

Abstract
Oncologic phase II trials that evaluate the activity of new therapeutic agents have evolved dramatically over the past 50 years. The standard approach beginning in the late 1960’s focused on individual studies that evaluated new anticancer agents against a wide range of both solid and hematopoietic malignancies often in a single ‘broad phase II trial’ that included hundreds of patients; such studies efficiently established the landscape for subsequent development of a specific drug with respect to likely disease focus, toxicity, dose and schedule. In the 1980’s and 1990’s emphasis on histological context drove an explosion in the number of individual phase II trials conducted; despite this increase in trial activity, investigations based on histology per se failed to improve the success rate of new agents brought to the clinic. Over the past 20 years, evolution toward a molecular drug development paradigm has demonstrably improved our ability to select patients more likely to benefit from systemic treatment; simultaneously, technological advances have permitted initial attempts at the rapid assignment of therapy based on pre-defined molecular characteristics of tumor or germline in broad-based master protocols that are inclusive of many diseases and molecularly-characterized disease subsets, akin to but much more sophisticated scientifically than the broad phase II platforms of the past.
Keywords: precision medicine, basket trial, bucket trial, master protocol, cancer, drug development, precision oncology, history of phase II trials, history of medicine
The introduction of routine molecular tumor characterization into oncologic treatment planning that began approximately 20 years ago has markedly enhanced both the specificity and efficacy of systemic cancer therapy.1 The rapid application of biological selection criteria, both genomic and protein-based, has improved treatment for patients with a variety of advanced cancers, and led to the first ‘histology agnostic’ FDA approvals of anticancer agents.2-7 Over the past decade, the launch of effective, molecularly targeted and immunotherapeutic molecules into oncologic practice has led to the discontinuation of nonspecific cytotoxic anticancer agent development.8
Application of the principles of precision medicine to cancer treatment9 relies on the measurement of a molecular characteristic in a specific patient’s tumor, a characteristic that suggests the potential utility of a specific molecularly-targeted therapeutic. This reorientation of the therapeutic paradigm, away from pleiotropic mechanisms of tumor cell killing, has advanced based on marked improvements in both biomarker discovery and validation, as well as the availability of sophisticated instrumentation capable of levels of diagnostic throughput that were inconceivable in the recent past. The field of precision oncology has also advanced, at least in part, through the development of several innovations in clinical trial design.10 The evolution of novel approaches to early phase precision cancer clinical trial development is a central topic of this review.11,12
In light of the early stage of development of such studies, the multiple operational challenges that they engender,13 and their modest clinical benefit to date,14 it seems reasonable to introduce this issue of the Cancer Journal by examining the historical precedents for the concept of the oncologic master protocol that includes basket, umbrella, and platform studies.15 Over the last ten years, such master protocols have been developed to provide an investigational framework for a coordinated evaluation of multiple therapeutic approaches in one or more molecularly-defined tumor types, with the goal of improving the efficiency of the cancer clinical trials process. Master protocols can be designed to provide sufficient information to support an application for new drug approval by the FDA, or, more frequently, to identify biomarker-selected drugs that can be more effectively predicted to be successful in the setting of a subsequent, definitive randomized study.
Evolution of the Oncologic Phase II Trial
For the most part, contemporary cancer clinical trials that fit under the rubric of a master protocol are designed to discover a signal of therapeutic activity in a group of patients selected by molecular subtype. These studies are most often not randomized trials and frequently employ an established drug dose and schedule defined previously in an early phase investigation.12 The goal of a master protocol is to develop an estimate of drug activity in the setting of a specific molecular characteristic of the cancer undergoing treatment.
Master protocols as a form of clinical investigation have developed through several evolutionary stages over the past 60 years. During the initial era of cancer clinical trials from the 1950s to the mid-1960s, the safety and modest efficacy of antimetabolites, alkylating agents, natural products, and corticosteroids were established, predominantly in hematopoietic malignancies.8 The number of trials conducted, predominantly with support from the National Cancer Institute (NCI), was small (Figure 1), but the notable development of combination programs for acute lymphocytic leukemia and early non-Hodgkin lymphoma trials demonstrated the feasibility of combining cytotoxic agents to produce modest, but definitive survival benefits in children and adults.16 The numbers of patients entered into these non-randomized studies varied considerably (from 10–20 to over 100) and included both dose-seeking and activity-dependent endpoints.
Figure 1.
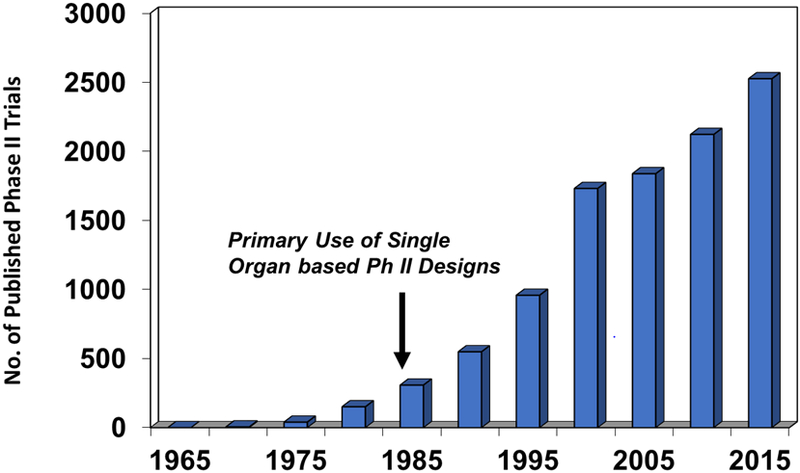
Number of published phase II cancer treatment trials from 1965 to 2015 found by searching the PubMed database.
