

Abstract
Hepatitis B virus infection (HBV) is a significant global health problem. Despite the success of universal hepatitis B vaccination in many countries, more than 350 million individuals worldwide are chronically infected and 15-40% of those will develop cirrhosis and/or hepatocellular carcinoma if left untreated. Available therapies for chronic hepatitis B (CHB) infection are effective at decreasing viremia and improving measured clinical outcomes, however, no single therapy is optimal. As such, alternative drug therapies and the investigation of their role in the management of CHB are warranted. Significant improvements in the understanding of the HBV life cycle, viral genomics, and virus-host interactions continue to lead to the development of novel viral targets and immune modulators. Currently, two major classes of agents are utilized in CHB: the interferons and the nucleos(t)ide analogues. Each agent has individual advantages and drawbacks. The development of specific antiviral therapy has led to the emergence of HBV drug-resistant strains that has limited the long-term therapeutic potential of available agents. This necessitates the development of new agents that target both wild-type and drug-resistant strains. Further understanding of the basic mechanisms and clinical nuances of drug therapy is warranted. As most novel therapies are in the earliest stages of clinical development and testing, in the near future, treatment will continue to be long-term and likely involve the use of combination therapies to prevent viral resistance. In this review, we will highlight the HBV life cycle and genome, focusing in on current and potential novel antiviral drug targets as well as the benefits and clinical challenges with these therapies.
Keywords: HBV, interferon, drug resistance, nucleoside/nucleotide analogues, lamivudine, entecavir, adefovir
INTRODUCTION
Infection with the hepatitis B virus (HBV) is a global health concern. Between 350-400 million people worldwide are chronically infected with HBV and 15 to 40% of those are at risk to progress to liver cirrhosis and/or hepatocellular carcinoma without intervening medical therapy [1]. An estimated 600,000 persons die each year due to the acute or chronic consequences of hepatitis B [2]. HBV is highly endemic in many countries, especially in Asia and Africa, and is common in immigrant populations from endemic countries [3-5]. Universal immunization against HBV has led to a dramatic reduction in the number of new cases, but a large number of HBV infected individuals suffer from chronic progressive liver disease leading to cirrhosis and its complications including portal hypertension, variceal hemorrhage and hepatocellular carcinoma [3]. Despite the availability of safe and effective vaccines, a 100% effective antiviral treatment is not yet available for patients with chronic HBV [6,7].
A better understanding of the HBV life cycle and pathogenesis of HBV studied in vitro and in vivo has led to the introduction of new oral antiviral therapies with expanded clinical indications. The approach to treatment of chronic HBV has dramatically changed over the last decade from interferon (IFN)-alpha based therapy to an evolving arsenal of novel oral antiviral agents. Currently, in the U.S., five oral agents are available for use for chronic HBV and several new therapies will likely soon be available [8,9]. For the clinician, rapid progression in the understanding of HBV infection, pathogenesis, and expanded treatment options have markedly increased the complexity of managing patients with chronic HBV. In determining appropriate therapy, physicians need an intimate understanding of the stage of disease, predictive factors of treatment response, potency of various agents, expected duration of treatment, the likelihood and consequences of the development of resistance, and patient preferences [10].
The overarching goal of treatment for HBV infection is to achieve a rapid cure without producing adverse events to the patient or inducing viral resistance. Only by targeting novel molecular mechanisms of the virus and infected cells at various stages of the HBV life cycle can we enhance current methods to better manage or treat chronic HBV infection. In this review, we will give a brief overview of the HBV and its life cycle focusing on potential targets for drug therapy. Will discuss current drug therapies available for chronic HBV infection and then focus on evolving drug targets and the agents currently being studied in clinical trials. In addition, an overview of potential therapeutic strategies against HBV will be provided. Management of drug resistance and treatment paradigm of CHB is discussed elsewhere [11,12].
HEPATITIS B VIRUS (FIGURE 1)
Fig. (1).
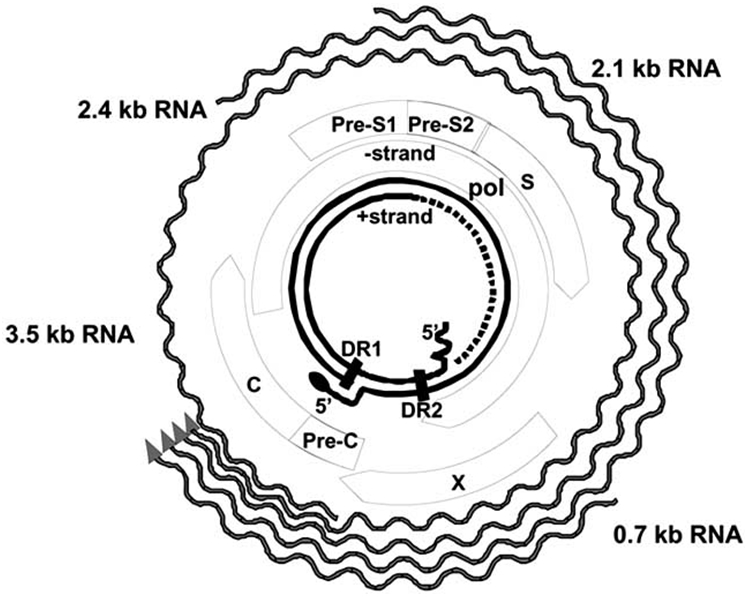
Hepatitis B virus genome structure is shown that includes C (core), Pre-C (pre-core), X (HBX protein), S (surface), Pre-S1, and Pre-S2, and the other structural elements.
HBV is small-enveloped virus belonging to the family Hepadnaviridae, which includes many related viruses whose natural hosts are avian and mammalian species. HBV is a DNA virus but replicates through an RNA intermediate. Hepadnaviruses have a preference for infecting hepatocytes, but small amounts of hepadnaviral DNA can be found in kidney, pancreas, and mononuclear cells. The exact consequences of extra-hepatic reservoirs of HBV remain uncertain [13-16]. HBV itself is considered to be a non-cytopathic virus and cellular injury appears to be largely immune-mediated. In infected individuals, the virus circulates in three distinct forms: 20-22 nm diameter spherical and filamentous lipoprotein particles and 40-42 nm diameter double-shelled infectious virions, also called Dane particles. The 20-22 nm lipoprotein HBsAg particles are produced by infected cells and contain only envelope glycoproteins and host derived lipids and significantly outnumber virions. On the other hand, the infectious mature virion, or Dane particle, is composed of an outer lipid bilayer that contains viral envelope proteins and encapsulates a nucleocapsid core. A partially double-stranded viral DNA genome with cohesive overlapping 5’ ends maintained in a relaxed circular structure resides in the nucleocapsid core [17,18]. DNA sequencing of infected individuals has revealed the presence of eight major genotypes (A-H), each with a distinct geographic distribution [19]. HBV genotypes have been shown to influence disease severity, clinical outcomes, risk of hepatocellular carcinoma, and response to therapy [20-24]. Much is still to be learned regarding the role of HBV genotype on clinical prognosis and choice of treatment.
HBV LIFE CYCLE (FIGURE 2)
Fig. (2).
