

SUMMARY
HCC and NAFLD are growing epidemics that seem to be interrelated and pose an important burden to public health. Obesity, T2DM, and NAFLD clearly augment the risk of HCC via multiple pathways, many of which are common to all 3 conditions. Resultant dysregulation of adipocytokines, oxidative stress, and insulin resistance contribute to a proinflammatory milieu that fosters the development of HCC via many oncogenic pathways. Mounting evidence that cirrhosis is not a requisite for tumor development in NASH should make us take pause and urge for strategies to identify such patients at an early stage. Furthermore, effective chemopreventive strategies are urgently needed to temper the increase of HCC in patients with obesity, T2DM, and NAFLD.
Keywords: Nonalcoholic steatohepatitis, Cirrhosis, Noncirrhotic, Insulin resistance, Hepatocellular carcinoma, Obesity, Diabetes, Chemoprevention
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is now the most common liver disease in the United States, with more than 80 million Americans affected. It represents a spectrum of diseases ranging from isolated hepatic steatosis (IHS) to steatosis in association with inflammation and cellular injury—the progressive subtype of NAFLD referred to as nonalcoholic steatohepatitis (NASH). NASH is often but not always associated with varying degrees of fibrosis that can develop into cirrhosis and all of its associated complications.1,2 The prevalence of the disease parallels the epidemic of the meta-bolic syndrome, namely obesity, type 2 diabetes mellitus (T2DM) and its other manifestations worldwide. It is no surprise that the mechanisms underlying the meta-bolic syndrome, insulin resistance, and obesity are also important in the development of both IHS and NASH.3
Hepatocellular carcinoma (HCC), known to occur most commonly in the setting of cirrhosis, is also increasing in incidence, and is now the second leading cause of cancer deaths worldwide.4 Historically, the risk factors for HCC in the United States have primarily included alcoholic cirrhosis, hepatitis B virus (HBV) infection, hemo-chromatosis, and hepatitis C virus (HCV) infection. Because of its link to the HCV epidemic, HCC is considered the fastest growing cause of cancer mortality overall in the United States as well.5 However, the clinical landscape of HCV is changing rapidly into a future where cure is not only probable, but will be nearly universal for those who have access to therapy. No doubt, this will reduce the development of HCV-related HCC in the future. Results of recent studies demonstrate that HCC is more prevalent in the setting of obesity and insulin resistance, and may occur in NAFLD patients without cirrhosis.4 Therefore, if the incidence of obesity, diabetes, and NASH continues to increase, and the HCV-related association decreases with effective treatment strategies, NAFLD could become the most common cause of HCC in the United States and other developed countries. Indeed, in a recent retro-spective cohort study that evaluated trends in HCC etiology among adult liver transplant recipients from 2002 to 2012, the number of patients undergoing liver trans-plant for HCC secondary to NASH increased by nearly 4-fold, whereas the number of patients with HCC secondary to HCV increased by only 2-fold.6 During that same 10-year period, the prevalence of NASH-related HCC increased steadily, becoming the second leading etiology of HCC-related liver transplant in the United States (increasing from 8.3% in 2002 to 10.3% in 2007 and to 13.5% in 2012).
To clarify these relationships, this review discusses the pathophysiologic mecha-nisms that underpin the close relationship between obesity, insulin resistance, NAFLD, and the progression to HCC, both in the presence and absence of cirrhosis.
OBESITY, DIABETES, NONALCOHOLIC FATTY LIVER DISEASE, AND NONALCOHOLIC STEATOHEPATITIS AS RISK FACTORS FOR HEPATOCELLULAR CARCINOMA
The results of several studies have elucidated the relative risk of several disease pro-cesses that are implicated in the pathogenesis of HCC. In a population-based study, authors analyzed 6991 cases from the Surveillance, Epidemiology, and End Results– Medicare databases that link cancer registry data and Medicare enrollment during the period from1994 to 2007.7 The authors estimated the population-attributable fractions (PAFs), that is, the proportions of cases that can be attributed to specific risk factors. They found that T2DM and/or obesity had the greatest PAF (36.6%), followed by alcohol-related disorders (23.5%), HCV (22.4%), HBV (6.3%), and rare genetic disorders (3.2%; Fig. 1). Although the relative risk of HCV for HCC incidence (39.9) was higher than the relative risk of T2DM and/or obesity (2.47), the high prevalence of T2DM and obesity in the United States accounted for more cases of HCC than HCV. However, the exact PAF of NAFLD remains to be determined, because NAFLD cases per se were not identified in the Surveillance, Epidemiology, and End Results database. Although the study was limited to age groups 68 years or older, it repre-sented about 25% of the general population of the United States.7
Fig. 1.
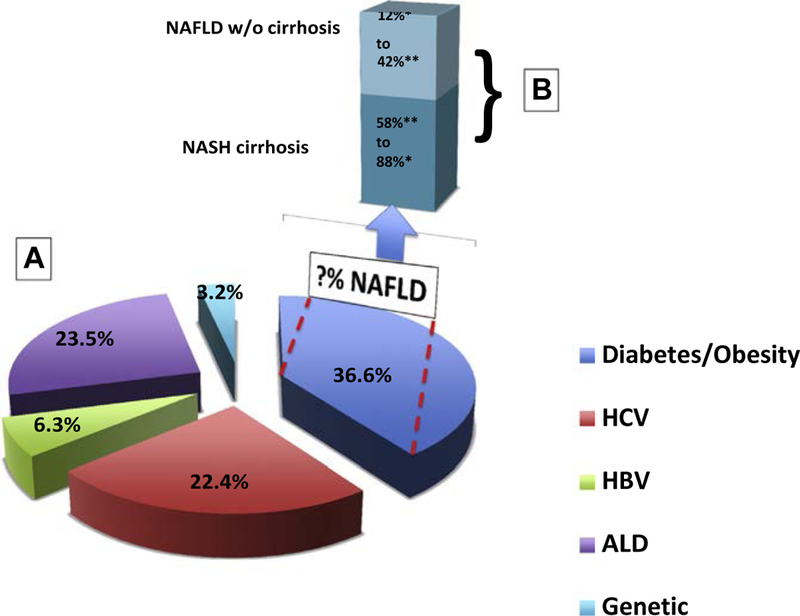
Risk factors for hepatocellular carcinoma (HCC) incidence in the United States. (A) Risk factors of HCC in the United States (presented as percentage). In a study by Welzel and colleagues of 6991 cases from the Surveillance, Epidemiology, and End Results (SEER)–Medicare databases from 1994 to 2007 the authors estimated the PAFs, that is, the proportions of cases that can be attributed to specific risk factors. The PAF owing to type 2 diabetes mellitus (T2DM) and/or obesity was the highest (36.6%), followed by alcohol-related disorders (23.5%), HCV (22.4%), HBV (6.3%), and rare genetic disorders (3.2%). Although the contribution of NAFLD is not exactly known, it is thought to contribute to the majority of the obesity and diabetes cases. (B) A breakdown of cases of HCC occurring in the setting of NAFLD in the presence or absence of cirrhosis to HCC. ALD, alcoholic liver disease; HBV, hepatitis B virus; HCV, hepatitis C virus; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis. (Data from [A] Welzel TM, Graubard BI, Quraishi S, et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am J Gastroenterol 2013;108(8):1314–21; [B] *Hashimoto E, Yatsuji S, Tobari M, et al. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroen-terol 2009;44 Suppl 19:89–95; and **Mittal S, Sada YH, El-Serag HB, et al. Temporal trends of non-alcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs pop-ulation. Clin Gastroenterol Hepatol 2014;146(5):S–917.)