From the mid-1960s through the late 1970s, phase II trials designed to determine therapeutic activity expanded significantly, and basic rules defining the activity thresholds for new cytotoxics appropriate for further testing were defined by Gehan and colleagues.17 The intellectual framework for many of the trials conducted during this era was grounded on cell kinetic studies in murine leukemia models suggesting that disease eradication based on combinations of cytotoxic agents was, indeed, possible.8 However, the boundary between pilot and later-stage clinical trials in this era was loosely demarcated. It was not until the mid-1960s that the Eastern Cooperative Group defined three phases of cancer clinical trials, including phase II screening investigations designed to test drugs for activity in a variety of human tumors.16
When a novel compound emerged from the National Cancer Institute’s murine screening program, it was quite common at that time to initiate a “broad phase II” trial of that agent in which patients with a wide variety of both solid tumor and hematologic malignancies were treated. One notable example of such a study was a trial of the anthracycline antibiotic doxorubicin conducted by the Southwest Oncology Group.18 A single trial of 472 patients defined the encouraging activity of doxorubicin for patients with advanced breast, prostate, bladder, and endometrial cancer, non-Hodgkin lymphoma, soft tissue sarcoma, and small cell lung cancer, established the standard intermittent dosing schedule for the drug, and characterized all of the intrinsic toxicities of the agent (on the heart, mucosae, and bone marrow). The study was submitted for publication within 24 months of trial initiation. Other pertinent examples of phase II screening trials that established the clinical activity levels and toxicity profiles of novel anticancer cytotoxic agents across a histologically broad range of cancers are shown in Table 1. In each case, based on these screening studies, the agent in question was evaluated further in subsequent trials for specific histologies.
Table 1.
Cancer Drug Development Employing Broad Phase II Trials
Despite the efficiency of the broad phase II trial as a screening tool, the cancer clinical trials field entered a third phase of clinical trial designs beginning in the early 1980s. It became standard dogma that the biologic activity of cancer therapeutics was substantively controlled by histologic context; and thus, investigators concluded that phase II trials should be conducted in a histologically-specific manner.19 As shown in Figure 1, the demise of the broad phase II trial led to “a growth industry” of single drug/single disease trials that would permit “a detailed description of patient characteristics within disease categories.”20 Phase II trials from the 1980s through the mid-1990s conformed to specific two stage designs.21,22 They were only occasionally randomized and enrolled patients who had ‘measurable disease’ usually determined by radiologic evaluation and, if possible, who had been exposed to minimal prior therapy. Two trials in every major disease were conducted, each with adequate numbers of patients to estimate an objective response rate to a particular drug.
Despite attempts to standardize entry criteria, carefully define accrual requirements to ensure sufficient numbers of patients objectively evaluable for response, and refine the statistical input into phase II trial development, an historical analysis of phase II clinical trial outcomes conducted by the NCI in this time frame makes clear the limits of empirical cancer drug development.23 Of 83 new cytotoxic agents developed during this period, 11 novel agents that were not analogs of compounds already known to possess clinical utility demonstrated response rates that were quite modest by standards of the times. As a consequence, there was a growing sense that the limits of cytotoxic drug discovery had been reached.16 Not unexpectedly, the fallacy of depending solely on histological specificity was also becoming clear by the late 1980s, both in then-current murine drug screening models as well as in clinical trials. Increasingly, investigators discussed the potential for molecular rather than histological drug targeting.24 Concurrently, the regulatory and administrative burdens of activating large numbers of disease-specific phase II trials for every agent or combination were becoming clear. The growing knowledge of the molecular (oncogenic) characteristics driving cancer progression propelled the beginnings of a remarkable transition across cancer drug discovery efforts in academia and industry focusing on translating cancer biology into therapeutics.
Development of the Precision Oncology Paradigm: One gene, One Drug, One Disease 2000–2010
The beginnings of what we now refer to as precision oncology comprised a series of well-known discoveries that, contemporaneously, appeared remarkable to both physicians and the lay public. Now, they are well-known stories to most practicing oncologists. These advances were noteworthy but also, in some ways, unpredictable and uniformly never sudden. Here, we discuss several case studies that highlight important issues in the early history of precision medicine, which focus on the development of agents targeting single molecular alterations critical to the pathogenesis of specific tumor histologies.
Labeling any development as representing “the first” in the history of precision oncology is a fraught task. Rather, the development of trastuzumab for the treatment of HER2 overexpressing breast cancer is an important turning point, illustrating how the introduction of targeted therapies began to change the natural history of specific cancers. The overexpression of HER2, a tyrosine kinase receptor which activates multiple signal transduction pathways to regulate cell growth, was identified in the late 1980s by Dennis Slamon and colleagues at the University of California, Los Angeles.25,26 Between 1987 and 1992, multiple investigators determined that HER2 overexpression could result in tumorigenesis, garnering interest in the protein as a potential therapeutic target.27-29 A humanized HER2 antibody, engineered at Genentech in 1992,30 was quickly introduced into multiple phase I and phase II clinical trials, the latter of which focused on enrolling only patients with increased HER2 expression by immunohistochemistry.31
Despite modest activity in a placebo-controlled phase 3 study, with an objective response rate of 15% in the intention-to-treat population,32 trastuzumab was hailed by the popular press as “ushering in a new era of cancer treatment that attempts to target the very flawed genetic mechanisms that cause the disease.”33 While trastuzumab did not dramatically improve outcomes on its own, its role as an important adjunct to chemotherapy in the metastatic,34 neoadjuvant,35 and adjuvant36 settings has been established. The identification of HER2 as a valuable therapeutic target spurred additional work resulting in the development of trastuzumab emtansine, a novel antibody-drug conjugate targeting HER2,37 and pertuzumab, a monoclonal antibody binding a different epitope of HER2.38
Whereas demonstrable HER2 expression was initially evaluated as a prognostic biomarker, indicating poorer outcomes regardless of therapy, it has now become a predictive biomarker, indicative of a subset of patients likely to respond to HER2-directed therapy. Moreover, the natural history of HER2-positive breast cancer has changed, and patients may no longer be at a survival disadvantage compared to patients with HER2-negative disease. Still, acquired resistance to HER2-directed therapy remains common, and cardiac toxicity is a very real risk for patients treated with trastuzumab.39