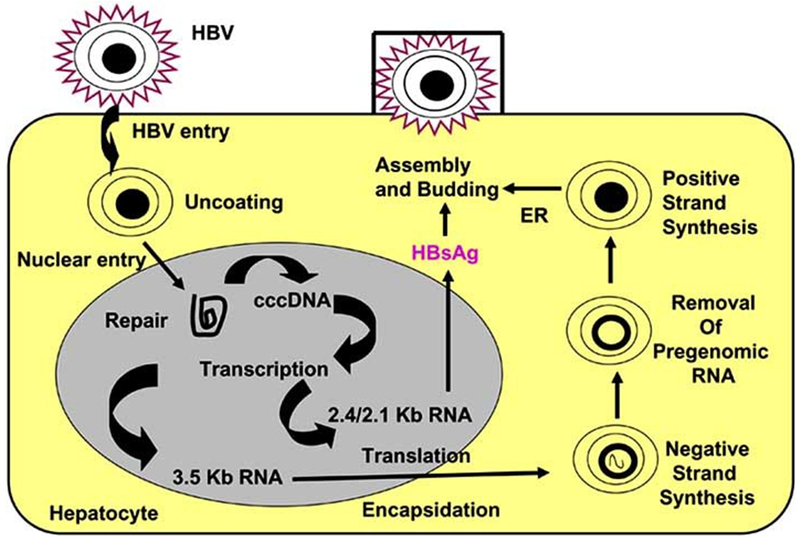
Hepatitis B virus life cycle: HBV enters hepatocyte through an unknown receptor. After uncoating, DNA is imported into the nucleus and transcription occurs. The cccDNA integrates with host DNA. The pregenomic RNA encapsidation occurs in the cytoplasm. Reverse transcription leads to negative followed by positive strand synthesis. Subsequently, viral assembly with HBsAg occurs in the cytoplasm followed by budding and secretion into the blood.
The HBV life cycle starts when the infectious viral particles attach and enter the host through an interaction of the host cell membrane and viral envelope proteins. Due to absence of a reliable human in vitro culture model for studying viral entry, the exact targets and mechanisms for viral entry are unknown. However, studies in duck hepatitis B virus models have enabled researchers to better understand the early events of infection [25]. In duck models, carboxy-peptidase D has been shown to be important in viral entry [26]. The PreS1 domain has also been repeatedly shown to be necessary for receptor binding and initiation of infection in a susceptible cell [27]. After viral entry, the particle uncoats and the genomic DNA is transferred to the cell nucleus by mechanisms that have not yet been completely elucidated [27-29]. Once the viral genome enters the nucleus, the partially double-stranded relaxed circular viral DNA is converted into covalently closed circular DNA (cccDNA) by an incompletely understood mechanism [28].
HBV cccDNA is the unit responsible for the persistent infection of hepatocytes. The nucleus based cccDNA allows for stable production of progeny that is not lost during cell division [28,30]. It is present in 5-50 copies per cell. It persists throughout the course of chronic HBV, even in those with serologic evidence of viral clearance [9]. Viral replication later occurs through a genomic RNA intermediate which functions as the template for reverse transcription. The reverse transcriptase lacks a proofreading function which results in a higher mutation rate than other DNA viruses. This mutation rate is estimated to be 1010-11 point mutations per day [31].
Four sets of mRNAs are transcribed from the cccDNA encoded by four main genes by a series of long overlapping reading frames in which several genes use the same DNA to encode viral proteins [9,27]. The four viral genes are surface, core, polymerase, and X. The preS-S (presurface-surface) region of the genome encodes the three viral surface antigens [18]. The three envelope genes PreS1, PreS2 and S encode for the large, middle and small envelope proteins, respectively. The core gene encodes for core antigen (HBcAg) and the precore gene encodes for the envelope antigen (HBeAg). The polymerase gene (pol) encodes for the large multi-functional polymerase protein with functions necessary for packaging and DNA replication. The X gene encodes for the X protein which is involved in host-cell signal transduction and can affect host and viral gene expression [18,30]. Host RNA polymerase II mediates transcription of viral mRNA. Genomic RNA allows for the core and promoter synthesis as well as sub-genomic RNAs that have specific messenger functions for the translation of the envelope proteins and the X protein which occurs in the cytoplasm [9,32].
Next, viral RNA is transported to the cytoplasm where translation yields the viral proteins. Nucleocapsids are then assembled and during this process a single pregenomic RNA is encapsidated in a reaction involving core as well as pol proteins [27]. The viral pol initiates encapsidation by binding to the 5’ encapsidation signal, epsilon, on the pregenomic RNA. The pol-epsilon complex is then encapsulated by the core protein to form the nucleocapsid. Once viral RNA is encapsidated, reverse transcription begins and two DNA strands are synthesized sequentially. HBV DNA polymerase reverse transcribes the pregenomic RNA into a negative-strand DNA. This negative strand DNA then becomes the template for positive-strand synthesis. Several host proteins appear to be involved in replication which occurs through complicated intramolecular and intermolecular interactions [33]. Concurrent with DNA synthesis, through a poorly understood mechanism, the nucleocapsid undergoes maturation and interaction occurs with the S protein to initiate viral assembly in the endoplasmic reticulum. The S protein is synthesized in the endoplasmic reticulum, where monomer aggregates form and bud into the lumen as lipoprotein HBsAg particles which then undergo further glycosylation in the endoplasmic reticulum and Golgi apparatus [18,34]. Some nucleocapsids bearing the mature genome are transported to the nucleus where they are converted back to cccDNA, however, most cores bud into small intracellular membranes containing the viral envelope proteins which contain the viral L, M, and S surface antigens which also undergo post-translational modification prior to being exported from the cell [34].
GOALS OF TREATMENT
Due to the integration of the virus into the host genome, HBV cannot be completely eradicated and cure is difficult to achieve [35]. The ideal antiviral agent would cause viral eradication with HBsAg conversion to anti-HBs, but this is rarely achieved with the currently available therapies [17]. Therefore, therapeutic endpoints against HBV infection are based on virologic, biochemical, and histologic responses.
The main clinical goal of therapy is to decrease HBV DNA levels in the serum to an undetectable level (using a PCR based assay), referred as complete virologic response [36]. This is usually associated with a biochemical response with a decrease in serum ALT and AST levels to within normal levels. Both complete virologic and biochemical response leads to an improvement in liver histology. In patients with HBeAg positive disease, an additional endpoint of therapy is HBeAg loss and the appearance of anti-HBe. Although there is no consensus on when to stop therapy after HBeAg seroconversion, most experts favor continuing anti-HBV therapy for six months or longer after development of anti-HBe [37].
AVAILABLE HBV THERAPIES
Currently, seven agents are approved for use in the United States for chronic HBV: interferon-a2b, pegylated interferon-a2a, lamivudine, adefovir (ADV), entecavir (ETV), telbivudine (TBV), and most recently, tenofovir (TDF). Table 1. shows the relative efficacy of these agents in the treatment of hepatitis B. The IFNs, administered subcutaneously, are naturally occurring cytokines that induce immune activity that results in various antiviral properties. The five available oral therapies are in a class of medications called the nucleoside/nucleotide analogues.
Table 1.
Efficacy of Currently Available Therapies for Chronic Hepatitis B
| IFNa | PEGb | Lamivudine | ADV | ETV | TBV | TDF | |
|---|---|---|---|---|---|---|---|
| Duration | |||||||
| Weeks | 16-20 | 52 | 48 | 48 | 48 | 52 | 48 |
| Loss of serum HBV DNAc | |||||||
| HBeAg + | 37% | 25% | 44% | 21% | 67% | 60% | 69% |
| HBeAg − | 60-70% | 63% | 50-70% | 51% | 90% | 88% | 93% |
| Normalization of ALT | |||||||
| HBeAg + | 23% | 39% | 41-72% | 48% | 68% | 77% | 69% |
| HBeAg − | 60-70% | 38% | 50-70% | 72% | 78% | 74% | 77% |
| Loss of HBsAg | |||||||
| HBeAg + | 8% | 3% | 0-2% | 0% | 2% | <1% | 3% |
| HBeAg − | 1% | 0% | 0% | 0% | <1% | <1% | NA |
| Improvement in histology | |||||||
| HBeAg + | NA | 38% | 52-56% | 53% | 72% | 65% | 67% |
| HBeAg − | NA | 48% | 60% | 64% | 70% | 67% | 71% |
| HBeAg Loss | 33% | 32-37% | 17-21% | 12% | 21% | 26 | 21% |
IFNa- Interferon
PEGb- Pegylated Interferon
Loss of serum HBV DNAc- Using different assays
ADV- adefovir
ETV- entecavir
TBV- telbuvidine
TDF- tenofovir
NA- Not available
INTERFERONS
Standard-IFN was the first therapy used for treatment of chronic HBV infection and has been used for over the last two decades.
