

INTRODUCTION
The earliest mention of the term cardiorenal syndrome (CRS) came about from a 2004 National Heart, Lung, and Blood Institute Working Group conference evaluating the interaction between the heart and kidney.1 The term is used to commonly refer to the collective dysfunction of heart and kidneys resulting in a cascade of feedback mechanisms causing damage to both the organs. Previously proposed definitions of CRS stressed the effects a diseased heart on causing dysfunction of the kidney with heart failure (HF) being the prototypical cardiovascular disease leading to kidney dysfunction from CRS. However, with further understanding of the pathophysiology of the disorder, it is now recognized that either the heart or the kidney can be the primary source of insult. Although the term CRS is often used globally to address the pathophysiologic interaction between the two organs, a recent classification of CRS proposed by the 7th Acute Dialysis Quality Initiate consensus conference has divided the syndromes into those that are “cardiorenal” (Figure 1) referring to when cardiac dysfunction leads to kidney dysfunction and those that are “renocardiac” (Figure 2) referring to when primary kidney dysfunction leads to cardiac dysfunction.2,3 These syndromes are further classified based on their acuity and the presence of a systemic (non-cardiac, non-renal) illness that may play a role in the pathophysiology (Figure 3). However, often the causal relationship (cardiorenal vs. renocardiac) may not be ascertainable when risk factors like diabetes, hypertension and atherosclerosis affect the function of both organs in parallel leading to a common clinical picture. 4,5
Figure. 1.

Pathophysiological interaction in acute cardiorenal syndrome (type 1) and acute renocardiac syndrome (Type 3).
CVP= central venous pressure; IAP= intraabdominal pressure; ACEi= Angiotensin converter enzyme inhibitor; RAAS= Renin angiotensin aldosterone system; SNS= Sympathetic nervous system; RBF= Renal blood flow; GFR= Glomerular filtration rate; NO= Nitrous oxide; ROS= Reactive oxygen species.
Figure. 2.

Pathophysiology of chronic cardiorenal syndrome (type 2) and chronic renocardiac syndrome (Type 4).
CVP= central venous pressure; IAP= intraabdominal pressure; ACEi= Angiotensin converter enzyme inhibitor; RAAS= Renin angiotensin aldosterone system; SNS= Sympathetic nervous system; RBF= Renal blood flow; GFR= Glomerular filtration rate; NO= Nitrous oxide; ROS= Reactive oxygen species.
Figure 3.
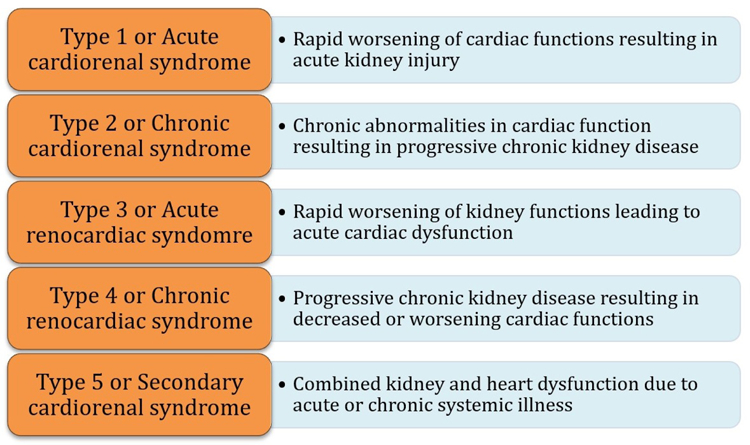
Types of Cardiorenal syndrome.
Given the aging population, longer cumulative exposure to common risk factors including hypertension, obesity, diabetes and vascular disorders, and advances in medical therapy, procedures and devices to assist patients with HF live longer, the prevalence of CKD and HF are likely to continue to rise. 6,7 It is hence important to understand the various mechanisms involved in the propagation of this syndrome. In this manuscript, the authors will review the role of the heart and kidney in the development of different CRS, and the interplay between the complex pathophysiologic pathways resulting in vicious cycle and end organ damage.
EVALUATING A CHANGE IN KIDNEY FUNCTION
Acute kidney injury (AKI) refers to an abrupt reduction of kidney function, resulting in the retention of the urea and other nitrogenous toxins, dysregulation of the electrolytes and retention of extracellular volume. Several consensus definitions of AKI have been established using serum creatinine and urine output to accurately identify the patients with AKI in clinical settings as well as in epidemiologic and outcomes studies. The Kidney Disease: Improving Global Outcomes (KDIGO) 8 definition is currently the most commonly used and is defined as follows:
Increase in serum creatinine by ≥0.3 mg/dl (≥26.5 micromol/l) in a 48 hours period, or
Increase in serum creatinine to ≥ 1.5 times baseline, which is known or presumed to have occurred within the prior seven days, or
Urine volume <0.5 ml/kg/hour for six hours
KDIGO criteria further stage AKI in to three categories as shown in Table 1. 8
Table 1.
KDIGO staging of Acute Kidney Injury
| Stage | Serum creatinine | Urine output |
|---|---|---|
| 1 | 1.5–1.9 times baseline or ≥ 0.3 mg.dL increase |
< 0.5 ml/kg.hr for 6–10 h |
| 2 | 2.0–2.9 mg/dL times baseline | < 0.5 ml/kg.hr for ≥ h |
| 3 | 3 times baseline or ≥ 4.0 mg/dL increase or initiation of RRT or in patients < 18 years a decrease in eGFR < 35 mL/min/1.73 m2 |
< 0.3 ml/kg.hr for ≥ 24 h or anuria for ≥ 12 h |
Abbreviations: eGFR, estimated glomerular filtration rate; RRT, renal replacement therapy.
Reprinted from Kidney International Supplements, Volume 2, Issue 2, Section 2: AKI Definition, Page 8. Copyright 2012. With permission from Elsevier.
However, there are concerns about the utility of serum creatinine as a biomarker to diagnose AKI in persons with HF. Volume overload in the setting of HF may lead to seemingly normal or even low creatinine values. Unmasking the effect of dilution is often mistaken for AKI leading to a decrease in dose or cessation of diuretics, which may be inappropriate.9 In fact, patients with acute decompensated HF (ADHF) that are left with residual congestion have increased mortality and risk of readmission, 10,11
The majority of patients with ADHF presents with signs and symptoms of fluid overload and is treated with diuretic therapy. 12 Up to 33% of patients may have an elevation in creatinine during treatment. 5 While this elevation can meet KDIGO criteria for AKI, the cardiology literature often refers to this as worsening renal function (WRF) because it is not clear there is evidence of kidney injury with every creatinine elevation and often the rise in creatinine is thought to be a hemodynamic effect. 7 Further confounding the issue is that a reduced muscle mass and protein intake and elevated levels of inflammation are common in advanced HF, and may be exacerbated by acute deteriorations in heart and kidney function and frequent hospitalizations. Alterations in these parameters may result in changes in serum creatinine leading to errors in estimated glomerular filtration rate (eGFR) thus negatively impacting care and outcomes for patients with HF and kidney disease.
CKD is defined by KDIGO as an abnormality in kidney function or structure that is present for > 3 months. The most common functional abnormality is an eGFR < 60 mL/min/1.73 m2 with or without the presence of persistent kidney damage (albumin to creatinine ratio (ACR) of >30 mg/g, urine sediment abnormalities, tubular dysfunction, history of kidney transplant).13 Recent KDIGO guidelines have classified CKD based on cause, GFR and degree of albuminuria (A1 < 30mg/g; A2 30–300mg/g; A3 > 300mg/g).