In other studies, a direct relationship between obesity and HCC has been demonstrated. Available data suggest that obesity increases the risk of HCC 1.5- to 4-fold (Table 1).8–10 In Danish and Korean studies, an association between obesity and an increased relative risk of HCC (1.9 and 1.56, respectively) was seen.9,11 Data from the United States also suggest that with obesity there is a significantly elevated relative risk of death from many cancers including HCC (4.52 for men; see Table 1).8 One large meta-analysis included 7 cohort studies in 5037 overweight subjects (body mass index of 25–30 kg/m2) and 10 studies in 6042 obese subjects (body mass index of 30 kg/m2). The authors found that, compared with normal weight people, HCC risk increased 17% in those who were overweight and 89% in those who were obese.12 Furthermore, in a study from Japan that looked at the recurrence of HCC after ablation therapy in NASH patients, the authors found that increased visceral fat was an indepen-dent risk factor for recurrence of HCC at 3 years (75.1% vs 43.1% with low visceral fat).13 The mechanism through which obesity leads to increased HCC is thought to be related both to a direct effect of obesity on insulin resistance and the perpetuation of a proinflam-matory milieu, as well as via its role in the development and progression of liver disease in patients with NAFLD. In a 2008 review of studies of the epidemiology of NAFLD, Lazo and Clark14 found that, on average, 76% (range, 33%–99%) of obese patients undergoing bariatric surgery are reported to have steatosis and 37% (range, 9.8%–72.5%) NASH.
Table 1.
Risk of obesity, diabetes, and NAFLD on incidence of HCC
| Author, Year | Location | Cases | Study Type | Risk Measurement |
Reported Risk |
|---|---|---|---|---|---|
| Obesity | |||||
| Moller et al,9 1994 | Denmark | 43,965 | Case control | RR | 1.9 |
| Wolk et al,10 2001 | Sweden | 28,129 | Case control | SIR | 2.4 |
| Calle et al,8 2003 | USA | 90,000 | Prospective | RR | 1.68 (F), 4.52 (M) |
| Oh et al,11 2005 | Korea | 781,283 | Prospective | RR | 1.56 |
| Larsson et al,12 2007 | NA | 11,079 | Meta-analysis | RR | 1.89 |
| Diabetes | |||||
| Adami et al,15 1996 | Sweden | 153,852 | Case control | SIR | 4.1 |
| Wideroff et al,17 1997 | Denmark | 109,581 | Case control | SIR | 4 (M), 2.1 (F) |
| El-Serag et al,16 2004 | USA | 824,263 | Case control | HRR | 2.16 |
| Yang et al,18 2011 | NA | NR | Meta-analysis | RR | 1.87 |
| Wang et al,111 2012 | NA | NR | Meta-analysis | RR | 2.31 |
| NAFLD/NASH | |||||
| Adams et al,26 2005 | USA | 420 | Prospective | Prevalence | 0.5% |
| Ekstedt et al,24 2006 | Sweden | 129 | Prospective | Prevalence | 2.3% |
| Rafiq et al,25 2009 | USA | 173 | Retrospective | Prevalence | 0% NAFLD and 2.8% NASH |
| Ascha et al,28 2010 | USA | 195 | Retrospective | Prevalence | 12.8% in NASH cirrhosis |
Abbreviations: CC, cryptogenic cirrhosis; F, female; HCC, hepatocellular carcinoma; HRR, hazard rate ratios; M, male; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; OR, odds ratio; RR, relative risk; SIR, standardized incidence ratio.
In a similar trend seen in many studies, T2DM was associated with a substantially increased risk of HCC (see Table 1). In a large cohort from Sweden consisting of 153,852 diabetics, the risk of HCC was increased 4-fold. Even after exclusion of other risk factors for HCC, such as cirrhosis and viral hepatitis, T2DM was associated with a 3-fold increased risk of HCC.15 In a large study from the Veterans Administration (VA) hospitals in the United States similar conclusions were drawn. Of 173,643 diabetic patients and 650,620 nondiabetic controls followed over 10 to 15 years, the incidence of HCC was increased more than 2-fold in diabetic patients, with the risk remaining significant even after the exclusion of patients with other causes of liver disease, including NAFLD.16 One limitation of these studies is that other causes of liver disease, such as alcohol and viral hepatitis, could have been underestimated because diag-nosis identification codes were used to identify patients. In a large Danish study of 109,581 patients hospitalized for T2DM, the standardized incidence ratio for HCC was 4 in men and 2.1 in women.17 Two large meta-analyses in 2011 confirmed that the relative risks of T2DM for the development of HCC were 1.87 and 2.31, respec-tively.18,19 Most recently, a 2014 meta-analysis of 21 studies that included a total of 9767 HCC patients evaluated the association of T2DM with overall and disease-free survival in HCC.20 In this study the pooled hazard ratios were 1.46 for overall survival and 1.57 for disease-free survival for patients with DM. Although it is possible that the increased HCC risk associated with diabetes seen in these studies may be mediated through the development of NAFLD, the presence of multiple pathogenic mechanisms common to obesity, insulin resistance and NAFLD21 suggests that this link may not be mediated through NAFLD per se.12
Data from several retrospective and prospective studies demonstrate a direct rela-tionship between NASH and HCC. Retrospective results suggest that between 4% and 27% of NASH patients develop HCC at one point 22,23; however, the true prevalence of HCC across the whole spectrum of NAFLD remains unknown. In prospective studies, the reported prevalence of HCC was 0% to 0.5% in patients with IHS, and as high as 12.8% in those with NASH.24–26 In a cross-sectional study of 4406 HCC patients, 59% had NAFLD/NASH, 36% T2DM, and 22% HCV infection.27 In this context, it is noteworthy that patients with HCV cirrhosis develop HCC at a higher rate than those with NAFLD/NASH. Ascha and colleagues28 followed 195 patients with NASH cirrhosis and 315 patients with HCV cirrhosis for 3.2 years and identified HCC in 12.8% and 20.3% of patients in the NASH and HCV groups, respectively. Here, the yearly cumula-tive incidence of HCC was 2.6% in patients with NASH-associated cirrhosis, whereas it was 4.0% in those with HCV-associated cirrhosis. Bhala and colleagues29 studied patients with advanced fibrosis and cirrhosis, including 247 owing to NASH and 264 owing to HCV, who were followed for a mean of 85.6 months. They found that NAFLD patients had a lower incidence of HCC compared with those with HCV (2.4% vs 6.8%; P<.03). In comparison to Ascha and associates’ study,28 Bhala and colleagues attributed the lower incidence of HCC in both their NASH and HCV populations to potential differences in population risk factors and the fact that only those with biopsy-proven NASH were included in the Ascha and colleagues study.
NASH is increasing in importance as an indication for liver transplantation (LT) and may exceed HCV as an indication numerically over the next decade. Interestingly, although HCC may occur more commonly in patients with HCV cirrhosis, the rate at which patients are transplanted for HCC in the setting of NASH is increasing rapidly. Indeed, in a recent retrospective cohort study that evaluated trends in HCC as an etiology among adult LT recipients from 2002 to 2012, the authors found that the num-ber of patients undergoing LT for HCC secondary to NASH increased by nearly 4-fold, whereas the number of LT patients with HCC secondary to HCV increased by only 2.5- fold in the Model of End-stage Liver Disease era of LT listing.6 As noted, during that 10-year period, NASH-related HCC increased steadily, becoming the second leading etiology of HCC-related LT in the United States.