IFNs exert their effect by binding to IFN receptors on cell membranes activating a cascade of secondary messengers. This initiates multiple complex mechanisms which results in the suppression of viral protein synthesis, degradation of viral mRNA, prevention of viral infection of cells, enhancement of antigen presentation by HLA I and II to the immune system, activation of natural killer and other immune cells, and increased cytokine production [38]. The main advantage of IFN use over nucleoside analogues is the possibility of immune mediated viral clearance and the absence of viral resistance. A meta-analysis of 15 randomized controlled trials revealed that IFN was superior to placebo in HBeAg-positive patients [39]. The rate of HBeAg loss, undetectable viral DNA and the rate of HBsAg loss in IFN treated patients compared to controls were 33% versus 12%, 37% versus 17%, and 8% versus 2%, respectively [39].
The conjugation of the IFN-α molecule with polyethylene glycol, which decreases the renal excretion of IFN, thus prolonging its half-life and allowing for once weekly dosing intervals has considerably improved patient compliance and have replaced thrice weekly standard-IFN for the treatment of CHB. In HBeAg-positive patients, treatment with peg-IFN was found superior to conventional IFN [40,41]. Three large randomized controlled trials have confirmed the improved efficacy of peg-IFN over lamivudine in HBeAg-positive and negative patients [42-44]. HBV genotype appears to predict response to peg-IFNs, with a higher HBeAg seroconversion rate in patients with genotypes A > B > C > D [40,44,45]. In HBeAg-positive patients with CHB, the rate of HBsAg loss after 1 year of treatment with Peg-IFN α-2b stratified by genotype is 14% for genotype A, 9% for genotype B, 3% for genotype C, and 2% for genotype D [46]. About a third of patients achieve HBeAg loss after 52 weeks of Peg-IFN α-2b therapy. Long term follow-up over a period of 3 years after stopping treatment shows that durability of HBeAg and HBsAg loss is also higher in genotype A. Sustained HBeAg and HBsAg loss in genotype A vs. non-A genotype after Peg-IFN α-2b is 96% vs. 76% and 58% vs. 11%, respectively. The exact role of genotype before embarking therapy is currently in a state of evolution and is an area on intense research.
HBV POLYMERASE INHIBITORS: NUCLEOSIDE AND NUCLEOTIDE ANALOGUES
As described earlier, hepadnavirus polymerase plays an important role in genome replication. Similar to the HIV virus, viral reverse transcriptase (or HBV polymerase) is a good target for inhibiting viral replication. Nucleos(t)ide analogues are chemically synthesized selective competitive inhibitors of HBV polymerase. These agents are incorporated into the viral DNA strand, resulting in chain termination. Nucleos(t)ide analogues may interfere with the synthesis of the negative DNA strand by reverse transcription, synthesis of the positive DNA strand, and possibly cccDNA formation in newly infected cells [47]. These agents are orally administered and therefore, adherence to the prescribed treatment regimen is better than IFN-based therapy. However, the drawback with these agents as compared to finite therapy with IFN-based regimen is that therapy is usually required for several years. Prolonged use of these agents is challenging due to the development of drug resistance.
LAMIVUDINE
Lamivudine was the first oral agent approved for the management of HBV. Lamivudine is rapidly absorbed after ingestion and it is phosphorylated in hepatocytes acquiring its antiviral properties [48]. It acts by terminating viral DNA synthesis and competitively inhibiting the viral polymerase. A randomized placebo-controlled trial involving Chinese patients who were treated with lamivudine or placebo daily for 52 weeks revealed that lamivudine led to virological, biochemical, and histological response [49]. However, long-term follow-up of patients treated for 2 to 4 years noted limitations of lamivudine monotherapy as demonstrated by the emergence of viral resistance after 8 months of therapy and eventually involving 76% of HBeAg-positive patients [50]. The development of resistance is lower for HBeAg negative disease. Lamivudine has been safely used in patients with cirrhosis and has been shown to decrease the risk of hepatic decompensation and hepatocellular carcinoma [51].
Resistance develops when mutations occur at M204I/V within the YMDD motif of the HBV gene encoding for polymerase. Resistance is more likely to occur in patients with high baseline levels of HBV DNA. Clinically, the emergence of drug resistance is usually associated with rise in HBV DNA levels that is followed by a flare in serum. Therefore, it is critical that patients be followed at regular intervals despite an initial favorable response to lamivudine therapy. Overall, lamivudine is an inexpensive and safe medication, however the early development and high rates of viral resistance limit its utility for long-term use, especially in HBeAg positive patients with high baseline HBV DNA levels.
ADEFOVIR
Adefovir (ADV) was the second nucleos(t)ide analogue licensed for use in the United States. It is an oral adenosine nucleotide analogue that acts by selectively inhibiting HBV polymerase. Efficacy with ADV therapy at 10mg per day was first shown in HBeAg-positive patients where 48 weeks of therapy showed significant normalization of ALT, suppression of HBV DNA, and rates of HBeAg seroconversion compared to placebo [44]. The rates of clearance of HBeAg and HBV DNA are lower with ADV compared to lamivudine, but biochemical and histological responses are similar. A study with HBeAg-negative patients treated for 144 weeks showed continued long term therapy revealed a durable response but discontinuation of treatment led to a rebound in HBV DNA levels [52]. The ADV-resistance mutations rtN236T and rtA181V were identified in 5.9% of patients after 144 weeks and 29% of patients after 5 years of therapy, although a more recent long-term follow-up study showed 5 year mutation rates of 20% in HBeAg-positive patients [46,53,54]. This high rate of resistance limits ADV use as stand-alone therapy in HBeAg-positive patients. However, it may be used as first-line therapy in individuals with HBeAg-negative CHB with HBV DNA levels below 107 copies/ml. Higher doses of ADV have greater potency, but also have higher rates of nephrotoxicity, the major limiting side effect of this therapy [55].
ENTECAVIR
Entecavir (ETV) is a cyclopentyl guanosine nucleoside analogue that blocks both the priming and elongation steps of viral replication, which results in potent inhibition of HBV DNA polymerase [56]. It was approved for use in the United States in 2005. ETV (0.5 mg/day) was shown to be more potent than lamivudine (100 mg/day) in a 48-week randomized-controlled trial in 648 nuleoside-naïve patients with HBeAg-negative CHB. The rate of normalization of ALT, undetectable serum HBV DNA levels, and histologic response in ETV vs. lamivudine group in this study were 78% vs. 71%, 90% vs. 72%, and 70% vs. 61%, respectively.
In HBeAg-positive patients, HBeAg seroconversion rates between lamivudine vs. ETV were not statistically significant but ETV treated patients had higher rates of normalization of serum ALT, reduction in HBV DNA levels, and histologic improvement as compared to lamivudine treated patients. In patients with lamivudine resistant mutations, 1mg daily ETV is the preferred dose as it is more potent than 0.5 mg daily ETV. The rate of development of ETV resistance after 2 years of therapy is higher in patients with lamivudine resistance vs. lamivudine naïve patients (9% vs. 0%) [57-59]. In nucleoside-naïve patients, continued treatment through 96 weeks has yet to reveal any evidence of ETV resistance and preliminary data from 5 years of treatment shows that emergence of resistance is only 1.2% [59]. In addition, ETV use in lamivudine-refractory HBeAg-positive patients has shown a continued clinical benefit through 96 weeks with a safety profile comparable to lamivudine [60].
