit alpha, mitochondrial. MT-CO1 = mitochondrially encoded cytochrome c oxidase subunit 1. SDH8 = succinate dehydrogenase subunit B. NDUFB8 = NADH dehydrogenase (Ubiquinone) 1 beta subcomplex, 8. VDAC1 = Voltage-dependent anion-selective channel 1. PPLA=Cardiac phospholamban. kDa = kilodalton. Scanned band images were quantitated by densitometry. Data presented as group mean ± SE. Two-way ANOVA used for all statistical analyses. *, P < 0.05 vs. genotype- and pretreatment-matched air controls. +, P < 0.05 vs. pretreatment- and exposure-matched Nrf2+/+ mice. §, P < 0.05 vs. genotype- and exposure-matched PBS group. (C) Aliquots of pooled lung nuclear protein (5 μg) were incubated with an end-labeled oligonucleotide probe containing antioxidant response element (ARE) consensus sequence, and gel shift analysis determined total ARE binding. Nuclear proteins (5 μg) from PBS/hyperoxia-Nrf2−/− mice were run as a negative control. Nuclear proteins were subjected for Western blot analysis using Nrf2-specific antibody and images were quantified. *, P < 0.05 vs. pretreatment-matched air controls. §, P < 0.05 vs. PBS/hyperoxia group. (D) Aliquots of pooled lung nuclear protein (5 μg) were incubated with an end-labeled oligonucleotide probe containing NF-κB consensus sequence, and gel shift analysis determined total NF-κB binding. Two shifted bands (arrow heads) indicate total DNA-NF-κB complex. Specific activity for p65 NF-κB subunit was quantified using a transcription factor ELISA. Nuclear proteins from PBS/hyperoxia-Nrf2−/− mice were used for reaction with cold probes (20 pmol addition of oligonucleotide) and for no antibody control to verify the reaction specificity. *, P < 0.05 vs. genotype- and pretreatment-matched air controls. +, P < 0.05 vs. pretreament- and exposure-matched Nrf2+/+ mice. §, P < 0.05 vs. genotype- and exposure-matched PBS group.
Western blot analyses determined expression of OXPHOS complex, PPLA, and VDAC1 (Fig. 7B). Representative proteins from mitochondrial complexes I (NDUFB8), IV (MT-CO1 encoded by mitochondral genome), and V (ATP5A) subunits were increased by SFN and hyperoxia in Nrf2+/+ mice while succinate dehydrogenase complex, subunit B (SDHB) in complex II was increased by SFN but decreased after hyperoxia in Nrf2+/+ mice. SFN effects on these OXPHOS subunits were relatively marginal in Nrf2−/− mice.
Effect of SFN on redox transcription factors
Nuclear ARE binding activity and translocated Nrf2 were increased by hyperoxia exposure in Nrf2+/+ mice at 3 days after hyperoxia while SFN pretreatment reduced nuclear Nrf2 level compared to PBS, probably due to the lowered oxidative stress and injury by SFN (Fig. 7C). Lung NF-κB level was determined to be a key molecule of the transcriptome networks influenced by SFN (Fig. E4A). Total NF-κB-DNA binding activity and specific p65 NF-κB activity was increased by hyperoxia in both genotypes, with higher DNA binding activity in PBS-treated Nrf2−/− mice than PBS-treated Nrf2+/+ mice (Fig. 7D). SFN significantly decreased NF-κB-DNA binding activity in Nrf2+/+ mice but not in Nrf2−/− mice after hyperoxia, indicating Nrf2-dependent SFN inhibition of NF-κB activity (Fig. 7D).
Functional roles of SFN-inducible ARE antioxidant enzymes
ARE-bearing Nqo1 and Gpx2 were among downstream antioxidant enzymes elevated by both hyperoxia and SFN. Hyperoxia-induced increase in lung BAL neutrophils was significantly greater (48–72 h) in Nqo1−/− mice and significantly lower (72 h) in mice fed with NQO1-inducing DMF diet (Nqo1+/+DMF), compared to Nqo1+/+ mice (Fig. 8A). Numbers of BAL epithelial cells were significantly increased by hyperoxia (48–72 h) in Nqo1−/− mice, but not in Nqo1+/+ and Nqo1+/+DMF mice (Fig. 8A). BAL neutrophil (48 h) and epithelial cell (48–72 h) numbers were significantly higher in Gpx2−/− mice than in Gpx2+/+ mice after hyperoxia (Fig. 8B).