HEPATOCELLULAR CARCINOMA IN NONALCOHOLIC FATTY LIVER DISEASE WITHOUT CIRRHOSIS
The fact that fibrosis and cirrhosis are not necessary for the development of HCC in NASH patients indicates that obesity, insulin resistance, and the proinflammatory milieu of NASH may mediate carcinogenesis directly. Evidence of the development of HCC in noncirrhotic patients continues to accumulate in case reports or case series (Table 2).29–52 In a recent study analyzing 1419 HCC cases that were related to NASH (120 cases), HCV (1013), and alcohol (286) in the VA system, cirrhosis was present in only 58.3% of NASH-related HCC cases.51 The researchers also found less robust adherence to HCC surveillance in NASH patients than in patients with HCV or alco-holic liver disease, and attributed the high rate of incidence of HCC in NASH without cirrhosis (41.7%) to the lower rate of surveillance, because there is currently no recom-mendation for surveillance in this population. There is also the incorrect perception that NASH patients without cirrhosis are unlikely to develop HCC. Interestingly, despite the lack of surveillance and resulting early detection, the 1-year survival rate did not differ among the different etiologies of HCC.53
Table 2.
Reports of HCC incidence in NAFLD/NASH without cirrhosis
| Study, Year | No. of Cases |
Average Age (y) |
Gender | Fibrosis | Comorbidities | Tumor |
|---|---|---|---|---|---|---|
| Zen et al,30 2001 | 1 | 62 | F | Pericellular | DM | Multifocal |
| Orikasa et al,31 2001 | 1 | 67 | F | Bridging | DM | Solitary |
| Bencheqroun et al,32 2004 | 1 | 68 | M | F2 | DM | Solitary |
| Bullock et al,33 2004 | 2 | 64/74 | M | None (F0) | DM, HTN, obesity | Solitary |
| Gonzalez et al,34 2004 | 1 | 73 | M | F1 | DM, HTN, obesity | Solitary |
| Cuadrado et al,35 2005 | 1 | 71 | M | Portal | DM, obesity | Solitary |
| Sato et al,36 2005 | 1 | 64 | M | Bridging | Obesity, dyslipidemia | Solitary |
| Hai et al,37 2006 | 1 | 72 | M | F2 | DM | Solitary |
| Ichikawa et al,38 2006 | 2 | 60/66 | M/F | F2–F3 | DM, obesity | Solitary |
| Hashizume et al,39 2007 | 3 | 54/72/82 | M | F1–F3 | DM, HTN, obesity | Solitary (2), Multifocal (1) |
| Guzman et al,40 2008 | 3 | 45/57/70 | M (1), F (2) | F0 | DM, HTN, obesity, dyslipidemia | Multifocal |
| Chagas et al,41 2009 | 1 | 65 | M | F1 | Obesity, dyslipidemia | Multifocal |
| Paradis et al,42 2009 | 16 | NR | M (16) | F0–F3 | DM, HTN, obesity, dyslipidemia | NR |
| Kawada et al,43 2009 | 6 | 59–81 | M (3), F (3) | F2–F3 | DM, HTN, obesity, dyslipidemia | Solitary |
| Hashimoto et al,44 2009 | 4/34 (12%) | NR | M, F | F1–F2 | NR | NR |
| Takuma et al,45 2010 | 7 | 67–75 | M (3), F (3) | F1–F3 | DM, HTN, obesity, dyslipidemia | Solitary (5), multifocal (2) |
| Tokushige et al,46 2010 | 10 | NR | NR | F1 (1), F2 (3), F3 (10) | NR | NR |
| Yasui et al,47 2011 | 43 | NR | M (77%) | F1 (10), F2 (15), F3 (18) | DM, HTN, obesity, dyslipidemia | Solitary (72%) |
| Ikura et al,49 2011 | 1 | 72 | M | F1 | HTN | Solitary |
| Ertle et al,52 2011 | 10 | 69 | M (89%) | F0–F3 | DM, HTN, obesity, dyslipidemia | NR |
| Mittal et al,51 2014 | Z50 | NR | NR | NR | DM, HTN, obesity, dyslipidemia | NR |
Interesting insights into possible areas of further research can be derived from NASH–HCC studies in which potential differences between NASH- and HCV- associated HCC are suggested. In the Mittal and colleagues’51 analysis of the VA population previously discussed, the authors noted that patients with NASH more often had fewer alpha-fetoprotein–secreting tumors. The authors speculated that less aggressive tumor biology in NASH-related HCC could be partially explained by this finding, but it could also be related to the short period of survival rate estimation of only 1 year. A Japanese group also reported lower alpha-fetoprotein synthesis in NASH-associated HCC (35.3%) compared with 69.6% of tumors in patients with HCV.48 Furthermore, they found that 52.9% of patients with NASH-related HCC had elevated des-gamma-carboxy prothrombin, a biomarker of HCC, compared with 41.3% of patients with HCV-related HCC.46 This result suggests that the tumor markers and cancer biology in NASH-related HCC may differ from HCV-related HCC, and should be investigated further.
Results of several studies support the possibility that NASH-related HCC may behave differently in certain subpopulations. Limited available data suggest that risk factors for the development of NASH without cirrhosis include older age, male gender, and the metabolic syndrome (see Table 2). Indeed, HCC was even reported to develop in patients with the metabolic syndrome and features of IHS without steatohepatitis or fibrosis.40 In a study of 87 Japanese NASH patients with HCC, Yasui and colleagues47 found that 56% of patients were noncirrhotic. The authors stratified the data by gender and noted that men developed HCC at a less advanced stage of liver fibrosis than women. Hashimoto and colleagues44 examined 34 cases of NASH-related HCC and found that there was a prevalence of advanced age, male gender, obesity, and T2DM; 12% of the patients had stage 1 or 2 fibrosis and 88% had advanced fibrosis (stage 3–4). These HCC patients tended to be older, male, and to have the metabolic syndrome. Although there are currently no recommendations to initiate HCC screening in noncirrhotic NASH, mounting data suggest that it should be a concern, particularly for older men with the metabolic syndrome. Inexpensive and reliable methods of HCC detection are needed to capture reliably patients at risk for HCC before the develop-ment of cirrhosis. Revisiting the surveillance guidelines may be warranted.
PATHOGENESIS OF HEPATOCELLULAR CARCINOMA IN OBESITY, DIABETES, AND NONALCOHOLIC FATTY LIVER DISEASE
Although the precise pathogenesis by which obesity and insulin resistance foster the development of HCC is not defined clearly, several mechanisms could operate in the setting of NAFLD. The inflammatory milieu associated with obesity and insulin resis-tance is characterized by abundant oxidative stress, activation of the unfolded protein response (UPR), and other inflammatory processes, including activation of the innate immune system. Many of these pathways could play a major role in the tumorigenicity and development of DNA damage, which provide a favorable setting for the develop-ment of HCC.53 Insulin resistance and obesity promote an aberrant adipocytokine pro-file, including increased interleukin-6, leptin, and tumor necrosis factor (TNF)-a and decreased adiponectin, which seems to contribute to increased cellular proliferation, angiogenesis, inhibition of apoptosis, and worsening insulin resistance.54,55
Insulin Resistance
In a prospective study of 6237 French men, hyperinsulinemia was associated with an approximately 3-fold increased risk for HCC.56 In addition to its role in glucose and lipid metabolism, insulin has pleotropic effects that regulate inflammatory and other pathways. Insulin-like growth factor-1 (IGF-1) and insulin receptor substrate-1 (IRS-1), an important substrate of IGF-1, are downstream targets of insulin that are crucial to cellular proliferation (Fig. 2).57,58 Human HCC cells overexpress both IGF-159 and IRS-1.60 IRS-1–mediated signals may act as survival factors and protect against transforming growth factor b1–induced apoptosis in HCC, which may contribute to hepatic oncogenesis.61 In addition, IRS-1 can promote hepatocyte proliferation via mitogen-activated protein kinase and phosphatidylinositol–3 kinase (PI3K), important pathways in HCC development.61 Interestingly, cirrhotic patients with HCC and impaired glucose metabolism that causes postprandial hyperin-sulinemia have accelerated HCC growth.62 The PI3K/phosphatase and tensin homo-log (PTEN)/Akt axis is a key regulator of critical cellular functions such as insulin and other growth factor signaling, glucose and lipid homeostasis, and apoptosis.63
Fig. 2.
