

Abstract
The combination of next-generation sequencing and advanced computational data analysis approaches has revolutionized our understanding of the genomic underpinnings of cancer development and progression. The coincident development of targeted small molecule and antibody-based therapies that target a cancer’s genomic dependencies has fuelled the transition of genomic assays into clinical use in patients with cancer. Beyond the identification of individual targetable alterations, genomic methods can gauge mutational load, which might predict a therapeutic response to immune-checkpoint inhibitors or identify cancer-specific proteins that inform the design of personalized anticancer vaccines. Emerging clinical applications of cancer genomics include monitoring treatment responses and characterizing mechanisms of resistance. The increasing relevance of genomics to clinical cancer care also highlights several considerable challenges, including the need to promote equal access to genomic testing.
Increased levels of precision are being achieved in the clinical care received by patients with cancer by including cancer genomics in diagnostic medicine. Over the past 8 years, the application of massively parallel or next-generation sequencing (NGS) to large-scale cancer genomics discovery projects has revealed extraordinary new information about the underlying genomic drivers of cancer development and progression across multiple anatomical locations. NGS and various analytical approaches are now being introduced into clinical practice to better inform the clinical care of patients with cancer. In this Review, various aspects of the clinical translation of cancer genomics are considered. In particular, genomics-based assays are increasingly being used to guide the selection of the most appropriate targeted therapies for patients according to the genomes of their tumours and nonmalignant cells, and the findings of various studies have demonstrated that these assays provide a clinical benefit in terms of improved patient outcomes. Genomics-based assays also have the potential to inform the use of immunotherapeutic agents, thus broadening their potential clinical applicability. The resulting ‘big data’ obtained from preclinical discovery and clinical application should improve data mining efforts and further improve our understanding of cancer vulnerabilities, enable data integration approaches that combine genomic and clinical data, and improve our ability to predict the most effective therapies for patients in a scalable manner. However, several challenges exist to both the successful clinical implementation of NGS assays and the analysis and management of the data they provide, including pertinent aspects of data privacy and data sharing (Box 1) that are important to acknowledge. All of these aspects can influence the progress of genomics-guided cancer medicine and ultimately will determine the extent of integration of genomics into clinical care.
Key points
Genomic assays that enable the characterization of the somatic and germline defects in individual tumour samples are increasingly being used in clinical diagnostics as a means of identifying therapeutic options.
Many technical and cost-associated considerations have a role in decision-making processes regarding the implementation of cancer genomics assays into clinical practice.
Genomic methods can reveal individual targetable alterations, mutational load, complex mutation signatures, and tumour-specific antigens, which might inform the utilization of targeted therapies, immune-checkpoint inhibitors, and personalized anticancer vaccines.
The occurrence of shared targetable alterations across diverse tumour types has prompted new paradigms in the application of genomic profiling and the design of clinical trials.
These assays increasingly provide information that is pertinent to clinical cancer care, although several important attendant challenges surround their implementation.
NGS assays of cancer samples
Challenges and considerations.
The application of NGS technologies to the characterization of human tumours has provided unprecedented opportunities to understand the biological basis of different cancer types, develop targeted therapies and interventions, discover genomic biomarkers of drug response and resistance, and to guide clinical decision-making regarding the treatment of patients1,2. Furthermore, the versatility of NGS assays in addition to the diversity of upstream sample preparation methodologies has enabled the characterization of cancer genomes, transcriptomes, and epigenomes3. NGS can reveal sequence mutations, small insertions and deletions, copy number alterations, structural rearrangements, and loss of heterozygosity in tumour DNA samples. Sequencing of tumour-derived RNA enables the identification of differentially expressed genes, gene fusions, small RNAs, aberrantly spliced isoforms, and allele-specific expression patterns. Chemical modifications of DNA and/or histones, and changes in higher-order chromatin structure can also be mapped with increasing levels of precision. The algorithmic analysis of data from multiple NGS-based assays, in addition to the intrinsic genetic complexity of cancer, poses a major challenge to the clinical interpretation of NGS data. Not only does every class of alteration require a distinct computational approach for detection, but widespread copy number alterations and intratumoural heterogeneity (as observed in multiclonal tumours) might additionally lead to reduced mutant allele frequencies and, therefore, decreased levels of detection sensitivity. As such, NGS approaches used in cancer diagnostics typically demand a high depth of sequence coverage in order to increase the likelihood of detecting mutations that occur in a small fraction of cancer cells4.
Sample-specific challenges.
Technical challenges related to the quantity and quality of tumour tissue samples can further complicate the NGS-based characterization of cancer genomes. While attempts to sequence the genomes of nonmalignant cells benefit from ample sources of fresh cells, such as peripheral blood and buccal swabs, tumour samples often provide only small amounts of genetic material, particularly when collected as biopsy samples or fine-needle aspirates. Such samples might also harbour low levels of tumour cellularity owing to the infiltration of non-malignant cells (such as immune cells, stromal cells, or vascular endothelial cells), leading to a loss of signal from somatic mutations and therefore a loss of sensitivity of detection. Additionally, although analyses of fresh-frozen tumour specimens can yield high-quality nucleic acids, most tumours continue to be processed and stored as formalin-fixed, paraffin-embedded (FFPE) tissue blocks. This tissue preservation process was established long ago to enable the histopathological analysis and room temperature archiving of clinical specimens. However, the formalin fixation process typically leads to the fragmentation and chemical modification of DNA5,6, which might affect the achievable depth of coverage and can elevate certain types of false positive results owing to DNA damage. Early NGS research initiatives, including The Cancer Genome Atlas, mitigated against these issues by restricting their focus to large, high-quality, high-purity, frozen tumour samples7,8. However, clinical sequencing assays in molecular pathology laboratories must best utilize whatever FFPE specimens are available from a given patient no matter how small or impure these specimens might be. Fortunately, advances in sample preparation methodologies have led to reductions in the minimum DNA input requirements and have made FFPE samples more amenable to NGS analysis9,10. Several research groups have examined the effects of many pre-analytical factors on the performance and accuracy of NGS-based mutation profiling in an effort to maximize the reproducibility, sensitivity, and specificity of these assays11,12. Despite these technical challenges, the findings of validation studies have reaffirmed that NGS-based methodologies provide highly robust and reproducible data, even when quality-controlled FFPE-derived DNA is being assayed, and that NGS-based assays satisfy the reproducibility and accuracy requirements of regulatory guidelines and those of agencies governing clinical laboratories, such as the Clinical Laboratory Improvement Amendments (CLIA) and the College of American Pathologists (CAP)13–17.
NGS-specific challenges.
When to pursue a comprehensive versus a more-targeted approach to characterization is a key consideration regarding the use of NGS-based diagnostic cancer assays. Comprehensive approaches such as whole-genome, whole-exome, and whole-transcriptome sequencing provide ample opportunity for discovery and can reveal the full spectrum of oncogenic alterations in a given tumour10,18,19. However, these approaches typically have higher computational requirements and longer turnaround times, therefore also incurring higher costs than their more-targeted alternatives. Thus, owing to these practical considerations, analyses involving smaller gene panels that encompass dozens to hundreds of genes with established clinical or biological relevance to cancer are a preferred strategy of many molecular pathology laboratories15,20. By enabling high-throughput testing in a manner that is compatible with the analysis of low-quality specimens and limited amounts of input DNA, targeted panels can be used to screen larger populations of patients who might potentially benefit from the detection of clinically actionable mutations. Furthermore, the deeper sequence coverage enabled by targeted sequencing increases the sensitivity for critical mutations in the setting of heterogeneous (multiclonal) or low-purity tumour samples. Nevertheless, whole-genome and/or whole-exome sequencing approaches enable the detection of greater total numbers of mutations and thus provide a more accurate indication of intratumoural heterogeneity and the presence of specific mutation signatures and of tumour-specific neoantigens, which all might be clinically informative21,22. However, the clinical significance of the various additional alterations revealed by these more-comprehensive approaches is often unknown. Similar trade-offs in breadth versus depth of sequencing and throughput also exist for RNA sequencing-based approaches, especially following the availability of targeted RNA sequencing applications for the detection of gene fusions and for the profiling of small panels of cancer-associated genes23.
