

Abstract
Liver diseases negatively impact the quality of life and survival of patients, and often require liver transplantation in cases that progress to organ failure. Understanding the cellular and molecular mechanisms of liver development and pathogenesis has been a challenging task, in part for the lack of adequate cellular models directly relevant to the human diseases.
Recent technological advances in the stem cell field have shown the potentiality of induced pluripotent stem cells (iPSC) and liver organoids as the next generation tool to model in vitro liver diseases. Hepatocyte-like cells and cholangiocyte are currently being generated from skin fibroblasts and mononuclear blood cells reprogrammed into iPSC and have been successfully used for disease modeling, drug testing and gene editing, with the hope to be able to find application also in regenerative medicine. Protocols to generate other liver cell types are still under development, but the field is advancing rapidly. On the other end, liver cells can now be isolated from liver specimens (liver explants or liver biopsies) and cultured in specific conditions to form polarized 3D organoids. The purpose of this review is to summarize all these recent technological advances and their potential applications but also to analyze the current issues to be addressed before the technology can reach its full potential.
Keywords: Liver development, Hepatocytes, Cholangiocytes, Stem cells, 3D culture, Regenerative medicine
1. Introduction
The liver is a complex organ, which is structurally and functionally heterogeneous, and comprises different cell types [1], including epithelial cells (i.e hepatocytes and cholangiocytes), Kupffer cells (KC), circulating monocytes, mesenchymal cells (i.e hepatic stellate cells) and sinusoidal endothelial cells. The main epithelial component is represented by hepatocytes, which account for > 60% of the total liver cells and are primarily responsible for the liver’s metabolic activity, detoxification, protein secretion and bile production. The other major epithelial component is represented by biliary epithelial cells (cholangiocytes) that line the intrahepatic and extrahepatic bile ducts and are involved in the transport and modification of the primary bile produced by the hepatocytes [1,2].
Because of its complexity and its central homeostatic role the liver is also the target of a wide range of diseases. In spite of recent extraordinary advances in the treatment of a number of liver diseases, there are unmet needs that still need to be filled in many congenital and acquired conditions that may progress to end-stage liver failure. A better understanding of the cellular and molecular pathobiological mechanisms responsible for liver diseases is needed to provide more effective treatments. The currently available liver cellular and animal models have significantly helped to decipher mechanisms of liver development and pathogenesis, but they still have some limitations. One of the major drawbacks is represented by the limited accessibility to fresh human liver tissue, the restricted availability of protocols for isolation of primary cell populations, and the difficulties in culturing these cells in vitro. Explants from liver transplants or low quality grafts unused for transplant have been, until recently, the major source of human liver cells. On the other hand, available liver cell lines are either cancer-derived or immortalized with limited functions compared to primary cells in vitro and might not reflect normal physiological responses [3,4]. In addition, animal models often fail to recapitulate the disease phenotype or to predict drug toxicity due to inter-species anatomo-physiological differences [5].
All these restrictions called for the development of better and more efficient human experimental models in vitro, and are at the basis of the newly available technology of induced pluripotent stem cells (iPSCs) and the efforts in generating several protocols for their differentiation into liver cells. In addition to iPSCs, the recent discovery of 3D organoids that can be generated from adult liver cells offers another approach to model liver disease in vitro. In this review, we will discuss how iPSC and organoid technologies can be applied to the study of liver pathobiology, with their pros and flaws.
2. Liver development in a dish: iPSC
Most of the genes and pathways involved in the process of liver development have been elucidated in the last decade through the use of animal models, in particular transgenic mice [6–9]. These notions have recently been used in the lab to generate liver cells in a “dish”.
The liver originates from the endoderm, one of the three germ layers formed during embryonic development [10]. Cells of the endoderm are committed to a specific differentiation fate as a consequence of distinct transcription factor expression [11]. In the mouse, around embryonic day E8.25, endoderm cells receive specific extracellular signals that induce their specification to the hepatic fate. A necessary player in this process is FGF [12]. BMP-2 and BMP-4 cooperate with FGF during the hepatic specification [13]. After hepatic endoderm, the next step is the specification of the liver diverticulum lined by endoderm cells called hepatoblasts. This happens at E9 in the mouse embryo and at day 22 in humans. Hepatoblasts proliferate forming a tissue bud and subsequently migrate away from the endoderm epithelium invading the septum transversum where they keep proliferating with further expansion of the liver [10]. A complex network of growth factors is active during this process and is involved in hepatoblasts proliferation. Some of the major components of this network are HGF, TGF-β and FGF10 that are essential for hepatoblasts proliferation [14].
Wnt-signaling (WNT) and β-catenin are also involved during liver development, even though WNT signaling is very complex and has a different role depending on the developmental stage. In early stages of development, WNT signaling has to be repressed anteriorly to preserve foregut identity and promote liver specification, while in the later stages of development WNT is necessary to promote hepatoblasts proliferation and biliary differentiation [15]. Interestingly, retinoic acid signaling is important for liver bud growth and hepatoblasts proliferation [16]. This signaling is regulated by the zinc finger transcription factor Wilms’ tumor suppressor gene (WT1), expressed in mesodermal cells [16]. Once hepatoblasts have reached a certain developmental stage they acquire the potential to differentiate either into the hepatocyte or cholangiocyte lineage [17]. This process is directed by the NOTCH signaling pathway through interaction with its ligand JAGGED, present on mesenchymal cells in the periportal areas [18]. NOTCH activation inhibits hepatoblast differentiation into hepatocytes, but propels their differentiation into cholangiocytes inducing expression of SOX9, one of the most specific markers of biliary cells [19]. Following the lineage segregation stage, hapatocytes and cholangiocytes go through a process of maturation with the acquisition of a specific morphology and physiologic function that will continue until after birth [7,10,17].
The advance in stem cell technology has resulted in the ability to reprogram somatic cells into pluripotent stem cells (iPSC), thus providing a unique platform for manufacturing cells from patients with different genetic diseases [20,21]. Using iPSCs makes it possible to recapitulate in a dish the developmental steps leading to a specific tissue, by sequentially adding to the culture a cocktail of growth factors that correspond to the pathways active in vivo as described above for the liver. Moreover, iPSCs can be easily derived from skin fibroblasts or most recently from mononuclear white blood cells or urine samples and in theory, be then differentiated into a variety of cell types [22,23]. Their limitless ability to self-renew and their differentiation potential, allow the production of large amounts of specific cell types affected for example by a genetic disease, or perform gene editing protocols. As iPSCs retain the same genetic background of their donors, they provide an attractive alternative for disease model and drug discovery when the traditional models are inadequate [20]. In the next section, we will describe the different strategies used to induce differentiation of iPSCs into liver cells.
2.1. Hepatocytes from iPSCs
Several in vitro protocols to differentiate iPSCs into hepatocyte-like cells (HLC) are available to date; they share a common approach to stepwise differentiation induction but present specific differences [24–26]. Some earlier protocols are based on the generation of aggregates of iPSCs in suspension, defined as embryoid bodies (EBs), and their subsequent differentiation into hepatocytes using a cocktail of growth factors and specific matrices [27–29]. However, these protocols have low efficiency of differentiation and result in mixed populations of cells that spontaneously arise in culture, thus increasing the variability between different differentiation attempts.