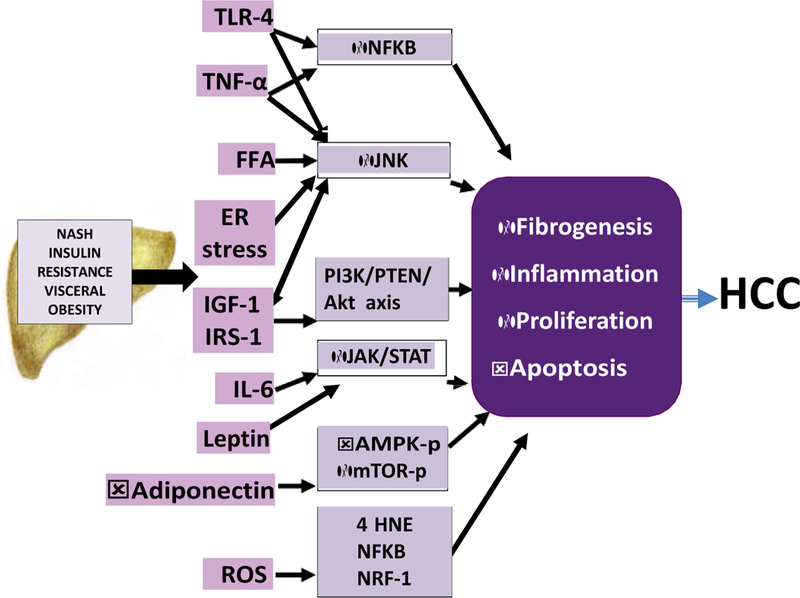
Potential pathways linking NASH to HCC. The inflammatory milieu associated with obesity, insulin resistance and NASH leads to the activation of multiple pathways that impair insulin signaling, induce oxidative stress, ER stress and other inflammatory processes. These in turn lead to the activation of oncogenic signaling pathways such as PI3K/PTEN/Akt, JAK/STAT, NF-kB, mTOR, 4HNE and NRF-1. In concert, these pathways promote cellular proliferation, increased angiogenesis, decreased apoptosis that can then foster the development of HCC. 4 HNE, trans-4-hydroxy-2-nonenal; AMPK-p, activated protein kinase; ER, endoplasmic reticu-lum; FFA, free fatty acids; IGF-1, insulin-like growth factor-1; IL-6, interleukin-6; IRS-1, insulin receptor substrate-1; mTOR-p, mammalian target of rapamycin complex 1; NF-kB, nuclear factor kappa B; NRF-1, nuclear factor erythroid 2-related factor; PI3k, phosphatidylinositol-3 kinase; PTEN, PI3K/phosphatase and tensin homolog; TLR, Toll-like receptors; TNF-a, tumor necrosis factor-a.
PTEN acts as a phosphoinositide phosphatase which terminates PI3K-propagated signaling.63 Thus, PTEN is a tumor suppressor that is dysregulated in obesity, insulin resistance, and T2DM, thus offering another mechanism through which NAFLD, T2DM, and insulin resistance could promote tumor growth in the setting of HCC.63
c-Jun amino-terminal kinases
The c-Jun amino-terminal kinases (JNKs) are associated with development of NASH and hepatic carcinogenesis.64–67 Free fatty acids, reactive oxygen species and TNF-a are increased in the setting of hyperinsulinemia and are activators of JNK1, which in turn phosphorylates IRS-1 (see Fig. 1). JNK1 activation and the subsequent phosphorylation of IRS-1 are very important factors in obesity-induced insulin resis-tance and increased hepatic inflammation and apoptosis; JNK2 has been shown not to play a role.64,65,68 Puri and colleagues64 have shown that the extent of JNK activation is associated with the level of histologic activity in NASH patients. Several studies have described an important role of JNK1 in the development of HCC.66,67,69 Chang and col-leagues66 demonstrated enhanced JNK1 activation in 55% of human HCC samples compared with non-HCC liver tissue. JNK1 plays an important role in the development of HCC, partially through activation of the mammalian target of rapamycin mTOR com-plex 1, which is involved in several human cancers.70,71
Endoplasmic Reticulum Stress
Emerging data suggest that endoplasmic reticulum (ER) stress plays an important role in NASH and HCC.72–74 The ER regulates protein synthesis and folding for a variety of cellular processes. Various stressors, such as nutrient or glucose deprivation, viral infections, disrupted calcium homeostasis, protein glycosylation, and excess fatty acids, can promote the accumulation of unfolded or misfolded proteins in the ER lumen and activation of an adaptive response called the UPR, which can result in salvage or further injury and apoptosis, depending on the trigger and milieu.75–77 JNK is a downstream target of the inositol-requiring enzyme-1, a branch of the UPR that has been shown to perpetuate inflammatory processes and insulin resistance.76 Both ER stress and the subsequent activation of the UPR have been found to play a role in HCC.78 Although the exact mechanism is not understood completely, it is thought to be owing to the effect of the activation of extracellular signal-regulated pro-tein kinase (ERK) which is highly expressed in HCC via PIK13 activation. ER stress also activates the nuclear factor erythroid 2-related factor 2 and transcription factor-6, both of which regulate the small heterodimer partner nuclear receptor, leading to its sup-pression. This in turn activates cyclin D1 and hence increases tumorigenicity and he-patocyte proliferation.79,80
Insulin resistance is also associated with increased oxidative stress. The generation of reactive oxygen species leads to upregulation of proinflammatory cytokines such as TNF-a. This upregulation then can promote tumor growth via both anti-apoptotic action and further upregulation of proinflammatory cytokines through activation of nu-clear factor-kappa B (NF-kB).81 Oxidative stress may favor tumorigenesis via inflam-mation and dysregulated cell proliferation; however, it may also directly induce cancer-promoting gene mutations. Trans-4-hydroxy-2-nonenal (4HNE), a product of lipid peroxidation, is important in cancers that are caused by mutations of the p53 gene (see Fig. 2).82 The p53 pathway targets include wild-type p53 activated fragment (p21WAF), a cyclin-dependent kinase inhibitor and the growth arrest DNA damage gene, GADD45, a p53-regulated and DNA damage-inducible protein, and the 14–3-3 protein, which plays a role in G2/M arrest.83 Nuclear respiratory factor-1 (Nrf-1) is an essential transcription factor in the prevention of oxidative injury. Nrf-1 knockout mice develop NASH and progress to HCC owing to increased oxidative stress.84 In a human study, Maki and colleagues85 showed a higher incidence and recurrence of HCC in patients with high levels of 8-Oxo-2’-deoxyguanosine (8-OHdG) and 4HNE in liver tissue, suggesting that oxidative stress may influence HCC carcinogenesis or tumor biological behavior.