TELBIVUDINE
Telbivudine (TBV) is a ß-L-nucleoside analogue of thymidine with specificity for hepadnaviruses. It is highly specific and selective inhibitor of both HBV first and second-strand DNA synthesis, targeting the viral DNA polymerase [9]. TBV was approved for use in the United States in 2006. A double-blind, randomized-controlled, phase III trial including CHB patients randomized to either TBV 600 mg daily or lamivudine 100 mg daily showed that TBV was superior to lamivudine in terms of mean reduction in serum HBV DNA levels, complete virologic response, and reduced rates of HBV drug resistance [61]. Adverse events reported with TBV include a class specific elevation of creatine phosphokinase levels and a few reported cases of transient myopathy. TBV has lower viral resistance than lamivudine, but it has significant resistance with one-year rates of 5.0% and 2.3%, in HBeAg-positive and HBeAg-negative patients. Results from the GLOBE Trial indicated that 96 week cumulative rates of virologic breakthrough for HBeAg-positive and HBeAg-negative patients were 21.6% and 8.6%, respectively [62]. TBV is a potent and safe antiviral agent for HBV but is associated with a high rate of resistance and TBV-resistant mutations are cross-resistant with lamivudine. It may be considered in CHB patients who are treatment-naïve and have low levels of serum HBV DNA levels (<107 IU/ml) and elevated serum ALT levels [63].
TENOFOVIR
Tenofovir disoproxil fumarate, an acyclic nucleoside analogue, is the prodrug of tenofovir (TDF). It has been available in the United States for treatment of HIV, but was recently approved for use in chronic HBV infection in mid-2008. Its molecular structure is closely related to ADV and it is directly incorporated into viral DNA causing inhibition of DNA polymerase. Despite its chemical similarities to ADV, it has been shown to have greater efficacy at HBV DNA suppression, histological improvement, and higher rates of HBsAg loss than ADV in HBeAg-positive patients treated for 48 weeks [64]. 72 week data showed that 89% of HBeAg-positive patients continued on TDF had HBV DNA viral suppression and 78% of patients who did not achieve complete viral suppression with ADV did so 24 weeks after switching to TDF [65]. In another recent study in patients with HBeAg-negative HBV, TDF was also superior to ADV in HBV viral suppression, improvement in histological score and viral suppression (71% vs. 49%) [66]. Recent 72 week data revealed that 98% of patients continued on TDF had serum HBV DNA <400 copies/mL, and 94% of patients who did not achieve complete viral suppression did so after 24 weeks of TDF therapy [67]. TDF was well tolerated in all of the above studies without evidence of significant renal toxicity and no resistance to TDF has been detected to date.
EMERGING NUCLEOS(T)IDE ANALOGUES IN HBV
There several new agents are in the various stages of clinical drug development. Table 2 gives a snap shot of pipeline of drugs that are in various stages of clinical trials.
Table 2.
HBV Nucleoside/Nucleotide Analogues in Various Stages of Clinical Development
| Drug | Phase | Company |
|---|---|---|
| Lamivudine (3TC) | Approved | GSK |
| Hepsera (adefovir dipivoxil) | Gilead | |
| Entecavir | BMS | |
| Tenofovir DF | Gilead | |
| Telbivudine (L-dT) | Idenix | |
| Emtricitabine (−FTC) | Phase III | Gilead |
| Clevudine (L-FMAU) | Gilead/Triangle | |
| Elvucitabine (⎬-L-Fd4C) | Phase II | Achillion |
| Valtorcitabine (val-L-dC) | Idenix | |
| Amdoxovir (DAPD) | Triangle | |
| Racivir ([+/−]-FTC) | Pharmasset | |
| LB80380 | LG Life Sciences | |
| Alamifovir (Purine nucleotide analogue) | Phase I | Lilly/Mitsubishi |
| MIV 210 (FLG prodrug) | Medivir/GSK | |
| Hepavir B (PMEA prodrug) | Ribapharm | |
| ⎬-L-FddC | Pre-clinical | Biochem/GSK |
EMTRICITABINE
Emtricitabine is a nucleoside analogue that is structurally closely related to lamivudine and blocks HBV polymerase in a similar manner. It is approved for use in the United States for HIV infection, but not currently approved for HBV. Studies indicate that it is effective in both HBeAg-positive and HBeAg-negative patients. Emtricitabine was shown to induce virologic response and histologic improvement at 48 weeks [68,69]. Emtricitabine shares a resistance profile with lamivudine and at 48 weeks 9% to 16% of patients developed resistance-conferring mutations [69]. The high rate of resistance effectively limits its use as monotherapy. It is currently available in combination with TDF for the treatment of HIV. Future studies to assess the efficacy of combination in CHB are warranted.
CLEVUDINE
Clevudine is a pyrimidine nucleoside analogue with a ß-L-configuration that acts in a different manner than other nucleoside analogues. It suppresses HBV replication by competitive inhibition via binding to the catalytic site of HBV polymerase resulting in inhibition of positive-strand DNA synthesis [70]. A multi-center, randomized phase II study comparing 10, 30 and 50 mg clevudine once daily in nucleoside naive patients for 12 weeks revealed that clevudine was able to suppress HBV DNA, cause biochemical response, and was well tolerated without serious adverse events. However, six patients had genomic changes with viral rebound concerning for the early development of resistance [71]. Another 12-week study in HBeAg-positive patients also revealed that 30 mg was the optimal dose and noted virologic and biochemical improvements compared to placebo [72]. A trial using clevudine 30 mg daily in HBeAg-negative patients for 24 weeks showed potent and sustained antiviral effect without evidence of viral resistance [73]. 48-week data revealing persistent viral suppression was recently presented but longer term treatment data is needed before recommendations for clevudine use can be determined [74].
PRADEFOVIR MESYLATE
Pradefovir mesylate, formerly known as remofovir, is a prodrug active metabolite of ADV that is preferentially metabolized into its active form in the liver, resulting in liver targeting while decreasing kidney exposure [75]. The lower concentrations in kidney result in a potentially lower risk of nephrotoxicity than with adefovir. The efficacy, pharmacokinetics, and safety of pradefovir were studied in a phase II dose finding trial comparing use pradefovir to ADV 10 mg per day. At 24 weeks HBV DNA responses were greater with pradefovir 30 mg than with ADV with the greatest reduction at 30 mg daily [76].
LB80380
LB80380 is a potent oral nucleotide prodrug that is chemically similar to ADV and TDF. It has been shown in vitro to have antiviral activity against HBV. LB80380 is rapidly converted to its parent drug LB80331 in the liver and intestine. LB80331 is further metabolized to LB80317, a nucleotide analogue of guanosine monophosphate that inhibits viral replication following incorporation into viral DNA. It has been shown in preclinical trials to have an excellent preclinical safety profile including lower potential for renal toxicity than ADV. A randomized placebo-controlled phase I/II clinical study of LB80380 for four weeks was completed with a 12 week follow-up period. HBV DNA suppression of −3.02 to −3.80 log10 copies/ml for doses ranging from 30 mg to 240 mg daily was noted. LB80380 has also been shown to be effective against YMDD mutants, in both in vitro and in vivo studies which may establish its clinical niche. Data from further trials of LB80380 for efficacy in patients with lamivudine resistant HBV are pending [77].
PMEO-DAPym
PMEO-DAPym, a novel acyclic pyrimidine analogue, has been assessed in vitro. Promising cell culture results revealed that most drug resistant HBV mutants, including multi-drug resistant strains, remained sensitive to PMEO-DAPym. Therefore this agent deserves further investigation for the treatment of HBV infection, especially given the need for drugs that do not share cross resistance [78].