Fig. 8. Role of Sulforaphane-responsive antioxidant enzymes in hyperoxia-induced lung injury.
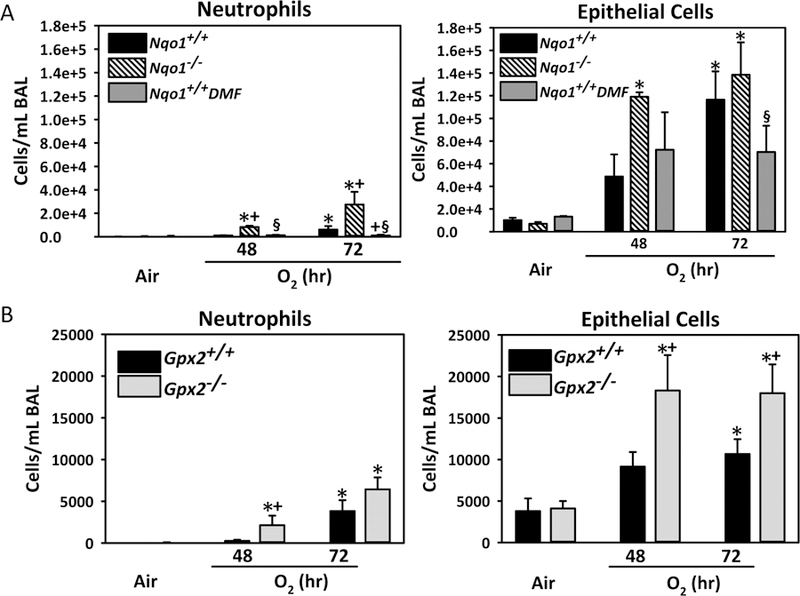
(A) Numbers of neutrophils and epithelial cells found in bronchopulmonary lavage (BAL) fluid from wild-type (Nqo1+/+), dimethyl fumarate (DMF)-fed wild-type (Nqo1+/+DMF), and Nqo1−/− mice after exposure to air and O2 (48–72 h). Data presented as group mean ± SE (n = 3/group for air, n = 5/group for 48 h O2, n = 7/group for 72 h O2). (B). Numbers of neutrophils and epithelial cells found in BAL fluid from Gpx2+/+ and Gpx2−/− mice after exposure to air and O2 (48–72 h). Data presented as group mean ± SE (n = 3/group for air, n = 6/group for 48 h O2, n = 10/group for 72 h O2). Two-way ANOVA used for statistical analyses. *, P < 0.05 vs. genotype and air controls. +, P < 0.05 vs. exposure-matched wild-type mice. §, P < 0.05 vs. exposure-matched Nqo1−/− mice.
DISCUSSION
We report in the current study that antioxidant SFN decreased susceptibility of mouse lungs to oxidant-induced ALI, and Nrf2 contributed to SFN-mediated protection. Genome-wide transcriptome analysis suggested that SFN may supplement substrates for mitochondrial function and energy metabolism in an Nrf2-dependent manner to enable the lung to reduce oxidant injury while Nrf2-deficiency may limit metabolic substrates and thus impair the mitochondria-mediated cellular processes. Our novel findings in vivo demonstrated prevention of ALI-like disorder by SFN that may have important clinical implications and provided molecular aspects of pulmonary SFN action.