Following the adoption of targeted NGS panels for tumour profiling by various clinical and research laboratories, a proliferation of different strategies involving a variety of different technologies has occurred, including the development of gene-content and computational analysis pipelines for the detection and reporting of clinically relevant somatic mutations. Selected regions of interest can be enriched from genomic DNA using two main methods: amplicon capture and hybridization capture. Amplicon capture, involving multiplexed amplification using PCR, enables deep-coverage sequencing from small quantities of input DNA with a rapid turnaround time for high-sensitivity mutation calling in a limited number of genes or mutational ‘hotspot’ regions16. By contrast, hybridization capture, which involves annealing of complementary DNA or RNA probes to target regions, can scale to larger numbers of genes than can be analyzed using amplicon capture or even up to the whole exome24–26. Hybridization-capture panels can also provide more-precise assessments of copy number alterations by adding evenly spaced probes across each chromosome and can enable the detection of known structural rearrangements by including probes that correspond to specific non-coding sequences where genomic breakpoints tend to occur20. The appeal of large hybridization-based panels has been driven by the desire to detect multiple classes of sequence and/or structural alterations, the presence of rare and common alleles across entire genes (rather than just in hot spots), and complex genomic features such as hypermutation and/or microsatellite instability in a single, uniform assay. Ultimately, decisions regarding the breadth, content, and detection methodologies of targeted NGS panels must be made by individual clinical laboratories. These decisions are often focused on the definition of clinical actionability. Considerations of throughput, turnaround time, and cost per assay, as well as the interpretability of increasingly complex results in the clinical context, are all of equal importance. Finally, insurance reimbursement for these assays is an ever-evolving issue that must be considered carefully.
Sequencing a nonmalignant comparator.
The inclusion of patient-matched nonmalignant DNA is an important component of many large sequencing panels and of all comprehensive assay types. Nonmalignant comparators are typically obtained from peripheral blood samples or occasionally derived from nonmalignant tissue samples such as skin cells, buccal swabs, or fibroblasts. This matched nonmalignant DNA serves as a control to help distinguish somatic mutations from inherited germline variants. In the absence of a nonmalignant DNA sample, variants identified from tumour sequencing must be filtered and prioritized according to databases of recurrent somatic and germline variations, thus increasing the likelihood of false positive and/or false negative mutation calls in platforms extending beyond well-characterized mutation hotspots21,27. Furthermore, matched sequencing of nonmalignant DNA samples can lead to the direct identification of pathogenic germline variants that increase cancer susceptibility in patients’ family members and, in some scenarios, might also influence the chances of a response to certain therapies, such as poly(ADP-ribose) polymerase (PARP) inhibitors28–30. With the increasing availability of evidence that germline susceptibility is more prevalent than previously understood across very large cohorts of both paediatric and adult patients with cancer29,31,32, the inclusion of a germline comparator in the clinical assay is becoming imperative, although this approach is not without attendant challenges. The possibility of incidental findings relating to an inherited susceptibility to cancer and/or other diseases has led institutions to consider different strategies concerning genetic counselling both before and after testing and to establish appropriate mechanisms for returning results in a manner that protects patients’ autonomy, privacy, and wellbeing33–35. Finally, when blood is used as the source of nonmalignant DNA, mutations indicative of clonal haematopoiesis can be detected. The clinical significance of such mutations remains an important unanswered question both for patients with cancer and for individuals without cancer36–40.
Computational analysis challenges.
Beyond important considerations about assay design and scope, the corresponding decisions regarding the computational data analysis pipeline that must accompany the NGS assay are similarly important. In general, as the NGS data that can be routinely generated increase in both complexity and scope (from panels of genes to the entire genome), the different types of genomic alterations that might be detected and, accordingly, the numbers of different algorithmic examinations of the data required to identify each type become equally more complex. Similarly, the time required to thoroughly review and interpret each variant of each type according to its veracity and implicated therapeutic, diagnostic, or prognostic implications also increases. One ramification of these questions of assay scope and likely turnaround time has led to variations in the implementation of NGS testing at different cancer treatment centres. For example, some laboratories aim to return information from the NGS assay in a clinically relevant time frame so that any information relating to either treatment, diagnosis, or prognosis can be considered by the oncologist as a component of medical evidence for determining the treatment of that patient. Alternatively, other centres aim to investigate the primary diagnostic biopsy sample or resection sample in a more relaxed time frame, in which any treatment-relevant information is included in the patient’s medical record and then taken into consideration if and/or when the patient develops a recurrent or metastatic cancer. In the latter approach, the relaxed time frame might better permit an increased scale of testing and appreciates two facts of cancer care: most patients are not eligible for alternatives to the standard-of-care approach until treatment with that standard fails and they have relapsed disease, and routinely obtaining a biopsy sample from a patient presenting with metastatic cancer that can help determine new options for the patient is often very difficult. Thus, having data from the primary diagnosis can provide an important source of evidence to guide future treatment decisions. In patients with a biopsy sample of a relapsed tumour available, of course, this is an optimal assay sample for the purposes of identifying a specific alternative treatment approach, but more rapid turnaround would be required for this information to be clinically utilized.
Challenges in variant interpretation.
NGS assays designed to explore the mutation status of cancer-associated genes beyond specific hotspot mutation loci reveal variants whose implications, either for the resulting protein function or the response to targeted therapy, have not yet been characterized. Obviously, sharing data on the type and frequency of these variants, once known, is imperative to moving the field forward. Data sharing is particularly important in improving the treatment of patients with rare cancer types, including those with paediatric cancers. Genomic data are most valuable in patients when the genes and variants identified are linked with clinical data, including the type and number of previous treatments, previous responses to treatment and their duration, and other clinicopathological characteristics41–43. As such, proper consent must be obtained from all patients before their data can be made publicly available as fully anonymized information, including outlining the mechanism of data release and the planned efforts to maintain data privacy. Further challenges remain in terms of storage and presentation of these large data sets in a fashion that enables big-data queries, permits cross-comparisons, and otherwise facilitates easy access to specific data by researchers and clinicians (Box 1).
RNA as an analyte.
An increasing appreciation that assaying RNA from the tumour (in addition to DNA) can provide information that is important to clinical decision-making is an emerging complication in the spectrum of NGS assays. In particular, focused RNA-based assays designed to detect established gene-fusion partners are being implemented owing to both the difficulties associated with accurately identifying gene-fusion partners in DNA (breakpoints can often occur in different introns) and the increasing numbers of available small-molecule inhibitors that inhibit the resulting fusion proteins. Similarly, in tumours with a high mutational load, data from RNA sequencing can help to identify the mutated genes that are driving tumour progression or, more generally, can be interpreted to evaluate the biology of the tumour itself in terms of the cancer subtype, aggressiveness, potential to metastasize, and other biological aspects. However, the use of RNA-based assays (in addition to DNA-based assays) adds complexity, analysis time, and costs. Furthermore, because RNA is a more labile molecule than DNA, it requires a higher level of care during handling, including the use of preservation methods and quality assessments before conducting an assay.