The current differentiation strategies include a stepwise induction of definitive endoderm, generation of hepatic progenitor cells and final maturation to “hepatocyte-like cells” by addition of specific cocktails of growth factors in culture [30]. Compared to the earlier culture conditions as EBs, these protocols have led to a high efficiency of differentiation and the production of iHLC that express albumin, CK18, cytochrome p450 enzymes, alpha-1-antitrypsin (A1AT), and asialoglycoprotein receptor1. Several functional assays including LDL uptake, albumin secretion, urea metabolism, glycogen production, inducible cytochrome P450 activity and in vivo transplantation have shown that iHLC derived with the different protocols acquire functions similar to primary human hepatocytes [24,26,31,32]. However, the persistent expression of AFP and lack or reduced activity of more mature cytochrome P450 isoforms such as CYP3A4 and CYP2A6 suggest that iPSC-derived hepatocytes remain somehow less differentiated than adult hepatocytes [25].
The fetal profile of iPSC-derived hepatocytes has triggered several studies aimed at improving their maturation and functionality [33]. In this regard, small molecules and chemicals (e.g. CHIR99021, DMSO and dexamethasone) have been introduced in culture to increase efficiency and reproducibility of hepatocyte differentiation and avoid the use of growth factors [34–36]. Despite all these efforts, additional and precise modifications to the available protocols are still necessary to fully exploit the potential of hepatocytes derived from iPSCs.
Other strategies are based on the use of artificial scaffolds and extracellular matrices to generate 3D cultures that reproduce the liver environment or the cross-talk with other cells by co-culture with endothelial cells or mesenchymal cells [37–39].
2.2. Cholangiocytes from iPSCs
The interest in developing protocols for the differentiation of iPSCs into cholangiocytes has recently increased [40,41]. In 2014 Dianat et al. were the first to describe a stepwise differentiation protocol, similar to those followed for hepatocytes, for the generation of cholangiocytes based on the temporal exposure to specific growth factors. The majority of the experiments were carried out using human embryonic stem cells and the cell line HepaRG, although an iPSCs line was used to confirm the applicability of the protocol [42]. Their approach is based on the generation of a monolayer of hepatoblasts that are subsequently cultured on collagen coated plates and exposed to various growth factors including HGH, EGF, IL-6 and sodium taurocholate to obtain mature cholangiocytes. Gene expression profiling of derived cholangiocytes confirmed that they activate specific pathways involved in biliary differentiation. When cultured in 3D conditions, cholangiocytes were able to form cysts and tubule-like structures with apico-basal polarity and to actively transport rhodamine, a substrate for the surface glycoprotein multidrug resistance protein 1 (MDR1), into their lumen. However, the applicability of this protocol to model biliary diseases was not investigated [42].
Similarly, De Assuncao and colleagues were able to differentiate cholangiocytes from iPSCs, with the introduction of SHH and the NOTCH ligand Jagged-1 during the hepatic endoderm specification, and the addition of TGFβ in the final stages of cholangiocyte maturation [43]. Using RNA sequencing the authors confirm several aspects of biliary development throughout every stage of their differentiation protocol with the limitation that the maturation level reached by iPSC-derived cholangiocytes is compared to those of cholangiocyte cell lines (i.e H69 and NHC) rather than freshly isolated biliary cells. The main innovation of this work was that iPSC-cholangiocytes derived with their protocol, were able to engraft the mouse liver and form new duct-like structures when transplanted using retrograde intra-biliary infusion in a mouse liver [43].
Two more recent studies followed the above ones, focusing on the generation of iPSC-derived cholangiocytes in three-dimensional conditions and with an application to model biliary diseases [33,44].
Sampaziotis and colleagues introduced a combination of fibroblast growth factor (FGF)-10 along with activin and retinoic acid to induce differentiation of hepatoblasts into early cholangiocyte-like cells or cholangiocytes progenitors (CPs) [33]. To promote further maturation, CPs were grown in three-dimensional conditions using matrigel, thus allowing the formation of cystic organoids and branching tubular structures organized in a single epithelial layer with a lumen inside. These structures express both mature and fetal biliary markers and exhibit a range of functions similar to the native biliary epithelium such as increased proliferation in response to VEGF, secretion of Rhodamine 123 through MDR1, active export of the fluorescent bile acid cholyllysyl-fluorescein (CLF) from the lumen and GGT and ALP activities [33].
Ogawa and colleagues followed a similar approach with the addition of a co-culture step to stimulate the NOTCH signaling pathway [44]. Chimeric aggregates of hepatoblasts and OP9 stromal cells, known to express different NOTCH ligands, were cultured in a mixture of collagen and matrigel. Under these conditions and following exposure to HGF, EGF and TGFβ, the cells organized in cyst and duct structures containing a single layer of epithelial-like cells with an internal lumen and expressing cholangiocytes markers with apico-basal polarity. Interestingly the formation of the cysts was abolished in the presence of gamma secretase inhibitor (GSI), a NOTCH pathway antagonist.
Both these protocols were applied to model biliary diseases (i.e ADPKD and cystic fibrosis) and will be discussed in detail [44].
As described in the studies above, cholangiocytes can be generated from iPSC in monolayer or in 3D cultures of organoids/spheroids. Three-dimensional approaches are based on the suspension of hepatoblasts or cholangiocyte-progenitor cells in matrigel that allows the formation of 3D structures that recapitulate the spatial organization of the organ. However, the spheroid configuration limits the accessibility of the apical domain of the structure, and the presence of matrigel introduces undefined variables in the culture [45]. On the other hand, a cell monolayer gives access to the entire cell surface but limits a polarized organization and function of the cells.
Based on these premises our group has focused on optimizing the current culture methods to increase the biological properties of cholangiocytes derived from iPSC and develop a functional polarized epithelium [46]. iPSC-derived cholangiocytes were cultured on porous filter supports, to mimic in vivo apical-basal polarity and to prime their functional maturation. In these culture conditions, cholangiocytes grow apical primary cilia and acquire a tight barrier function as confirmed by the measurement of the trans-epithelial resistance (TER) (over 1000 Ω·cm2) and the expression of tight junction proteins (i.e ZO-1) restricted at the apical cell junctions. Secretory signaling functions, such as response to the hormone secretin when administered basolaterally, and c-AMP dependent apical secretion of fluid, further confirmed cholangiocytes maturation. Furthermore, polarized monolayers of iPSC-cholangiocytes maintain their functions when expanded and sub-cultured for several passages. This is an important step forward to reduce the cost of differentiation and the inter-variability among different experiments [46].
Contrary to hepatocytes, that are still lacking proof of full maturation, it appears that iPSC-derived cholangiocytes better reproduce the biological properties of the adult tissue, possibly suggesting a default fate program toward cholangiocyte in the differentiation process. These results highlight the enormous potential of using iPSC as a system to model in vitro biliary diseases, an application that will be further discussed in a separate paragraph.