Genetic polymorphisms in GSTM1 and GSTT1, which are Nrf2-inducible enzymes that convert SFN into SFN-GSH, affected SFN excretion and plasma levels as well as excretion of xenobiotic conjugates in humans who took SFN-rich dietary supplement (Egner et al., 2014; Gasper et al., 2005). This indicated correlation of NRF2 with bioavailability and efficacy of SFN. SFN efficacy has been widely investigated in murine models of various disorders (Dinkova-Kostova and Kostov, 2012), and Nrf2-dependent roles of SFN, particularly in inflammatory and infectious airway diseases, were reported using Nrf2+/+ and Nrf2−/− mice. Inhaled arsenic-induced lung injury was Nrf2-dependently inhibited by SFN (Zheng et al., 2012). We previously reported that orally administered SFN significantly lowered respiratory syncytial virus load and lung inflammation in Nrf2+/+ but not in Nrf2−/− mice (Cho et al., 2009). SFN also decreased bacterial growth on co-cultured alveolar macrophages isolated from chronic obstructive pulmonary disorder patients and inhibition of an Nrf2-regulated scavenger receptor decreased the phagocytic effects of SFN (Harvey et al., 2011). Airway hyperresponsiveness and inflammation by chlorine gas was also resolved by sulforaphane in Nrf2+/+ mice but not in Nrf2−/− mice (Ano et al., 2017). In a ovalbumin and cigarette smoke model of refractory asthma, sulforaphane restored steroid therapy sensitivity through activation of histone deacetylase 2 in an Nrf2-dependent manner (Sakurai et al., 2018).
Importantly, we determined that SFN markedly increased transcription of genes involved in mitochondrial respiration and energy metabolism and cardiovascular-muscle function in Nrf2+/+ lungs, but not in Nrf2−/− lungs. Transcription of mitochondrial OXPHOS complex, transporters/symporters, and TCA cycle enzymes was coordinately increased with transcription of enzymes in fatty acid β-oxidation (FAO), glycolysis, and amino acid degradation pathways which provide the OXPHOS chain with metabolic substrates and electrons (Fig. 9). SFN-mediated induction of the transcriptome for mitochondria and metabolism was manifest basally and expression of these genes was consistently greater in Nrf2+/+ lungs than in Nrf2−/− lungs after hyperoxia. Hyperoxia may cause cell- or region-specific changes in mitochondrial function and energy metabolism as predicted by focal changes of VDAC1 in Nrf2+/+ lungs. SFN also induced transcripts in mitochondrial biogenesis and homeostasis, Ppargc1a and Ppargc1b for mitochondrial reactive oxygen species (ROS) removal/metabolism and Mfn2 and Mtfp1 for mitochondrial fusion/fission. Overall, we postulate that SFN fortifies mitochondrial functions to protect lungs against oxidative insult. Recent studies have underscored a role for Nrf2 in induction of genes for FAO, NADPH generation, ATP synthesis, and mitophagy (Tebay et al., 2015). Nrf2 also increased mitochondrial biogenesis and autophagy in rodent lung and other tissues under stressed conditions via induction of heme oxygenase-1, superoxide dismutase 2 (SOD2), and PGC-1α (Chang et al., 2015). In animal models with high glucose availability or in cancer cells, Nrf2 redirected glucose metabolism from aerobic pyruvate/TCA cycle into anabolic pentose-phosphate pathway (Chartoumpekis et al., 2015).
Fig. 9. Potential mechanisms of Nrf2-dependent sulforaphane action in lung protection.
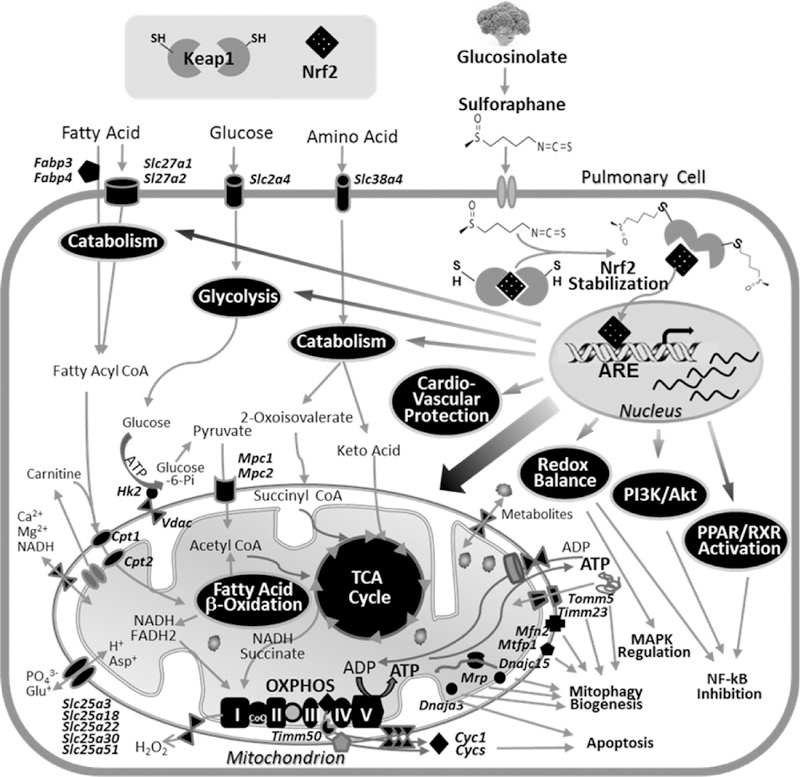