Expanding assay scopes.
Importantly, the scope of clinical utility of NGS-based cancer assays is beginning to expand substantially. Initially, these tests revealed the presence of therapeutic targets in an era in which any cancer in any anatomical location had some chance of carrying pathogenic alterations in one or more proteins for which targeted therapies might be available, either as FDA-approved agents or in clinical trials. The information provided by NGS has become more valuable to therapy-related decision making, as we now better understand both the co-occurrence and mutual exclusivity of mutations in genes associated with cancer (FIG. 1). With data from increasing numbers of correlative studies available that link mutational profiles with outcomes, the prognostic value of data from an NGS-based assay will continue to increase. For example, mutational load is associated with a response to immune-checkpoint inhibition44–46, and a high mutational load can indicate the presence of inherited or somatically acquired DNA mismatch repair defects, which also has therapeutic implications. However, attendant challenges to the use of NGS-based cancer diagnostics with an expanded scope also exist.
Fig. 1. Clinical utility of genomic assays in cancer care.
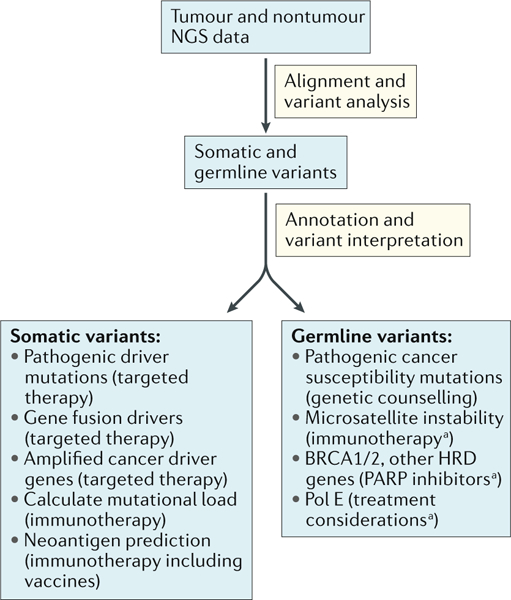
Following the introduction of next-generation sequencing (NGS)-based assays, and reflecting rapid progress in cancer biology and therapeutics, clinical NGS-based diagnostic assays have achieved clinical utility owing to the ability of their results to direct therapeutic decision-making. BRCA, breast cancer susceptibility protein; HRD, homologous recombination deficiency; PARP, poly(ADP-ribose) polymerase; Pol E, DNA polymerase-ε. aCan be germ line or somatic alterations and have the same treatment indication regardless of the origins of the alteration.
Clinical utility of NGS assays
Identifying therapeutic targets.
Large-scale tumour sequencing efforts have revealed the identity of new genomic aberrations that drive tumour growth and progression, and these driver mutations can increasingly be inhibited clinically using an expanding repertoire of molecularly targeted therapies2. Precision oncology entered a new era nearly two decades ago with the development and approval of two targeted therapies: trastuzumab to treat patients with HER2-amplified metastatic breast cancer and imatinib to treat patients with BCR–ABL-fusion-positive chronic myelogenous leukaemia (CML)47,48. Currently, FDA-approved, genotype-directed therapies are available for many different tumour types. Inhibitors of EGFR, ALK, and ROS1 are routinely administered to patients with non-small-cell lung cancer (NSCLC) harbouring genomic alterations in one of those genes49–51. BRAF mutations, which occur in approximately half of all patients with cutaneous melanoma, can be targeted by multiple inhibitors of the MAPK signalling pathway52–54. Furthermore, in patients with metastatic colorectal cancer, EGFR-directed therapies such as cetuximab and panitumumab are specifically administered to patients without oncogenic mutations in KRAS and NRAS55. As the portfolio of targeted therapies expands, the appropriate administration of these therapies and the design of clinical trials to test the efficacy of new drugs requires rapid and sensitive approaches in order to profile targetable genomic alterations in all relevant tumour types. By creating databases of genes and their alterations present in tumours with different anatomical locations, clinical trial accrual processes can be projected with a higher level of precision, thus making the testing and approval of new therapies more efficient.
Complexity aspects.
The diversity and rapidly evolving landscape of clinically relevant mutations means that molecular profiling platforms must also be both comprehensive and flexible in order to incorporate all classes of genomic alterations and new targets following the emergence of new knowledge. Hotspot panels are widely used for the detection of recurrent actionable mutations in EGFR, BRAF, and/or other genes, although broader, hybridization-capture-based panels are better suited for the detection of amplifications, deletions, and chromosomal rearrangements. Rearrangements that produce kinase gene fusions, in particular, are an important class of targetable alteration56. These fusions, while once associated exclusively with haematological malignancies (such as BCR–ABL-positive CML), are also now commonly found in several types of solid tumours, as exemplified by the presence of fusions containing ALK, RET, or ROS1 in patients with NSCLC57. Data from screening efforts have demonstrated that the genes encoding certain targetable kinases, such as FGFR2, BRAF, and NTRK1/2/3, are expressed in fusions across many different tumour types with different partner genes, thus underscoring the need for methods that enable the detection of multiple different rearrangements, including distinct configurations, to ensure that all therapeutically actionable fusions can be detected56,58,59. Panel-based tumour sequencing approaches have also led to the discovery of new targetable alterations such as MET exon 14-skipping splice site mutations, which reduce the ubiquitin-mediated degradation of MET, confer sensitivity to crizotinib, and occur in ~3% of all NSCLCs60,61. More-comprehensive approaches, such as whole-exome and whole-genome sequencing, provide the greatest level of discovery potential and are a preferred approach in certain clinical contexts, such as paediatric cancers62,63, although the costs and complexity of such analyses currently limit their utility in widespread clinical testing.
Shared alterations.
Molecular profiling of tumour samples has been increasingly deployed across a wide range of cancers, and the results have reaffirmed the ubiquitous presence of particular driver alterations across many different histologically defined tumour types56,64,65. BRAFV600E mutations, which have historically been associated with melanoma, are found at meaningful frequencies in patients with NSCLC, colon cancer, thyroid cancer, multiple myeloma, glioma, or pancreatic cancer. HER2 amplifications, once thought to be exclusive to HER2-positive breast cancers, are now considered relatively common in patients with oesophagogastric cancer, breast cancer, bladder cancer, or endometrial cancer as a result of large-scale discovery genomics. Furthermore, NTRK fusions have been reported in more than a dozen different tumour types66. These alterations can all be targeted therapeutically, and therefore, broad mutation profiling regardless of lineage is necessary to ensure that both patients with common and those with rare cancer types have the opportunity to receive the optimal available treatment. Drugs that have received FDA approval for use in a particular tumour type, such as the BRAF inhibitors vemurafenib and dabrafenib in patients with melanoma, can be prescribed off-label for patients with different cancer types harbouring the same target lesions. However, access to these therapies and their reimbursement by insurance companies is often limited in patients with such non-standard indications. Furthermore, drug efficacy can be difficult to measure and track in patients when administered on an ad hoc basis. To address these challenges and establish an evidence base for the delivery of genomically guided targeted therapy in the future, ASCO launched the Targeted Agent and Profiling Utilization Registry (TAPUR), which started recruiting patients in 2016, providing patients with specific targetable alterations free access to their appropriate commercially available targeted agent with the aim of establishing a broad dataset of clinical outcomes.
Clinical trial evolution.