While albuminuria is often thought in the context of CKD, this is not always the case in patients with CRS. In the setting of HF and CRS, albuminuria may not be a product of CKD, but actually the effects of cardiac dysfunction.14–16
EPIDEMIOLOGY
Impairment in kidney function is common in HF patients and is associated with worse clinical outcomes than in persons without impaired kidney function. In the Acute Decompensated Heart Failure National Registry [ADHERE] which included > 105,000 patients admitted with ADHF, 91% of patients had some degree of renal dysfunction, with 64% having CKD stage 3 or higher. Patients with more severe renal dysfunction had worse in-hospital clinical outcomes (need for mechanical ventilation, admission to an intensive care unit, cardiopulmonary resuscitation, new-onset dialysis), greater length of hospital stay, and in-hospital mortality. Overall, eGFR was found to be an independent predictor of mortality. 17 In a meta-analysis of acute and chronic HF populations, the overall prevalence of CKD was 49% (with higher prevalence in acute HF [53%] vs. chronic HF [42%]). AKI was seen in 23–35% of patients. Both CKD and WRF were associated with significantly increased mortality risk. 18
In addition to the lack of a consensus definition of AKI in the setting of HF, the clinical significance of these changes has recently been brought into question. Specifically, the context in which renal function changes needs to be considered when deciding on whether it is truly a deleterious state. Recent studies have demonstrated that AKI occurring in response to diuretic treatment associated with symptomatic improvement and signs of decongestion appears to be associated with improved outcomes whereas AKI without appropriate response to therapy is associated with worse outcomes. 19–21
Contrary to this, and what one would think is a clinical improvement, other studies have shown improvement in renal function (IRF) in ADHF actually has worse outcomes. In a study of 900 patients admitted with ADHF, 31.4% of the population experienced IRF and had an increased mortality relative to the rest of the cohort.22 Similar results were seen in the Diuretic Optimization Strategies Evaluation [DOSE], which was a multicenter, randomized, double blind, and placebo-controlled trial of diuretic strategies in ADHF patients. Analysis comparing the changes in renal function (stable, IRF, WRF) showed that patients with IRF compared with the rest of the cohort had a higher composite outcome of death, rehospitalization, and an emergency visits (HR 2.46, 95% CI 1.54–3.93, P <. 001).23
Serum creatinine has remained the most widely measured marker of kidney function but evaluates the function of the glomerulus and does not necessarily reflect tubular function or injury. This has led to a number of novel biomarkers being evaluated in the diagnosis of AKI, specifically from tubular damage. 24,25 A recent study looked at a panel of kidney tubular injury biomarkers including neutrophil gelatinase-associated lipocalin (NGAL), N-acetyl-[beta]-D-glucosaminidase (NAG), and kidney injury molecule 1 (KIM-1) in ADHF patients with and without WRF. 26 Kidney tubular injury biomarkers did not appear to have an association with WRF in the context of aggressive diuresis of patients with ADHF. This data suggests that the elevations in creatinine seen in the course of diuresis during ADHF, may not be AKI, thus bringing into the question the sensitivity of serum creatinine to diagnose ‘true kidney injury’. Indeed, therapies such as ultrafiltration for HF have fallen out of favor, given the seeming lack of benefit and increased risk of AKI (using serum creatinine)27, but whether this reflects true tubular damage needs to be confirmed using these novel biomarkers.
MECHANISM OF DISEASE PROCESS
Some of the difficulties in defining, researching, and treating CRS stem from the fact that multiple different pathophysiologic processes are involved (hemodynamic, hormonal, inflammatory). Thus, though it is more simplistic to consider individual pathophysiologic processes, these individual processes must be recognized as one portion of a larger multifaceted and complex pathophysiology (Table 2). Additionally, the significance and impact of each process varies depending on clinical status.
Table 2.
Summary of various mechanisms of cardiorenal syndrome
| Mechanism | Mediator | End-organ Outcome | |
|---|---|---|---|
| Heart | Kidney | ||
| • Increased central venous and intra-abdominal pressures | • Increased salt/water retention • Activation of RAAS/SNS |
• Acute/chronic HF • Adverse remodeling of heart and lungs |
• Renal venous congestion • Reduced GFR |
| • Reduced cardiac output and cardiac index | • Peripheral vasodilation/reduced vascular resistance • Reduced perfusion pressure |
• Activation of RAAS/SNS detrimental to heart • Cardiac ischemia from reduced perfusion |
• Reduced renal perfusion • Renal ischemia |
| • Neurohormonal dysregulation ◦ RAAS activation ◦ SNS activation ◦ Adenosine/AVP |
• Impaired baroreceptor reflexes • Increased renin secretion • Increased Ang II secretion • Increased aldosterone secretion • Increased ET-1 expression • Oxidative stress |
• Myocyte hypertrophy, left ventricular dysfunction • Proinflammation, profibrotic effect • Hypertension |
• Arteriolar vasoconstriction • Reduced GFR • Enhanced reabsorption of sodium/water • Proinflammation, profibrotic effect |
| • Oxidative stress | • Increased reactive oxygen species formation • Ang II–enhanced NADPH-oxidase activity • Uremic toxin–mediated cytokine release |
• Left ventricular hypertrophy • Accelerated atherosclerosis • Endothelial dysfunction • Inflammation • Fibrosis |
• Endothelial dysfunction • Accelerated atherosclerosis • Inflammation • Interstitial fibrosis |
| • Inflammatory mediators | • TNF-α • TWEAK • Members of IL-1 family • IL-6 • CRP |
• Atherosclerosis • Inflammation • Left ventricular dysfunction • Cardiac hypertrophy • Myocardial cell death • Fibrosis |
• Inflammation • Fibrosis • Atherosclerosis • Glomerular damage by mesangial cell apoptosis |
| • Renal failure–disturbances | • PBUTs (indoxyl sulfate, p-cresyl sulfate) • Chronic inflammatory cytokines • Oxidative stress • FGF-23 • Calcium/phosphate-mediated inflammation • Anemia |
• Endothelial dysfunction • Atherosclerosis • Left ventricular dysfunction • Cardiac hypertrophy |
• Atherosclerosis • Inflammation • Increased interstitial and perivascular fibrosis |
Abbreviations: Ang II, angiotensin II; AVP, arginine vasopressin; CRP, C-reactive protein; ET-1, endothelin-1; FGF-23, fibroblast growth factor-23; GFR, glomerular filtration rate; IL, interleukin; PBUTs, protein-bound uremic toxins; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system; TNF-α, tumor necrosis factor alpha; TWEAK, tumor necrosis factor alpha–related weak inducer of apoptosis.
1. Role of central venous and intraabdominal pressure
Elevated intraabdominal pressure (IAP) can result in intraabdominal hypertension (IAH) and abdominal compartment syndrome (ACS) in severe cases. IAH is defined as unrelenting elevated IAP of ≥ 12 mmHg and IAP > 20 mmHg defines ACS. 28 IAP elevations are traditionally seen and discussed in the context of surgical complications but are now increasingly recognized as an important pathophysiologic contribution to the CRS.
ADHF results in volume overload and increased central venous pressure (CVP). To maintain blood flow through the vascular system an adequate pressure gradient is required across the capillary network. Elevated venous pressures attenuate the gradient for forward blood flow across the renal vasculature resulting in sluggish flow and causing congestion, glomerular dysfunction and a decrease in urine output. Various studies have demonstrated that elevated IAP results in reduced GFR and renal plasma flow29,30 and that an elevated CVP is significantly associated with decreased kidney function.
In a study of 40 patients with ADHF, 60% of patient had elevated IAP. Patients with elevated IAP (≥8 mmHg) at baseline had higher serum creatinine levels compared with those with normal IAP (2.3 ± 1.0 mg/dl vs. 1.5 ± 0.8 mg/dl, p = 0.009, respectively). Interestingly, intensive medical therapy resulted in significant reduction in right-and left-sided filling pressures and an improvement in cardiac index (CI); these hemodynamic improvements did not correlate with improvements in renal function or IAP. However, changes in IAP did correlate with changes in renal function. This disconnect between hemodynamics and IAP likely explains why a subset of patients have deterioration in renal function despite improvement in hemodynamics since they have a persistent increase in IAP at follow up. 31
While elevated IAP and IAH have an important role in renal dysfunction, elevations in CVP have also been shown to closely correlate with renal function. A retrospective study conducted on patients who underwent right heart catheterization showed that an increased CVP (>6 mmHg) was associated with impaired renal function as well as a strong and independent predictor of all-cause mortality. 32 Similarly, another study of 145 patients with ADHF found that CVP were higher in persons who developed WRF compared to those who did not (18 ± 7 mm Hg vs. 12 ± 6 mm Hg, p < 0.001). 33 In addition, the mean baseline CI was actually higher in subjects who developed WRF suggesting that CVP may be more closely associated with eGFR than CI. Furthermore, a study of 196 patients with HF showed that tricuspid regurgitation was independently associated with lower GFR. 34 Significant tricuspid regurgitation can impair venous return and reflux blood into the renal-hepatic system.