The development of imatinib for the treatment of chronic myeloid leukemia (CML) was also painted as a miracle by the press and by patients who learned of a pill that might forestall the development of accelerated phase disease and likely death.40 This was understandable. The phase I study of imatinib, a small molecule inhibitor of the BCR-ABL tyrosine kinase, identified no maximally tolerated dose (MTD), and 53 of 54 patients achieved a complete hematologic response;41 based on these results as well as those of three phase II studies, imatinib was granted accelerated approval by the FDA in 2001. The 72 days required for FDA review was the fastest agency approval in the history of anticancer agent development.42 Compared to the 30 years of research required to identify the role of the BCR/ABL fusion oncogene in the pathogenesis of CML, this was a remarkably short time frame, an element of the imatinib story that is easy to overlook.43
The history of imatinib has been unusual in many ways, one of which is the durability of the responses produced by the drug. Resistance to therapy does develop and has spurred the introduction of second- and third-line agents. Still, patients with CML who are treated with BCR-ABL tyrosine kinase inhibitors can expect to live near-normal lifespans.44 The introduction of these drugs has altered the natural history of CML to the extent that the field is now exploring the potential of therapy discontinuation.45
However, the development of targeted therapies has rarely been so straightforward. Our understanding of the role of a putative molecular characteristic in oncogenesis may change over time, thus complicating efforts to target it therapeutically. The case of the epidermal growth factor receptor (EGFR) in lung cancer is instructive. Initial work on EGFR, which was known to play an important role in modulating proliferative cell signaling, was based on its overexpression in multiple tumor types. In one study, EGFR was highly expressed in over 90% of NSCLC tumor specimens examined.46 Despite this observation, phase I trials of gefitinib, an inhibitor of the EGFR tyrosine kinase, were notable for the modest number of clinical responses observed,47-49 while the two phase II studies of gefitinib, which randomized patients to one of two dose levels, identified response rates of 9–19%.50,51 Furthermore, a retrospective analysis of tumor specimens from these two phase II trials found no relationship between EGFR expression (as determined by immunohistochemistry) and clinical response.52 Still, gefitinib received accelerated approval by the FDA in 2003.53
Several groups of investigators sought to further characterize responders by analyzing tumor specimens from patients entered on the phase II studies as well as those treated on expanded access programs. They identified several subgroups of patients with higher response rates: Japanese patients (compared to non-Japanese patients),51 women,50,54 never smokers,55,56 and patients with adenocarcinoma histologies.54-56 In definitive studies done simultaneously by two groups at Harvard and a third group at Memorial Sloan Kettering and Washington University, the EGFR gene was sequenced in tumor specimens from gefitinib responders and non-responders. The tumors of almost all patients who had experienced a clinical response to gefitinib were characterized by mutations in the tyrosine kinase domain of EGFR, definitively establishing mutated EGFR as both the molecular target for gefitinib and a predictive biomarker for response.56-58
While EGFR-targeted therapies have become a success story in lung cancer, resistance to treatment is inevitable, and studies over the last ten years have focused on the development of second and third generation EGFR inhibitors which specifically target mechanisms of resistance to first generation inhibitors.59-63 Currently, the effort to characterize mechanisms of resistance to third generation inhibitors is ongoing with the aim of developing therapies that target these alterations or prevent their emergence.64
While resistance to targeted therapies may be due to so-called “on-target” molecular alterations, such as additional EGFR mutations, it often results from compensatory mechanisms, especially when a target is one member of a signaling pathway. Although the BRAF inhibitors vemurafenib and dabrafenib both improved progression free survival (PFS) in patients with untreated BRAFV600E mutated advanced melanoma compared to chemotherapy in two phase 3 studies, responses were short-lived, with a median PFS of just over 5 months for both drugs.65,66 Additional pharmacodynamic studies performed on patient tumor specimens from these and other trials found that acquired resistance to BRAF inhibitors was frequently associated with up-regulation of signaling in the MAPK pathway.67-69 As a result, combinations of BRAF and MEK inhibitors were studied in multiple large phase 3 trials and were found to improve both PFS and overall survival (OS) compared to the use of BRAF inhibitors alone;70-73 these combinations now comprise standard of care therapy for patients with BRAF-mutated melanoma. These cases clearly demonstrate the limitations of the one gene, one drug, one disease model, and have led to recent efforts to identify promising combinations of targeted therapies.74
The Rapid Expansion and Concomitant Limitations of “Tumor Profiling”
In 2019, a patient’s tumor may undergo high throughput massively parallel DNA and RNA sequencing (often referred to as next generation sequencing, or NGS) over a matter of two or three weeks to identify potential therapeutic targets.75,76 Our ability to do so has developed over the last fifteen years, not only shortening the length of time required for such studies, but expanding the depth and character of available genomic analyses. Whereas technologies in the late 2000s and early 2010s were limited to PCR-based evaluations of mutational “hotspots” and immunohistochemical evaluations of protein expression, whole exome and whole genome sequencing is regularly performed on the tumors of patients treated at tertiary cancer centers. The recent announcement from the Centers for Medicare and Medicaid Services that Medicare will cover next-generation tumor profiling of patients with advanced cancer, coupled with FDA approval of the Foundation One NGS platform, means that such profiling is now accessible to patients treated in the community.77
Genomic and proteomic analyses now have the capacity to detect a variety of aberrations including point mutations, insertion and deletion mutations (indels), copy number alterations, chromosomal rearrangements and gene fusions, DNA methylation patterns, transcript levels, and levels of protein expression.76 Analysis of a patient’s tumor is typically paired with evaluation of matched normal cells, most often from a buccal swab or peripheral blood, to distinguish somatic aberrations found only in a tumor from germline abnormalities.78 More recently, examinations of circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) have been investigated as a means of dynamically and non-invasively assessing tumor burden as well as evaluating the changing genomic landscape of a tumor throughout a patient’s treatment, with an eye to better understanding mechanisms of drug resistance.79-81 While these circulating tumor materials may offer additional insights into the mutational load at metastatic sites compared to the primary tumor,82 the quantity of tumor material is often small and the specificity of such assays compared to analysis of the primary specimen itself is under active investigation.83