The occurrence of the same or similar targetable alterations across multiple cancer types has also prompted the development of novel clinical trial designs in order to systematically test the efficacy of drugs across different lineages. In one such study design, so-called basket trials, patient eligibility is defined on the basis of the presence of a specific genetic alteration rather than a specific tumour type67 (FIG. 2a). Basket trials involve single, molecularly targeted drugs and are ideally suited for mutations occurring at a low frequency across multiple different cancer types, in which individual disease-specific studies are unlikely to accrue enough patients to provide a statistically robust evaluation of responsiveness. Such studies have emerged as an efficient way to expand access to specific targeted therapies for patients harbouring the appropriate alterations, particularly in patients with rare cancers, for whom clinical trials have historically not been widely available. Results of several such studies have also revealed the importance of tumour type in conditioning responses to targeted therapies68,69. These trials are in contrast to ‘umbrella’ trials, which involve multiple cohorts and are designed to test different targeted agents in patients with distinct mutations in a single cancer type, in which cohort assignment is determined on the basis of molecular profiling70–72 (FIG. 2b). The US National Cancer Institute (NCI) has established an ambitious study, the Molecular Analysis for Therapy Choice (NCI-MATCH) initiative, which is designed to broaden access to clinical trials and targeted therapies for patients receiving treatment outside of the largest academic medical centres73. NCI-MATCH functions as an umbrella of basket trials, involving centralized molecular profiling using a standardized hotspot panel in participating diagnostics laboratories in order to assign patients to one of >30 treatment arms. Other initiatives, such as the Novartis Signature Trial Program, enable the rapid activation of ‘n of one’ studies at treatment centres at which patients with specific alterations identified following molecular profiling are being treated.
Fig. 2. Clinical trial designs invoking cancer genomics assays.
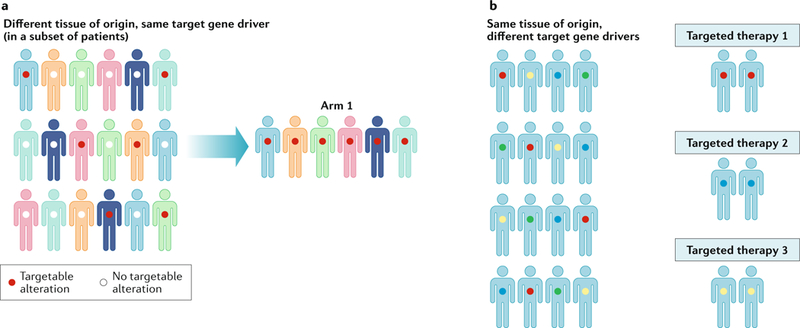
Two basic clinical trial designs for the testing of genomically targeted cancer therapies have emerged. a | Basket trials place all patients with tumours expressing the same genomic target into the same ‘basket, enabling patients to receive a matched targeted therapy. b | Umbrella trials involve the investigation of multiple targeted therapies and the enrolment of specific groups of patients into different trials according to their tumour genotype. In both types of trials, a next-generation sequencing assay that enables the detection of many targetable alterations can provide information enabling patients to be included into one of multiple trial cohorts available within a cancer centre. Figure adapted from REF143, NRC Research Press, CC-BY-4.0 (https://creativecommons.org/licenses/by/4.0/).
Challenges of acquired resistance.
The application of molecularly targeted therapies in patients with genomically matched tumours has led to remarkable initial responses, although the co-occurrence of other alterations and other biological factors might cause innate drug resistance in certain subsets of patients. Furthermore, patients with metastatic cancers nearly always eventually develop acquired resistance following prolonged treatment with targeted therapy74. Secondary genomic alterations in the target itself might render it insensitive to inhibition, and activation of downstream or bypass effectors can separately lead to pathway reactivation in the presence of the drug. The identification of on-target secondary resistance mutations in BCR–ABL1 in patients with CML and EGFR in patients with NSCLC following administration of imatinib or EGFR inhibitors, respectively, has led to the development of more-potent second-generation and third-generation inhibitors75–77.
Genomics-based analyses of tumour biology have revealed many novel mechanisms of acquired resistance through two main approaches. First, by comparing sequencing data from cohorts of patients with pretreated metastatic cancer, particularly from prospectively sequenced patient populations, with data from patients with untreated primary tumours, mutations might be found to be substantially enriched in those with metastatic disease. For example, ligand-binding domain mutations in the gene encoding the oestrogen receptor (ESR1) were first identified owing to their high frequency in metastatic breast cancers following antioestrogen therapy relative to a virtual absence of such mutations in primary tumours78,79. Second, by sequencing paired pretreatment and post-treatment biopsy samples obtained at different times from the same patient, mutational profiles can be directly compared in order to identify novel acquired mutations. Paired sequencing of BRAF-mutant melanoma biopsy samples before and after treatment with RAF inhibitors has revealed a broad spectrum of acquired mutations in MEK1 and other members of the MAPK signalling pathway80–82. In a comparison of a pretreatment sample of PI3KCA-mutant breast cancer with samples from multiple resistant lesions from the same patient following treatment with a PI3Kα small-molecule inhibitor (alpelisib), only PTEN alterations were found to mediate treatment resistance, with different types of PTEN-inactivating alterations occurring at different metastatic sites83. Serial sampling, followed by genomic analysis of cell-free tumour-derived DNA, is increasingly being used to detect the emergence of resistance mutations in samples of plasma, urine, and other bodily fluids. Knowledge of the spectrum of acquired resistance mutations associated with individual targeted therapies is critical to the development of therapy combinations that can delay the onset of acquired resistance in patients. Several important vignettes exist regarding the mechanisms of acquired resistance, although our overall understanding of such mechanisms is quite poor. This dearth of knowledge, as discussed previously, largely reflects an inability to obtain samples of treatment-resistant disease following relapse. Certain cancer centres have invested in the rapid autopsy approaches required in an attempt to begin to elucidate the mechanisms of acquired resistance in greater detail, although such efforts remain quite rare.
Challenges to implementation.
The current proliferation of therapies targeting the specific mutations and downstream pathways that drive cancer progression has made keeping track of the clinical implications of genomic alterations arising in their patients’ tumours increasingly difficult for both oncologists and other clinical practitioners. Many academic cancer centres have convened molecular tumour boards that draw on expertise from several diverse medical specialities and from experts in cancer genomics to collectively discuss individual patients and make treatment recommendations on the basis of both genomic and clinical features19,84. However, this approach cannot easily be upscaled to accommodate the increasing numbers of patients undergoing prospective clinical tumour sequencing. To this end, several groups have attempted to curate biological and clinical information from expert recommendations, clinical guidelines, and scientific literature into databases to provide support to clinicians and enable optimal treatment decisions to be made85–88. These knowledge bases can account for distinct functions and implications of different alterations in the same gene or for the presence of the same alteration in different cancer types. Furthermore, these resources must be updated regularly to incorporate emerging knowledge of the clinical relevance of particular alterations and the rapidly changing availability of data from genomically matched clinical trial cohorts. Owing to the sheer volume of information that must be captured, some groups have employed crowdsourcing methodologies to ensure that evidence statements and their accompanying annotations are as comprehensive and accurate as possible87. Ultimately, no single knowledge base is entirely complete, nor can the effects and implications of interactions among co-occurring mutations be readily inferred. Nevertheless, these resources are crucial for the proper interpretation and utilization of complex genomic data obtained from prospective clinical sequencing.