EMERGING THERAPIES - NOVEL DRUG TARGETS IN VARIOUS STAGES OF DEVELOPMENT
Cytidine Deaminases
APOBEC3G
Human APOBEC3G (apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G) is a cellular cytidine deaminase displaying a broad range of antiretroviral properties. APOBEC3G inhibits HBV production by interfering with its replication without hypermutating the majority of HBV genomes. APOBEC3G has also been shown to have a broad antiviral activity on a wide variety of viruses which includes HIV, lentiviruses, murine leukemia virus, and human T-cell leukemia virus type 1. It has been shown that HBV is highly vulnerable to the editing activity of endogenous human deaminase and suggests that APOBEC3G could contribute to innate anti-HBV host responses. More study is needed prior to clinical applicability [79,80].
Heteroaryldihydropyrimidines
Heteroaryldihydropyrimidines (HAPs) are a newly recognized class of antivirals inhibiting the production of HBV virions in tissue culture models. One molecule, methyl 4-(2-chloro-4-fluorophenyl)-6-methyl-2-(pyridin-2-yl)-1,4-dihydropyrimidine-5-carboxylate (HAP-1) was shown to bind to the HBV core protein altering the kinetics of assembly, and gives rise to aberrant structures incompatible with a functional capsid resulting in its degradation. The kinetics of this interaction and its possible clinical applicability are now being studied [81,82]. Bay 41-4109 is the most clinically advanced agent of the HAP family. In vitro studies in cell culture have shown HBV DNA and HBcAg inhibition in a dose-dependent manner by the disassembly of the core capsids rendering them dysfunctional. No testing in humans has been reported, but Bay 41-4109 is a promising anti-HBV candidate if proven safe in future animal testing [83,84].
Phenlypropenamides
The phenlypropenamides are a group of compounds that inhibit HBV encapsidation. The phenylpropenamide derivative, AT-61, showed a specific anti-HBV activity that appeared to interfere with the encapsidation process [85]. A related compound, AT-130, was found to be a more potent inhibitor of HBV replication and both agents have also been shown to be effective against lamivudine-resistant HBV mutants but have not been shown to be more potent than lamivudine monotherapy [86]. These agents have been shown to affect the packaging of the pregenomic RNA into nucleocapsids and may play a clinical role in combination therapy with nucleos(t)ide analogues.
Helioxanthin Analogues
Helioxanthin and its derivatives are new anti-HBV agents which have been shown to inhibit HBV DNA, HBV RNA, and viral protein expression. Laboratory derivative 5-4-2 and a natural helioxanthin analogue (HE-145), which is isolated from Taiwania cryptomerioides, a coniferous tree native to Asia, suppressed HBV replication by post-transcriptional down-regulation of necessary transcription factors thus diminishing HBV promoter activity, blocking viral gene expression, and HBV replication [87-89]. Recently, HE-145 was shown to suppress transcriptional complex formation in the HBV core promoter without binding to the HBV core promoter or DNA directly [88]. Again, these compounds hold promise and further investigation is anticipated.
Glucosidase inhibitors
Glycosylation of the HBV envelope proteins in the endoplasmic reticulum is required in various steps of the viral life cycle. Thus, endoplasmic reticulum glucosidase inhibitors been developed and demonstrate anti-HBV and anti-hepatitis C properties by inhibiting viral morphogenesis and infectivity by preventing proper folding of envelope glycoproteins. One glucosidase inhibitor, celgosivir, is currently being evaluated in clinical trials against hepatitis C infection. In vitro data suggests encouraging anti-HBV activity that warrants further study [90].
Peptide Inhibitors
Peptide aptamers, a new class of intracellular inhibitors has been shown to block the function of target proteins, especially during morphogenesis [91]. In the duck model, the peptide aptamer, PA34, specifically binds to the duck HBV core protein, strongly blocking HBV replication by inhibiting viral capsid formation by initiating intracellular redistribution of its target protein preventing assembly of the complete HBV virion [92].
Viral Entry Inhibitors
Specific inhibition of virus entry is an attractive therapeutic concept to control acute and chronic viral infections and in HIV infection this has been accomplished with the interference of virus entry using a gp41 protein-derived peptide, enfuvirtide, which prevents fusion of the virus with the host cellular membrane. A recent in vivo study has demonstrated that acylated HBV preS-derived lipopeptides targeting an envelope protein component can prevent interaction of HBV with its cellular receptor. Potential clinical applications of HBV preS-derived lipopeptides include post-exposure prophylaxis, preventing vertical transmission during birth, prevention of re-infection after liver transplantation, or possibly in chronic HBV infection in combination with established therapies [93].
Gene Therapy
The future of anti-viral therapy is dependent upon improving understanding of the HBV replication that will help in discovery of newer therapeutic agents utilizing nucleic acid–based interventions. We will discuss briefly the role of gene therapy in this section including antisense oligodeoxyribonucleic acids (ODNs), ribozymes, and short interfering RNAs (siRNAs).
ODNs
ODNs are antisense compounds that attach to pre-mRNA or mRNA and form RNA-DNA duplexes, which can be degraded by the ribonuclease H (RNase H). The effect of ODNs is partly via antisense mechanisms and partly via stimulation of Toll-like receptors (TLRs) [94]. This stimulatory property of TLRs leads to activation of cytokine cascade that is thought to be beneficial in clearing hepatotropic viruses [95]. However, this enthusiasm is balanced by the risks related to immune activation especially in an immunocompromised host. Cytokine release may itself lead to hepatic injury. Therefore, safer ODNs that lack the immune stimulation effects are needed to afford specific inhibition without collateral damage.
Ribozymes
Synthetic ribozymes are now being developed as therapeutic agents to block infection with viruses such as HBV [96]. Nuclease-resistant ribozymes can be used to target HBV RNA and hold promise in targeted therapy against HBV. Ribozymes cause hydrolysis of the phosphodiester bond by binding to the mRNA with their two arms attached to the complementary sequences that flank the cleavage site [97]. The delivery and stability of ribozymes are a significant barrier that need to be overcome before clinical application [98].
SiRNA
SiRNAs are short (21- to 23-nucleotide) chains of dsRNAs, which cause a robust sequence-specific silencing of the target gene. Long dsRNAs lead to non-specific stimulation of interferon signaling but the short dsRNA fragments do not stimulate IFN. Therefore, short dsRNA are specific and more suitable for specific gene targeting.
After entry into the cell, dsRNA is cleaved by an RNase helicase, into small interfering RNAs that are transported by the dsRNA-binding protein R2D2 onto protein complexes forming RNA-induced silencing complexes (RISCs). RISCs incorporate a single strand of siRNA into the complex and cleave mRNAs containing complementary sequences leading to silencing of the target gene. In vitro and in vivo studies have shown that siRNAs and also short hairpin (sh) RNA can both efficiently silence HBV transcription and replication [99,100]. Therapeutic targeting using shRNA led to a precipitous decline in viral replication and a parallel reduction of HBV core antigen levels in hepatocytes [101, 102]. SiRNA may be one of the most powerful agents that may lead to sustained viral suppression if safe delivery methods can be developed and biodegradation of these agents can be checked [102].
Immunomodulatory Agents
The success of IFN-based regimens in the treatment of HBV has led to development of alternative immuno-modulatory therapies for the management of CHB. All these agents work by activation of host immune response in either non-specific or HBV-specific CD4+ T helper and CD8+ cytotoxic lymphocyte mediated manner [103]. The nonspecific modalities include the use of the TLRs, thymosin, the various interferons and other cytokines; whereas the specific modalities include the use of therapeutic vaccines, dendritic cell (DC) and cytotoxic T-lymphocyte (CTL)-based treatment.
Thymosin
Thymosin alpha-1 (Talpha1) is a synthetic polypeptide that has been shown to exhibit anti-viral effects by induction of a Th1 response. Numerous studies have been conducted to test its efficacy and safety in the management of CHB. Although several trials have shown improvement in ALT levels and suppression of HBV DNA in patients with CHB, the effect was modest compared with that of interferon or lamivudine [104-106].