2.3. Non-parenchymal cells (NPCs) from iPSCs
Non-parenchymal liver cells including Kupffer cells (KC), hepatic stellate cells (HSC) and sinusoidal endothelial cells (LSEC), represent an essential cellular component to reproduce liver function in vitro. Primary cultures of NPCs are quite a challenge since their isolation relies on the availability of surgical specimens and a non-specific activation of these cells when cultured in vitro might be an issue. Therefore, the generation of mature NPCs cells from iPSCs would be an ideal solution to overcome all these issues.
Only one group so far has developed a protocol for the generation of LSECs and HSCs from iPSCs [47]. To obtain mature LSEC the authors followed an already established protocol for the induction of iPSCs into mesoderm, and the generation of progenitor cells positive for the endothelial marker CD34. These cells morphologically resemble endothelial cells and can be expanded in culture for multiple passages maintaining high expression levels of the LSEC progenitor markers FLK1, CD34, CD31, CDH5, STAB1 and LYVE1. To induce further maturation, these progenitors cells are cultured in presence of TGFβ inhibitor and hypoxic conditions that promote the acquisition of mature endothelial morphology and expression of F8, STAB2 and FCGR2B [47].
Also HSC originate from the mesoderm. The same authors used in this case the surface marker ALCAM to select HSC progenitor cells that are then further differentiated into mature HSC by inhibiting the Rho signaling pathway with Y27632. Under these culture conditions cells acquire mature HSC morphology, expression of specific mature markers such as NGFR, LRAT and HGF and are able to store Vitamin A as suggested by the presence of Vitamin A droplets [47]. Despite the successful results in co-culture experiments with hepatocytes, further studies to confirm the maturation of these cells and the efficacy of this system for disease modeling and drug therapy will be necessary.
3. Modeling of genetic and complex liver diseases
iPSCs can be generated directly from patients with specific diseases and their differentiation program allows to recapitulate in vitro the progression of the disease, thus making them an ideal tool to model liver diseases, better if genetic.
Several groups have applied iPSCs technology to model liver diseases and have provided proof of principle of its efficacy in understanding the pathophysiology of a broad range of disorders. An example is the work from Rashid et al. in which hepatocytes are derived from iPSC of patients with different inherited metabolic disorders (i.e A1AT deficiency, familial hypercholesterolemia and Glycogen Storage Disease Type 1a (GSD1a)). In this work, the authors were able to reproduce in vitro key cellular aspects of the three different diseases such as the retention of polymers of A1AT in the ER, the impaired ability to incorporate LDL, and the accumulation of intracellular glycogen [48]. In a different study, Zhang and colleagues demonstrate that iPSC-derived hepatocytes from a patient with Wilson’s disease, present with altered cytoplasmic localization of the liver transporter protein ATP7B and defects in copper export, accurately reproducing the disease phenotype [49].
As for the other epithelial component of the liver, three studies show that iPSC- derived cholangiocytes can model in vitro different genetic cholangiopathies. Sampaziotis et al. used iPSC-derived organoids from a patient with polycystic liver disease (PLD) to validate in vitro the effect of Ocreotide, a drug already used in clinic to reduce cyst size in PLD patients [50].
Cystic fibrosis (CF) cholangiopathy is an autosomic recessive disease caused by mutation of the gene coding for cystic fibrosis transmembrane conductance regulator (CFTR), a chloride channel that regulates fluid secretion in cholangiocytes. Both Sampaziotis and Ogawa have shown that cholangiocytes derived from iPSC of a patient carrying the common CFTR mutation ΔF508 reproduce in vitro the secretory defect that can be partially corrected by the use of clinically approved small corrector molecules.
More interestingly, in our recent study, using iPSC-derived polarized cholangiocyte monolayers to model human CF liver disease (ΔF508 mutation), we have identified aberrant innate immune pathways that in addition to the secretory defect, are crucial in the development of liver disease in CF [46]. CF human cholangiocytes show an increase NF-kB activation in response to LPS followed by an aberrant production of inflammatory mediators. We have identified the persistent activation of the Src family of tyrosine kinases (SFK), as a pathogenetic event in the inflammatory response of CF cholangiocytes. Interestingly, inhibition of Src in combination with the FDA approved CFTR potentiators/correctors mentioned above, was able to recover several cholangiocyte funtions (i.e secretion, barrier function, changes in cytoskeletal structure) [46]. This study is a proof that disease modeling by using iPSC represents the next generation tool to investigate pathogenetic mechanism of human diseases.
More recently, Guan and colleagues have used 3D organoids composed of hepatocytes and cholangiocyte-like cells organized as a layer of epithelia surrounding the lumen of bile duct-like structures, to study the effect of Jagged1 gene mutations responsible for Alagille syndrome (ALGS) [51]. ALGS cultures fail to develop tubular structures and mainly consist of vesicles lined by hepatocytes, recapitulating the disease phenotype. Moreover, also mRNA expression of NOTCH2 and its target genes Hey1 and HES1 is reduced while JAG1 mRNA decreases significantly during the different developmental stages [51].
All the genetic diseases modeled so far are monogenic diseases. Complex liver disorders also represent a major challenge in the field, since not only the genetic component is involved but also environmental factors and multiple cell types can influence the disease phenotype. Primary biliary cirrhosis (PBC) and Primary Sclerosing Cholangitis (PSC) represent an example of complex liver disorders whose etiology and pathophysiology remain still unclear [41,52]. iPSCs-based systems could represent an optimal in vitro tool to study the complex cellular interactions in co-culture systems with different cells derived from the same patient (i.e immune and mesenchimal cells). In addition, the comparison with other cell culture systems (i.e 3D organoids) could help discriminate the impact of the genetic component versus its environmental counterpart on the pathogenesis of these diseases. Studies on PBC and PSC are eagerly awaited.
3.1. Drug toxicity
Drugs or their intermediary metabolites can have a toxic effect on the liver, making drug induced liver injury (DILI) the leading cause of acute liver failure in the United States alone and the major reason for drug withdrawal from the market [53]. The search of in vitro models that could predict liver toxicity has been so far a challenge. Primary hepatocytes are considered the “gold standard” for drug metabolism and drug toxicity screenings, but their availability is still dependent on human donors and whether their phenotype and metabolic function are instable in long-term culture conditions, which that vary from batch to batch.
The use of iPSCs-derived hepatocytes for drug toxicity screenings has recently become an attractive tool and indeed several groups have shown their ability to distinguish between hepatotoxins and liver safe compounds [54–56]. Holmgren and colleagues have used iPSCs derived hepatocytes as a system to study long-term toxicity of hepatotoxic compounds, an unmet need of the pharmaceutical industry that is not possible with primary human hepatocyte cultures. The authors show that indeed iPSC-derived hepatocytes have time-dependent increased sensitivity to amiodarone, aflatoxin B1, and troglitazone [57].
In another recent study, hepatocytes derived from iPSC of a patient with familial hypercholesterolemia were used to screen 2340 already existing drugs for repurposing. Thanks to this study the authors have been able to identify cardiac glycosides as a potential therapeutic target for the treatment of the disease [58].