Leptin
Leptin, a product of the obese (ob) gene, is a regulator of food intake and energy expenditure via hypothalamic-mediated mechanisms.86 The importance of leptin in angiogenesis, hematopoiesis, and lipid and carbohydrate metabolism as well as in immune systems, has been demonstrated recently.86,87 Furthermore, leptin has a direct effect on hepatic stellate cells and is likely to play an important role in the pro-gression of liver fibrosis in NASH.87 The leptin receptor has 6 isoforms, of which obRB is the most important. Leptin levels are increased in obese individuals, suggesting a state of leptin resistance.86 Suppressors of cytokine signaling and insulin resistance are associated with leptin resistance, with higher levels of leptin found in NASH patients and in patients with HCC.88,89 In a human study, the expression of leptin and its receptor in both adjacent nontumor liver tissue and HCC tissue was explored using immunohistochemical staining. It was found that adjacent nontumorous liver tissue expressed higher levels of leptin and its receptor than the HCC tissue.89 The authors suggested that leptin might act as an endocrine growth factor that stimulates hepatocytes toward the initiation and progression of HCC.
Available evidence suggests that leptin mediates its effect through the Jak2/Stat 3 pathway, which in turn augments cyclin D1 protein expression, leading to cellular pro-liferation and thereby the development of HCC.90 In addition, leptin inhibits apoptosis by inhibiting transforming growth factor-b1. Therefore, leptin may mediate HCC onco-genesis via stimulation of proliferation and inhibition of proapoptotic pathways. Inter-estingly, these pathways also lead to increased methionine adenosyl transferase (MAT) 2A and MAT2b gene expression, which are known to play a role in hepatocyte proliferation.91 Finally, leptin promotes fibrogenesis by stimulating hepatic stellate cells. Stellate cells are critical mediators of fibrogenesis and angiogenesis via multiple pathways including ERK/Akt, nuclear factor kappa D (NF-kD), and hypoxia-inducible factor 1, each of which has been associated with the development of HCC.87,92
S-Adenosylmethionine
Recently, a role for abnormal levels of S-adenosylmethionine (SAMe) in the develop-ment of NASH and HCC has been proposed.93 SAMe is the principal biological methyl donor made in all mammalian cells. The liver plays a central role in the homeostasis of SAMe.93,94 SAMe is endogenously produced from methionine and adenosine triphos-phate by MAT.93 MAT has 2 subunits, a1 and a2, encoded by MAT1A and MAT2A. MAT1A is expressed mostly in differentiated liver and encodes the a1 subunit, whereas MAT2A encodes for the catalytic subunit a2, which is distributed widely. Mice deficient in MAT1A develop NASH and HCC. Deletion of MAT1A may impair very-low-density lipoprotein (VLDL) assembly, leading to the synthesis of small, lipid-poor VLDL particles and decreased secretion of triglycerides. Additionally, low SAMe levels promote proinflammatory cytokine release and development of NASH.93 Several abnormal pathways have also been identified in MAT1A-knockout mice that can contribute to HCC formation, including (1) a decrease in apurinic/apyr-imidinic endonuclease activity, which leads to DNA instability and malignant transfor-mation, (2) increased liver kinase B1 activity, which induces the activation of 50 adenosine monophosphate-activated protein kinase (AMPK) and enhanced hepatocyte proliferation, (3) leptin signaling, which induces MAT2A and MAT2b genes via activation of these survival pathways and promotes enhanced cell proliferation (the MAT2A-encoded protein induces leptin’s mitogenic response by raising intracellular SAMe levels, leading to polyamine biosynthesis and growth), and (4) activation of the ERK pathway, which is highly expressed in HCC.93
These pathways suggest that the tumorigenic process is closely related to insulin resistance, oxidative stress and changes in adiponectin and leptin levels which pre-cede the development of fibrosis. Thus, it is plausible that HCC development in NASH patients without cirrhosis is the result of the activation of these pathways.
Toll-Like Receptors
TLRs are a family of pattern-recognition receptors that recognize pathogen-associated molecular patterns and endogenous components that result from cell death, known as damage-associated molecular patterns, and activate the innate immune system.95–97 Ten members of the TLR family have been identified. They play a role in ligand recog-nition after which a signal via myeloid differentiation factor (MyD)88 leads to activation of NF-kB and production of proinflammatory cytokines, including TNF-a and IL-6.96,98 The MyD88-dependent pathways activate JNK signaling as well. TLR4 may play a specific role in HCC progression by increasing proliferation, preventing apoptosis, and increasing production of cytokines (TNF-a and interleukin-6).99,100
Potential Chemoprevention of Hepatocellular Carcinoma in Obesity and Nonalcoholic Fatty Liver Disease
There is currently no effective chemoprevention to decrease the incidence of HCC, irrespective of disease etiology, with the possible exceptions of the treatment of HBV and HCV infection.101 With soaring numbers of NAFLD/NASH patients and the incremental increase in HCC, chemopreventive agents are crucially needed. Indeed, with increasing evidence that HCC can develop in NASH without cirrhosis, the lack of clear recommendations for surveillance in this population and the absence of effec-tive treatments for NASH is concerning. Preliminary data point to statins, metformin, and SAMe as potentially effective chemopreventive compounds. Clinical trials are urgently needed to evaluate methods to prevent or hinder the development of HCC in patients with NASH.
Statins
In previous studies, a beneficial effect of statins in reducing the risk of HCC has been shown.102–104 Multiple mechanisms have been proposed from animal studies, including (1) statin inhibition of cell proliferation via inhibition of v-myc avian myelocy-tomatosis viral oncogene homolog (MYC) protein phosphorylation which seems to play a role in liver tumorigenicity (atorvastatin blocked MYC and suppressed tumor initiation in an animal model of MYC-induced HCC and in human cell lines of HCC105), (2) induction of apoptosis (simvastatin was found to induce apoptosis selectively in living cancer cells but not in normal cells106,107), and (3) inhibition of angiogenesis.107
In a randomized trial of 91 patients with HCC who were treated with local therapy followed by randomization to either 40 mg of atorvastatin or placebo, survival was increased in the atorvastatin group (18 vs 9 months; P 5 .006).104 In a matched case-control study conducted within a VA cohort of patients with T2DM, the incidence of HCC was reported at least 6 months after entry in the cohort.103 Statin use was recorded by searching filled prescriptions. The study identified 1303 HCC cases and 5212 controls and found a significant reduction in the incidence of HCC (range, 25%–40%) in statin-treated diabetic patients. This beneficial effect was evident in pa-tients with and without cirrhosis. This study was limited by many confounding vari-ables, including in particular that patients with T2D have lipid abnormalities and that NAFLD was not identified per se given the lack of a diagnostic code for NAFLD in the VA system. In a recent nationwide case-control study from Sweden, 3994 HCC patients treated with statins (determined from the national drug registry) were compared with 19,970 controls matched by age and sex.102 In this study, the odds ratio for HCC among statin users was 0.88, suggesting a modest effect of statins on decreasing the risk of HCC.102 Although these data are intriguing, they have important limitations and the true effect of statins on chemoprevention in HCC will need to be ascertained through carefully conducted, randomized, controlled trials.