2. Role of cardiac output and cardiac index
Initially, much of the progressive decline in renal function observed with HF was thought to be secondary to poor renal perfusion from a reduced cardiac output. The pathophysiologic theory is inadequate renal blood flow or perfusion pressure prompts renin release by the juxtaglomerular cells of the afferent arterioles because of a low flow state in the ascending limb of the loop of Henle and the pressure-sensing baroreceptors. This leads to:
the retention of sodium,
increased vascular congestion, and
further worsening of renal function due to renal afferent arteriolar vasoconstriction.
In an animal study, an impaired response to an acute sodium load in rats with left ventricular dysfunction secondary to healed myocardial infarction demonstrated a model of circulatory impairment. 35 In theory, by augmenting contractility, heart rate, and CI, inotropes can lead to a short-term improvement in urine output, mental status, and other clinical indicators of organ perfusion. However, investigations suggest that this concept is very limited and management of patients with CRS based solely on the low-flow theory does not lead to improved outcomes. This is supported by findings from Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness [ESCAPE] study, a trial evaluating hemodynamically guided management of ADHF (using a pulmonary artery catheter) versus usual clinical care. 36 From the 433 individuals admitted with ADHF, the investigators found no correlation between baseline renal function and CI. Furthermore, improvement in CI did not result in improved renal function, prevention of death, or prevention of rehospitalization. An important caveat of the ESCAPE trial was patients were excluded if in cardiogenic shock or investigators felt invasive hemodynamic monitoring was clinically indicated.
Counter to these findings though, a more recent study of patients with acute cardiogenic shock did find an association between decreased CI and AKI. 37 This population was excluded from the ESCAPE trial, and these results suggest that in patients with an acute severe decline in cardiac output or a markedly depressed cardiac output, a low forward flow pathophysiologic state contributes to CRS. Despite this theory of low forward flow, treatment with inotropic agents in various groups of patients with HF and AKI did not change the clinical outcomes and further endorses the fact that CRS pathophysiology and management is far more complicated than previously thought. 38,39 Thus, in the acute setting, the influence of CI is variable and may contribute in the most severe forms of ADHF, but likely does not play a significant role in the majority of patients.
While these studies looked at ADHF, data is more limited in CHF. One study did evaluate a large cohort of patients undergoing right heart catheterization without a clear discrimination of acuity or stability of HF status. In this study, CI was found to correlate with renal function, but was not the sole contributing hemodynamic process. 32 As discussed above, central venous pressure is other dominant hemodynamic factor influencing renal function.
3. Role of neurohormonal dysregulation
The renin angiotensin aldosterone system (RAAS) plays an important role in the progression of kidney damage and worsening of HF. 40 In patients with HF, neurohormonal mechanisms are activated to restore tissue perfusion. Additionally in HF, over activity of the sympathetic nervous system (SNS) due to impaired baroreceptor reflexes results in increased renin release from the juxtamedullary cells of the kidneys. 41 Renin synthesis is also influenced by the hydrostatic pressure sensed at glomerular afferent arterioles, and the reduced quantity of chloride delivered to macula densa. 42An elevation in renin leads to increased production of angiotensin II (Ang II) that has multiple maladaptive systemic effects on the heart, vasculature, and kidneys. In the kidneys, AngII causes renal efferent arteriolar vasoconstriction and an increased fraction of renal plasma flow filtered across the glomerulus. This result in an increased peritubular oncotic pressure and reduced hydrostatic pressure causing enhanced reabsorption of sodium in the proximal tubules. Ang II has a direct stimulating effect on proximal tubule sodium-bicarbonate co-transporters and apical sodium hydrogen exchangers, through which solute is proximally reabsorbed independent of the GFR. 43 Ang II also promotes the aldosterone mediated reabsorption of sodium in the distal tubules 42 and increases the expression of endothelin-1 (ET-1) in the kidney. 44 ET-1 is a potent vasoconstrictor, pro-inflammatory and pro-fibrotic peptide and leads to pathological changes resulting in kidney injury. 45
Ang II type 1 receptors (AT1) are also found in the heart. In animal models, stimulation of AT1 receptors results in cardiac myocyte hypertrophy through paracrine release of transforming growth factor-β1 and ET-1 from the cardiac fibroblast. 46 Ang II causes vascular smooth muscle contraction via the AT1 receptors. Furthermore, Ang II mediates the oxidative stress via reactive oxygen species formation in the heart and kidney tissue leading to inflammation and hypertension. 47 In patients with HF, left ventricular dysfunction causes activation of SNS in an effort to maintain the perfusion through mechanisms such as increased contractility, lustitropy, and systemic vasoconstriction. 48
Adenosine is released in response to increased sodium load in the distal tubule and via adenosine type 1 receptors in the proximal tubule and afferent arterioles; it mediates constriction of afferent arterioles and reduction of renal blood flow and GFR. Additionally, activation of adenosine type 2 receptors induces the release of renin and enhances sodium reabsorption at the proximal tubule and reduces diuresis. 49 Efficacy of adenosine type 1 receptor antagonist in CRS is controversial. The results of PROTECT (A placebo-controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for patients hospitalized with ADHF to assess treatment effect on congestion and renal function) trial showed that rolofylline group did not meet the primary (dyspnea improvement) or secondary (death, cardiovascular or renal rehospitalization, or persistent renal impairment) outcomes. Thus, further clinical studies are needed to determine the utility of adenosine A1 receptor antagonist in CRS population. 50
Arginine vasopressin (AVP) is a nonapeptide synthesized in the hypothalamus, stimulated in response to serum osmolality. It has effects on glomerular hemodynamics, arterial blood pressure, and non-hemodynamic renal mechanisms. Patients with ADHF often have an activation of AVP release. AVP causes water retention via vasopressin V2 (V2) receptors in the collecting duct. Studies have shown that elevated AVP levels contribute to the progression of CKD. 51,52 The renal hemodynamic effects of AVP may be due to its effects on the RAAS. AVP could potentially stimulate renin secretion directly via activation of V2 receptors or indirectly through reduction in sodium concentration at the macula densa. 51 Plasma AVP has been found to be elevated in patients with left ventricular dysfunction without overt clinical HF and has been associated with poor outcomes. 53,54
4. Role of oxidative stress
Oxidative stress is defined as an imbalance between oxidants and antioxidants resulting in excessive accumulation of former leading to cellular injury. 55 Reactive oxygen species (ROS) are generated as by-products of cellular metabolism, primarily in the mitochondria. 56 Oxidative stress ensues when formation of ROS surpasses the body’s antioxidative processing ability resulting in the accumulation of ROS leading to cellular damage, endothelial dysfunction and progression of atherosclerosis.