Genetic factors or specific polymorphisms that modulate cytochrome P450 (CYP) activity can affect drug sensitivity among different individuals. Using iPSCs bearing different polymorphisms identified in a patient, Takayama and colleagues generated hepatocyte-like cells and demonstrated that a single nucleotide polymorphism in the cytochrome subunit CYP2D6 is able to create a higher resistance to tamoxifen, which is metabolized into a toxic metabolite, and also a higher susceptibility to Desipramine, which is instead detoxified by CYP2D6 [59].
All this data indicate that the essential drug-metabolizing machinery is present in iPSCs derived hepatocytes. However, the lack of expression of more mature isoforms of CYP450 when compared to primary hepatocytes still represents an obstacle that needs to be tackled in order to exploit the full potential of HLC as a reproducible and renewable platform for modeling hepatotoxicity and DILI.
3.2. Gene editing
The development of a gene-editing platform has enabled us to introduce changes in the DNA with extreme precision. Zinc finger nucleases (ZNFs), transcription activator-like effector nucleases (TALENs) and most recently CRISPR/Cas9 offer the possibility to correct mutations that cause a disease, to restore the expression of genes that are absent and to remove deleterious genes or viral genomes [60–62]. As for the liver this platform could be applied to treat monogenic inherited disorders, complex genetic disorders but also to study pathogens that exclusively target human hepatocytes such as hepatitis B virus. iPSCs are directly derived from the patient and maintain the same genetic background of the individual, thus representing the ideal tool for gene editing applications and to test the effects of specific mutations in genes whose function is still unknown. After gene correction the healthy isogenic cell lines can be differentiated into the specific liver cell and ideally be used for gene-replacement therapies.
As a proof of concept this technology has found applicability in the correction of mutations in the gene SERPINA1 in iPSC derived from patients with alpha-1 antitrypsin deficiency. Two separated studies show that hepatocyte-like cells (HLC) derived from the corrected iPSC restored their function and reduced the expression of intracellular AAT to levels comparable to normal controls [63,64]. Zhang et al., in a different study have used a self-inactivating viral vector to correct a specific point mutation in the ATP7B gene responsible for Wilson’s disease. HLC derived from the corrected iPSC restored the functional defect in vitro [49]. Using the CRISPR/Cas9 technology Guan et al. were able to correct the genetic defect in iPSCs derived from patients with Alagille syndrome, by reverting a mutation in Jag1, and recovering the disease phenotype. Interestingly, they also introduced the same mutation in a normal control and show that the mutation is indeed responsible for the phenotype [51].
Taken together these exciting results indicate that gene therapy in the clinic and autologous cell replacement therapies could be a potential resource in the future for targeting liver disorders.
3.3. iPSC derived liver cells: what are we still missing?
This review of the different protocols to generate hepatic cells from iPSCs indicates that iPSCs are an excellent system to model liver disease in vitro and represent an enormous step forward in the liver field where the only potential source of primary cells has been surgical samples derived from patients undergoing liver transplant. iPSCs can be successfully differentiated into hepatocytes and cholangiocytes, and can be used to study the mechanisms of human disease and development, high-throughput screening of new therapeutic compounds and in the future for cell replacement therapies (Table 1). On this end, several efforts are already being made to develop clinical grade cells compatible with good manufacturing practices (GMPs) for regenerative medicine purposes (Table 2). However, there are still several issues that need to be addressed before iPSCs derived hepatic cells can reach their full potential. As for hepatocytes the major issue is represented by their immature phenotype even though different steps are being made toward solving this issue with the introduction of co-culture 3D systems, bioengineered scaffolds and extracellular matrices. Another important aspect to address is the limited ability of hepatocyte-like cells to repopulate the liver of animal models for future cell replacement therapies. Interestingly the possibility to achieve direct reprogramming of fibroblasts into hepatic cells in vivo offers a new alternative and hope for the treatment of liver fibrosis but several aspects need to be improved to maximize the efficacy of this technique.
Table 1.
Summary of advantages and limitations of iPSC and liver organoid technology and their applicability for in vitro studies.
| Model | Advantages | Limitations | Applications |
|---|---|---|---|
| iPSC-derived liver cells | • Unlimited source of patient-derived cells | iPSC-hepatocyte like cells | • Disease modeling |
| • Same genetic background of the patient | • Persistent expression of fetal markers | • High throughput drug screenings | |
| • Non-invasive derivation from skin fibroblasts, blood cells and urine | • Lack of more mature cyt p450 isoforms | • Gene editing for correction therapy | |
| • Recapitulate the different steps of liver development | • Limited ability to repopulate the liver of animal models | • Replacement therapies | |
| iPSC-derived cholangiocytes | |||
| • Limited accessibility to the apical domain when in spheroid configuration | |||
| Liver Organoids | • 3D spatial organization | • Monolayer maturation only when polarized | |
| • Genetic and epigenetic signature of the patient | • Homogeneous population of cells that requires further differentiation | ||
| • Maintain genetic stability in culture | • Need of matrigel matrix for 3d aggregation | ||
| • Cryopreservation | • Lack of studies in co-culture systems | ||
| • Bio-banking |
Table 2.
Possible applications of iPSC/liver organoid technologies for liver diseases treatment based on the current literature.
| Liver disease | Cell model | In vitro disease modeling | Potential drug testing | Gene editing and autologous cell replacement |
|---|---|---|---|---|
| A1AT | iPSC | Yes | Mutation correctors | Yes |
| Wilson’s disease | iPSC | Yes | Yes | |
| PFIC | iPSC/liver organoids | Yes | Mutation correctors | Yes |
| Cystic fibrosis liver disease (CFLD) | iPSC/liver organoids | Yes | Mutation correctors, Src kinase inhibitors, TLR4/NF-kB inhibitors, nuclear receptor agonists (i.e PPARγ) | Yes |
| MDR3 deficiency | Yes | Mutation correctors | Yes | |
| PSC | iPSC/liver organoids | Yes | nuclear receptor agonists (i.e FXR, PPARα, NF-kB inhibitors, therapeutic bile acids | No |
| PBC | iPSC/liver organoids | Yes | nuclear receptor agonists (i.e FXR, PPARα, NF-kB inhibitors, therapeutic bile acids | No |
| BA | iPSC/liver organoids | Yes | none | Engineered biliary tissue replacement |
| AGS | iPSC/liver organoids | Yes | none | Yes |
| Polycystic liver diseases (ADPKD, ADPLD, ARPKD, CD, CHF) | iPSC/liver organoids | Yes | Somatostatin analogs, PPARγ agonists, VEGF signaling inhibitors, mTOR inhibitors, anti-fibrotic drugs, NF-kB inhibitors | No |
While hepatocytes represent the major component of the liver, it is important not to forget the rest of the cells that contribute to the complex liver architecture. For this reason, the possibility to generate cholangiocytes from iPSCs has provided scientists with an efficient in vitro tool to study biliary diseases and evaluate potential therapeutic targets, improving our understanding of cholangiopathies. Most importantly the introduction of biliary epithelial cells should be considered when attempting to generate 3D structures or bioengineered livers.