These advances have enabled the detection of low frequency mutations and have fostered the development of many new targeted therapies. However, complex challenges remain. Reports from extensive sequencing efforts may list the molecular aberrations identified in a patient’s tumor, but only a minority at best may be “targetable” with approved or experimental agents. Moreover, such reports may neglect to describe the allelic frequency of such aberrations or distinguish between driver mutations – those which induce tumorigenesis – and passenger mutations, which are not themselves pathogenic.76,84,85 Tumor heterogeneity is well established, and analyzed specimens may not represent the dominant phenotype. While driver mutations are commonly present in early clones, tumor evolution, especially in response to therapeutic pressures, may abet the emergence of clones that do not carry the driver mutation or that express subclonal alterations which are not sensitive to targeted therapy approaches.86,87 More broadly, efforts to target tumor molecular aberrations have been hampered by the almost universal development of on- or off-target resistance.84 This has led to a recent focus on evaluating combinations of agents to delay the emergence of such resistance, for example by inhibiting a signaling pathway at more than one level.70
Co-Development of Drugs with Predictive and Pharmacodynamic Biomarkers
As precision oncology moves beyond the “one gene, one drug, one disease” approach, it is critical that drugs and biomarkers are developed together. In the past, such efforts have often been retrospective; the efficacy of cetuximab for KRAS wildtype colorectal cancers88-90 and of gefitinib for EGFR mutant non-small cell lung cancers (NSCLC)57,58 were determined by tissue analysis of responders and non-responders to these agents in unselected clinical trials. The co-development of drugs and biomarkers is not straightforward and runs the risk of excluding populations that might benefit from an agent, especially if it has more than one target. However, potential benefits include more efficient and less costly drug development.79
Predictive biomarkers present in a tumor indicate likelihood of response to a given therapy, which may or may not target the molecular aberration in question. They may be used to select patients who will benefit from a given therapy or, as in the case of KRAS mutations and cetuximab, indicate those who will not.91 Preclinical investigation of new agents should include the development of putative predictive biomarkers in vitro and in vivo; functional testing using patient-derived xenograft (PDX) models, organoids, and other novel approaches may predict for patient sensitivity to targeted agents.92 After a recommended phase II dose is identified in a phase I trial, expansion cohorts can be utilized to enrich for patients whose tumors express the biomarker of interest to preliminarily evaluate efficacy and further explore toxicity. Prior to a phase II study, additional biomarker validation can be performed using patient samples from the dose escalation portion of the trial. If the predictive value of the biomarker is unclear, later phase trials can enroll patients whose tumors may or may not express the biomarker and stratify analysis by presence or absence of that biomarker.93,94 Expansion cohorts and biomarker enriched arms on phase II trials can be added or removed based on evolving estimates of benefit, with early stopping rules for efficacy and toxicity.94
While much research in precision oncology in the last twenty years has focused on the development of predictive biomarkers, particularly the presence of single mutations or genetic alterations, it is imperative that the development of pharmacodynamic (PD) biomarkers be pursued with equal vigor. Recent efforts to enhance the range of applicable PD assays will ensure that targets are not only identified as potentially predictive biomarkers, but that drugs interacting with these targets do so with greater specificity. PD biomarkers may impart information about proof-of-mechanism (is the drug engaging its intended target?), proof-of-concept (does engaging the target result in changed tumor biology, such as apoptosis?), an agent’s biologically effective dose, and mechanisms of resistance. They may assess a range of molecular events, including protein phosphorylation, proliferation, apoptosis, cell cycle regulation, and epigenetic regulation.80
The approach to demonstrating a targeted agent’s mechanism of action has also changed over the recent past. A preclinically validated mechanism of action can often now be validated in patients in multiple steps. Primary PD biomarkers assess the engagement of a drug with its presumed target, while secondary PD biomarkers examine its downstream effect, for example on signaling pathways. Tertiary PD biomarkers evaluate the effect of an agent on cell biology, such as its impact upon cell cycle progression or apoptosis.95 Ideally, PD biomarkers from all three categories should be incorporated into drug development, although this is not always possible. Increasingly, multiplex assays are being developed to measure multiple biomarkers simultaneously on the same tumor specimen. While this approach requires additional target validation, it may increase the efficiency and reliability of biomarker development. Groups of related biomarkers may be interpreted together, decreasing the chance that a single errant biomarker measurement will bias interpretation of drug effect.96,97
PD biomarkers can and should play a role in the determination of the ideal dose and schedule of a novel therapy. Dose-finding efforts for nonspecific cytotoxic chemotherapies have traditionally been based on a putative dose-response relationship, seeking to identify the maximum tolerated dose (MTD) as the recommended dose for further study. This approach is less relevant with respect to evaluating targeted therapies, however, as they may not have a straightforward dose-response or dose-toxicity relationship. For example, there may be minimal toxicity until a target is saturated, after which there could be extensive toxicity.98 In the case of very effective agents like imatinib, clinical efficacy may be observed well before the MTD is reached.99 For targeted therapies, investigators have increasingly worked to identify the biologically effective dose, defined as the minimum dose and schedule of an agent required to engage the target consistently to produce antitumor activity.95 Doing so requires the investigator to establish the relationship between plasma drug concentration and the primary PD biomarker in question, which provides confidence that plasma concentration may be used as a proxy for tissue concentration.95
Effective PD biomarker development is complex; challenges include tumor specimen quality, tumor heterogeneity, assay reproducibility and standardization, and tissue acquisition and analysis with respect to dosing.96 Phase 0 trials, in which a non-toxic dose of a novel agent is given to a small number of patients with the aim of utilizing tumor biopsies to analyze the agent’s pharmacodynamic effects, provide a means of evaluating the properties of a drug and developing PD biomarkers in patients without the concomitant toxicity seen in phase I clinical trials.100 For example, the knowledge gained from a phase 0 study of veliparib was used to determine a biologically effective dose which has since been evaluated in multiple phase II studies of the drug.95,101 While PD biomarker development may be expensive and time-consuming, it is also costly to conduct large, unsuccessful clinical trials on unselected patient populations; hence, PD endpoints should be incorporated routinely into the development of targeted agents.