Oxidative stress in the setting of CRS can be triggered by ischemic injury, venous congestion (which causes circumferential wall stress in the endothelial cell membrane), and inflammation. 57,58 Most of the adenosine triphosphate (ATP) is produced from fatty acid oxidation in the heart, but in the setting of HF there is a shift from fatty acid oxidation to glycolysis in myocytes leading to myocardial ATP production decreasing by 30–40%. The energy deficiency is compensated by glycolysis but it is insufficient to meet the energy needs in HF leading to low threshold for hypoxemia, apoptosis, and cell death. Furthermore, due to reduced mitochondrial oxidative metabolism of fatty acid oxidation, there is accumulation of free fatty acids in myocytes, leading to lipotoxicity. 59 In a study, patients admitted with ADHF on admission who subsequently developed AKI were studied for markers of oxidative stress (IL-6, myeloperoxidase, nitric oxide, copper/zinc superoxide dismutase, and endogenous peroxidase). Results demonstrated significantly heightened presence of dual oxidative stress markers, in patients who developed CRS type 1.60
In addition to the deleterious effects of volume expansion and hemodynamics, RAAS and SNS activation also plays an important role in amplifying the oxidative stress in HF and CKD patients. Ang II has a deleterious effect by activating NADPH-oxidase promoting oxidative injury by producing ROS causing mitochondrial dysfunction. 61 The enhanced NADPH-oxidase activity has been demonstrated in endothelial cells, renal tubular cells and cardiac myocytes. 62–64
Patients with advance CKD and end-stage renal disease (ESRD) have some factors such as, uremic toxins and dialysate solutions utilized in renal replacement therapy that could lead to increased synthesis and release of proinflammatory cytokines, oxidative stress, immune system dysregulation leading to carotid artery intima-media thickness (marker of early stage of atherosclerosis) and left ventricular hypertrophy. 65,66 ESRD patients have higher cardiovascular morbidity and mortality that could not be explained by classic cardiac risk factors; thus, perhaps oxidative stress, endothelial dysfunction, and hyperhomocysteinemia might be playing an additive role in these patients. 67
5. Role of inflammatory mediators
Both CKD and HF are states of heightened chronic inflammation, resulting in the generation of pro-inflammatory biomarkers that play a crucial role in tissue damage to both organs leading to cell death and fibrosis. Important triggers that initiate and propagate the inflammatory cascade include the activation of SNS and RAAS, venous congestion, ischemia, and oxidative stress. Pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and TNF-α related weak inducer of apoptosis (TWEAK), members of interleukin-1 (IL-1) family, and interleukin-6 (IL-6) have been associated with HF as well as CKD. In kidneys, TNF-α and IL-6 promote accumulation of inflammatory cells in the interstitium by increasing monocyte chemoattractant proteins expressions. TNF-α also results in glomerular damage by mesangial cell apoptosis. 68 Some of these biomarkers are prognostic for all-cause mortality in HF patients, such as soluble ST2, which is a member of the IL-1 family. 69,70 Similarly, in CKD, IL-6 correlates well with progression of disease and also predicts the mortality. 71 It has also been shown that levels of these pro-inflammatory markers are higher in persons with CKD 72 and those on dialysis. 73
C-Reactive protein (CRP), an acute phase reactant, has been shown to contribute in the pathogenesis of atherosclerosis through a variety of mechanisms. CRP activates the complement system and is widely distributed in early atherosclerotic lesions. 74,75 CRP is a potent stimulator of tissue factor production (a potent procoagulant) by monocytes and this effect is further augmented in the presence of inflammatory mediators. 76 A study of 4269 individuals hospitalized with ADHF, patients with CRP in the fourth quartile (≥ 9.6 mg/L) were independently associated with higher all-cause mortality (adjusted hazard ratio, 1.68) within 120 days after discharge. 77 In hemodialysis patients high CRP levels predict left ventricular dysfunction, cardiac hypertrophy, and mortality. 78,79 These inflammatory proteins are not simply inert markers of disease activity but rather play an active and complex role in the pathophysiology of CRS.
6. Role of renal failure associated disturbances
Protein-bound uremic toxins (PBUT) are currently an emerging area of interest due to their potential association with cardiovascular disease. 80,81 Indoxyl sulphate (IS) and p-crestyl sulphate (PCS) are the two most extensively studied PBUT that have demonstrated a role in the pathogenesis and progression of CRS. In normal kidneys, both are cleared through tubular secretion. Experimental studies have demonstrated detrimental effect of IS and PCS through alteration of oxidative stress, endothelial dysfunction and atherosclerosis. Both have been associated with nephrotoxicity, decreased endothelial proliferation, and impaired wound repair suggesting their role in progression of CKD. 82–85 A study using a nephrectomy CKD mouse model revealed effects of PCS on cardiac cells including increased apoptosis, increased interstitial and perivascular fibrosis, and a reduction in left ventricular diastolic function. Oxidative stress was implicated in PCS-induced changes in the heart muscle. 86 Furthermore, IS also augments oxidative stress in kidney and heart leading to cardiorenal fibrosis. 87–89 A study of 139 CKD patients demonstrated that IS was a powerful predictor of overall and cardiovascular mortality after adjusting for confounders. 90 Though there has been evidence suggesting a negative effect of PBUT on heart, kidney and vascular cells, further research is needed to better understand the role of PBUT in the pathophysiology of CRS.
Fibroblast growth factor-23 (FGF23), a hormone produced in the bone that controls phosphate and vitamin D metabolism by the kidney, is a strong predictor of adverse cardiovascular outcomes in patients with CKD and ESRD. Elevated FGF-23 has been associated with left ventricular hypertrophy (LVH) and mortality in advance CKD patients. 91 While it has been suggested that FGF23 may induce myocardial hypertrophy through a direct effect on cardiac myocytes, this remains debated due to the absence of alpha-klotho receptors (which mediate FGF23 action) in the heart. Other data also suggests that FGF23 may directly depress myocardial contractility and ventricular relaxation, cause hypertrophy and increase risk of arrhythmias, by altering calcium trafficking. 92,93
7. Role of anemia
Anemia is common in patients with advance CKD and HF with the majority of these patients have anemia of chronic disease. The prevalence of anemia in CRS has been reported to vary from 5% to 55% with anemia reported to be an independent predictor of mortality. 94–96 In the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure) registry with over 48,000 patients, 51.2% had mild anemia (Hb<12.1 g/dl) and 25% were moderately to severely anemic (hemoglobin levels of 5 to 10.7 g/dl) 97
In CKD patients, anemia is associated with:
cognitive impairment,
poor quality of life,
progression of kidney disease,
cardiovascular comorbidities, and
higher mortality.98
In a multicenter survey of 5222 patients with CKD, 47.7% were found to be anemic (hemoglobin level ≤ 12 g/dl). 99
There are several ways anemia contributes to the pathophysiology of CRS. Lack of oxygen supply to a heart that is already under stress or kidney that is already damaged may cause ischemic insults that can results in progressive cell death in both the organs. Red blood cells contain many antioxidants, and therefore anemia may results in increased oxidative stress. 100 Anemia can cause tissue ischemia and peripheral vasodilation, which leads to activation of SNS, RAAS as well as release of ADH resulting in vasoconstriction, salt and water retention, and chronic renal venous congestion that leads to progressive nephron loss and interstitial fibrosis. Chronic anemic state also results in left ventricular hypertrophy and myocardial cell death from ischemia and necrosis. 101,102
Although correction of anemia in CHF patients with erythropoiesis-stimulating agents (ESA) results in improved outcomes (reduced hospitalization, improved new york heart association class, 6-minute walk test, and quality of life) but normalization of the hemoglobin levels may not result in favorable outcomes. Trials targeting a higher level of hemoglobin levels (≥ 13 g/dL) surprisingly were associated with higher rate of adverse events. 103,104 The trial to reduce cardiovascular events with aranesp therapy [TREAT] was a randomized, double blind, placebo-controlled study with over 4000 patients. 105 The use of darbepoetin alfa in patients with diabetes, CKD, and moderate anemia who were not undergoing dialysis to achieve a hemoglobin level of approximately 13 g/dl did not reduce the risk of death or a cardiovascular event or a renal event. There was an increased risk of fatal or nonfatal stroke in patients assigned to darbepoetin alfa group. The RED-HF (The reduction of events by darbepoetin alfa in heart failure) was a randomized, double blind trial with 2278 patients with systolic heart failure and mild to moderate anemia (hemoglobin level, 9.0 to 12.0 g/dl). 106 Patients were randomized to either darbepoetin alfa (to achieve a hemoglobin target of 13 g/dl) or placebo. There was no difference in the primary outcome (death from any cause or hospitalization for worsening HF).
Anemia plays an important role in the pathophysiology of CRS and management of anemia is complex especially in patients with CKD and CHF. The main unanswered question is the range of hemoglobin levels to target in this population, targets based on CKD guidelines (10 to 12 g/dl) or higher (12 to 13 g/dl) but less than 13 g/dl (as trials with hemoglobin level of 13 or higher were associated with negative outcomes) is still unknown.