Dietary sulforaphane is known to stabilize cytoplasmic Nrf2 through Keap1 binding to inhibit Nrf2 ubiquitination for its proteasomal degradation. Stabilized Nrf2 may exert transcriptional activation of antioxidant response element (ARE)-responsive genes involved in redox regulation but also in fatty acid, glucose, and amino acid catabolism including enzymes and cellular transporters. Transcription activation then results in provision of metabolic substrates for mitochondrial machinery, tricarboxylic acid (TCA) cycle, fatty acid β-oxidation (FAO), and oxidative phosphorylation (OXPHOS) complex as well as mitochondrial symporters and transporters. Nrf2 may also directly upregulate ARE-bearing genes that encode these mitochondrial apparatus and mitochondrial quality control processes (i.e., mitophagy, biogenesis). Enhanced cyctochrome C (Cyc1) from OXPHOS is known to cause apoptosis, and ARE-responsive vasodilation and cardioprotective genes may protect pulmonary hypertension and vascular remodeling. Overall, sulforaphane-Nrf2 responses enrich mitochondrial bioenergetics to produce ATP on cellular demand but also facilitate other mitochondrial functions for cellular homeostasis against oxidative stress. Pathway analyses indicated that these Nrf2-dependently regulated lung genes are associated with pathways including peroxisome proliferator-activated receptor/retinoid X receptor (PPAR/RXR) activation, phosphatidylinositol 3- kinase (PI3K)/Akt signaling, which may modulate NF-κB and MAPK signaling cascade. Proteins and cellular processes predicted to be modulated by SFN-Nrf2 axis are highlighted in black.
Mitochondria DNA copy number is known to vary constantly depending on energy demands (Clay Montier et al., 2009). Different from genomic DNA, the mitochondrial genome is vulnerable to ROS damage probably in part due to the lack of histone protection and effective DNA repair mechanisms (Tatarenkov and Avise, 2007), and the copy number of mitochondrial DNA may be altered by damage, resulting in mitochondrial dysfunction. It has been therefore considered as a critical component of overall mitochondrial health, and has been related to aging and disease conditions such as cancer, cardiovascular diseases, neurodegenerative disorders, and infertility (Ashar et al., 2017; Yue et al., 2018). We did not find significant changes in pulmonary mitochondrial genome by hyperoxia or SFN at 3 days of hyperoxia exposure. Therefore, SFN action on mitochondria in the current system may be mainly through transcriptional activation of genomic DNA-encoded mitochondrial proteins. While Nrf2 and hyperoxia effects on mitochondrial DNA was not significant at the current time-frame of the experiments, further studies are warranted at early period of lung injury development by hyperoxia
Functional roles of Nrf2 in mitochondrial metabolism have been strongly supported in mice with altered Nrf2 expression (i.e., Nrf2-deficient, Keap1-knockdown). Proteomics approaches found differential hepatic lipid metabolism and mitochondrial enzymes in Nrf2+/+ and Nrf2−/− mice, and potential AREs were elucidated in Vdac1, ornithine aminotransferase (Oat), and Atp5a1 (Abdullah et al., 2012). Nrf2-deficiency lowered basal mitochondrial membrane potential and suppressed O2 consumption, FAO, and OXPHOS in various tissues and cells (Ludtmann et al., 2014). Augmented mitochondrial functions and upregulation of FAO genes were found in Keap1-knockdown mice after caloric restriction (Kulkarni et al., 2013; Ludtmann et al., 2014). Mitochondrial morphology and integrity change by H2O2 in rat cardiomyocytes were Nrf2-dependently preserved (Strom et al., 2016). Enhanced mitochondrial and total ROS production were apparent in Nrf2-deficient cortical slice and glio-neuronal co-culture, and the authors attributed these changes to the limited substrates and impaired activity of complex I in Nrf2−/− systems (Kovac et al., 2015). Nrf2-deficient neurons also had altered NADH and flavin adenine dinucleotide homeostasis coupled with reduced ATP production (Holmstrom et al., 2013). Nrf2-mediated mitochondrial quality control (copy number increase, mitophagy and biogenesis gene induction) was also linked with resolution of lung bacterial inflammation (Athale et al., 2012; Chang et al., 2015).