Despite the promise of precision oncology approaches, only a subset of patients have tumours harbouring specific actionable alterations for which compelling clinical data exist that can directly influence the choice of therapy. Many other additional recurrent driver mutations are currently considered ‘undruggable’, and other potentially viable target alterations remain incompletely characterized. Furthermore, the definition of ‘actionability’ used to assess the clinical utility of molecular profiling varies widely across different studies and institutions. Reports published in the past 2 years from various initiatives using tumour sequencing panels to prospectively identify targetable mutations across different cancer types suggest that between 30% and 94% of patients harbour actionable mutations41,56,89,90. This disparity largely reflects different interpretations of the clinical significance of specific mutations, which range from those with compelling clinical evidence available in that they can be directly targeted using FDA-approved or investigational therapies to those with more speculative evidence of effectiveness based upon the results of pre-clinical studies. Furthermore, only a minority of patients harbouring actionable mutations are currently enrolled in genomically matched clinical trial cohorts on the basis of alterations detected using clinical sequencing56,90–96. This fact might reflect shortcomings in the availability and/or geographical accessibility of relevant trials, as well as a lack of physician and/or patient awareness of these studies. However, these trial enrolment rates do not take into account patients who receive FDA-approved therapies on the basis of their genomic alterations or those who might eventually enrol in a clinical trial following disease progression on standard treatment protocols. The development of new agents inhibiting novel targets and the proliferation of basket clinical trials designed to enrol patients harbouring a targetable somatic alteration in any one of multiple tumour types will inevitably further expand the clinical utility of tumour genomic profiling.
The utility of tumour mutational profiling extends beyond the identification of individual actionable mutations that predict a response to targeted therapies. The presence of specific mutational signatures and other complex genomic features might also be used to inform clinical decisions97,98. Tumour mutational load has emerged as a proposed biomarker that tends to correlate with clinical benefit from immune-checkpoint inhibitors44–46. The presence of a specific mutational signature, microsatellite instability (MSI), has also been identified as a predictor of a response to immune-checkpoint inhibition, leading the FDA to approve the anti-programmed cell death protein 1 (PD-l)-antibody pembrolizumab, for use in patients with MSI-high solid tumours regardless of histology or anatomical location99,100. This is the first ever approval of a cancer therapy based solely on the presence of a genomic biomarker, irrespective of the tissue of origin. The presence of other complex signatures, including loss of function of genes involved in homologous recombination, resulting in homologous recombination deficiencies, might predict a response to PARP inhibitors, another targeted anticancer therapy101–103. Moreover, tumour sequencing might also provide information that is important for diagnosis or prognosis and/or reveal alterations suggesting a lack of a response to particular interventions, uncover occult germline cancer susceptibility alleles that necessitate follow-up genetic counselling, establish the clonal relatedness of distinct lesions, and facilitate disease monitoring after treatment. For these reasons, and owing to the increased availability of testing options, tumour genomic profiling has become a mainstay of precision oncology and a necessary component of cancer care delivery.
Immunotherapeutic decision-making
As momentum supporting the inclusion of genomic profiling as a mainstay of evidence-based precision oncology has grown, newer uses for genomic assays have emerged that consider the data in light of cancer immunology and vulnerability to the associated therapies. In addition to the relationship between mutational load and sensitivity to immune-checkpoint inhibition, genomics can also have more-nuanced roles in guiding the use of immunotherapy. In particular, these newer, precision medicine-based applications of immunog-enomics have their foundations in cancer immunology studies conducted in the mid-to-late 1980s by several groups. These early efforts sought to uncover the mechanism that could explain several intriguing experimental and clinical observations, including why mice with actively growing carcinogen-induced tumours removed by surgery that were subsequently challenged with cells from their original cancers did not regrow their cancers and why certain patients with cutaneous melanomas, in rare cases, had spontaneous disease regression with-out treatment. Using what were then considered novel approaches, such as positional cloning and immunological assays such as enzyme-linked immunospot (ELISPOT) or dextramer-based flow cytometry assays, cancer immunologists demonstrated that tumour cells express the products of mutated genes and that these proteins act as tumour-specific mutant antigens (TSMAs) or neoantigens, which are capable of eliciting an antitumour immune response104–106. Neoantigens, by definition, are specifically encoded by the tumour genome, wherein an alteration in tumour DNA causes a change in the amino acid sequence, resulting in a peptide that presents a stimulus to the patient’s immune system (that is identified as non-self). These early efforts identified TSMAs from both mouse models and from human melanoma cells, although the time and effort required, at the time, was not scalable to the identification of TSMAs in multiple patients, and as such, treatment approaches to targeting specific TSMAs were not pursued in clinical trials. Renewed interest in these early investigations has emerged in the past few years with the advent of NGS technologies and more-advanced computational predictors of neoantigens.
Even as early efforts to characterize frequently mutated genes in cancers were emerging in the mid-2000s, Vogelstein and Allison107, among other authors, predicted that the altered proteins identified in breast and colorectal cancers produce, on average, ten and seven neoantigens, respectively, with predicted binding to HLA-A*0201, that are putative targets for immune manipulation to elicit tumour cell destruction. Shortly thereafter, the emergence of NGS and computational approaches that compared tumour DNA sequences with those obtained from nonmalignant cells in an unbiased fashion greatly facilitated the discovery of all somatic alterations in tumour DNA and revealed the proportions of tumour cells carrying each specific mutation (such as the founder clone versus subclones)108. Coincidentally, neural network-based algorithms emerged that provided computational predictions of the binding affinities of the predicted mutant peptides to different HLA molecules, thus permitting evaluations of the implications of the modified sequences for immune recognition109–112. These algorithms were informed by experimental data on the affinity of different HLAs for specific peptides, and as these data have expanded to include more HLA alleles, so has the accuracy and breadth of the predictions.
In current practice, exome-capture assays and analysis pipelines that compare NGS data from tumours with exome-sequencing data from nonmalignant cells can also yield predicted neoantigens using secondary analysis through a neoantigen-prediction algorithm such as NetMHC (FIG. 3). This type of analysis requires not only the predicted amino acid sequence-changing peptides (translated from DNA sequencing) as input but also knowledge of the specific HLA haplotypes of the patient. HLA haplotypes can be determined using a conventional clinical sequencing assay but are frequently identified using specialized analyses of exome sequencing data from a nonmalignant tissue sample113–115. Analysis pipelines that facilitate neoantigen prediction based upon predicted mutations through output of neoantigen peptides and their predicted binding affinities for different HLA molecules have also been published (such as pVACseq116 and Vaxrank117), and the IEDB website provides a range of data analysis programmes, some of which enable neoantigen prediction. Importantly, established driver mutations are rarely also strong neoantigens, emphasizing that exome sequencing will most likely provide the most comprehensive picture of the neoantigen repertoire of a given tumour. As might be anticipated, tumour types in patients with a history of repeat exposures to potent carcinogens such as ultraviolet-associated melanomas and smoking-associated NSCLCs and urothelial carcinomas, as well as cancers that emerge as a consequence of mismatch-repair defects, all have higher numbers of neoantigens than cancers of other aetiologies by virtue of their elevated mutation load. However, comparisons of RNA sequencing data designed to evaluate the expression of predicted mutations indicate that a substantial percentage of mutations predicted from analysis of DNA are in genes that are not transcribed as RNA (typically >50% in highly mutated tumours) and are, therefore, unlikely to be biologically relevant118. This observation emphasizes the importance of including data from tumour RNA sequencing as a component of downstream evaluation of neoantigen expression despite the various clinical challenges and additional costs associated with this type of assay.
Fig. 3. NGS-based neoantigen discovery.