8. Pathogenesis of Type 5 CRS
Type 5 CRS (CRS-5) occurs when an overwhelming systemic disease process results in damage to heart and kidney simultaneously (Figure 4). Based on pathophysiology and severity of the disease process, CRS-5 has been distinguished into four stages: hyperacute, acute, subacute, and chronic. Systemic diseases that can result in CRS-5 are sepsis, connective tissue disorders such as lupus, sarcoidosis, amyloidosis, and cirrhosis. Injury to the kidney and heart is often mediated by pro-inflammatory cytokines, complement factors, and RAAS activation, which are often the common end pathway for other forms of CRS. For instance, in sepsis, increased renal vascular resistance and early rise in pro-inflammatory cytokines (IL-6) and oxidative stress can lead to organ damage. 107,108 Sepsis also results in autonomic nervous system dysfunction, and activation of RAAS. The plethora of effects of sepsis on the function of various organs, including heart and kidney, makes it very difficult to differentiate between the effects of sepsis itself and the effect of inter-organ cross-talk. Moreover, management of sepsis can contribute to the development of CRS-5. Fluid resuscitation can result in tissue edema and IAH, increase venous congestion and reduced renal perfusion. Iodinated contrast agents and certain drugs can results in myocardial depression and nephrotoxicity resulting in development and/or worsening of CRS-5. In chronic inflammatory and autoimmune disease processes, the concomitant damage to both the organs and ongoing cross-talk between the heart and kidney leads to a similar pathophysiologic mechanisms as discussed in other types of CRS. 109
Figure. 4.

Pathophysiological interactions in cardiorenal syndrome (type 5).
SVR= Systemic vascular resistance; ROS= Reactive oxygen species; DIC= Disseminated intravascular coagulation; SNS= Sympathetic nervous system; RAAS= Renin angiotensin aldosterone system.
FUTURE DIRECTIONS
Despite an improved understanding of the different pathophysiological process involved in the development of CRS, therapies at improving outcomes in this population have only met with minimal success. One possibility for this may be the inability to accurately diagnose AKI using conventional biomarkers such as creatinine. Whether the incorporation of more novel filtration markers such as cystatin C, beta-2 microglobulin, beta trace protein will provide use with better estimates of kidney function needs to be evaluated further in the setting of HF. Further, based on emerging evidence, there is a need to incorporate more sensitive, and perhaps specific tubular markers of injury, inflammation and repair into the renal endpoints of HF clinical trials. 110
CONCLUSION
Pathophysiology of the various types of CRS is complex and challenging. Given the rising burden of the disease, adverse clinical outcomes and high impact on mortality, early diagnosis of the syndrome is crucial. To improve the survival and morbidly associated with the disease, better understanding of various aspects of pathophysiology is needed to better mange these patients. CRS-5 is a complex and challenging condition to diagnose as the time and sequence of dysfunction of heart and kidney are dictated by the underlying cause and nature of condition.
KEY POINTS.
Cardiorenal syndrome (CRS) is a term commonly refers to the collective dysfunction of heart and kidneys resulting in cascade of feedback mechanisms resulting in damage to both the organs.
Multiple mechanisms (hemodynamic, neurohormonal, inflammatory, and oxidative stress) are involved in the pathophysiology of CRS.
In CRS, renin angiotensin aldosterone system and sympathetic nervous system activation leads to salt avidity and volume overload.
Venous congestion and elevated intraabdominal pressure plays an important role in the pathophysiology of CRS.
The role of creatinine and novel biomarkers for diagnosing kidney disease in the setting of heart failure needs to be further evaluated.
SYNOPSIS.
Cardiorenal syndrome commonly refers to the collective dysfunction of heart and kidney resulting in a cascade of feedback mechanism causing damage to both the organs and is associated with adverse clinical outcomes. The pathophysiology of cardiorenal syndrome is complex, multifactorial, and dynamic. Improving the understanding of disease mechanisms will aid in developing targeted pharmacologic and non-pharmacologic therapies for the management of this syndrome. This review will discuss the various mechanisms involved in the pathophysiology of the cardiorenal syndrome.
Acknowledgments
However, this work was supported by NIDDK grant K23 DK114556 to Pranav Garimella and T32DK104717 to Ujjala Kumar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT
There are no financial conflicts of interest to disclose.
References:
- 1.Cardio-Renal Connections in Heart Failure and Cardiovascular Disease | National Heart, Lung, and Blood Institute (NHLBI). https://www.nhlbi.nih.gov/events/2004/cardio-renal-connections-heart-failure-and-cardiovascular-disease. Accessed January 15, 2019.
- 2.House AA, Anand I, Bellomo R, et al. Definition and classification of Cardio-Renal Syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrology Dialysis Transplantation 2010;25(5):1416–1420. [DOI] [PubMed] [Google Scholar]
- 3.Ronco C, McCullough P, Anker SD, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. European heart journal 2009;31(6):703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berl T, Henrich W. Kidney-Heart Interactions: Epidemiology, Pathogenesis, and Treatment. CJASN 2006;1(1):8–18. doi: 10.2215/CJN.00730805 [DOI] [PubMed] [Google Scholar]
- 5.Ronco C, House AA, Haapio M. Cardiorenal syndrome: refining the definition of a complex symbiosis gone wrong. Intensive care medicine 2008;34(5):957. [DOI] [PubMed] [Google Scholar]
- 6.Health, United States, 2017, With Special Feature on Mortality :87 https://www.cdc.gov/nchs/data/hus/hus17.pdf. [PubMed]
- 7.Damman K, Tang WHW, Testani JM, McMurray JJV. Terminology and definition of changes renal function in heart failure. Eur Heart J 2014;35(48):3413–3416. doi: 10.1093/eurheartj/ehu320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khwaja A KDIGO clinical practice guidelines for acute kidney injury. Nephron Clinical Practice 2012;120(4):c179–c184. [DOI] [PubMed] [Google Scholar]
- 9.Testani JM, McCauley BD, Chen J, Shumski M, Shannon RP. Worsening Renal Function Defined as an Absolute Increase in Serum Creatinine Is a Biased Metric for the Study of Cardio-Renal Interactions. Cardiology 2010;116(3):206–212. doi: 10.1159/000316038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrosy AP, Pang PS, Khan S, et al. Clinical course and predictive value of congestion during hospitalization in patients admitted for worsening signs and symptoms of heart failure with reduced ejection fraction: findings from the EVEREST trial. Eur Heart J 2013;34(11):835–843. doi: 10.1093/eurheartj/ehs444 [DOI] [PubMed] [Google Scholar]
- 11.Lala A, McNulty SE, Mentz RJ, et al. Relief and Recurrence of Congestion During and After Hospitalization for Acute Heart Failure: Insights From Diuretic Optimization Strategy Evaluation in Acute Decompensated Heart Failure (DOSE-AHF) and Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARESS-HF). Circ Heart Fail 2015;8(4):741–748. doi: 10.1161/CIRCHEARTFAILURE.114.001957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fonarow GC, Stough WG, Abraham WT, et al. Characteristics, Treatments, and Outcomes of Patients With Preserved Systolic Function Hospitalized for Heart Failure: A Report From the OPTIMIZE-HF Registry. Journal of the American College of Cardiology 2007;50(8):768–777. doi: 10.1016/j.jacc.2007.04.064 [DOI] [PubMed] [Google Scholar]