Metformin
Although metformin may not have significant efficacy in the treatment of NASH itself, it may have a role in decreasing the risk of HCC. In several studies, insulin sensitizers in patients with T2DM may have reduced the risk of HCC.108–114 Metformin has an anti-tumor effect in HCC through the suppression of the mTOR pathway which occurs via 2 mechanisms. The first mechanism involves inhibition of mitochondrial oxidative phos-phorylation leading to AMPK activation and subsequent mTOR pathway suppression; the second mechanism works via glycemic control leading to inhibition of IGF-R and thus inactivation of the mTOR pathway in insulin-responsive cancers.115 A large Italian case control study compared 610 HCC patients treated with metformin with 618 un-treated cirrhotic patients and 1696 controls.109 Metformin use was associated with a decreased incidence of HCC in diabetic patients (odds ratio, 0.15). Given the complex relationship between T2DM, HCC, and cirrhosis and the fact that this was a retrospec-tive case control study, a definitive conclusion cannot be drawn from this research. Two recent meta-analyses included 10 studies with 22,650 cases of HCC in 334,307 patients with T2DM. Metformin use was associated with a 50% reduction in the incidence of HCC, whereas sulfonylurea and insulin use increased HCC inci-dence by 62% and 161%, respectively.111 Similarly, another metaanalysis of 17 case-controlled studies and 32 cohort studies of HCC reported a combined relative risk estimate of 2.31 for HCC among diabetic patients. In these studies, sulphonylur-eas and treatment with insulin increased the risk of HCC. The authors attributed this increased risk to the effects of hyperinsulinemia on stimulating oncogenesis. In another study, survival in 135 patients with early stage HCC who underwent radiofrequency ablation was assessed; of the 53 diabetic patients, 21 were treated with metformin and 32 were not. The authors found that the survival rate after ablation was less in diabetic patients compared with nondiabetic patients (1 year, 82.8% vs 93.9%; 3 years, 55.1% vs 80.2%; 5 years, 41.3% vs 64.7%; P 5 .004) and that survival was further decreased in diabetics who were not on metformin (hazard ratio, 0.24; P 5 .02) compared with those who were.112 Large, randomized trials are needed to better understand the role of metformin or other antidiabetic drugs in HCC chemoprevention.
S-Adenosylmethionine
Lu and colleagues116 examined the effect of SAMe for chemoprevention of HCC in animal models. Here, SAMe led to inhibition of HCC progression and showed proap-optotic and antiangiogenic properties. Because of the potential direct effect of SAMe on HCC pathogenesis and its proapoptotic and antiangiogenic properties,93,117 there is an ongoing phase II clinical trial using SAMe as a chemopreventive agent in HCC patients with hepatitis C in the setting of NAFLD.
KEY POINTS.
Obesity, type 2 diabetes mellitus (T2DM), and insulin resistance are strongly associated with nonalcoholic fatty liver disease (NAFLD) and increased incidence of hepatocellular carcinoma (HCC).
HCC incidence is increasing as NAFLD becomes the most common cause of liver disease.
HCC can develop in NAFLD patients without cirrhosis so cancers may be missed given the high prevalence of NAFLD and the limitations of current screening strategies.
Activation of pathways that promote inflammation, insulin resistance, angiogenesis, and cellular proliferation seen in these diseases promote the development of HCC.
Clinical studies to prevent the development of HCC in patients with obesity, T2DM or NAFLD are critically needed.
Acknowledgments
Financial Support: USC Research Center of Liver Diseases, P30 DK48522. Dr M.E. Rinella has provided consulting services for Abbvie, Fibrogen, Takeda, and NGM pharmaceuticals. Dr M. Noureddin has nothing to disclose.
REFERENCES
- 1.Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged popula-tion utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 2011;140(1):124–31. [DOI] [PubMed] [Google Scholar]
- 2.Rinella ME, Loomba R, Caldwell SH, et al. Controversies in the diagnosis and management of NAFLD and NASH. Gastroenterol Hepatol (N Y) 2014;10(4): 219–27. [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol 2003;98(9): 2042–7. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molec-ular carcinogenesis. Gastroenterology 2007;132(7):2557–76. [DOI] [PubMed] [Google Scholar]
- 5.Seefe LB. Introduction: the burden of hepatocellular carcinoma. Gastroenter-ology 2004;127(5 Suppl 1):S1–4. [DOI] [PubMed] [Google Scholar]
- 6.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carci-noma in the U.S. Hepatology 2014;59(6):2188–95. [DOI] [PubMed] [Google Scholar]
- 7.Welzel TM, Graubard BI, Quraishi S, et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am J Gastroenterol 2013;108(8):1314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calle EE, Rodriguez C, Walker-Thurmond K, et al. Overweight, obesity, and mor-tality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003;348(17):1625–38. [DOI] [PubMed] [Google Scholar]
- 9.Moller H, Mellemgaard A, Lindvig K, et al. Obesity and cancer risk: a Danish record-linkage study. Eur J Cancer 1994;30A(3):344–50. [DOI] [PubMed] [Google Scholar]
- 10.Wolk A, Gridley G, Svensson M, et al. A prospective study of obesity and cancer risk (Sweden). Cancer Causes Control 2001;12(1):13–21. [DOI] [PubMed] [Google Scholar]
- 11.Oh SW, Yoon YS, Shin SA. Effects of excess weight on cancer incidences depending on cancer sites and histologic findings among men: Korea National Health Insurance Corporation Study. J Clin Oncol 2005;23(21):4742–54. [DOI] [PubMed] [Google Scholar]
- 12.Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer 2007;97(7):1005–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohki T, Tateishi R, Shiina S, et al. Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut 2009;58(6):839–44. [DOI] [PubMed] [Google Scholar]
- 14.Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis 2008;28(4):339–50. [DOI] [PubMed] [Google Scholar]
- 15.Adami HO, Chow WH, Nyren O, et al. Excess risk of primary liver cancer in patients with diabetes mellitus. J Natl Cancer Inst 1996;88(20):1472–7. [DOI] [PubMed] [Google Scholar]
- 16.El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004;126(2):460–8. [DOI] [PubMed] [Google Scholar]
- 17.Wideroff L, Gridley G, Mellemkjaer L, et al. Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in Denmark. J Natl Cancer Inst 1997;89(18):1360–5. [DOI] [PubMed] [Google Scholar]
- 18.Yang WS, Va P, Bray F, et al. The role of pre-existing diabetes mellitus on hepa-tocellular carcinoma occurrence and prognosis: a meta-analysis of prospective cohort studies. PLoS One 2011;6(12):e27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang WM, Xu Y, Yang XR, et al. Prognostic role of diabetes mellitus in hepato-cellular carcinoma patients after curative treatments: a meta-analysis. Hepato-biliary Pancreat Dis Int 2011;10(4):346–55. [DOI] [PubMed] [Google Scholar]
- 20.Wang YG, Wang P, Wang B, et al. Diabetes mellitus and poorer prognosis in hepatocellular carcinoma: a systematic review and meta-analysis. PLoS One 2014;9(5):e95485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 2003;37(5):1202–19. [DOI] [PubMed] [Google Scholar]
- 22.Ratziu V, Bonyhay L, Di Martino V, et al. Survival, liver failure, and hepatocellular car-cinoma in obesity-related cryptogenic cirrhosis. Hepatology 2002;35(6):1485–93. [DOI] [PubMed] [Google Scholar]
- 23.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer 2009;115(24):5651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekstedt M, Franzen LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006;44(4):865–73. [DOI] [PubMed] [Google Scholar]