Novel Methods for Evaluating Targeted Therapies Using “Master Protocols”
While the widespread adoption of tumor sequencing has resulted in a proliferation of data, a persistent question has been how to best exploit this data to improve patient outcomes.85 In the early 2010s, efforts focused on analyzing tissue from exceptional responders to novel therapies102 and N of 1 trials, in which a patient’s response to a therapy that had been “matched” to a specific tumor alteration was compared to that patient’s response to a previous standard of care agent.94 Many cancer centers have developed molecular tumor boards in which a patient’s tumor profiling data is discussed and a recommendation is made for a standard therapy (either on- or off-label) or a clinical trial.6,75
In efforts to evaluate the concept of precision oncology more broadly, several academic cancer centers have conducted studies examining the concept of matching patients to targeted therapies based on tumor profiling. While in several cases the proportion of patients who could be assigned a matched standard or investigational therapy was small, these studies demonstrated that a precision oncology approach was feasible.103 Moreover, several studies found that patients treated on trials utilizing a molecularly-matched agent had improved outcomes compared to those who did not, although these studies were not randomized.104-107
SHIVA, the first randomized trial of precision oncology as an approach, was a phase II study that randomized patients with multiple tumor histologies to receive either 1) one of 11 molecularly targeted agents based on the presence or absence of aberrations in the hormone receptor, PI3K/AKT/mTOR, and RAF/MEK pathways or 2) physician’s choice of standard therapy.108 While no difference in PFS was observed, the study used only a limited range of targeted therapies and did not account for differing levels of evidence regarding the relevance of each patient’s pathway aberration. Such data, which may implicate a mutation as a known driver with therapeutic implication but may also identify a variation without any known pathogenic consequence, must be considered when designing precision oncology trials. In one study of 860 patients with metastatic lung adenocarcinoma, 37% of whom received a targeted therapy guided by molecular profiling of their tumor, the level of evidence supporting the use of that therapy for the patient’s particular mutation was predictive of clinical response.109
The development of master protocols to organize the testing of patients with multiple tumor histologies and/or tumor molecular aberrations creates an infrastructure with which a variety of hypotheses may be tested simultaneously, and tumors organized by genomic or other “omic” changes, in addition to/or instead of histology. Master protocols have been defined as “coordinated efforts to evaluate more than one or two treatments in more than one patient type or disease within the same overall trial structure.”15 Classically, master protocols have been described as falling into one of two categories: basket studies, which seek to treat patients across multiple histologies whose tumors share the same alteration, and umbrella studies, which assign patients with one tumor type to one of several therapies based on tumor profiling data.15 While such studies may be registration trials in intent, they are more often signal-finding trials intended to identify potentially interesting therapies worthy of further study in certain patient populations.
Several basket studies have led to FDA approvals for new indications of drugs already approved for use in different settings. A study of vemurafenib in patients with advanced BRAFV600 mutated malignancies (exclusive of melanoma) found an overall response rate (ORR) of 42% of patients with NSCLC and 29% of those with anaplastic thyroid cancer.110 While few or no responses were observed in multiple other tumor types, this data led to FDA approvals for dabrafenib in patients with V600E-mutated NSCLC111 and anaplastic thyroid cancer.112 A phase II study of pembrolizumab, an anti-programmed death 1 (PD-1) antibody, in patients with mismatch repair-deficient and proficient tumors demonstrated the susceptibility of these tumors to immune checkpoint inhibition.113 A subsequent phase II study of pembrolizumab in patients with 12 tumor types whose tumor demonstrated mismatch repair deficiency found a 53% response rate across histologies114 and led in 2017 to the first tissue agnostic approval of a drug by the FDA.115
Umbrella trials have, to date, primarily served as exploratory signal-finding studies. They often operate using adaptive designs, wherein new arms may be added based on new evidence or removed based on lack of response, and patients may be assigned to a therapy based on an algorithm that utilizes evolving data to account for that patient’s likelihood of response.13 The BATTLE studies, which assigned patients with advanced NSCLC to treatment arms based on molecular profiling of their tumors, demonstrated the feasibility of obtaining on-trial biopsies in heavily pretreated patients and using molecular characterization to assign patients to treatment arms in real time.116,117
The terms “basket trial” and “umbrella trial” are useful heuristics but may not adequately describe all large platform precision oncology trials.12 For example, the CREATE study examined crizotinib in patients with multiple tumor types and alterations in both ALK and MET genes.118 The ongoing NCI-MATCH (Molecular Analysis for Therapy Choice) study uses on-study biopsies to assign patients with any histology to a broad range of therapies based on their tumor molecular alterations.119 Both basket and umbrella trials may treat patients with multiple histologies whose tumors harbor multiple mutations or other alterations; such ‘hybrid’ designs conform to the overall concept of a master protocol while demonstrating the evolving definitions of umbrella or basket trials.12
Beyond terminology, master protocols present numerous challenges. They are time-consuming, require significant coordination among multiple stakeholders, and can be costly.15 The Lung Master Protocol, or Lung-MAP, an umbrella study of patients with advanced NSCLC, is the result of a complex public-private partnership including the National Cancer Institute, the Foundation for the National Institutes of Health, SWOG, and Friends of Cancer Research, among others.120 Due to their complexity, master protocols provide a difficult format for sponsors hoping to achieve registration and create significant work for regulatory officials and institutional review boards faced with numerous amendments.13 They are rarely randomized, making it difficult to draw conclusions about the efficacy of an agent.12 Questions also remain about the statistical design of master protocols. For example, some basket studies are designed as a series of Simon two-stage studies, treating each arm as a separate trial for statistical purposes, whereas others allow aggregation of data from similar arms, which permits investigators to deem a therapy effective earlier.121
Despite these challenges, master protocols offer many opportunities for both patients and investigators. They enable patients with rare cancers to participate more readily in clinical trials and may lead to new therapeutic options. For example, the phase I/II basket study of larotrectinib which ultimately led to the drug’s tissue agnostic FDA approval in 2018122 found an ORR of 75% in patients with infantile fibrosarcoma, a rare tumor which often results in limb amputation.7 They efficiently group patients with heterogeneous tumors (histologically, molecularly, or both), are adaptable, and enable large collaborations, centralizing the work of designing and administering a clinical trial.13,15 They also can provide access to laboratories performing validated assessments of specific, treatment-defining molecular alterations in patients’ tumors. Once established, furthermore, the infrastructure for these trials can speed the screening of new therapeutic agents across a wide range of both common and understudied malignancies—in a fashion similar to, but much more sophisticated than, the “broad phase II” clinical trial platforms of the past.