4. 3D culture systems and liver organoids
As highlight by the magazine The Economist in “The year of the organoid-The World in 2016”, another recent advance of the stem cell technology is the possibility to generate organoids. Organoids are based on the three-dimensional culture systems of stem cells from several different sources (iPSC, embryonic stem cells, adult stem cells and tissue-derived stem cells). The use of a 3D system offers the advantage to better preserve the cell-to cell contacts that allow cells to directly interact with each other and self-aggregate to reproduce the cellular organization of the organ. The majority of these methods are based on the use of matrigel matrix, a solubilized basement membrane preparation extracted from the Engelbreth-Holm-Swarm (EHS) mouse sarcoma [65] that is the major hint to induce maturation. In past years, several protocols have been developed for the generation of organoids cultures from iPSC-derived liver cells and from adult liver tissue.
Two separate studies have used 3D culture conditions and matrigel to differentiate cholangiocytes derived from iPSC. Both studies were able to recapitulate the in vivo organization of cholangiocytes, as a single epithelial layer with a lumen and the expression of biliary specific markers [33] [44]. The culture in 3D conditions was improving cholangiocytes maturation but, in both cases, the end-point 3D structures were not propagated in culture.
Another interesting study from Song et al. showed a system to induce 3D cell aggregates of iPDC-derived hepatocytes and stromal cells just using a microwell platform without any kind of matrix. These aggregates are then encapsulated in alginate capsule and transplanted into the peritoneal cavity of immune competent mouse therefore inducing further maturation of the hepatocytes [66].
Liver organoids have also been generated from adult somatic tissue-resident stem cells. In this specific case, they are derived directly from the liver tissue of the patient and represent an ideal tool for disease modeling, to test the efficacy and toxicity of drug compounds and they could offer an autologous source of cells for transplantation [67].
Based on the previous methodology developed by Clevers group, for the generation of Lgr5+ intestinal organoids, Broutier et al. have developed a system to obtain liver organoids from EpCam+ ductal cells from normal or diseased human livers [67]. These cells, when grown in matrigel and in low attachment plate in the presence of EGF, HGF, FGF and RSPO1, organize in 3D organoids with biliary progenitor identity that can possibly be differentiated toward the hepatic lineage. Contrary to iPSC-derived organoids, the advantage of this system is that tissue organoids can be cultured for long term (> 1 year) still maintaining their genetic stability, can be cryopreserved and easily recovered after thawing, simplifying the process of cell banking [67].
In another recent study liver organoids were generated from the extrahepatic portion of the biliary tree (ECOs) [68]. Cells isolated by brushing or scraping the bile duct luminal surface grow in matrigel with a similar cocktail of specific growth factors and generate organoid structures. Interestingly, ECOs have the capacity to repopulate tubular polyglycolic acid (PGA) scaffolds, and still maintain their functionality and marker expression, thus representing a bioengineered tissue resembling the biliary epithelium. When transplanted in vivo in a mouse, whose midportion of the common bile duct (CBD) is removed, these bioengineered ducts are able to achieve biliary reconstruction. The ability of ECOs to replace a damaged CBD is a promising result for future regenerative medicine applications to manage liver diseases [68].
Organoids derived from the liver tissue are composed by a homogeneous population of cells rather than by multiple cell types and eventually they would require further differentiation toward the hepatocytes or cholangiocyte fate.
An attempt to generate organoids that mimic the human liver bud with multiple cell types has been otherwise done by Takebe and colleagues [69]. In this approach, the starting cell population is represented by iPSC that are differentiated into hepatic endoderm in a 2D monolayer. Subsequently, hepatic cells are co-cultured with human umbilical vein endothelial cells (HUVEC) and human mesenchymal stem cells (MSC) on matrigel coated plates. Under these conditions, the cells self-assemble to generate 3D clusters defined as liver buds since they resemble the human liver bud stage during liver development. When transplanted ectopically in NOD/SCID mice, the liver buds connect with the host vessels and become vascularized, improving their maturation and showing the presence of human specific metabolites in the serum of the mice. Excitingly, when transplanted in the mesentery, they improve survival in a TK-NOG mouse model of gancyclovir-induced liver failure [69].
Although a limitation is represented by the expression profile of the liver buds, that resembles more a fetal rather than an adult liver, these results still support the potential applicability of this model for drug responsiveness and future replacement therapies.
4.1. Liver organoids: what are we still missing?
Organoids are a unique tool to study human development and disease since they can recapitulate the original organ structure and perform specific tissue functions (Table 1). They also have the potential to be used for drug testing and toxicity, since the human liver metabolizes drugs in a different manner than other animals. Last but not least, they could offer an unlimited source of cells for cell replacement therapies since they maintain their genetic stability in culture (Table 2). When we refer to liver tissue organoids, however, some limitations still need to be addressed and kept in mind (Table 1).
First, the need of an extracellular matrix that drives the cells to aggregate into a 3D structure. So far, most of the organoids cultures, including liver ones, require for their 3D assembly, Matrigel or other animal derived type of matrices, whose composition is not defined and introduces undefined variables when adapting culture conditions. This poses a limitation for their potential use in regenerative medicine and other clinical applications. Further efforts will be required toward the development of new bioengineered synthetic platforms that allow the self-aggregation of liver cells in 3D structures without losing their fate specification. The liver is a relatively new field in the world of organoids, however similar issues are being addressed also in other fields (i.e intestine, pancreas) [45,70] and will certainly be an example to overcome this limitation.
A second limitation is that liver organoids do not fully recapitulate multiple cell types present in the liver and their level of maturation. Although, the original study in which the methodology has been developed, shows the possibility of differentiating liver organoid toward an hepatocytic or a biliary cell fate [67], no other studies have followed using this system for similar applications. The actual phenotype of liver organoids is more close to biliary cells and unless better conditions are defined to differentiate these cells toward mature hepatocytes, their applicability for example for drug testing or cell replacement could be limited.
What we are instead eagerly missing is the generation of co-culture systems to study the cross-talk of the epithelial component of liver-derived organoids with other cell types of the liver (i.e mesenchymal cells, immune cells, endothelial cells). Once again the intestinal field is more advance on this task. Indeed several studies have shown that it is possible to co-culture intestinal organoids with different type of immune cells. By culturing innate lymphoid cells (ILCs), Lindemans et al. have identified the mechanism initiated by immune cells to promote epithelial regeneration mediated by IL-22 [71]. Interestingly a study from another group added even more complexity to a similar system by including a bacterial component to the co-culture and showing that a specific strain of Lactobacillus can stimulate IL-22 secretion by lymphocytes of the lamina propria and therefore exert a protective effect for the intestinal barrier [72].
5. Conclusions
The search for in vitro systems to model liver physiology and pathophysiology has been a long and still incomplete quest. We have witnessed a considerable improvement in the last decade thanks to the opportunity to derive liver cells from iPSC and to the generation of liver-tissue organoids (Fig. 1). As reviewed in this article these novel technologies have covered important gaps in the field, such as the availability of a larger amount of cells derived from disease livers and their relatively high stability in cultures. iPSC and liver organoids have certainly shown to be excellent tools to model monogenic diseases and together with novel technologies of genome editing they will soon find applicability for personalized medicine. We still have no proof that iPSC derived liver cells can model more complex diseases since they are not derived from the diseased environment. However, this limitation could be overcome by using co-culture system that mimic the cross-talk of epithelial cells with the immune and mesenchymal component. On this respect, the field is rapidly evolving and we can benefit from the experience with other organs (i.e intestine, pancreas).