Mitochondria sense intracellular Ca2+ concentration for homeostasis, which is particularly critical in metabolically active myocardium and airway/vascular smooth muscles. Mitochondrial dysfunction is prominent in myocardial hypertrophy or cardiomyopathy, and a null mutation in adenine nucleotide translocator-1 (ANT1 or SLC25A4) was associated with the disease (Strauss et al., 2013). Mitochondrial compromise is also characteristic in pulmonary artery hypertension which is closely related to cardiac pathogenesis. Mitochondrial ROS and diffusible metabolites have been proposed to activate NLRP3 inflammasome and TLR9 leading to vascular remodeling and pulmonary hypertension (Sutendra and Michelakis, 2014). We reported that cardio-muscular Ca2+-pump regulator Pln, blood pressure-lowering Nppa, and natriuretic peptide-activating Corin were markedly induced by SFN only in wild-type mice, suggesting Nrf2-dependent SFN action on cardiovascular tone. Supporting this concept, Strom et al. (Strom et al., 2016) found increased sensitivity of mitochondria to swelling by Ca2+ in Nrf2−/− hearts compared to Nrf2+/+ hearts. A glucoraphanin-containing diet also ameliorated hypertension and atherosclerotic changes in spontaneously hypertensive, stroke-prone rats (Lopes et al., 2015). Taken together with potential AREs that we found in these genes, SFN may protect the cardiovascular system models through Nrf2-dependent induction of vasodilating and cardio-muscular Ca2+ handing genes.
In conclusion, results suggest a preventive role for SFN in development of ALI-like phenotypes in adult mice. These results may have important implications for the prevention of ALI in clinical settings where sepsis, blunt force trauma, and other disorders may lead to ALI or acute respiratory distress syndrome (e.g.(Marzec et al., 2007)). Molecular mechanisms underlying SFN action may be in part attributed to enhancing pulmonary mitochondrial integrity and dynamics thus diminishing oxidant toxicity. Differential transcriptome changes in Nrf2-sufficient and -deficient lungs imply that SFN action is primarily via Nrf2. NF-κB or MAPKs, central to multiple molecular networks of the Nrf2-dependently changed genes by SFN, may also be critical signal transducers in response to SFN. Our studies support the emerging concept of Nrf2-mitochondria linkage, particularly in association with potential intervention in asthma, ALI/ARDS, and chronic obstructive distress syndrome where transfer of stem cell or stem cell mitochondria to airway cells attenuated mitochondrial dysfunction and reduced pulmonary inflammation (Ahmad et al., 2014; Islam et al., 2012; Li et al., 2018; Morrison et al., 2017). While we examined effects of systemic SFN in the lung only at the peak of hyperoxic injury, additional examination of early molecular events and extra pulmonary (e.g., heart, liver) effects will add to our understanding of SFN mechanisms. Finally, it is important to note there is increasing evidence of genotoxicity (e.g., DNA mutation, DNA strand break, or DNA adduct formation) by broccoli extracts or SFN (Latte et al., 2011; Sestili et al., 2010) which warrants investigation of undesirable, off-target effects of SFN.
Supplementary Material
HIGHLIGHTS.
Sulforaphane prevented murine acute lung injury in an Nrf2-dependent manner.
Pulmonary sulforaphane activity was featured by linking mitochondria and Nrf2.
Potential ARE motifs were enriched in sulforaphane-induced energy metabolism genes.
Enhanced mitochondrial biogenesis conveyed airway defense against oxidant disorders.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the NIEHS, National Institutes of Health, Department of Health and Human Services. The authors thank. Mr. Herman Price for coordinating hyperoxia exposures at the NIEHS Inhalation Facility under contract to Alion Science and Technology, Inc. Microarray analysis was performed at the NIEHS Microarray Core, and Ms. Carolyn Favaro and Ms. Isabel Lea in the National Toxicology Program submitted array data to GEO and NIEHS CEBS. Drs. Richard S. Paules and Alison Harrill at the NIEHS provided excellent critical review of the manuscript.