- 25.Rafiq N, Bai C, Fang Y, et al. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol 2009;7(2):234–8. [DOI] [PubMed] [Google Scholar]
- 26.Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005;129(1): 113–21. [DOI] [PubMed] [Google Scholar]
- 27.Sanyal A, Poklepovic A, Moyneur E, et al. Population-based risk factors and resource utilization for HCC: US perspective. Curr Med Res Opin 2010;26(9): 2183–91. [DOI] [PubMed] [Google Scholar]
- 28.Ascha MS, Hanouneh IA, Lopez R, et al. The incidence and risk factors of he-patocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010;51(6):1972–8. [DOI] [PubMed] [Google Scholar]
- 29.Bhala N, Angulo P, van der Poorten D, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collabora-tive study. Hepatology 2011;54(4):1208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zen Y, Katayanagi K, Tsuneyama K, et al. Hepatocellular carcinoma arising in non-alcoholic steatohepatitis. Pathol Int 2001;51(2):127–31. [DOI] [PubMed] [Google Scholar]
- 31.Orikasa H, Ohyama R, Tsuka N, et al. Lipid-rich clear-cell hepatocellular carci-noma arising in non-alcoholic steatohepatitis in a patient with diabetes mellitus. J Submicrosc Cytol Pathol 2001;33(1–2):195–200. [PubMed] [Google Scholar]
- 32.Bencheqroun R, Duvoux C, Luciani A, et al. Hepatocellular carcinoma without cirrhosis in a patient with nonalcoholic steatohepatitis. Gastroenterol Clin Biol 2004;28(5):497–9. [DOI] [PubMed] [Google Scholar]
- 33.Bullock RE, Zaitoun AM, Aithal GP, et al. Association of non-alcoholic steatohe-patitis without significant fibrosis with hepatocellular carcinoma. J Hepatol 2004; 41(4):685–6. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez L, Blanc JF, Sa Cunha A, et al. Obesity as a risk factor for hepatocel-lular carcinoma in a noncirrhotic patient. Semin Liver Dis 2004;24(4):415–9. [DOI] [PubMed] [Google Scholar]
- 35.Cuadrado A, Orive A, Garcia-Suarez C, et al. Non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma. Obes Surg 2005;15(3):442–6. [DOI] [PubMed] [Google Scholar]
- 36.Sato K, Ueda Y, Ueno K, et al. Hepatocellular carcinoma and nonalcoholic stea-tohepatitis developing during long-term administration of valproic acid. Virchows Arch 2005;447(6):996–9. [DOI] [PubMed] [Google Scholar]
- 37.Hai S, Kubo S, Shuto T, et al. Hepatocellular carcinoma arising from nonalco-holic steatohepatitis: report of two cases. Surg Today 2006;36(4):390–4. [DOI] [PubMed] [Google Scholar]
- 38.Ichikawa T, Yanagi K, Motoyoshi Y, et al. Two cases of non-alcoholic steatohepatitis with development of hepatocellular carcinoma without cirrhosis. J Gastroenterol Hepatol 2006;21(12):1865–6. [DOI] [PubMed] [Google Scholar]
- 39.Hashizume H, Sato K, Takagi H, et al. Primary liver cancers with nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 2007;19(10):827–34. [DOI] [PubMed] [Google Scholar]
- 40.Guzman G, Brunt EM, Petrovic LM, et al. Does nonalcoholic fatty liver disease predispose patients to hepatocellular carcinoma in the absence of cirrhosis? Arch Pathol Lab Med 2008;132(11):1761–6. [DOI] [PubMed] [Google Scholar]
- 41.Chagas AL, Kikuchi LO, Oliveira CP, et al. Does hepatocellular carcinoma in non-alcoholic steatohepatitis exist in cirrhotic and non-cirrhotic patients? Braz J Med Biol Res 2009;42(10):958–62. [DOI] [PubMed] [Google Scholar]
- 42.Paradis V, Zalinski S, Chelbi E, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a patholog-ical analysis. Hepatology 2009;49(3):851–9. [DOI] [PubMed] [Google Scholar]
- 43.Kawada N, Imanaka K, Kawaguchi T, et al. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J Gastroenterol 2009;44(12): 1190–4. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto E, Yatsuji S, Tobari M, et al. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol 2009;44(Suppl 19):89–95. [DOI] [PubMed] [Google Scholar]
- 45.Takuma Y, Nouso K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J Gastroenterol 2010; 16(12):1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tokushige K, Hashimoto E, Yatsuji S, et al. Prospective study of hepatocellular carcinoma in nonalcoholic steatohepatitis in comparison with hepatocellular carcinoma caused by chronic hepatitis C. J Gastroenterol 2010;45(9):960–7. [DOI] [PubMed] [Google Scholar]
- 47.Yasui K, Hashimoto E, Komorizono Y, et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin Gastroenterol Hepatol 2011;9(5):428–33 [quiz: e50]. [DOI] [PubMed] [Google Scholar]
- 48.Wakai T, Shirai Y, Sakata J, et al. Surgical outcomes for hepatocellular carcinoma in nonalcoholic fatty liver disease. J Gastrointest Surg 2011;15(8): 1450–8. [DOI] [PubMed] [Google Scholar]
- 49.Ikura Y, Mita E, Nakamori S. Hepatocellular carcinomas can develop in simple fatty livers in the setting of oxidative stress. Pathology 2011;43(2):167–8. [DOI] [PubMed] [Google Scholar]
- 50.Arase Y, Kobayashi M, Suzuki F, et al. Difference in malignancies of chronic liver disease due to non-alcoholic fatty liver disease or hepatitis C in Japanese elderly patients. Hepatol Res 2012;42(3):264–72. [DOI] [PubMed] [Google Scholar]
- 51.Mittal S, Sada YH, El-Serag HB, et al. Temporal trends of non-alcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin Gastroenterol Hepatol 2015;13(3):594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ertle J, Dechene A, Sowa JP, et al. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer 2011;128(10):2436–43. [DOI] [PubMed] [Google Scholar]
- 53.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10(11):656–65. [DOI] [PubMed] [Google Scholar]
- 54.Grundy SM, Brewer HB Jr, Cleeman JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Associa-tion conference on scientific issues related to definition. Circulation 2004;109(3): 433–8. [DOI] [PubMed] [Google Scholar]
- 55.Harrison SA. Liver disease in patients with diabetes mellitus. J Clin Gastroenterol 2006;40(1):68–76. [DOI] [PubMed] [Google Scholar]
- 56.Balkau B, Kahn HS, Courbon D, et al. Hyperinsulinemia predicts fatal liver cancer but is inversely associated with fatal cancer at some other sites: the Paris prospective study. Diabetes Care 2001;24(5):843–9. [DOI] [PubMed] [Google Scholar]
- 57.Moore MA, Park CB, Tsuda H. Implications of the hyperinsulinaemia-diabetes-cancer link for preventive efforts. Eur J Cancer Prev 1998;7(2):89–107. [PubMed] [Google Scholar]
- 58.Kaburagi Y, Yamauchi T, Yamamoto-Honda R, et al. The mechanism of insulin-induced signal transduction mediated by the insulin receptor substrate family. Endocr J 1999;(46 Suppl):S25–34. [DOI] [PubMed] [Google Scholar]
- 59.Kim SO, Park JG, Lee YI . Increased expression of the insulin-like growth factor I (IGF-I) receptor gene in hepatocellular carcinoma cell lines: implications of IGF-I receptor gene activation by hepatitis B virus X gene product. Cancer Res 1996; 56(16):3831–6. [PubMed] [Google Scholar]
- 60.Tanaka S, Mohr L, Schmidt EV, et al. Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology 1997;26(3):598–604. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka S, Wands JR. Insulin receptor substrate 1 overexpression in human hepatocellular carcinoma cells prevents transforming growth factor beta1-induced apoptosis. Cancer Res 1996;56(15):3391–4. [PubMed] [Google Scholar]
- 62.Saito K, Inoue S, Saito T, et al. Augmentation effect of postprandial hyperinsuli-naemia on growth of human hepatocellular carcinoma. Gut 2002;51(1):100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peyrou M, Bourgoin L, Foti M. PTEN in liver diseases and cancer. World J Gastroenterol 2010;16(37):4627–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008; 134(2):568–76. [DOI] [PubMed] [Google Scholar]
- 65.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature 2002;420(6913):333–6. [DOI] [PubMed] [Google Scholar]