Fig. 1.
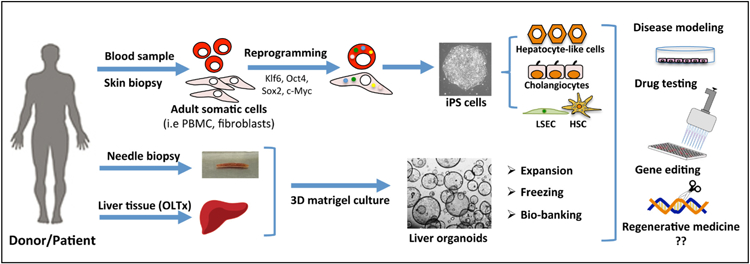
Generation of liver cells from iPSC and liver organoids and their applications. The scheme represents an overview of iPSC and organoid technology in relation to liver diseases. iPSC are generated with non-invasive procedures from patients and healthy donors and can be differentiated in liver cells (i.e hepatocyte-like cells, cholangiocytes, LSEC and HSC). Liver organoids are isolated from explanted liver tissues or from needle biopsies. Both technologies can be used for disease modeling, drug testing, genome editing and hopefully in the future for regenerative medicine.
Encouraging results come from the use of iPSC and organoids for drug testing. The continuous effort of bioengineers to create biomaterials and extracellular matrix able to improve the maturation of these cells will allow the generation of “clinical grade” platform in substitution of primary or immortalized cell cultures.
Still an open question is the applicability of these technologies to regenerative medicine. The promises are an increase number of cells available, no need for immunosuppression, cell autologous correction of mutations by gene editing. The concerns are that a low efficiency of engraftment with a lack of functional stability and the critical question about their long-term safety. These are issues that are currently being addresses and will require the combine effort of biologists, bioengineers and clinicians. The development of bioengineer platforms aim to preserve the environmental cues necessary for cells to grow will accelerate the process.
Overall, iPSC derived liver cells and liver organoids represent certainly two different and possibly complementary approaches to model in vitro a tissue, and as long as their limitations are understood and properly addressed in the experimental plan, we foresee that they will soon reach clinical applicability.
Acknowledgement
The authors would like to thank Leslie R. Martinez for editorial assistance.
Funding
This work was supported by the National Institute of Health (RO1DK096096, RO!DK079005, RO1DK101528, DK034989 Silvio O. Conte Digestive Diseases Research Core Center); by Partners Seeking a Cure Foundation and a grant from Connecticut Innovations (16-RMA-YALE-26).
Abbreviations:
- FGF
Fibroblasts growth factor
- BMP2–4
Bone Morphogenetic Protein 2–4
- HGF
Hepatocyte growth factor
- TGF-β
Transforming growth factor beta
- SOX9
SRY-box 9
- CK18
Keratin, type I cytoskeletal 18
- LDL
Low-density lipoprotein
- EGF
epidermal growth factor
- AFP
alpha-feto protein
- DMSO
Dimethyl sulfoxide
- IL-6
Interleukin 6
- GGT
Gamma-glutamyltransferase
- SHH
Sonic hedgehog
- NHC
Normal human cholangiocytes
- VEGF
Vascular endothelial growth factor
- ALP
alkaline phosphatase
- ADPKD
autosomal dominant polycystic kidney disease
- cAMP
Cyclic adenosine monophosphate
- ZO-1
zonula occludens-1
- LPS
lipopolysaccharides
- FLK1
Fetal liver kinase 1
- ALCAM
Activated Leukocyte cell adhesion molecule
- CD34,CD31
Hematopoietic progenitor cell antigen CD34, CD31
- CDH5
Cadherin-5
- STAB1
Stabilin-1
- LYVE
Lymphatic vessel endothelial hyaluronan receptor 1
- FCGR2B
Low affinity immunoglobulin gamma Fc region receptor II-b
- NGFR
Tumor necrosis factor receptor superfamily member 16
- LRAT
Lecithin retinol acyltransferase
- Y27632
Rock inhibitor
- ATP7B
Copper-transporting ATPase 2
- NF-kB
nuclear factor kappa-light-chain-enhancer of activated B cells
- HEY1
Hairy/enhancer-of-split related with YRPW motif protein 1
- HES1
hairy and enhancer of split-1
- FDA
U.S. Food and Drug Administration
- LGR5
leucine-rich repeat-containing G-protein coupled receptor 5
- EpCAM
epithelial cell adhesion molecule
- RSPO1
R-spondin 1
- IL-22
interleukin 22
Footnotes
This article is part of a Special Issue entitled: Animal Models in Liver Disease edited by Peter Fickert and Martin Wagner.
Transparency document
The Transparency document associated with this article can be found, in online version.