Cytokines
Type 1 IFN has been consistently shown to block HBV viral replication. In addition, IFN-y also inhibits HBV replication via activation of natural killer cells within the liver. IFNs primarily work through the activation of the Jak/STAT signaling cascade that leads to the expression of interferon-stimulated genes (ISGs), which are required for their anti-viral effects [107].
Interferon-y
Although IFN-y has been shown to suppress HBV replication in vitro and in vivo models clinical trials did not suggest any benefit over standard IFN-based regimen [107].
Interferon-A
Interferon-lambda is a newly recognized cytokine that blocks HBV replication by stimulation of ISGs through a totally independent pathway [108]. Clinical drug development of this cytokine is attractive and may provide added benefit in achieving viral eradication.
Interleukins
IL-2 is a potent T-cell stimulator and has been shown to downregulate HBV gene expression by a post-transcriptional pathway [109]. IL-2 has been used in the management of HIV but has limited efficacy in patients with CHB [110, 111]. In future, rIL-2 may be considered as an adjunctive therapy to prime other forms of immuno-modulatory therapies such as therapeutic vaccination.
IL-12 and IL-18 both inhibit HBV replication in in vitro models but did not show any clinical efficacy in CHB patients [112-114]. Role of currently available cytokines except type 1 IFN are limited in current management of CHB.
Dendritic Cell Vaccination
Dendritic cells are antigen-presenting cells, which have the ability to engulf viruses and migrate to the regional lymph nodes where they undergo maturation and present foreign protein peptides to MHC molecules leading to stimulation of naïve T cells. These activated T cells release IFN-y, IL-2, IL-12, and IL-18 and in turn cause inhibition of HBV replication. HBV is known to evade immune surveillance and integrates with host DNA without evoking an immune response. Patients with CHB are thought to have a defect in dendritic cell function that can be overcome by treatment with tumor necrosis factor alpha [115]. Therefore, we believe that dendritic cell vaccination may prove to be an important adjunctive therapy among the various anti-HBV treatment modalities [116].
Toll Like Receptor Ligands
TLRs are key mediators of innate immune system. TLRs recognize viruses based upon their structural specificity and activate phagocytes and DCs to trigger an immune response. Induction of cytokines via this immune activation has been shown to suppress HBV replication [117]. Therefore, TLRs may be useful in the treatment of CHB in future.
Vaccination
Therapeutic vaccines have been used in smaller clinical trials in humans with mixed results. The use of these agents is based upon the principal that HBV immune evasion is due to poor HBV-specific T-cell responses. Thus, utilization of these vaccines is thought to stimulate HBV-specific T-cell responses [118]. In a phase I efficacy study of an HBV DNA vaccine in ten HBV chronic carriers, non-responders to current antiviral therapy, a potent HBV-specific T-cell response was noted [119]. Out of the ten patients enrolled in the study, five showed reduction in HBV DNA levels and one patient had HBsAg loss. Another clinical trial showed the efficacy of lamivudine monotherapy versus combination of lamivudine 100 mg once daily and HBsAg vaccine 20 μg administered once every 2 weeks for 12 doses with 12 months of post-treatment follow-up [120]. Combination therapy was better than lamivudine alone in achieving HBeAg seroconversion and HBV DNA suppression to undetectable levels. Further studies are needed to better define the role of therapeutic vaccination in the treatment of CHB.
Cytotoxic T Lymptocyte Based Therapy
A lipopeptide-based vaccine containing one CTL epitope from the HBV core region has been shown to induce a HBV-specific CTL response in healthy volunteers to levels comparable to those observed during acute HBV infection [121,122]. A follow-up phase II trial in CHB patients did not show any therapeutic efficacy to this approach [123]. Although immunomodulatory therapies are attractive, they are far from being ready for direct clinical application.
CONCLUSION
As described, HBV has a complex life cycle with unique viral-host interactions that affords for great potential when considering targets for therapeutic agents. However, despite the extensive knowledge of HBV gained through the use of animal models, the lack of a reliable human in vitro culture model poses a distinct challenge to understand all aspects of the viral life cycle and its cellular host interactions. Although several agents are available for use in the United States, but there are distinct challenges with these therapies including the emergence of drug resistance, the timing and duration of therapy, costs, and the lack of long-term safety and efficacy data. The primary obstacles to available therapies are the poor HBsAg seroconversion rates and the development of drug resistance. Preventing HBV antiviral drug resistance and appropriate management when resistance occurs has become the major focus in the management of CHB. Similar to the management of HIV, there is great hope that resistance may be prevented with the use of two or more nucleos(t)ide agents in combination. Combination therapy is actively being studied and in the near future, this is expected to become the standard clinical approach. However more data is needed before formal recommendations on combination therapies can be made.
Significant advances in the field of HBV virology over the last 20 years has led to the preclinical testing of multiple novel HBV drug targets, gene therapies, and immune modulators. In the near future, it is expected that scientific advances will enable new culture models to be developed which will largely expand our current knowledge of HBV. Only then will newer and more potent and safer agents be designed and tested.
ACKNOWLEDGEMENT
This research was funded with the support of the UCSD Digestive Diseases Research Development Center, U.S. PHS grant #DK080506.
ABBREVIATIONS
- HBV
Hepatitis B virus infection
- CHB
Chronic hepatitis B
- ADV
Adefovir
- ALT
Alanine aminotransferase
- cccDNA
Covalently closed circular DNA
- ETV