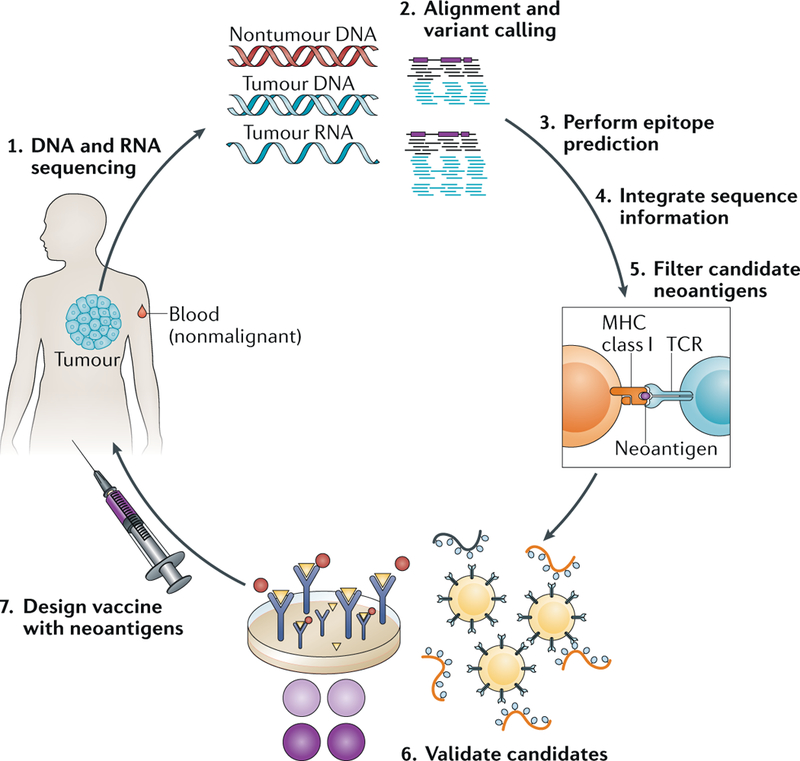
Neoantigen discovery is pursued using next-generation sequencing (NGS) data from comparisons of tumour DNA with nontumour genomic DNA obtained from the same patient to identify the presence of somatic variants with the potential to alter amino acid sequences in the resulting protein. As illustrated in step 1, tumour and nontumour hybrid-capture exome NGS data are generated, as are RNA sequencing data from the tumour isolate. In step 2, computational predictions of the presence of somatic variants are made using the appropriate algorithms, and these predictions are parsed into the resulting novel peptides, along with the calculated HLA haplotypes, and then evaluated by a neoantigen predictor (step 3). These processes are followed by sequence integration (step 4), which culls the potential neoantigen list using cross-comparisons with RNA sequencing data, including evidence of expression, and finally, quality filtering steps (step 5) eliminate known false positive results (such as hypothetical proteins) to produce a final neoantigen prediction list. Neoantigens can be validated using enzyme-linked immunospot (ELISPOT) assays or other approaches (step 6), although this validation step is not always pursued, especially when few candidates exist or a short turnaround time is essential. The design of antitumour vaccines can involve many different approaches, including long (~20 mer) or short (8–11 mer) peptides, RNA-based or DNA-based vaccines, or dendritic cell vaccines. MHC, major histocompatibility complex; TCR, T cell receptor. Figure is adapted from images courtesy of J. Hundal, Washington University School of Medicine in St Louis, MO, USA, and K Campbell, Washington University in St Louis, MO, USA.
Predictions of neoantigen expression can enable a more refined estimation of mutational load, although the results of a multi-omics study involving patients with urothelial carcinomas treated with the anti-programmed cell death 1 ligand 1 (PD-L1) antibody atezolizumab, did not support a statistical correlation between either mutational load or neoantigen load and a response to treatment119. Similarly, the findings of other studies have demonstrated that immunohistochemical evaluations of PD-L1 expression also do not provide an unequivocal biomarker indicative of a response to anti-PD-1 or anti-PD-L1 antibodies120. These results support the notion that a multi-component predictor of response to immune-checkpoint inhibition will be required to enable clinicians to confidently select patients who are most likely to benefit from these agents.
The design of personalized immunotherapies, such as antitumour vaccines, is another potential clinical application of neoantigen prediction. In these approaches, the neoantigens that emerge from computational predictions with the highest binding affinities for either class I or class II HLA molecules have high levels of RNA and/or peptide expression, conform to various other user-defined criteria, and can be used to construct a patient-specific vaccine of several possible types. Namely, vaccines can be based on DNA, RNA, or protein, and each specific method has strengths and weaknesses in terms of scalability of manufacture, ease of delivery, costs, efficacy, and other important factors. Vaccines can also use patient-derived immune components such as dendritic cells, as has been demonstrated in patients with melanoma118. The findings of two studies published in July 2017 demonstrate the successful use of personalized anticancer vaccines using long peptide-based121 and RNA-based poly-neoepitope122 vaccine methods in patients with melanoma. In these studies, some patients received a combination of the vaccine and immune-checkpoint inhibition, which, as predicted by preclinical studies in mouse models123, had a synergistic effect.
RNA sequencing data can also be evaluated to provide both informative measures of immune cell activity and important metrics of monitoring after immunotherapy. These applications range from simple measures, such as quantifications of the expression of immune-checkpoint proteins in the tumour and/or tumour microenvironment that might be more reliable than immunohistochemical evaluations, to the more complex characterization of the infiltrating immune cell types found in a tumour mass124,125. RNA sequencing data can also enable the characterization of B cell126 and T cell repertoires127–129. When performed both before and after vaccination, the resulting comparison indicates the extent of vaccine-induced T cell expansion and diversity118.
Future perspectives
The use of clinical genomics in cancer care is gaining traction as the utility of the assays increases. As might be imagined in this rapidly progressing field, several genomic applications are currently under active development and might further expand the clinical utility of genomic assays in the future. A prime example involves the development and extension of assays designed to profile DNA isolated from samples obtained using minimally invasive procedures, such as sampling of blood plasma or other bodily fluids (sputum, urine, or cerebrospinal fluid). Liquid biopsy sampling takes advantage of the fact that cell death in actively growing tumours leads to the release of tumour cell-derived DNA into the circulation and into other fluids that come into contact with organs130. Cell-free DNA (cfDNA) collected in this fashion might be used for tumour mutation profiling, genomic monitoring of response to therapy, and identifying emerging mechanisms of resistance to therapy, thereby providing highly sensitive and specific indicators to guide clinical care and decision-making processes131–133 (FIG. 4). Nevertheless, considerable challenges must be overcome before liquid biopsy applications are likely to entirely replace tumour profiling and imaging approaches in the clinical assessment of patients with cancer. Most notably, the fraction of tumour-derived DNA present in blood plasma samples is typically much lower than that present in tumour tissues, thus indicating a need for modified sample preparation methods and much deeper sequence coverage to achieve sufficient sensitivity for low-frequency mutations that might approach a frequency similar to that of background sequencing errors. This issue is especially pertinent for patients with early stage disease, as studies have shown that both tissue site and tumour stage lead to differential levels of cfDNA in the circulation134. Furthermore, while several successful examples have been reported135,136, the clinical use of liquid biopsies for the reliable detection of minimal residual disease and the diagnosis of cancer during the early stages of disease are likely to prove even more challenging. Unique molecular indexing approaches to sequence library construction have been adapted for this purpose, and this method enables the suppression of base-calling errors via the generation of collapsed error-free consensus sequences137–139. However, this approach requires ultra-high-depth sequencing to produce a sufficient number of replicate reads from each cfDNA fragment and might, therefore, become prohibitively expensive without reductions in panel size and/or sequencing costs. Additional technological improvements, enabling liquid biopsy samples to be analyzed with increased levels of sensitivity, will likely be required, or indeed, non-sequencing-based approaches to such analysis (such as droplet-digital PCR) might also provide benefits, among others, in terms of cost and sensitivity. Comparisons with the results of conventional imaging-based investigations will be important to establishing the utility and clinical benefit of liquid biopsy approaches in patients with cancer.