References
- [1].Malarkey DE, Johnson K, Ryan L, Boorman G, Maronpot RR, New insights into functional aspects of liver morphology, Toxicol. Pathol 33 (2005) 27–34. [DOI] [PubMed] [Google Scholar]
- [2].Ishibashi H, Nakamura M, Komori A, Migita K, Shimoda S, Liver architecture, cell function, and disease, Semin. Immunopathol 31 (2009) 399–409. [DOI] [PubMed] [Google Scholar]
- [3].Gerets HH, Tilmant K, Gerin B, Chanteux H, Depelchin BO, Dhalluin S, Atienzar FA, Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins, Cell Biol. Toxicol 28 (2012) 69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guo L, Dial S, Shi L, Branham W, Liu J, Fang JL, Green B, Deng H, Kaput J, Ning B, Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes, Drug Metab. Dispos 39 (2011) 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mariotti V, Strazzabosco M, Fabris L, Calvisi DF, Animal models of biliary injury and altered bile acid metabolism, Biochim. Biophys. Acta 1864 (2018) 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Strazzabosco M, Fabris L, Development of the bile ducts: essentials for the clinical hepatologist, J. Hepatol 56 (2012) 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fabris L, Cadamuro M, Libbrecht L, Raynaud P, Spirli C, Fiorotto R, Okolicsanyi L, Lemaigre F, Strazzabosco M, Roskams T, Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development, Hepatology 47 (2008) 718–728. [DOI] [PubMed] [Google Scholar]
- [8].Morell CM, Fabris L, Strazzabosco M, Vascular biology of the biliary epithelium, J. Gastroenterol. Hepatol 28 (Suppl. 1) (2013) 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ober EA, Lemaigre FP, Development of the liver: insights into organ and tissue morphogenesis, J. Hepatol 68 (2018) 1049–1062. [DOI] [PubMed] [Google Scholar]
- [10].Lemaigre FP, Mechanisms of liver development: concepts for understanding liver disorders and design of novel therapies, Gastroenterology 137 (2009) 62–79. [DOI] [PubMed] [Google Scholar]
- [11].Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS, Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control, Genes Dev 10 (1996) 1670–1682. [DOI] [PubMed] [Google Scholar]
- [12].Jung J, Zheng M, Goldfarb M, Zaret KS, Initiation of mammalian liver development from endoderm by fibroblast growth factors, Science 284 (1999) 1998–2003. [DOI] [PubMed] [Google Scholar]
- [13].Rossi JM, Dunn NR, Hogan BL, Zaret KS, Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm, Genes Dev 15 (2001) 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Berg T, Rountree CB, Lee L, Estrada J, Sala FG, Choe A, Veltmaat JM, De Langhe S, Lee R, Tsukamoto H, Crooks GM, Bellusci S, Wang KS, Fibroblast growth factor 10 is critical for liver growth during embryogenesis and controls hepatoblast survival via beta-catenin activation, Hepatology 46 (2007) 1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McLin VA, Rankin SA, Zorn AM, Repression of Wnt/beta-catenin signaling in the anterior endoderm is essential for liver and pancreas development, Development 134 (2007) 2207–2217. [DOI] [PubMed] [Google Scholar]
- [16].Wang Z, Dolle P, Cardoso WV, Niederreither K, Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives, Dev. Biol 297 (2006) 433–445. [DOI] [PubMed] [Google Scholar]
- [17].Si-Tayeb K, Lemaigre FP, Duncan SA, Organogenesis and development of the liver, Dev. Cell 18 (2010) 175–189. [DOI] [PubMed] [Google Scholar]
- [18].Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ, Notch signaling controls liver development by regulating biliary differentiation, Development 136 (2009) 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, Stanger BZ, Jacquemin P, Pierreux CE, Clotman F, Lemaigre FP, Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9, Gastroenterology 136 (2009) 2325–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ellis J, Bhatia M, iPSC technology: platform for drug discovery, Point Clin. Pharmacol. Ther 89 (2011) 639–641. [DOI] [PubMed] [Google Scholar]
- [21].Sterneckert JL, Reinhardt P, Scholer HR, Investigating human disease using stem cell models, Nat. Rev. Genet 15 (2014) 625–639. [DOI] [PubMed] [Google Scholar]
- [22].Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R, Reprogramming of human peripheral blood cells to induced pluripotent stem cells, Cell Stem Cell 7 (2010) 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA, Generation of human induced pluripotent stem cells from urine samples, Nat. Protoc 7 (2012) 2080–2089. [DOI] [PubMed] [Google Scholar]
- [24].Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA, Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells, Hepatology 51 (2010) 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, Song X, Guo Y, Zhao Y, Qin H, Yin X, Wu C, Che J, Lu S, Ding M, Deng H, Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells, Cell Res 19 (2009) 1233–1242. [DOI] [PubMed] [Google Scholar]
- [26].Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, Weber A, Vallier L, Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development, Hepatology 51 (2010) 1754–1765. [DOI] [PubMed] [Google Scholar]
- [27].Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N, Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers, Mol. Med 6 (2000) 88–95. [PMC free article] [PubMed] [Google Scholar]
- [28].Lavon N, Yanuka O, Benvenisty N, Differentiation and isolation of hepatic-like cells from human embryonic stem cells, Differentiation 72 (2004) 230–238. [DOI] [PubMed] [Google Scholar]
- [29].Schwartz RE, Linehan JL, Painschab MS, Hu WS, Verfaillie CM, Kaufman DS, Defined conditions for development of functional hepatic cells from human embryonic stem cells, Stem Cells Dev 14 (2005) 643–655. [DOI] [PubMed] [Google Scholar]
- [30].Gerbal-Chaloin S, Funakoshi N, Caillaud A, Gondeau C, Champon B, Si-Tayeb K, Human induced pluripotent stem cells in hepatology: beyond the proof of concept, Am. J. Pathol 184 (2014) 332–347. [DOI] [PubMed] [Google Scholar]
- [31].Hannan NR, Segeritz CP, Touboul T, Vallier L, Production of hepatocyte-like cells from human pluripotent stem cells, Nat. Protoc 8 (2013) 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH, Baharvand H, Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells, Stem Cell Rev 6 (2010) 622–632. [DOI] [PubMed] [Google Scholar]
- [33].Sampaziotis F, Cardoso de Brito M, Madrigal P, Bertero A, Saeb-Parsy K, Soares FA, Schrumpf E, Melum E, Karlsen TH, Bradley JA, Gelson WT, Davies S, Baker A, Kaser A, Alexander GJ, Hannan NR, Vallier L, Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation, Nat. Biotechnol 33 (2015) 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shan J, Schwartz RE, Ross NT, Logan DJ, Thomas D, Duncan SA, North TE, Goessling W, Carpenter AE, Bhatia SN, Identification of small molecules for human hepatocyte expansion and iPS differentiation, Nat. Chem. Biol 9 (2013) 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Siller R, Greenhough S, Naumovska E, Sullivan GJ, Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells, Stem Cell Reports 4 (2015) 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tahamtani Y, Azarnia M, Farrokhi A, Sharifi-Zarchi A, Aghdami N, Baharvand H, Treatment of human embryonic stem cells with different combinations of priming and inducing factors toward definitive endoderm, Stem Cells Dev 22 (2013) 1419–1432. [DOI] [PubMed] [Google Scholar]
- [37].Baptista PM, Vyas D, Moran E, Wang Z, Soker S, Human liver bioengineering using a whole liver decellularized bioscaffold, Methods Mol. Biol 1001 (2013) 289–298. [DOI] [PubMed] [Google Scholar]
- [38].Gieseck RL 3rd, Hannan NR, Bort R, Hanley NA, Drake RA, Cameron GW, Wynn TA, Vallier L, Maturation of induced pluripotent stem cell derived hepatocytes by 3D-culture, PLoS One 9 (2014) e86372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, Bhatia SN, Invert molding for scalable control of tissue microarchitecture, Nat. Commun 4 (2013) 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Strazzabosco M, Fabris L, Spirli C, Pathophysiology of cholangiopathies, J. Clin. Gastroenterol 39 (2005) S90–S102. [DOI] [PubMed] [Google Scholar]