LIST OF ABBREVIATIONS
- Aco2
aconitase 2, mitochondrial
- AIN
AIN-76A diet
- ALI
acute lung injury
- ANT1
adenine nucleotide translocator-1
- ARDS
acute respiratory distress syndrome
- ARE
antioxidant response element
- Atp
ATP synthase
- BAL
bronchoalveolar lavage
- CCL
Chemokine (C-C motif) ligand
- CEBS
Chemical Effects in Biological Systems
- Cox
cytochrome c oxidase
- ddPCR
droplet digital PCR
- DMF
dimethyl fumarate
- FAO
fatty acid β-oxidation
- Gpx2
glutathione peroxidase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GST
glutathione-S-transferase
- H. pylori
Helicobacter pylori
- HRP
horseradish peroxidase
- Idh3a
isocitrate dehydrogenase 3 (NAD+) alpha
- ILK
integrin-linked kinase
- IPA
Ingenuity Pathway Analysis
- Keap1
Kelch-like ECH-associated protein 1
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- Mmp3
matrix metalloproteinase 3
- Mpc
mitochondrial symporter
- Mt
mitochondria
- MT-CO1
mitochondrially encoded cytochrome c oxidase subunit 1
- Muc5ac
mucin 5, subtypes A and C
- NAC
N-acetylcysteine
- Nduf
NADH dehydrogenase (Ubiquinone)
- mt-Nd6
NADH dehydrogenase 6, mitochondrial
- Nox4
NADPH oxidase 4
- Nppa
natriuretic peptide type A
- NQO1
NAD(P)H:quinone oxidoreductase
- Nrf2
NF-E2 related factor 2
- 100% O2 or O2
hyperoxia
- Oat
ornithine aminotransferase
- OXPHOS
oxidative phosphorylation
- Pln, PPLA
phopholamban
- PPAR
peroxisome proliferator activated receptor
- Ppargc1a, PGC-1α
peroxisome proliferator activated receptor, gamma, coactivators
- PWM
position weight matrix
- RIPA
radioimmunoprecipitation assay
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- SBE
standardized broccoli sprout extract
- SDHB
Succinate dehydrogenase complex, subunit B
- Selp
selectin, platelet
- SFN
sulforaphane
- Slc7a11
solute carrier family 7 member 11
- SOD
superoxide dismutase
- Sord
sorbitol dehydrogenase
- TBARS
thiobarbituric acid reactive substances
- Tlr
toll like receptor
- VDAC1
voltage-dependent anion-selective channel protein 1
Footnotes
This article has an Online Data Supplement.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- Abdullah A, Kitteringham NR, Jenkins RE, Goldring C, Higgins L, Yamamoto M, Hayes J, Park BK, 2012. Analysis of the role of Nrf2 in the expression of liver proteins in mice using two-dimensional gel-based proteomics. Pharmacol. Rep PR 64, 680–697. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, et al. , 2014. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 33, 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano S, Panariti A, Allard B, O’Sullivan M, McGovern TK, Hamamoto Y, Ishii Y, Yamamoto M, Powell WS, Martin JG, 2017. Inflammation and airway hyperresponsiveness after chlorine exposure are prolonged by Nrf2 deficiency in mice. Free. Radic. Biol. Med 102, 1–15. [DOI] [PubMed] [Google Scholar]
- Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, Mychaleckyj JC, Taylor KD, Coresh J, Rotter JI, et al. , 2017. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol 2, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA, 2012. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free. Radic. Biol. Med 53, 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson AS, Tubbs E, Mecham B, Chacko S, Nenonen HA, Tang Y, Fahey JW, Derry JMJ, Wollheim CB, Wierup N, et al. , 2017. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med 9, eaah4477. [DOI] [PubMed] [Google Scholar]
- Begleiter A, Leith MK, Thliveris JA, Digby T, 2004. Dietary induction of NQO1 increases the antitumour activity of mitomycin C in human colon tumours in vivo. Br. J. Cancer 91, 1624–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Vousden KH, 2007. p53: new roles in metabolism. Trends. Cell. Biol 17, 286–291. [DOI] [PubMed] [Google Scholar]
- Bricker GV, Riedl KM, Ralston RA, Tober KL, Oberyszyn TM, Schwartz SJ, 2014. Isothiocyanate metabolism, distribution, and interconversion in mice following consumption of thermally processed broccoli sprouts or purified sulforaphane. Mol. Nutr. Food. Res 58, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Reynolds C, Brooker A, Talalay P, Fahey JW, 2015. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res 16, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnowski J, Hanske L, Schumacher F, Glatt H, Platz S, Rohn S, Blaut M, 2015. Glucosinolates Are Mainly Absorbed Intact in Germfree and Human Microbiota-Associated Mice. J. Agric. Food. Chem 63, 8418–8428. [DOI] [PubMed] [Google Scholar]