Fig. 4. Liquid biopsy assays enable the monitoring of genomic alterations present in circulating tumour DNA.
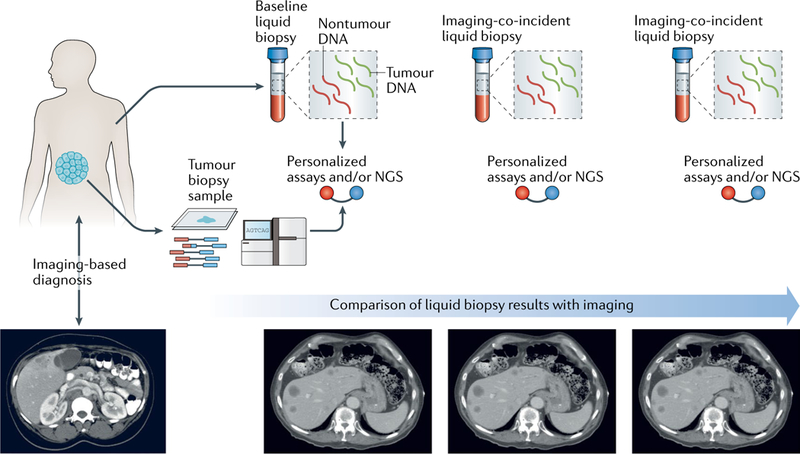
The findings of genomic characterizations comparing tumour and nontumour DNA can inform the design and use of liquid biopsy-based approaches. These directed assays enable the detection and comparison of somatic mutations present in circulating tumour DNA (ctDNA) during treatment and over time, in comparison with a baseline tumour DNA sample taken after diagnosis and before tumour resection. Liquid biopsy results can then be compared to those obtained using conventional imaging-based approaches to disease monitoring for the potential to detect recurrent disease or emerging therapy resistance (only when specific resistance-conferring genotypes are known). NGS, next-generation sequencing. Figure is adapted from image courtesy of N. Rosenfeld, Cancer Research UK Cambridge Institute, UK, and D. Tsui, Memorial Sloan Kettering Cancer Center, NY, USA.
The likelihood of progress in clinical genomics is affected by several attendant challenges. First, the current clinical implementation of cancer genomics has largely been limited to academic tertiary cancer centres that provide cutting-edge clinical cancer care. In the USA, only a small minority of patients with cancer receive treatment at these centres. In fact, approximately 80% of patients with cancer in the USA are treated locally at community hospitals where the practicing oncologists have little or no access to advanced genomic testing, let alone the training or expertise required to successfully interpret the results of such assays in determining treatment options for their patients. This reality effectively limits the access of most patients to the clinical benefits that genomic tests can provide and creates a vexing problem that has yet to be solved. Second, big data analytics-based efforts that combine genomic data with treatment and outcomes data across multiple studies have the ability to transform the treatment of the disease, although this potential is limited when data are not broadly shared. Data sharing has emerged as a cornerstone of the NCI Blue Ribbon Panel report from the Cancer Moonshot Initiative, as advocated in 2016 by Vice President Biden140. In response to the call for broader data sharing, several big-data initiatives such as the NIH Genomic Data Commons, the AACR Project GENIE, and the Global Alliance for Genomics and Health have been established in an attempt to centralize and standardize cancer genomic data and accelerate progress towards identifying improved therapeutic strategies41,141,142. Challenges clearly remain in harmonizing the genomic results obtained using different sequencing platforms with unique bioinformatics pipelines and filtering criteria, although optimal data sharing will ultimately be essential for the identification of novel genomic targets and predictive biomarkers. We anticipate that these efforts, along with the innovations described above, will serve to further establish a critical role of genomics-based techniques in the diagnosis and treatment of patients with cancer.
Conclusions
In conclusion, genomics-based assays are increasingly being utilized as a component of evidence-based diagnosis that can inform the care of patients with cancer. Emerging applications will expand the use of N GS-based diagnostics in cancer medicine, although such applications are likely to create attendant challenges that will require innovative approaches to ensure reproducibility, expand access, and educate providers.
Box 1. Obstacles to data sharing and data mining.
Data sharing is crucial to enabling comprehensive data mining, evaluation of results, and the compilation of data to ensure the statistical significance of genomic findings, although considerable obstacles exist that will likely influence the ability to share and mine data, as illustrated here. These initiatives fall into three main categories: technology-associated obstacles, clinical data-associated obstacles, and practical or legal obstacles. Broadly speaking, the technology-related aspects of data sharing are made difficult by the incredible rate of improvement in next-generation sequencing (NGS) and computational analysis methods over the past 10 years. The obstacles to the sharing and mining of clinical data reflect the mismatch in sophistication between our ability to generate genomic data and our inability to store large amounts of clinical data in an accessible form owing to an absence of guidelines, shared language elements, and the other complications we have described. Various practical and legal obstacles can also pose substantial challenges to data sharing and reflect the need for a standardized language of consent and highlight the lack of consensus on how best to approach data privacy measures. NLP, natural language processing; WGS, whole-genome sequencing.

NLP, natural language processing; WGS, wholegenome sequencing
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
IEDB Analysis Resource, Epitope Prediction and Analysis Tools: http://tools.immuneepitope.org/main/
NCI-MATCH Trial (Molecular Analysis for Therapy Choice): https://www.cancer.gov/about-cancer/treatment/clinicaltrials/nci-supported/nci-match
NIH Genomic Data Commons: https://gdc.cancer.gov
The Global Alliance for Genomics and Health: https://www.ga4gh.org/
The Novartis signature trial programme: http://www.trials.novartis.com/en/clinical-trials/us-oncology/oncology/signature/about/
References
- 1.Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Hyman DM, Taylor BS & Baselga J Implementing genome-driven oncology. Cell 168, 584–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyerson M, Gabriel S & Getz G Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 11, 685–696 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Griffith M et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 1, 210–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Do H & Dobrovic A Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 61, 64–71 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Williams C et al. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am. J. Pathol. 155, 1467–1471 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagle N et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2, 82–93 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Allen EM et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 20, 682–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Kateb H, Nguyen TT, Steger-May K & Pfeifer JD Identification of major factors associated with failed clinical molecular oncology testing performed by next generation sequencing (NGS). Mol. Oncol. 9, 1737–1743 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goswami RS et al. Identification of factors affecting the success of next-generation sequencing testing in solid tumors. Am. J. Clin. Pathol. 145, 222–237 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Cottrell CE et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 16, 89–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lih C-J et al. Analytical validation of the next-generation sequencing assay for a nationwide signal-finding clinical trial: molecular analysis for therapy choice clinical trial. J. Mol. Diagn. 19, 313–327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng DT et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh RR et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 15, 607–622 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Jennings LJ et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 19, 341–365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laskin J et al. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb. Mol. Case Stud. 1, a000570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roychowdhury S et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. TranslMed. 3, 111ra121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frampton GM et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garofalo A et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 8, 79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Rooij N et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levin JZ et al. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 10, R115 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnirke A et al. Solution hybrid selection with ultralong oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albert TJ et al. Direct selection of human genomic loci by microarray hybridization. Nat. Methods 4, 903–905 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Hodges E et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 39, 1522–1527 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Jones S et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl Med. 7, 283ra53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrader KA et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2, 104–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin KY & Kraus WL PARP inhibitors for cancer therapy. Cell 169, 183 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Susswein LR et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 18, 823–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandelker D et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA versus guideline-based germline testing. JAMA 318, 825–835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green RC et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15, 565–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johns AL et al. Lost in translation: returning germline genetic results in genome-scale cancer research. Genome Med. 9, 41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray SW et al. Oncologists’ and cancer patients’ views on whole-exome sequencing and incidental findings: results from the CanSeq study. Genet. Med. 18, 1011–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie M et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young AL, Challen GA, Birmann BM & Druley TE Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 12484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coombs CC et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 10.1158/2159-8290.CD-17-0151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakravarty D et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 10.1200/P0.17.00011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Allen EM et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slamon DJ et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Druker BJ et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Mok TS et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Kwak EL et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw AT et al. Crizotinib in R0S1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 371, 1963–1971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman PB et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larkin J et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 371, 1867–1876 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Robert C et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 372, 30–39 (2015). [DOI] [PubMed] [Google Scholar]