- 13.Levin A, Stevens PE, Bilous RW, et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Supplements 2013;3(1):1–150. [Google Scholar]
- 14.Jackson CE, Solomon SD, Gerstein HC, et al. Albuminuria in chronic heart failure: prevalence and prognostic importance. The Lancet 2009;374(9689):543–550. doi: 10.1016/S0140-6736(09)61378-7 [DOI] [PubMed] [Google Scholar]
- 15.Ninomiya T, Perkovic V, de Galan BE, et al. Albuminuria and Kidney Function Independently Predict Cardiovascular and Renal Outcomes in Diabetes. J Am Soc Nephrol 2009;20(8):1813–1821. doi: 10.1681/ASN.2008121270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Astor BC, Hallan SI, Miller ER, Yeung E, Coresh J. Glomerular filtration rate, albuminuria, and risk of cardiovascular and all-cause mortality in the US population. Am J Epidemiol 2008;167(10):1226–1234. doi: 10.1093/aje/kwn033 [DOI] [PubMed] [Google Scholar]
- 17.Heywood JT, Fonarow GC, Costanzo MR, et al. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail 2007;13(6):422–430. doi: 10.1016/j.cardfail.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 18.Damman K, Valente MAE, Voors AA, O’Connor CM, van Veldhuisen DJ, Hillege HL. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J 2014;35(7):455–469. doi: 10.1093/eurheartj/eht386 [DOI] [PubMed] [Google Scholar]
- 19.Marco Metra, Beth Davison, Luca Bettari, et al. Is Worsening Renal Function an Ominous Prognostic Sign in Patients With Acute Heart Failure? Circulation: Heart Failure 2012;5(1):54–62. doi: 10.1161/CIRCHEARTFAILURE.111.963413 [DOI] [PubMed] [Google Scholar]
- 20.Khan NA, Ma I, Thompson CR, et al. Kidney Function and Mortality among Patients with Left Ventricular Systolic Dysfunction. JASN 2006;17(1):244–253. doi: 10.1681/ASN.2005030270 [DOI] [PubMed] [Google Scholar]
- 21.Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic Importance of Early Worsening Renal Function Following Initiation of Angiotensin Converting Enzyme Inhibitor Therapy in Patients with Cardiac Dysfunction. Circ Heart Fail 2011;4(6):685–691. doi: 10.1161/CIRCHEARTFAILURE.111.963256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Testani JM, McCauley BD, Chen J, Coca SG, Cappola TP, Kimmel SE. Clinical Characteristics and Outcomes of Patients With Improvement in Renal Function During the Treatment of Decompensated Heart Failure. Journal of Cardiac Failure 2011;17(12):993–1000. doi: 10.1016/j.cardfail.2011.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.BRISCO MA, ZILE MR, HANBERG JS, et al. Relevance of Changes in Serum Creatinine During a Heart Failure Trial of Decongestive Strategies: Insights From the DOSE Trial. J Card Fail 2016;22(10):753–760. doi: 10.1016/j.cardfail.2016.06.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCullough PA, Kellum JA, Haase M, et al. Pathophysiology of the cardiorenal syndromes: executive summary from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol 2013;182:82–98. doi: 10.1159/000349966 [DOI] [PubMed] [Google Scholar]
- 25.Cruz DN, Schmidt-Ott KM, Vescovo G, et al. Pathophysiology of cardiorenal syndrome type 2 in stable chronic heart failure: workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol 2013;182:117–136. doi: 10.1159/000349968 [DOI] [PubMed] [Google Scholar]
- 26.Tariq Ahmad, Keyanna Jackson, Rao Veena S, et al. Worsening Renal Function in Patients With Acute Heart Failure Undergoing Aggressive Diuresis Is Not Associated With Tubular Injury. Circulation 2018;137(19):2016–2028. doi: 10.1161/CIRCULATIONAHA.117.030112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bart BA, Goldsmith SR, Lee KL, et al. Ultrafiltration in Decompensated Heart Failure with Cardiorenal Syndrome. New England Journal of Medicine 2012;367(24):2296–2304. doi: 10.1056/NEJMoa1210357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the international conference of experts on intra-abdominal hypertension and abdominal compartment syndrome. I. Definitions. Intensive care medicine 2006;32(11):1722–1732. [DOI] [PubMed] [Google Scholar]
- 29.Bradley SE, Bradley GP. THE EFFECT OF INCREASED INTRA-ABDOMINAL PRESSURE ON RENAL FUNCTION IN MAN 1. Journal of Clinical Investigation 1947;26(5):1010–1022. doi: 10.1172/JCI101867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalfino L, Tullo L, Donadio I, Malcangi V, Brienza N. Intra-abdominal hypertensionand acute renal failurein critically ill patients. Intensive care medicine 2008;34(4):707–713. [DOI] [PubMed] [Google Scholar]
- 31.Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? Journal of the American College of Cardiology 2008;51(3):300–306. [DOI] [PubMed] [Google Scholar]
- 32.Damman K, Deursen VM van, Navis G, Voors AA, Veldhuisen DJ van, Hillege HL. Increased Central Venous Pressure Is Associated With Impaired Renal Function and Mortality in a Broad Spectrum of Patients With Cardiovascular Disease. Journal of the American College of Cardiology 2009;53(7):582–588. doi: 10.1016/j.jacc.2008.08.080 [DOI] [PubMed] [Google Scholar]
- 33.Mullens W, Abrahams Z, Francis GS, et al. Importance of Venous Congestion for Worsening of Renal Function in Advanced Decompensated Heart Failure. Journal of the American College of Cardiology 2009;53(7):589–596. doi: 10.1016/j.jacc.2008.05.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeder MT, Holst DP, Kaye DM. Tricuspid Regurgitation Contributes to Renal Dysfunction in Patients With Heart Failure. Journal of Cardiac Failure 2008;14(10):824–830. doi: 10.1016/j.cardfail.2008.07.236 [DOI] [PubMed] [Google Scholar]
- 35.Hostetter TH, Pfeffer JM, Pfeffer MA, Dworkin LD, Braunwald E, Brenner BM. Cardiorenal hemodynamics and sodium excretion in rats with myocardial infarction. American Journal of Physiology-Heart and Circulatory Physiology 1983;245(1):H98–H103. doi: 10.1152/ajpheart.1983.245.1.H98 [DOI] [PubMed] [Google Scholar]
- 36.Binanay C, Califf RM, Hasselblad V, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA 2005;294(13):1625–1633. doi: 10.1001/jama.294.13.1625 [DOI] [PubMed] [Google Scholar]
- 37.Tarvasmäki T, Haapio M, Mebazaa A, et al. Acute kidney injury in cardiogenic shock: definitions, incidence, haemodynamic alterations, and mortality. European Journal of Heart Failure 2018;20(3):572–581. doi: 10.1002/ejhf.958 [DOI] [PubMed] [Google Scholar]
- 38.Bellomo R, Chapman M, Finfer S, Hickling K, Myburgh J. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet 2000;356(9248):2139–2143. doi: 10.1016/S0140-6736(00)03495-4 [DOI] [PubMed] [Google Scholar]
- 39.Lauschke A, Teichgräber UKM, Frei U, Eckardt K-U. ‘Low-dose’ dopamine worsens renal perfusion in patients with acute renal failure. Kidney International 2006;69(9):1669–1674. doi: 10.1038/sj.ki.5000310 [DOI] [PubMed] [Google Scholar]
- 40.Ferrario CM, Strawn WB. Role of the Renin-Angiotensin-Aldosterone System and Proinflammatory Mediators in Cardiovascular Disease. The American Journal of Cardiology 2006;98(1):121–128. doi: 10.1016/j.amjcard.2006.01.059 [DOI] [PubMed] [Google Scholar]
- 41.Kopp UC. Neural Control of Renin Secretion Rate Morgan & Claypool Life Sciences; 2011. https://www.ncbi.nlm.nih.gov/books/NBK57240/. Accessed January 3, 2019. [PubMed] [Google Scholar]
- 42.Harrison-Bernard LM. The renal renin-angiotensin system. Advances in Physiology Education 2009;33(4):270–274. doi: 10.1152/advan.00049.2009 [DOI] [PubMed] [Google Scholar]
- 43.Johnson MD, Malvin RL. Stimulation of renal sodium reabsorption by angiotensin II. Am J Physiol 1977;232(4):F298–306. doi: 10.1152/ajprenal.1977.232.4.F298 [DOI] [PubMed] [Google Scholar]