- 66.Chang Q, Zhang Y, Beezhold KJ, et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol 2009; 50(2):323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hui L, Zatloukal K, Scheuch H, et al. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest 2008;118(12):3943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schattenberg JM, Singh R, Wang Y, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 2006;43(1):163–72. [DOI] [PubMed] [Google Scholar]
- 69.Abdelmalek MF, Sanderson SO, Angulo P, et al. Betaine for nonalcoholic fatty liver disease: results of a randomized placebo-controlled trial. Hepatology 2009;50(6):1818–26. [DOI] [PubMed] [Google Scholar]
- 70.Fujishita T, Aoki M, Taketo MM. JNK signaling promotes intestinal tumorigenesis through activation of mTOR complex 1 in Apc(Delta716) mice. Gastroenterology 2011;140(5):1556–63.e6. [DOI] [PubMed] [Google Scholar]
- 71.Zhou L, Huang Y, Li J, et al. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol 2010;27(2):255–61. [DOI] [PubMed] [Google Scholar]
- 72.Rinella ME, Siddiqui MS, Gardikiotes K, et al. Dysregulation of the unfolded pro-tein response in db/db mice with diet-induced steatohepatitis. Hepatology 2011;54(5):1600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henkel A, Green RM. The unfolded protein response in fatty liver disease. Semin Liver Dis 2013;33(4):321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai R, Chen R, Li H. Cross-talk between PI3K/Akt and MEK/ERK pathways me-diates endoplasmic reticulum stress-induced cell cycle progression and cell death in human hepatocellular carcinoma cells. Int J Oncol 2009;34(6):1749–57. [DOI] [PubMed] [Google Scholar]
- 75.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306(5695):457–61. [DOI] [PubMed] [Google Scholar]
- 76.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005;115(10):2656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kharroubi I, Ladriere L, Cardozo AK, et al. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004;145(11): 5087–96. [DOI] [PubMed] [Google Scholar]
- 78.Vandewynckel YP, Laukens D, Geerts A, et al. The paradox of the unfolded protein response in cancer. Anticancer Res 2013;33(11):4683–94. [PubMed] [Google Scholar]
- 79.Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol 2003;38(5):605–14. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta 2011;1812(8):893–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004;431(7007):461–6. [DOI] [PubMed] [Google Scholar]
- 82.Hu W, Feng Z, Eveleigh J, et al. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002;23(11):1781–9. [DOI] [PubMed] [Google Scholar]
- 83.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20(15):1803–15. [DOI] [PubMed] [Google Scholar]
- 84.Xu Z, Chen L, Leung L, et al. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A 2005;102(11):4120–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maki A, Kono H, Gupta M, et al. Predictive power of biomarkers of oxidative stress and inflammation in patients with hepatitis C virus-associated hepatocel-lular carcinoma. Ann Surg Oncol 2007;14(3):1182–90. [DOI] [PubMed] [Google Scholar]
- 86.Kelesidis T, Kelesidis I, Chou S, et al. Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med 2010;152(2):93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52(5):1836–46. [DOI] [PubMed] [Google Scholar]
- 88.Dotsch J, Rascher W, Meissner U. New insights into leptin resistance by modifying cytokine receptor signal transduction. Eur J Endocrinol 2005;152(3):333–4. [DOI] [PubMed] [Google Scholar]
- 89.Wang XJ, Yuan SL, Lu Q, et al. Potential involvement of leptin in carcinogenesis of hepatocellular carcinoma. World J Gastroenterol 2004;10(17):2478–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fruhbeck G Intracellular signalling pathways activated by leptin. Biochem J 2006;393(Pt 1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramani K, Yang H, Xia M, et al. Leptin’s mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology 2008;47(2):521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275(4):2247–50. [DOI] [PubMed] [Google Scholar]
- 93.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev 2012;92(4):1515–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol 2013;12(2):183–9. [PMC free article] [PubMed] [Google Scholar]
- 95.Chen CJ, Kono H, Golenbock D, et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13(7):851–6. [DOI] [PubMed] [Google Scholar]
- 96.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140(6):805–20. [DOI] [PubMed] [Google Scholar]
- 97.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464(7285):104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48(1):322–35. [DOI] [PubMed] [Google Scholar]
- 99.Yu LX, Yan HX, Liu Q, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010;52(4):1322–33. [DOI] [PubMed] [Google Scholar]
- 100.Dapito DH, Mencin A, Gwak GY, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012;21(4):504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hosaka T, Suzuki F, Kobayashi M, et al. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology 2013;58(1):98–107. [DOI] [PubMed] [Google Scholar]
- 102.Bjorkhem-Bergman L, Backheden M, Soderberg Lofdal K. Statin treatment reduces the risk of hepatocellular carcinoma but not colon cancer-results from a nationwide case-control study in Sweden. Pharmacoepidemiol Drug Saf 2014;23(10):1101–6. [DOI] [PubMed] [Google Scholar]
- 103.El-Serag HB, Johnson ML, Hachem C, et al. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 2009;136(5):1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kawata S, Yamasaki E, Nagase T, et al. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br J Cancer 2001;84(7):886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cao Z, Fan-Minogue H, Bellovin DI, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011;71(6):2286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spampanato C, De Maria S, Sarnataro M, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol 2012;40(4):935–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao J, Jia WD, Li JS, et al. Combined inhibitory effects of celecoxib and fluvas-tatin on the growth of human hepatocellular carcinoma xenografts in nude mice. J Int Med Res 2010;38(4):1413–27. [DOI] [PubMed] [Google Scholar]
- 108.Singh S, Singh PP, Singh AG, et al. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol 2013;108(6):881–91 [quiz: 892]. [DOI] [PubMed] [Google Scholar]
- 109.Donadon V, Balbi M, Mas MD, et al. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int 2010;30(5):750–8. [DOI] [PubMed] [Google Scholar]
- 110.Donadon V, Balbi M, Valent F, et al. Glycated hemoglobin and antidiabetic strategies as risk factors for hepatocellular carcinoma. World J Gastroenterol 2010;16(24):3025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang P, Kang D, Cao W, et al. Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev 2012;28(2):109–22. [DOI] [PubMed] [Google Scholar]
- 112.Chen TM, Lin CC, Huang PT, et al. Metformin associated with lower mortality in diabetic patients with early stage hepatocellular carcinoma after radiofrequency ablation. J Gastroenterol Hepatol 2011;26(5):858–65. [DOI] [PubMed] [Google Scholar]
- 113.Lai SW, Chen PC, Liao KF, et al. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol 2012;107(1):46–52. [DOI] [PubMed] [Google Scholar]
- 114.Hassan MM, Curley SA, Li D, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010; 116(8):1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology 2013;58(2):810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lu SC, Ramani K, Ou X, et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology 2009; 50(2):462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anstee QM, Day CP. S-adenosylmethionine (SAMe) therapy in liver disease: a review of current evidence and clinical utility. J Hepatol 2012;57(5):1097–109. [DOI] [PubMed] [Google Scholar]