Overview of this Issue of the Cancer Journal
This issue of the Cancer Journal is an attempt to provide an overview of the evolving field of master protocol development in the service of precision oncology. Following the introductory chapter that traces the lineage of basket and umbrella trials from their modest beginnings as an attempt to efficiently screen new agents under development by the NCI in the 1970’s using an histology-inclusive approach known as the “broad phase II trial’, the administrative, regulatory, and biostatistical underpinnings of master protocol conduct are outlined. This is followed by a description of the workings of the molecular characterization laboratories that are at the heart of efforts to ‘match’ patient treatment to genomic features of a patient’s germline and/or tumor. Next, colleagues from the ECOG-ACRIN network group define the elaborate clinical trial and information technology platform that supports the ongoing NCI-MATCH study, which permitted the largest master protocol ever conducted in oncology to proceed using an evidence-based, automated patient assignment algorithm rather than relying upon an expert evaluation case by case. The subsequent contribution provides an overview of the entire NCI precision medicine clinical trials portfolio including the LUNG-MAP, ALCHEMIST, and MPACT trials. Investigators from the MD Anderson Cancer Center then describe their own major precision medicine platform and how it has been used to develop novel early phase clinical trials at their institution. Because a major initial stimulus for the development of the field of precision oncology was the demonstration that next generation tumor sequencing could drive the choice of effective systemic therapy in an individual patient, the NCI’s effort to understand the biology underlying ‘exceptional responders’ to both targeted and cytotoxic treatments is reviewed. Finally, future approaches to the field of precision oncology are described in terms of the efforts of plan for a new generation of master protocols that will inform the use of immunotherapy and combinations of targeted therapies in patients whose tumors are resistant to current standards of care.
Acknowledgments
Supported by: ZIA BC 011078; Phase 0/1 Clinical Trials from the National Cancer Institute
Contributor Information
Deborah B. Doroshow, Icahn School of Medicine at Mount Sinai.
James H. Doroshow, Division of Cancer Treatment and Diagnosis, National Cancer Institute, NIH.
References
- 1.Doroshow JH. Precision medicine in oncology In: DeVita VT, Lawrence TS, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology (11th ed). Philadelphia: Wolters Kluwer; 2019:186–96. [Google Scholar]
- 2.Sparano JA, Gray RJ, Makower DF, et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N Engl J Med 2018; 379:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw AT, Felip E, Bauer TM, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol 2017; 18:1590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–65. [DOI] [PubMed] [Google Scholar]
- 5.Khorashad JS, Kelley TW, Szankasi P, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood 2013; 121:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Editorial Board. Making precision oncology the standard of care. Lancet Oncology 2017; 18:835. [DOI] [PubMed] [Google Scholar]
- 7.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018; 378:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res 2008; 68:8643–53. [DOI] [PubMed] [Google Scholar]
- 9.Desmond-Hellmann S, Sawyers CL, Cox DR, Fraser-Liggett C. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington D.C.: National Academies Press; 2011. [PubMed] [Google Scholar]
- 10.Kummar S, Williams PM, Lih C-J, et al. Application of molecular profiling in clinical trials for advanced metastatic cancers. J Natl Cancer Inst 2015; 107: DOI: 10.1093/jnci/djv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willyard C “Basket studies” will hold intricate data for cancer drug approvals. Nat Med 2013; 19:655. [DOI] [PubMed] [Google Scholar]
- 12.Janiaud P, Serghiou S, Ioannidis JPA. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat Rev 2019; 73:20–30. [DOI] [PubMed] [Google Scholar]
- 13.Cecchini M, Rubin EH, Blumenthal GM, et al. Challenges with novel clinical trial designs: master protocols. Clin Cancer Res 2019; 25: 2049–2057. [DOI] [PubMed] [Google Scholar]
- 14.Eckhardt SG, Lieu C. Is precision medicine an oxymoron? JAMA Oncol 2018; DOI: 10.1001/jamaoncol.2018.5099. [DOI] [PubMed] [Google Scholar]
- 15.Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med 2017; 377:62–70. [DOI] [PubMed] [Google Scholar]
- 16.Keating P, Cambrosio A. Cancer on Trial: Oncology as a New Style of Practice. Chicago: The University of Chicago Press; 2012. [Google Scholar]
- 17.Gehan EA, Schneiderman MA. Historical and methodological developments in clinical trials at the National Cancer Institute. Stat Med 1990; 9:871–80. [DOI] [PubMed] [Google Scholar]
- 18.O’Bryan RM, Luce JK, Talley RW, Gottlieb JA, Baker LH, Bonadonna G. Phase II evaluation of adriamycin in human neoplasia. Cancer 1973; 32:1–8. [DOI] [PubMed] [Google Scholar]
- 19.Rubinstein L Phase II design: history and evolution. Chin Clin Oncol 2014; 3:48: DOI: 10.3978/j.issn.2304-3865.2014.02.02. [DOI] [PubMed] [Google Scholar]
- 20.Wittes RE, Marsoni S, Simon R, Leyland-Jones B. The phase II trial. Cancer Treat Rep 1985; 69: 1235–9. [PubMed] [Google Scholar]
- 21.Simon R How large should a phase II trial of a new drug be? Cancer Treat Rep 1987;71: 1079–85. [PubMed] [Google Scholar]
- 22.Simon R Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989; 10:1–10. [DOI] [PubMed] [Google Scholar]
- 23.Marsoni S, Hoth D, Simon R, Leyland-Jones B, De Rosa M, Wittes RE. Clinical drug development: an analysis of phase II trials, 1970–1985. Cancer Treat Rep 1987; 71:71–80. [PubMed] [Google Scholar]
- 24.Doroshow JH, Kummar S. Translational research in oncology−−10 years of progress and future prospects. Nat Rev Clin Oncol 2014; 11:649–62. [DOI] [PubMed] [Google Scholar]
- 25.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 26.Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244:707–12. [DOI] [PubMed] [Google Scholar]
- 27.Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci USA 1987; 84:7159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 1987; 237:178–82. [DOI] [PubMed] [Google Scholar]
- 29.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA 1992; 89:10578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA 1992; 89:4285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baselga J Clinical trials of herceptin® (trastuzumab). Eur J Cancer 2001; 37:18–24. [PubMed] [Google Scholar]