- 55.De Roock W et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 11, 753–762 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw AT, Hsu PP, Awad MM & Engelman JA Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 13, 772–787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stransky N, Cerami E, Schalm S, Kim JL & Lengauer C The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ross JS et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer. 138, 881–890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paik PK et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 5, 842–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frampton GM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 5, 850–859 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Downing JR et al. The Pediatric Cancer Genome Project. Nat. Genet. 44, 619–622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oberg JA et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med. 8, 133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kandoth C et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas RK et al. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 39, 347–351 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Amatu A, Sartore-Bianchi A & Siena S NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 1, e000023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cunanan KM et al. Basket trials in oncology: a trade-off between complexity and efficiency. J. Clin. Oncol. 35, 271–273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hyman DM et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 373, 726–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hyman DM et al. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 35, 2251–2259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Govindan R et al. ALCHEMIST trials: a golden opportunity to transform outcomes in early-stage non-small cell lung cancer. Clin. Cancer Res. 21, 5439–5444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Papadimitrakopoulou V et al. The BATTLE-2 study: a biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung cancer J. Clin. Oncol. 34, 3638–3647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Herbst RS et al. Lung Master Protocol (Lung-MAP)-a biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin. Cancer Res. 21, 1514–1524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abrams J et al. National Cancer Institute’s Precision Medicine Initiatives for the new National Clinical Trials Network. Am. Soc. Clin. Oncol. Educ. Book 2014, 71–76 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Garraway LA & Jänne PA Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2, 214–226 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Saglio G et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 362, 2251–2259 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Cortes JE et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 367, 2075–2088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jänne PA et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Toy W et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robinson DR et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wagle N et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 29, 3085–3096 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Allen EM et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 4, 94–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi H et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Juric D et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Ka inhibitor. Nature 518, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.LoRusso PM et al. Pilot trial of selecting molecularly guided therapy for patients with non-V600 BRAF-mutant metastatic melanoma: experience of the SU2C/MRA Melanoma Dream Team. Mol. Cancer Ther. 14, 1962–1971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Damodaran S et al. Cancer Driver Log (CanDL): catalog of potentially actionable cancer mutations. J. Mol. Diagn. 17, 554–559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Griffith M et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49, 170–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meric-Bernstam F et al. A decision support framework for genomically informed investigational cancer therapy. J. Natl Cancer Inst. 107, djv098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sholl LM et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wheler JJ et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 76, 3690–3701 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Beltran H et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 1 , 466–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meric-Bernstam F et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J. Clin. Oncol. 33, 2753–2762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schwaederle M et al. On the road to precision cancer medicine: analysis of genomic biomarker actionability in 439 patients. Mol. Cancer Ther. 14, 1488–1494 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Stockley TL et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 8, 109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schwaederle M et al. Precision oncology: the UC San Diego Moores Cancer Center PREDICT experience. Mol. Cancer Ther. 15, 743–752 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Tsimberidou A-M et al. Initiative for molecular profiling and advanced cancer therapy (IMPACT): an MD Anderson Precision Medicine Study. JCO Precis. Oncol. 10.1200/PO.17.00002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hause RJ, Pritchard CC, Shendure J & Salipante SJ Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 22, 1342–1350 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Le DT et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Le DT et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bryant HE et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 102.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 103.Robson M et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017). [DOI] [PubMed] [Google Scholar]
- 104.De Plaen E et al. Immunogenic (tum-) variants of mouse tumor P815: cloning of the gene of tum-antigen P91A and identification of the tum-mutation. Proc. Natl Acad. Sci. USA 85, 2274–2278 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Monach PA, Meredith SC, Siegel CT & Schreiber H A unique tumor antigen produced by a single amino acid substitution. Immunity 2, 45–59 (1995). [DOI] [PubMed] [Google Scholar]
- 106.van der Bruggen P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 (1991). [DOI] [PubMed] [Google Scholar]
- 107.Segal NH et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Mardis ER DNA sequencing technologies: 2006–2016. Nat. Protoc. 12, 213–218 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Lundegaard C et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–W512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nielsen M et al. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput. Biol. 4, e1000107 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rasmussen M et al. Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J. Immunol. 197, 1517–1524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peters B & Sette A Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6, 132 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shukla SA et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33, 1152–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Warren RL et al. Derivation of HLA types from shotgun sequence datasets. Genome Med. 4, 95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Szolek A et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hundal J et al. pVAC-Seq: a genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 8, 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rubinsteyn A, Hodes I, Kodysh J & Hammerbacher J Vaxrank: a computational tool for designing personalized cancer vaccines. Preprint at bioRxiv 10.1101/142919 (2017). [DOI] [Google Scholar]
- 118.Carreno BM et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Snyder A et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multiomic analysis. PLoS Med. 14, e1002309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diggs LP & Hsueh EC Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark. Res. 5, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ott PA et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 123.Gubin MM et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rooney MS, Shukla SA, Wu CJ, Getz G & Hacohen N Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mose LE et al. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with VDJer. Bioinformatics. 32, 3729–3734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cole C, Volden R, Dharmadhikari S, Scelfo-Dalbey C & Vollmers C Highly accurate sequencing of full-length immune repertoire amplicons using Tn5-enabled and molecular identifier-guided amplicon assembly. J. Immunol. 196, 2902–2907 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Robins H Immunosequencing: applications of immune repertoire deep sequencing. Curr. Opin. Immunol. 25, 646–652 (2013). [DOI] [PubMed] [Google Scholar]
- 129.Li B et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 48, 725–732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Diehl F et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Alix-Panabieres C & Pantel K Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 6, 479–491 (2016). [DOI] [PubMed] [Google Scholar]
- 132.Schwarzenbach H, Hoon DSB & Pantel K Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011). [DOI] [PubMed] [Google Scholar]
- 133.Tie J et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl Med. 8, 346ra92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bettegowda C et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci. TranslMed. 6, 224ra24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chaudhuri AA et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 7, 1394–1403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bianchi DW et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA 314, 162–169 (2015). [DOI] [PubMed] [Google Scholar]
- 137.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl Med. 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kennedy SR et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 9, 2586–2606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.[No authors listed.] Panel’s ‘Moonshot’ goals released. Cancer Discov. 6, 1202–1203 (2016). [DOI] [PubMed]
- 141.Siu LL et al. Facilitating a culture of responsible and effective sharing of cancer genome data. Nat. Med. 22, 464–471 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jensen MA, Ferretti V, Grossman RL & Staudt LM The NCI Genomic Data Commons as an engine for precision medicine. Blood 130, 453–459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gagan J & van Allen EM Next-generation sequencing to guide cancer therapy. Genome Med. 7, 80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]