- 44.Barton M, Shaw S, d’uscio LV, Moreau P, Lüscher TF. Angiotensin II Increases Vascular and Renal Endothelin-1 and Functional Endothelin Converting Enzyme Activityin Vivo:Role of ETAReceptors for Endothelin Regulation. Biochemical and Biophysical Research Communications 1997;238(3):861–865. doi: 10.1006/bbrc.1997.7394 [DOI] [PubMed] [Google Scholar]
- 45.Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. European Journal of Clinical Investigation 2006;36(s3):78–88. doi: 10.1111/j.1365-2362.2006.01689.x [DOI] [PubMed] [Google Scholar]
- 46.Gray MO, Long CS, Kalinyak JE, Li H-T, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-β1 and endothelin-1 from fibroblasts. Cardiovascular research 1998;40(2):352–363. [DOI] [PubMed] [Google Scholar]
- 47.Hitomi H, Kiyomoto H, Nishiyama A. Angiotensin II and oxidative stress. Current opinion in cardiology 2007;22(4):311–315. [DOI] [PubMed] [Google Scholar]
- 48.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The Sympathetic Nervous System in Heart Failure: Physiology, Pathophysiology, and Clinical Implications. Journal of the American College of Cardiology 2009;54(19):1747–1762. doi: 10.1016/j.jacc.2009.05.015 [DOI] [PubMed] [Google Scholar]
- 49.Hiroharu Funaya, Masafumi Kitakaze, Koichi Node, Tetsuo Minamino, Kazuo Komamura, Masatsugu Hori. Plasma Adenosine Levels Increase in Patients With Chronic Heart Failure. Circulation 1997;95(6):1363–1365. doi: 10.1161/01.CIR.95.6.1363 [DOI] [PubMed] [Google Scholar]
- 50.Massie BM, O’Connor CM, Metra M, et al. Rolofylline, an Adenosine A1−Receptor Antagonist, in Acute Heart Failure. New England Journal of Medicine 2010;363(15):1419–1428. doi: 10.1056/NEJMoa0912613 [DOI] [PubMed] [Google Scholar]
- 51.Bardoux P, Martin H, Ahloulay M, et al. Vasopressin contributes to hyperfiltration, albuminuria, and renal hypertrophy in diabetes mellitus: study in vasopressin-deficient Brattleboro rats. Proc Natl Acad Sci USA 1999;96(18):10397–10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres VE. Vasopressin in chronic kidney disease, an elephant in the room? Kidney Int 2009;76(9):925–928. doi: 10.1038/ki.2009.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francis GS, Benedict C, Johnstone DE, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990;82(5):1724–1729. [DOI] [PubMed] [Google Scholar]
- 54.Rouleau JL, Packer M, Moyé L, et al. Prognostic value of neurohumoral activation in patients with an acute myocardial infarction: effect of captopril. J Am Coll Cardiol 1994;24(3):583–591. [DOI] [PubMed] [Google Scholar]
- 55.Sies H Oxidative stress: oxidants and antioxidants. Experimental Physiology: Translation and Integration 1997;82(2):291–295. [DOI] [PubMed] [Google Scholar]
- 56.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. American Journal of Physiology-Lung Cellular and Molecular Physiology 2000;279(6):L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005 [DOI] [PubMed] [Google Scholar]
- 57.Colombo PC, Doran AC, Onat D, et al. Venous congestion, endothelial and neurohormonal activation in acute decompensated heart failure: cause or effect? Current heart failure reports 2015;12(3):215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubattu S, Mennuni S, Testa M, et al. Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress? International Journal of Molecular Sciences 2013;14(11):23011–23032. doi: 10.3390/ijms141123011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katz AM, Konstam MA. Heart Failure: Pathophysiology, Molecular Biology, and Clinical Management Lippincott Williams & Wilkins; 2012. [Google Scholar]
- 60.Virzì GM, Clementi A, de Cal M, et al. Oxidative Stress: Dual Pathway Induction in Cardiorenal Syndrome Type 1 Pathogenesis. Oxid Med Cell Longev 2015;2015. doi: 10.1155/2015/391790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimura S, Zhang G, Nishiyama A, et al. Role of NAD(P)H oxidase-and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 2005;45(5):860–866. [DOI] [PubMed] [Google Scholar]
- 62.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. Journal of molecular and cellular cardiology 2003;35(7):851–859. [DOI] [PubMed] [Google Scholar]
- 63.Chabrashvili T, Kitiyakara C, Blau J, et al. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2003;285(1):R117–R124. [DOI] [PubMed] [Google Scholar]
- 64.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. P22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin iiinduced hypertrophy in vascular smooth muscle cells. Journal of Biological Chemistry 1996;271(38):23317–23321. [DOI] [PubMed] [Google Scholar]
- 65.Granata S, Zaza G, Simone S, et al. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics 2009;10:388. doi: 10.1186/1471-2164-10-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Modaresi A, Nafar M, Sahraei Z. Oxidative Stress in Chronic Kidney Disease 2015;9(3):15. [PubMed] [Google Scholar]
- 67.Becker BN, Himmelfarb J, Henrich WL, Hakim RM. Reassessing the cardiac risk profile in chronic hemodialysis patients: a hypothesis on the role of oxidant stress and other non-traditional cardiac risk factors. JASN 1997;8(3):475–486. https://jasn.asnjournals.org/content/8/3/475. Accessed January 21, 2019. [DOI] [PubMed] [Google Scholar]
- 68.Radeke HH, Meier B, Topley N, Flöge J, Habermehl GG, Resch K. Interleukin 1-alpha and tumor necrosis factor-alpha induce oxygen radical production in mesangial cells. Kidney Int 1990;37(2):767–775. [DOI] [PubMed] [Google Scholar]
- 69.Tsutamoto T, Hisanaga T, Wada A, et al. Interleukin-6 Spillover in the Peripheral Circulation Increases With the Severity of Heart Failure, and the High Plasma Level of Interleukin-6 Is an Important Prognostic Predictor in Patients With Congestive Heart Failure. Journal of the American College of Cardiology 1998;31(2):391–398. doi: 10.1016/S0735-1097(97)00494-4 [DOI] [PubMed] [Google Scholar]
- 70.Wettersten N, Maisel AS. Biomarkers for Heart Failure: An Update for Practitioners of Internal Medicine. Am J Med 2016;129(6):560–567. doi: 10.1016/j.amjmed.2016.01.013 [DOI] [PubMed] [Google Scholar]
- 71.Barreto DV, Barreto FC, Liabeuf S, et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney International 2010;77(6):550–556. doi: 10.1038/ki.2009.503 [DOI] [PubMed] [Google Scholar]
- 72.Stenvinkel P, Ketteler M, Johnson RJ, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int 2005;67(4):1216–1233. doi: 10.1111/j.1523-1755.2005.00200.x [DOI] [PubMed] [Google Scholar]
- 73.Pereira BJ, Shapiro L, King AJ, Falagas ME, Strom JA, Dinarello CA. Plasma levels of IL-1 beta, TNF alpha and their specific inhibitors in undialyzed chronic renal failure, CAPD and hemodialysis patients. Kidney Int 1994;45(3):890–896. [DOI] [PubMed] [Google Scholar]
- 74.Torzewski J, Torzewski M, Bowyer DE, et al. C-Reactive Protein Frequently Colocalizes With the Terminal Complement Complex in the Intima of Early Atherosclerotic Lesions of Human Coronary Arteries. Arteriosclerosis, Thrombosis, and Vascular Biology 1998;18(9):1386–1392. doi: 10.1161/01.ATV.18.9.1386 [DOI] [PubMed] [Google Scholar]
- 75.Arici M, Walls J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: Is C-reactive protein the missing link? Kidney International 2001;59(2):407–414. doi: 10.1046/j.1523-1755.2001.059002407.x [DOI] [PubMed] [Google Scholar]
- 76.Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood 1993;82(2):513–520. http://www.bloodjournal.org/content/82/2/513. Accessed January 22, 2019. [PubMed] [Google Scholar]
- 77.Minami Y, Kajimoto K, Sato N, Hagiwara N. Effect of Elevated C-Reactive Protein Level at Discharge on Long-Term Outcome in Patients Hospitalized for Acute Heart Failure. The American Journal of Cardiology 2018;121(8):961–968. doi: 10.1016/j.amjcard.2017.12.046 [DOI] [PubMed] [Google Scholar]