- Chang AL, Ulrich A, Suliman HB, Piantadosi CA, 2015. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic. Biol. Med 78, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoumpekis DV, Wakabayashi N, Kensler TW, 2015. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism. Biochem. Soc. Trans 43, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Gladwell W, Yamamoto M, Kleeberger SR, 2013. Exacerbated airway toxicity of environmental oxidant ozone in mice deficient in Nrf2. Oxid. Med. Cell. Longev 2013, 254069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Imani F, Miller-DeGraff L, Walters D, Melendi GA, Yamamoto M, Polack FP, Kleeberger SR, 2009. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med 179, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Gladwell W, Marzec J, McCaw ZR, Bienstock RJ, Kleeberger SR, 2015. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid. Redox Signal 22, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR, 2002a. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Resir. Cell Mol 26, 175–182. [DOI] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Zhang LY, Kensler TW, Kleeberger SR, 2002b. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am. J. Resir. Cell Mol. Biol 26, 42–51. [DOI] [PubMed] [Google Scholar]
- Cho HY, and Kleeberger SR, 2015. Association of Nrf2 with airway pathogenesis: lessons learned from genetic mouse models. Arch. Toxicol 89, 1931–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR, 2005. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free. Radic. Biol. Med 38, 325–343. [DOI] [PubMed] [Google Scholar]
- Cho HY, van Houten B, Wang X, Miller-Degraff L, Fostel J, Gladwell W, Perrow L, Panduri V, Kobzik L, Yamamoto M, et al. , 2012. Targeted Deletion of Nrf2 Impairs Lung Development and Oxidant Injury in Neonatal Mice. Antioxid. Redox Signal 17, 1066–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, Ho E, 2011. Metabolism and Tissue Distribution of Sulforaphane in Nrf2 Knockout and Wild-Type Mice. Pharm. Res 28, 3171–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay Montier LL, Deng JJ, Bai Y, 2009. Number matters: control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 36, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Kostov RV, 2012. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med 18, 337–347. [DOI] [PubMed] [Google Scholar]
- Egner PA, Chen JG, Zarth AT, Ng DK, Wang JB, Kensler KH, Jacobson LP, Munoz A, Johnson JL, Groopman JD, et al. , 2014. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: results of a randomized clinical trial in China. Cancer Prev. Res 7, 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Holtzclaw WD, Wehage SL, Wade KL, Stephenson KK, Talalay P, 2015. Sulforaphane Bioavailability from Glucoraphanin-Rich Broccoli: Control by Active Endogenous Myrosinase. PLoS One 10, e0140963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Wehage SL, Holtzclaw WD, Kensler TW, Egner PA, Shapiro TA, Talalay P, 2012. Protection of humans by plant glucosinolates: efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res 5, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Zhang Y, Talalay P 1997, Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. U.S.A 94, 10367–10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furda A, Santos JH, Meyer JN, Van Houten B, 2014. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol 1105, 419–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper AV, Al-Janobi A, Smith JA, Bacon JR, Fortun P, Atherton C, Taylor MA, Hawkey CJ, Barrett DA, Mithen RF, 2005. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr 82, 1283–1291. [DOI] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, Biswal S, 2011. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med 3, 78ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber D, Li Z, Garcia-Lloret M, Wong AM, Lee TY, Thames G, Krak M, Zhang Y, Nel A, 2014. Sulforaphane-rich broccoli sprout extract attenuates nasal allergic response to diesel exhaust particles. Food Funct 5, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson CM, Matthay MA, 2018. Endothelial biomarkers in human sepsis: pathogenesis and prognosis for ARDS. Pulm. Circ 8, 2045894018769876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT, Abramov AY, 2013. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2, 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J, 2012. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nature Med 18, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW, Talalay P, 2013. Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Top. Curr. Chem 329, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Ng D, Carmella SG, Chen M, Jacobson LP, Munoz A, Egner PA, Chen JG, Qian GS, Chen TY, et al. , 2012. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 33, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov AY, 2015. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 1850, 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni SR, Armstrong LE, Slitt AL, 2013. Caloric restriction-mediated induction of lipid metabolism gene expression in liver is enhanced by Keap1-knockdown. Pharm. Res 30, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latte KP, Appel KE, Lampen A, 2011. Health benefits and possible risks of broccoli - an overview. Food Chem. Toxicol 49, 3287–3309. [DOI] [PubMed] [Google Scholar]
- Li X, Michaeloudes C, Zhang Y, Wiegman CH, Adcock IM, Lian Q, Mak JCW, Bhavsar PK, Chung KF, 2018. Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J. Allergy Clin. Immunol 141, 1634–1645 e1635. [DOI] [PubMed] [Google Scholar]
- Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM, 2015. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 66, 1240–1250. [DOI] [PubMed] [Google Scholar]
- Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT, 2014. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J 457, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, Aplenc R, Yamamoto T, Yamamoto M, Cho HY, et al. , 2007. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J 21, 2237–2246. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Howard JP, 2012. Progress in modelling acute lung injury in a pre-clinical mouse model. Eur. Respir. J 39, 1062–1063. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, Zimmerman GA, 2012. The acute respiratory distress syndrome. J. Clinic. Invest 122, 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, Krasnodembskaya AD, 2017. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med 196, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse EC, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, et al. , 2012. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 13, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. , 2015. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Morishima Y, Ishii Y, Yoshida K, Nakajima M, Tsunoda Y, Hayashi SY, Kiwamoto T, Matsuno Y, Kawaguchi M, et al. , 2018. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free. Radic. Biol. Med 129, 473–485. [DOI] [PubMed] [Google Scholar]
- Sestili P, Paolillo M, Lenzi M, Colombo E, Vallorani L, Casadei L, Martinelli C, Fimognari C, 2010. Sulforaphane induces DNA single strand breaks in cultured human cells. Mut. Res 689, 65–73. [DOI] [PubMed] [Google Scholar]
- Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. , 2013. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. U.S.A 110, 3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom J, Xu B, Tian X, Chen QM, 2016. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J 30, 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G, Michelakis ED, 2014. The metabolic basis of pulmonary arterial hypertension. Cell Metab 19, 558–573. [DOI] [PubMed] [Google Scholar]
- Tatarenkov A, Avise JC, 2007. Rapid concerted evolution in animal mitochondrial DNA. Proc. Biol. Sci 274, 1795–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD, 2015. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free. Radic. Biol. Med 88, 108–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, Holbrook JT, Criner G, Sethi S, Rayapudi S, Sudini KR, Sugar EA, Burke A, Thimmulappa R, Singh A, et al. , 2016. Lack of Effect of Oral Sulforaphane Administration on Nrf2 Expression in COPD: A Randomized, Double-Blind, Placebo Controlled Trial. PLoS One 11, e0163716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaka A, Fahey JW, Fukumoto A, Nakayama M, Inoue S, Zhang S, Tauchi M, Suzuki H, Hyodo I, Yamamoto M, 2009. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res 2, 353–360. [DOI] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. , 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Yang X, Ning P, Xi X, Yu H, Feng Y, Shao R, Meng X, 2018. A mitochondria-targeted ratiometric two-photon fluorescent probe for detecting intracellular cysteine and homocysteine. Talanta 178, 24–30. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Tao S, Lian F, Chau BT, Chen J, Sun G, Fang D, Lantz RC, Zhang DD, 2012. Sulforaphane prevents pulmonary damage in response to inhaled arsenic by activating the Nrf2-defense response. Toxicol. Appl. Pharmacol 265, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.