Entecavir
- HAPs
Heteroaryldihydropyrimidines
- HBV
Hepatitis B Vrus
- HBeAg
Hepatitis B e antigen
- HBsAg
Hepatitis B s Ag
- HBcAg
Hepatitis B c Ag
- IFN
Interferon
- TBV
Telbivudine
- TDF
Tenofovir
- ODN
Antisense deoxyribonucleotides
- siRNA
Short interfering RNA
- RISC
RNA-induced silencing complexes
- ISG
Interferon-stimulated genes
- DC
Dendritic cell
- CTL
Cytotoxic T-lymphocyte
REFERENCES
- [1].McMahon BJ Semin. Liver Dis, 2005, 25 Suppl 1, 3–8. [DOI] [PubMed] [Google Scholar]
- [2].WHO 2008; Vol. 2008, p World Health Organization; Department of Communicable Diseases Surveillance and Response. [Google Scholar]
- [3].Lau GK Clin. Liver Dis, 2001, 5, 361–79. [DOI] [PubMed] [Google Scholar]
- [4].Lavanchy D J. Viral Hepat, 2004, 11, 97–107. [DOI] [PubMed] [Google Scholar]
- [5].McQuillan GM; Coleman PJ; Kruszon-Moran D; Moyer LA; Lambert SB; Margolis HS Am. J. Public Health, 1999, 89, 14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Raney AK; Hamatake RK; Hong Z Expert Opin. Invest. Drugs, 2003, 12, 1281–95. [DOI] [PubMed] [Google Scholar]
- [7].Thermet A; Rollier C; Zoulim F; Trepo C; Cova L Vaccine, 2003, 21, 659–62. [DOI] [PubMed] [Google Scholar]
- [8].Hadziyannis SJ Expert Opin. Biol. Ther, 2006, 6, 913–21. [DOI] [PubMed] [Google Scholar]
- [9].Balsano C; Alisi A Curr. Med. Chem, 2008, 15, 930–9. [DOI] [PubMed] [Google Scholar]
- [10].Hoofnagle JH; Doo E; Liang TJ; Fleischer R; Lok AS Hepatology, 2007, 45, 1056–75. [DOI] [PubMed] [Google Scholar]
- [11].Pawlotsky JM; Dusheiko G; Hatzakis A; Lau D; Lau G; Liang TJ; Locarnini S; Martin P; Richman DD; Zoulim F Gastroenterology, 2008, 134, 405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Loomba R; Liang TJ Antivir. Ther, 2007, 12 Suppl 3, H33–41. [PubMed] [Google Scholar]
- [13].Marion PL Prog. Med. Virol, 1988, 35, 43–75. [PubMed] [Google Scholar]
- [14].Korba BE; Gowans EJ; Wells FV; Tennant BC; Clarke R; Gerin JL Virology, 1988, 165, 172–81. [DOI] [PubMed] [Google Scholar]
- [15].Barker LF; Maynard JE; Purcell RH; Hoofnagle JH; Berquist KR; London WT; Gerety RJ; Krushak DH J. Infect. Dis, 1975, 132, 451–8. [DOI] [PubMed] [Google Scholar]
- [16].Halpern MS; England JM; Deery DT; Petcu DJ; Mason WS; Molnar-Kimber KL Proc. Natl. Acad. Sci. USA, 1983, 80, 4865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Loomba R; Liang TJ Antivir. Ther, 2006, 11, 1–15. [PubMed] [Google Scholar]
- [18].Ganem D; Prince AM N. Engl. J. Med, 2004, 350, 1118–29. [DOI] [PubMed] [Google Scholar]
- [19].Kao JH J. Gastroenterol. Hepatol, 2002, 17, 643–50. [DOI] [PubMed] [Google Scholar]
- [20].Chu CJ; Hussain M; Lok AS Gastroenterology, 2002, 122, 1756–62. [DOI] [PubMed] [Google Scholar]
- [21].Liu CJ; Kao JH; Chen DS Liver Int, 2005, 25, 1097–107. [DOI] [PubMed] [Google Scholar]
- [22].Yang HI; Yeh SH; Chen PJ; Iloeje UH; Jen CL; Su J; Wang LY; Lu SN; You SL; Chen DS; Liaw YF; Chen CJ J. Natl. Cancer Inst, 2008, 100, 1134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Palumbo E Am. J. Ther, 2007, 14, 306–9. [DOI] [PubMed] [Google Scholar]
- [24].Wiegand J; Wedemeyer H; Finger A; Heidrich B; Rosenau J; Michel G; Bock CT; Manns MP; Tillmann HL Antivir. Ther, 2008, 13, 547–54. [PubMed] [Google Scholar]
- [25].Glebe D; Urban S World J. Gastroenterol, 2007, 13, 22–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Breiner KM; Urban S; Schaller H J. Virol, 1998, 72, 8098–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ganem DSR, In Field’s Virology; Knipe DH, PM, Ed.; Lippincott-Raven: Philadelphia, 2001, p 2923–2970. [Google Scholar]
- [28].Glebe D World J. Gastroenterol, 2007, 13, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kann M; Schmitz A; Rabe B World J. Gastroenterol, 2007, 13, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Locarnini S Semin. Liver Dis, 2004, 24 (Suppl 1), 3–10. [DOI] [PubMed] [Google Scholar]
- [31].Nowak MA; Bonhoeffer S; Hill AM; Boehme R; Thomas HC; McDade H Proc. Natl. Acad. Sci. USA, 1996, 93, 4398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tavis JE; Ganem D J. Virol, 1996, 70, 5741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu J; Toft DO; Seeger C EMBO J, 1997, 16, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bruss V World J. Gastroenterol, 2007, 13, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Locarnini S; Birch C J. Hepatol, 1999, 30, 536–50. [DOI] [PubMed] [Google Scholar]
- [36].Locarnini S; Hatzakis A; Heathcote J; Keeffe EB; Liang TJ; Mutimer D; Pawlotsky JM; Zoulim F Antivir. Ther, 2004, 9, 679–93. [PubMed] [Google Scholar]
- [37].Lok AS; McMahon BJ Hepatology, 2007, 45, 507–39. [DOI] [PubMed] [Google Scholar]
- [38].Dusheiko G; Antonakopoulos N Gut, 2008, 57, 105–24. [DOI] [PubMed] [Google Scholar]
- [39].Wong DK; Cheung AM; O’Rourke K; Naylor CD; Detsky AS; Heathcote J Ann. Intern. Med, 1993, 119, 312–23. [DOI] [PubMed] [Google Scholar]
- [40].Janssen HL; van Zonneveld M; Senturk H; Zeuzem S; Akarca US; Cakaloglu Y; Simon C; So TM; Gerken G; de Man RA; Niesters HG; Zondervan P; Hansen B; Schalm SW Lancet, 2005, 365, 123–9. [DOI] [PubMed] [Google Scholar]
- [41].Cooksley WG; Piratvisuth T; Lee SD; Mahachai V; Chao YC; Tanwandee T; Chutaputti A; Chang WY; Zahm FE; Pluck N J. Viral Hepat, 2003, 10, 298–305. [DOI] [PubMed] [Google Scholar]
- [42].Lau GK; Piratvisuth T; Luo KX; Marcellin P; Thongsawat S; Cooksley G; Gane E; Fried MW; Chow WC; Paik SW; Chang WY; Berg T; Flisiak R; McCloud P; Pluck N N. Engl. J. Med, 2005, 352, 2682–95. [DOI] [PubMed] [Google Scholar]
- [43].Cooksley WG Expert Opin. Pharmacother, 2005, 6, 1373–80. [DOI] [PubMed] [Google Scholar]
- [44].Marcellin P; Lau GK; Bonino F; Farci P; Hadziyannis S; Jin R; Lu ZM; Piratvisuth T; Germanidis G; Yurdaydin C; Diago M; Gurel S; Lai MY; Button P; Pluck N N. Engl. J. Med, 2004, 351, 1206–17. [DOI] [PubMed] [Google Scholar]
- [45].Chan HL; Leung NW; Hui AY; Wong VW; Liew CT; Chim AM; Chan FK; Hung LC; Lee YT; Tam JS; Lam CW; Sung JJ Ann. Intern. Med, 2005, 142, 240–50. [DOI] [PubMed] [Google Scholar]
- [46].Flink HJ; Hansen BE; Heathcote EJ; Feinman SV; Simsek H; Karayalcin S; Mach T; Leemans WF; de Man RA; Verhey E; Schalm SW; Janssen HL Am. J. Gastroenterol, 2006, 101, 2523–9. [DOI] [PubMed] [Google Scholar]
- [47].Balsano C Mini Rev. Med. Chem, 2008, 8, 307–18. [DOI] [PubMed] [Google Scholar]