- 32.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 1999; 17:2639–48. [DOI] [PubMed] [Google Scholar]
- 33.Roan S Weapon in the war on cancer. Los Angeles Times; 1998; Section 1. [Google Scholar]
- 34.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783–92. [DOI] [PubMed] [Google Scholar]
- 35.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673–84. [DOI] [PubMed] [Google Scholar]
- 36.Smith I, Procter M, Gelber RD, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet 2007; 369:29–36. [DOI] [PubMed] [Google Scholar]
- 37.Doroshow DB, LoRusso PM. Trastuzumab emtansine: determining its role in management of HER2+ breast cancer. Future Oncol 2018; 14:589–602. [DOI] [PubMed] [Google Scholar]
- 38.Gerratana L, Bonotto M, Bozza C, et al. Pertuzumab and breast cancer: another piece in the anti-HER2 puzzle. Expert Opin Biol Ther 2017; 17:365–74. [DOI] [PubMed] [Google Scholar]
- 39.Pernas S, Barroso‐Sousa R, Tolaney SM. Optimal treatment of early stage HER2-positive breast cancer. Cancer 2018; 124:4455–66. [DOI] [PubMed] [Google Scholar]
- 40.Druker BJ. Perspectives on the development of imatinib and the future of cancer research. Nat Med 2009; 15:1149–52. [DOI] [PubMed] [Google Scholar]
- 41.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344:1031–7. [DOI] [PubMed] [Google Scholar]
- 42.Cohen MH, Williams G, Johnson JR, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res 2002; 8:935–42. [PubMed] [Google Scholar]
- 43.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008; 112:4808–17. [DOI] [PubMed] [Google Scholar]
- 44.Bower H, Björkholm M, Dickman PW, Höglund M, Lambert PC, Andersson TM-L. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol 2016; 34:2851–7. [DOI] [PubMed] [Google Scholar]
- 45.Patel AB, Wilds BW, Deininger MW. Treating the chronic-phase chronic myeloid leukemia patient: which TKI, when to switch and when to stop? Expert Rev Hematol 2017; 10:659–74. [DOI] [PubMed] [Google Scholar]
- 46.Rusch V, Baselga J, Cordon-Cardo C, et al. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res 1993; 53:2379–85. [PubMed] [Google Scholar]
- 47.Ranson M, Hammond LA, Ferry D, et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol 2002; 20:2240–50. [DOI] [PubMed] [Google Scholar]
- 48.Herbst RS, Maddox A-M, Rothenberg ML, et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumors: results of a phase I trial. J Clin Oncol 2002; 20:3815–25. [DOI] [PubMed] [Google Scholar]
- 49.Baselga J, Rischin D, Ranson M, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol 2002; 20:4292–302. [DOI] [PubMed] [Google Scholar]
- 50.Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149–58. [DOI] [PubMed] [Google Scholar]
- 51.Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non–small-cell lung cancer. J Clin Oncol 2003; 21:2237–46. [DOI] [PubMed] [Google Scholar]
- 52.Bailey R, Kris M, Wolf M, et al. Gefitinib (‘Iressa’, ZD1839) monotherapy for pretreated advanced non-small-cell lung cancer in IDEAL 1 and 2: tumor response is not clinically relevantly predictable from tumor EGFR membrane staining alone. Lung Cancer 2003; 41: Suppl. 2: S71. [Google Scholar]
- 53.Cohen MH, Williams GA, Sridhara R, et al. United States Food and Drug Administration drug approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res 2004; 10:1212–8. [DOI] [PubMed] [Google Scholar]
- 54.Jänne PA, Gurubhagavatula S, Yeap BY, et al. Outcomes of patients with advanced non-small cell lung cancer treated with gefitinib (ZD1839, “Iressa”) on an expanded access study. Lung Cancer 2004; 44:221–30. [DOI] [PubMed] [Google Scholar]
- 55.Miller VA, Kris MG, Shah N, et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non–small-cell lung cancer. J Clin Oncol 2004; 22:1103–9. [DOI] [PubMed] [Google Scholar]
- 56.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101:13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497–500. [DOI] [PubMed] [Google Scholar]
- 58.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129–39. [DOI] [PubMed] [Google Scholar]
- 59.Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011; 17:1616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013; 19:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang JC-H, Ahn M-J, Kim D-W, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J Clin Oncol 2017; 35:1288–96. [DOI] [PubMed] [Google Scholar]
- 62.Goss G, Tsai C-M, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2016; 17:1643–52. [DOI] [PubMed] [Google Scholar]
- 63.Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017; 376:629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oxnard GR, Hu Y, Mileham KF, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol 2018; 4:1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–65. [DOI] [PubMed] [Google Scholar]
- 67.Paraiso KHT, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 2010; 102:1724–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012; 366:707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sosman JA, Pavlick AC, Schuchter LM, et al. Analysis of molecular mechanisms of response and resistance to vemurafenib (vem) in BRAFV600E melanoma. J Clin Oncol 2012; 30(15_suppl):8503. [Google Scholar]
- 70.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371:1877–88. [DOI] [PubMed] [Google Scholar]
- 71.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015; 386:444–51. [DOI] [PubMed] [Google Scholar]
- 72.Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol 2017; 28:1631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015; 372:30–9. [DOI] [PubMed] [Google Scholar]
- 74.Holbeck SL, Camalier R, Crowell JA, et al. The National Cancer Institute ALMANAC: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res 2017; 77:3564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med 2011; 3:111ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013; 153:17–37. [DOI] [PubMed] [Google Scholar]
- 77.CMS finalizes coverage of next generation sequencing tests, ensuring enhanced access for cancer patients. Available from: https://www.cms.gov/newsroom/press-releases/cms-finalizes-coverage-next-generation-sequencing-tests-ensuring-enhanced-access-cancer-patients
- 78.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 2015; 7:283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yap TA, Sandhu SK, Workman P, de Bono JS. Envisioning the future of early anticancer drug development. Nat Rev Cancer 2010; 10:514–23. [DOI] [PubMed] [Google Scholar]
- 80.Gainor JF, Longo DL, Chabner BA. Pharmacodynamic biomarkers: falling short of the mark? Clin Cancer Res 2014; 20:2587–94. [DOI] [PubMed] [Google Scholar]
- 81.Goldberg SB, Patel AA. Monitoring immunotherapy outcomes with circulating tumor DNA. Immunotherapy 2018; 10:1023–5. [DOI] [PubMed] [Google Scholar]