- [41].Strazzabosco M, Fiorotto R, Cadamuro M, Spirli C, Mariotti V, Kaffe E, Scirpo R, Fabris L, Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium, Biochim. Biophys. Acta 1864 (2018) 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dianat N, Dubois-Pot-Schneider H, Steichen C, Desterke C, Leclerc P, Raveux A, Combettes L, Weber A, Corlu A, Dubart-Kupperschmitt A, Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells, Hepatology 60 (2014) 700–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].De Assuncao TM, Sun Y, Jalan-Sakrikar N, Drinane MC, Huang BQ, Li Y, Davila JI, Wang R, O’Hara SP, Lomberk GA, Urrutia RA, Ikeda Y, Huebert RC, Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes, Lab. Investig 95 (2015) 684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, Grompe M, Keller G, Kamath BM, Ghanekar A, Directed differentiation of cholangiocytes from human pluripotent stem cells, Nat. Biotechnol 33 (2015) 853–861. [DOI] [PubMed] [Google Scholar]
- [45].Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordonez-Moran P, Clevers H, Lutolf MP, Designer matrices for intestinal stem cell and organoid culture, Nature 539 (2016) 560–564. [DOI] [PubMed] [Google Scholar]
- [46].Fiorotto R, Amenduni M, Mariotti V, Fabris L, Spirli C, Strazzabosco M, Src kinase inhibition reduces inflammatory and cytoskeletal changes in DeltaF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy, Hepatology 67 (2018) 972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Koui Y, Kido T, Ito T, Oyama H, Chen SW, Katou Y, Shirahige K, Miyajima A, An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells, Stem Cell Reports 9 (2017) 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L, Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells, J. Clin. Invest 120 (2010) 3127–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y, Pan Q, Liu X, Zychlinski D, Lu H, Tortorella MD, Schambach A, Wang Y, Pei D, Esteban MA, Rescue of ATP7B function in hepatocyte-like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin, Hum. Mol. Genet 20 (2011) 3176–3187. [DOI] [PubMed] [Google Scholar]
- [50].Sturges NC, Wikstrom ME, Winfield KR, Gard SE, Brennan S, Sly PD, Upham JW, A.C.M.O.T.A.R.E.S.T.f.C. Fibrosis, Monocytes from children with clinically stable cystic fibrosis show enhanced expression of toll-like receptor 4, Pediatr. Pulmonol 45 (2010) 883–889. [DOI] [PubMed] [Google Scholar]
- [51].Guan Y, Xu D, Garfin PM, Ehmer U, Hurwitz M, Enns G, Michie S, Wu M, Zheng M, Nishimura T, Sage J, Peltz G, Human hepatic organoids for the analysis of human genetic diseases, JCI Insight 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lazaridis KN, Strazzabosco M, Larusso NF, The cholangiopathies: disorders of biliary epithelia, Gastroenterology 127 (2004) 1565–1577. [DOI] [PubMed] [Google Scholar]
- [53].Kaplowitz N, Idiosyncratic drug hepatotoxicity, Nat. Rev. Drug Discov 4 (2005) 489–499. [DOI] [PubMed] [Google Scholar]
- [54].Medine CN, Lucendo-Villarin B, Storck C, Wang F, Szkolnicka D, Khan F, Pernagallo S, Black JR, Marriage HM, Ross JA, Bradley M, Iredale JP, Flint O, Hay DC, Developing high-fidelity hepatotoxicity models from pluripotent stem cells, Stem Cells Transl. Med 2 (2013) 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sirenko O, Hesley J, Rusyn I, Cromwell EF, High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells, Assay Drug Dev. Technol 12 (2014) 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Szkolnicka D, Farnworth SL, Lucendo-Villarin B, Storck C, Zhou W, Iredale JP, Flint O, Hay DC, Accurate prediction of drug-induced liver injury using stem cell-derived populations, Stem Cells Transl. Med 3 (2014) 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Holmgren G, Sjogren AK, Barragan I, Sabirsh A, Sartipy P, Synnergren J, Bjorquist P, Ingelman-Sundberg M, Andersson TB, Edsbagge J, Long-term chronic toxicity testing using human pluripotent stem cell-derived hepatocytes, Drug Metab. Dispos 42 (2014) 1401–1406. [DOI] [PubMed] [Google Scholar]
- [58].Cayo MA, Mallanna SK, Di Furio F, Jing R, Tolliver LB, Bures M, Urick A, Noto FK, Pashos EE, Greseth MD, Czarnecki M, Traktman P, Yang W, Morrisey EE, Grompe M, Rader DJ, Duncan SA, A drug screen using human iPSC-derived hepatocyte-like cells reveals cardiac glycosides as a potential treatment for hypercholesterolemia, Cell Stem Cell 20 (2017) 478–489 (e475). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Takayama K, Morisaki Y, Kuno S, Nagamoto Y, Harada K, Furukawa N, Ohtaka M, Nishimura K, Imagawa K, Sakurai F, Tachibana M, Sumazaki R, Noguchi E, Nakanishi M, Hirata K, Kawabata K, Mizuguchi H, Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 16772–16777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cho SW, Kim S, Kim JM, Kim JS, Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease, Nat. Biotechnol 31 (2013) 230–232. [DOI] [PubMed] [Google Scholar]
- [61].Porteus MH, Carroll D, Gene targeting using zinc finger nucleases, Nat. Biotechnol 23 (2005) 967–973. [DOI] [PubMed] [Google Scholar]
- [62].Sander JD, Joung JK, CRISPR-Cas systems for editing, regulating and targeting genomes, Nat. Biotechnol 32 (2014) 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L, Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells, Nature 478 (2011) 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, Liu JO, Deng C, Ye Z, Jang YY, Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells, Hepatology 57 (2013) 2458–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Orkin RW, Gehron P, McGoodwin EB, Martin GR, Valentine T, Swarm R, A murine tumor producing a matrix of basement membrane, J. Exp. Med 145 (1977) 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Song W, Lu YC, Frankel AS, An D, Schwartz RE, Ma M, Engraftment of human induced pluripotent stem cell-derived hepatocytes in immunocompetent mice via 3D co-aggregation and encapsulation, Sci. Rep 5 (2015) 16884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, Huch M, Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation, Nat. Protoc 11 (2016) 1724–1743. [DOI] [PubMed] [Google Scholar]
- [68].Sampaziotis F, Justin AW, Tysoe OC, Sawiak S, Godfrey EM, Upponi SS, Gieseck RL 3rd, de Brito MC, Berntsen NL, Gomez-Vazquez MJ, Ortmann D, Yiangou L, Ross A, Bargehr J, Bertero A, Zonneveld MCF, Pedersen MT, Pawlowski M, Valestrand L, Madrigal P, Georgakopoulos N, Pirmadjid N, Skeldon GM, Casey J, Shu W, Materek PM, Snijders KE, Brown SE, Rimland CA, Simonic I, Davies SE, Jensen KB, Zilbauer M, Gelson WTH, Alexander GJ, Sinha S, Hannan NRF, Wynn TA, Karlsen TH, Melum E, Markaki AE, Saeb-Parsy K, Vallier L, Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids, Nat. Med 23 (2017) 954–963. [DOI] [PubMed] [Google Scholar]
- [69].Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H, Vascularized and functional human liver from an iPSC-derived organ bud transplant, Nature 499 (2013) 481–484. [DOI] [PubMed] [Google Scholar]
- [70].Candiello J, Grandhi TSP, Goh SK, Vaidya V, Lemmon-Kishi M, Eliato KR, Ros R, Kumta PN, Rege K, Banerjee I, 3D heterogeneous islet organoid generation from human embryonic stem cells using a novel engineered hydrogel platform, Biomaterials 177 (2018) 27–39. [DOI] [PubMed] [Google Scholar]
- [71].Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY, Takashima S, Hua G, Martin ML, O’Rourke KP, Lo YH, Mokry M, Romera-Hernandez M, Cupedo T, Dow L, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MRM, Hanash AM, Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration, Nature 528 (2015) 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hou Q, Ye L, Liu H, Huang L, Yang Q, Turner JR, Yu Q, Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22, Cell Death Differ (2018). [DOI] [PMC free article] [PubMed]