- 78.Kim B-S, Jeon DS, Shin MJ, et al. Persistent Elevation of C-Reactive Protein May Predict Cardiac Hypertrophy and Dysfunction in Patients Maintained on Hemodialysis. AJN 2005;25(3):189–195. doi: 10.1159/000085585 [DOI] [PubMed] [Google Scholar]
- 79.Yeun JY, Levine RA, Mantadilok V, Kaysen GA. C-reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. American Journal of Kidney Diseases 2000;35(3):469–476. doi: 10.1016/S0272-6386(00)70200-9 [DOI] [PubMed] [Google Scholar]
- 80.Chinnappa S, Tu Y-K, Yeh YC, Glorieux G, Vanholder R, Mooney A. Association between Protein-Bound Uremic Toxins and Asymptomatic Cardiac Dysfunction in Patients with Chronic Kidney Disease. Toxins (Basel) 2018;10(12). doi: 10.3390/toxins10120520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lekawanvijit S Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal Syndrome. Toxins (Basel) 2018;10(9). doi: 10.3390/toxins10090352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin C-J, Liu H-L, Pan C-F, et al. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res 2012;43(6):451–456. doi: 10.1016/j.arcmed.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 83.Enomoto A, Takeda M, Tojo A, et al. Role of Organic Anion Transporters in the Tubular Transport of Indoxyl Sulfate and the Induction of its Nephrotoxicity. JASN 2002;13(7):1711–1720. doi: 10.1097/01.ASN.0000022017.96399.B2 [DOI] [PubMed] [Google Scholar]
- 84.Dou L, Bertrand E, Cerini C, et al. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney International 2004;65(2):442–451. doi: 10.1111/j.1523-1755.2004.00399.x [DOI] [PubMed] [Google Scholar]
- 85.Wu I-W, Hsu K-H, Lee C-C, et al. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant 2011;26(3):938–947. doi: 10.1093/ndt/gfq580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han H, Zhu J, Zhu Z, et al. p-Cresyl Sulfate Aggravates Cardiac Dysfunction Associated With Chronic Kidney Disease by Enhancing Apoptosis of Cardiomyocytes. J Am Heart Assoc 2015;4(6). doi: 10.1161/JAHA.115.001852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Owada S, Goto S, Bannai K, Hayashi H, Nishijima F, Niwa T. Indoxyl Sulfate Reduces Superoxide Scavenging Activity in the Kidneys of Normal and Uremic Rats. AJN 2008;28(3):446–454. doi: 10.1159/000112823 [DOI] [PubMed] [Google Scholar]
- 88.Fujii H, Nishijima F, Goto S, et al. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol Dial Transplant 2009;24(7):2089–2095. doi: 10.1093/ndt/gfp007 [DOI] [PubMed] [Google Scholar]
- 89.Lekawanvijit S, Krum H. Cardiorenal syndrome: acute kidney injury secondary to cardiovascular disease and role of protein-bound uraemic toxins. J Physiol 2014;592(Pt 18):3969–3983. doi: 10.1113/jphysiol.2014.273078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barreto FC, Barreto DV, Liabeuf S, et al. Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. CJASN 2009;4(10):1551–1558. doi: 10.2215/CJN.03980609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011;121(11):4393–4408. doi: 10.1172/JCI46122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Touchberry CD, Green TM, Tchikrizov V, et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab 2013;304(8):E863–873. doi: 10.1152/ajpendo.00596.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kao Y-H, Chen Y-C, Lin Y-K, et al. FGF-23 dysregulates calcium homeostasis and electrophysiological properties in HL-1 atrial cells. Eur J Clin Invest 2014;44(8):795–801. doi: 10.1111/eci.12296 [DOI] [PubMed] [Google Scholar]
- 94.Ezekowitz JA, McAlister FA, Armstrong PW. Anemia Is Common in Heart Failure and Is Associated With Poor Outcomes: Insights From a Cohort of 12 065 Patients With New-Onset Heart Failure. Circulation 2003;107(2):223–225. doi: 10.1161/01.CIR.0000052622.51963.FC [DOI] [PubMed] [Google Scholar]
- 95.Palazzuoli A, Antonelli G, Nuti R. Anemia in Cardio-Renal Syndrome: clinical impact and pathophysiologic mechanisms. Heart Fail Rev 2011;16(6):603–607. doi: 10.1007/s10741-011-9230-x [DOI] [PubMed] [Google Scholar]
- 96.Adams KF, Patterson JH, Oren RM, et al. Prospective assessment of the occurrence of anemia in patients with heart failure: Results from the Study of Anemia in a Heart Failure Population (STAMINA-HFP) Registry. American Heart Journal 2009;157(5):926–932. doi: 10.1016/j.ahj.2009.01.012 [DOI] [PubMed] [Google Scholar]
- 97.Young JB, Abraham WT, Albert NM, et al. Relation of Low Hemoglobin and Anemia to Morbidity and Mortality in Patients Hospitalized With Heart Failure (Insight from the OPTIMIZE-HF Registry). The American Journal of Cardiology 2008;101(2):223–230. doi: 10.1016/j.amjcard.2007.07.067 [DOI] [PubMed] [Google Scholar]
- 98.American Journal of Kidney Diseases. American Journal of Kidney Diseases 2006;47:S11–S15. doi: 10.1053/j.ajkd.2006.03.010 [DOI] [PubMed] [Google Scholar]
- 99.McClellan W, Aronoff SL, Bolton WK, et al. The prevalence of anemia in patients with chronic kidney disease. Current Medical Research and Opinion 2004;20(9):1501–1510. doi: 10.1185/030079904X2763 [DOI] [PubMed] [Google Scholar]
- 100.Grune T, Sommerburg O, Siems WG. Oxidative stress in anemia. Clin Nephrol 2000;53(1 Suppl):S18–22. http://europepmc.org/abstract/med/10746801. Accessed January 21, 2019. [PubMed] [Google Scholar]
- 101.Brezis M, Rosen S. Hypoxia of the Renal Medulla — Its Implications for Disease. New England Journal of Medicine 1995;332(10):647–655. doi: 10.1056/NEJM199503093321006 [DOI] [PubMed] [Google Scholar]
- 102.Denton KM, Shweta A, Anderson WP. Preglomerular and Postglomerular Resistance Responses to Different Levels of Sympathetic Activation by Hypoxia. JASN 2002;13(1):27–34. https://jasn.asnjournals.org/content/13/1/27. Accessed January 21, 2019. [DOI] [PubMed] [Google Scholar]
- 103.Singh AK, Szczech L, Tang KL, et al. Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. New England Journal of Medicine 2006;355(20):2085–2098. doi: 10.1056/NEJMoa065485 [DOI] [PubMed] [Google Scholar]
- 104.Drüeke TB, Locatelli F, Clyne N, et al. Normalization of Hemoglobin Level in Patients with Chronic Kidney Disease and Anemia. New England Journal of Medicine 2006;355(20):2071–2084. doi: 10.1056/NEJMoa062276 [DOI] [PubMed] [Google Scholar]
- 105.Pfeffer MA, Burdmann EA, Chen C-Y, et al. A Trial of Darbepoetin Alfa in Type 2 Diabetes and Chronic Kidney Disease. New England Journal of Medicine 2009;361(21):2019–2032. doi: 10.1056/NEJMoa0907845 [DOI] [PubMed] [Google Scholar]
- 106.Swedberg K, Young JB, Anand IS, et al. Treatment of Anemia with Darbepoetin Alfa in Systolic Heart Failure. New England Journal of Medicine 2013;368(13):1210–1219. doi: 10.1056/NEJMoa1214865 [DOI] [PubMed] [Google Scholar]
- 107.Bouglé A, Duranteau J. Pathophysiology of sepsis-induced acute kidney injury: the role of global renal blood flow and renal vascular resistance. Contrib Nephrol 2011;174:89–97. doi: 10.1159/000329243 [DOI] [PubMed] [Google Scholar]
- 108.Mehta RL, Rabb H, Shaw AD, et al. Cardiorenal syndrome type 5: clinical presentation, pathophysiology and management strategies from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol 2013;182:174–194. doi: 10.1159/000349970 [DOI] [PubMed] [Google Scholar]
- 109.Di Lullo L, Bellasi A, Barbera V, et al. Pathophysiology of the cardio-renal syndromes types 1–5: An uptodate. Indian Heart Journal 2017;69(2):255–265. doi: 10.1016/j.ihj.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hatamizadeh P, Fonarow GC, Budoff MJ, et al. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 2013;9(2):99–111. doi: 10.1038/nrneph.2012.279 [DOI] [PubMed] [Google Scholar]