- 82.Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017; 8:1324 DOI: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late stage human malignancies. Sci Transl Med 2014; 6:224ra24–224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med 2016; 375:1289–94. [DOI] [PubMed] [Google Scholar]
- 85.Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell 2017; 168:584–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017; 168:613–28. [DOI] [PubMed] [Google Scholar]
- 87.Rübben A, Araujo A. Cancer heterogeneity: converting a limitation into a source of biologic information. J Transl Med 2017;15: 190 DOI: 10.1186/s12967-017-1290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lièvre A, Bachet J-B, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006; 66:3992–5. [DOI] [PubMed] [Google Scholar]
- 89.Di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 2007; 96:1166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lièvre A, Bachet J-B, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26:374–9. [DOI] [PubMed] [Google Scholar]
- 91.Oldenhuis CN a. M, Oosting SF, Gietema JA, de Vries EGE. Prognostic versus predictive value of biomarkers in oncology. Eur J Cancer 2008; 44:946–53. [DOI] [PubMed] [Google Scholar]
- 92.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 2015; 15:747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garcia VM, Cassier PA, de Bono J. Parallel anticancer drug development and molecular stratification to qualify predictive biomarkers: dealing with obstacles hindering progress. Cancer Discov 2011; 1:207–12. [DOI] [PubMed] [Google Scholar]
- 94.Dienstmann R, Rodon J, Tabernero J. Optimal design of trials to demonstrate the utility of genomically-guided therapy: putting precision cancer medicine to the test. Mol Oncol 2015; 9:940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parchment RE, Doroshow JH. Pharmacodynamic endpoints as clinical trial objectives to answer important questions in oncology drug development. Sem Oncol 2016; 43:514–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kinders R, Ferry-Galow K, Wang L, Srivastava AK, Ji J (Jay), Parchment RE. Implementation of validated pharmacodynamic assays in multiple laboratories: challenges, successes, and limitations. Clin Cancer Res 2014; 20:2578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marrero A, Lawrence S, Wilsker D, Voth AR, Kinders RJ. Translating pharmacodynamic biomarkers from bench to bedside: analytical validation and fit-for-purpose studies to qualify multiplex immunofluorescent assays for use on clinical core biopsy specimens. Sem Oncol 2016; 43:453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hansen AR, Graham DM, Pond GR, Siu LL. Phase 1 trial design: is 3 + 3 the best? Cancer Control 2014; 21:200–8. [DOI] [PubMed] [Google Scholar]
- 99.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 2009; 101:708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kummar S, Rubinstein L, Kinders R, et al. Phase 0 clinical trials: conceptions and misconceptions. Cancer J 2008; 14:133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol 2009; 27:2705–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol 2015; 33:975–7. [DOI] [PubMed] [Google Scholar]
- 103.André F, Bachelot T, Commo F, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 2014; 15:267–74. [DOI] [PubMed] [Google Scholar]
- 104.Tsimberidou A-M, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 2012; 18:6373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014; 311:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sohal DPS, Rini BI, Khorana AA, et al. Prospective clinical study of precision oncology in solid tumors. J Natl Cancer Inst 2016; 108 Available from: https://academic.oup.com/jnci/article/108/3/djv332/2412397 [DOI] [PubMed] [Google Scholar]
- 107.Bedard PL, Oza A, Clarke B, et al. Molecular profiling of advanced solid tumors at Princess Margaret Cancer Centre and patient outcomes with genotype-matched clinical trials. Clin Cancer Res 2016; 22(1 Supplement):Abstract PR03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Le Tourneau C, Delord J-P, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 2015; 16:1324–34. [DOI] [PubMed] [Google Scholar]
- 109.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov 2017; 7:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 Mutations. New Engl J Med 2015; 373:726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.FDA grants regular approval to dabrafenib and trametinib combination for metastatic NSCLC with BRAF V600E mutation. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm564331.htm
- 112.FDA approves dabrafenib plus trametinib for anaplastic thyroid cancer with BRAF V600E mutation. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606708.htm
- 113.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017; 357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm560040.htm
- 116.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 2011; 1:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Papadimitrakopoulou V, Lee JJ, Wistuba II, et al. The BATTLE-2 Study: A biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol 2016; 34:3638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schöffski P, Wozniak A, Stacchiotti S, et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 ‘CREATE.’ Ann Oncol 2017; 28:3000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Conley BA, Doroshow JH. Molecular analysis for therapy choice: NCI MATCH. Sem Oncol 2014;41(3):297–9. [DOI] [PubMed] [Google Scholar]
- 120.Herbst RS, Gandara DR, Hirsch FR, et al. Lung master protocol (Lung-MAP)-a biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin Cancer Res 2015; 21:1514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology: a trade-off between complexity and efficiency. J Clin Oncol 2017; 35:271–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.FDA approves larotrectinib for solid tumors with NTRK gene fusions. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm626720.htm
- 123.Wilson WL, Schroeder JM, Bisel HF, Mrazek R, Hummel RP. Phase II study of hexamethylmelamine (NSC 13875). Cancer 1969; 23:132–6. [DOI] [PubMed] [Google Scholar]
- 124.Hoogstraten B, Gottlieb JA, Caoili E, Tucker WG, Talley RW, Haut A. CCNU (1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea, NSC-79037) in the treatment of cancer. Phase II study. Cancer 1973; 32:38–43. [DOI] [PubMed] [Google Scholar]
- 125.Haas CD, Coltman CA, Gottlieb JA, et al. Phase II evaluation of bleomycin. A Southwest oncology Group study. Cancer 1976;38:8–12. [DOI] [PubMed] [Google Scholar]
- 126.Weiss AJ, Metter GE, Nealon TF, et al. Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep 1977; 61:55–8. [PubMed] [Google Scholar]