- [48].Chang CN; Skalski V; Zhou JH; Cheng YC J. Biol. Chem, 1992, 267, 22414–20. [PubMed] [Google Scholar]
- [49].Lai CL; Chien RN; Leung NW; Chang TT; Guan R; Tai DI; Ng KY; Wu PC; Dent JC; Barber J; Stephenson SL; Gray DF N. Engl. J. Med, 1998, 339, 61–8. [DOI] [PubMed] [Google Scholar]
- [50].Lau DT; Khokhar MF; Doo E; Ghany MG; Herion D; Park Y; Kleiner DE; Schmid P; Condreay LD; Gauthier J; Kuhns MC; Liang TJ; Hoofnagle JH Hepatology, 2000, 32, 828–34. [DOI] [PubMed] [Google Scholar]
- [51].Lai CJ; Terrault NA Gastroenterol. Clin. North Am, 2004, 33, 629–54, x-xi. [DOI] [PubMed] [Google Scholar]
- [52].Hadziyannis SJ; Tassopoulos NC; Heathcote EJ; Chang TT; Kitis G; Rizzetto M; Marcellin P; Lim SG; Goodman Z; Ma J; Arterburn S; Xiong S; Currie G; Brosgart CL N Engl. J. Med, 2005, 352, 2673–81. [DOI] [PubMed] [Google Scholar]
- [53].Hadziyannis SJ; Tassopoulos NC; Heathcote EJ; Chang TT; Kitis G; Rizzetto M; Marcellin P; Lim SG; Goodman Z; Ma J; Brosgart CL; Borroto-Esoda K; Arterburn S; Chuck SL Gastroenterology, 2006, 131, 1743–51. [DOI] [PubMed] [Google Scholar]
- [54].Marcellin P; Chang TT; Lim SG; Sievert W; Tong M; Arterburn S; Borroto-Esoda K; Frederick D; Rousseau F Hepatology, 2008, 48, 750–8. [DOI] [PubMed] [Google Scholar]
- [55].Simsek H; Shorbagi A; Balaban Y; Tatar G J. Hepatol, 2008, 49, 464–5. [DOI] [PubMed] [Google Scholar]
- [56].Billich A Curr. Opin. Invest. Drugs, 2001, 2, 617–21. [PubMed] [Google Scholar]
- [57].Sherman M; Yurdaydin C; Sollano J; Silva M; Liaw YF; Cianciara J; Boron-Kaczmarska A; Martin P; Goodman Z; Colonno R; Cross A; Denisky G; Kreter B; Hindes R Gastroenterology, 2006, 130, 2039–49. [DOI] [PubMed] [Google Scholar]
- [58].Lai CL; Shouval D; Lok AS; Chang TT; Cheinquer H; Goodman Z; DeHertogh D; Wilber R; Zink RC; Cross A; Colonno R; Fernandes L N. Engl. J. Med, 2006, 354, 1011–20. [DOI] [PubMed] [Google Scholar]
- [59].Chang TT; Gish RG; Hadziyannis SJ; Cianciara J; Rizzetto M; Schiff ER; Pastore G; Bacon BR; Poynard T; Joshi S; Klesczewski KS; Thiry A; Rose RE; Colonno RJ; Hindes RG Gastroenterology, 2005, 129, 1198–209. [DOI] [PubMed] [Google Scholar]
- [60].Sherman M; Yurdaydin C; Simsek H; Silva M; Liaw YF; Rustgi VK; Sette H; Tsai N; Tenney DJ; Vaughan J; Kreter B; Hindes R Hepatology, 2008, 48, 99–108. [DOI] [PubMed] [Google Scholar]
- [61].Lai CL; Gane E; Liaw YF; Hsu CW; Thongsawat S; Wang Y; Chen Y; Heathcote EJ; Rasenack J; Bzowej N; Naoumov NV; Di Bisceglie AM; Zeuzem S; Moon YM; Goodman Z; Chao G; Constance BF; Brown NA N. Engl. J. Med, 2007, 357, 2576–88. [DOI] [PubMed] [Google Scholar]
- [62].Lai CL, Gane E; Hsu GW; Thongsawat S; Wang Y; Chen Y; Heathcote EJ; Rasenack J; Bzowej N; Naoumove N; Zeuzem S; Di Bisceglie AM; Chao GC Hepatology, 2006, 44, 222a. [Google Scholar]
- [63].Buti M Nat. Clin. Pract. Gastroenterol. Hepatol, 2008, 5, 494–5. [DOI] [PubMed] [Google Scholar]
- [64].Heathcote EJ, Gane E; DeMan R; Krastev Z; Germanidis G; Lee S; Flisiak R; Kaita K; Manns M; Kotzev I; Tchernev K; Buggisch P; Weilert F; Kurdas O; Shiffman ML; Trinh H Hepatology, 2007, 46, 861A.17668884 [Google Scholar]
- [65].Heathcote EJ Gane E; Gordon S Liver Int, 2008, 48, S32. [Google Scholar]
- [66].Marcellin P BM; Krastev Z; Germanidis G; Lee S; Flisiak R; Kaita K; Manns M; Kotzev I; Tchernev K; Buggisch P; Weilert F; Kurdas O; Shiffman ML; Trinh H Hepatology, 2007, 46, 80A. [Google Scholar]
- [67].Marcellin PJI; Habersetzer F Liver Int, 2008, 48, S26. [Google Scholar]
- [68].Lim SG; Ng TM; Kung N; Krastev Z; Volfova M; Husa P; Lee SS; Chan S; Shiffman ML; Washington MK; Rigney A; Anderson J; Mondou E; Snow A; Sorbel J; Guan R; Rousseau F Arch. Intern. Med, 2006, 166, 49–56. [DOI] [PubMed] [Google Scholar]
- [69].Gish RG; Trinh H; Leung N; Chan FK; Fried MW; Wright TL; Wang C; Anderson J; Mondou E; Snow A; Sorbel J; Rousseau F; Corey L J. Hepatol, 2005, 43, 60–6. [DOI] [PubMed] [Google Scholar]
- [70].Chong Y; Chu CK Bioorg Med. Chem. Lett, 2002, 12, 3459–62. [DOI] [PubMed] [Google Scholar]
- [71].Lim SG; Leung N; Hann HW; Lau GK; Trepo C; Mommeja-Marin H; Moxham C; Sorbel J; Snow A; Blum MR; Rousseau F; Marcellin P Aliment Pharmacol. Ther, 2008, 27, 1282–92. [DOI] [PubMed] [Google Scholar]
- [72].Lee HS; Chung YH; Lee K; Byun KS; Paik SW; Han JY; Yoo K; Yoo HW; Lee JH; Yoo BC Hepatology, 2006, 43, 982–8. [DOI] [PubMed] [Google Scholar]
- [73].Yoo BC; Kim JH; Kim TH; Koh KC; Um SH; Kim YS; Lee KS; Han BH; Chon CY; Han JY; Ryu SH; Kim HC; Byun KS; Hwang SG; Kim BI; Cho M; Yoo K; Lee HJ; Hwang JS; Lee YS; Choi SK; Lee YJ; Yang JM; Park JW; Lee MS; Kim DG; Chung YH; Cho SH; Choi JY; Kweon YO; Lee HY; Jeong SH; Yoo HW; Lee HS Hepatology, 2007, 46, 1041–8. [DOI] [PubMed] [Google Scholar]
- [74].Park SY; K. Y. O.; Cho HJ; Park GS; Seo EH; Bae RC; Choi HY J. Hepatol, 2008, 2, OL–097. [Google Scholar]
- [75].Reddy KR; Matelich MC; Ugarkar BG; Gomez-Galeno JE; DaRe J; Ollis K; Sun Z; Craigo W; Colby TJ; Fujitaki JM; Boyer SH; van Poelje PD; Erion MD J. Med. Chem, 2008, 51, 666–76. [DOI] [PubMed] [Google Scholar]
- [76].Lee KSL, S. G.; Chuang WL; Hwang SG; Cho M; Lai MY; Chao YC; Chang T-T; Han KH; Lee CM; Um SH; Yeon JE; Yang SS; Teo EK; Peng CY; Lin HH; Yang SS; Huo TI; Nguyen T; Chen TY; Hu KQ; Xu Y; Sullivan-Bólyai JZ J. Hepatol, 2006, 44, S274. [Google Scholar]
- [77].Yuen MF; Kim J; Kim CR; Ngai V; Yuen JC; Min C; Kang HM; Shin BS; Yoo SD; Lai CL Antivir. Ther, 2006, 11, 977–83. [PubMed] [Google Scholar]
- [78].Brunelle MN; Lucifora J; Neyts J; Villet S; Holy A; Trepo C; Zoulim F Antimicrob. Agents Chemother, 2007, 51, 2240–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Baumert TF; Rosler C; Malim MH; von Weizsacker F Hepatology, 2007, 46, 682–9. [DOI] [PubMed] [Google Scholar]
- [80].Izumi T; Shirakawa K; Takaori-Kondo A Mini Rev. Med. Chem, 2008, 8, 231–8. [DOI] [PubMed] [Google Scholar]
- [81].Stray SJ; Bourne CR; Punna S; Lewis WG; Finn MG; Zlotnick A Proc. Natl. Acad Sci USA, 2005, 102, 8138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bourne C, L. S., Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A J. Virol, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wu GY; Zheng XJ; Yin CC; Jiang D; Zhu L; Liu Y; Wei L; Wang Y; Chen HS J. Chemother, 2008, 20, 458–67. [DOI] [PubMed] [Google Scholar]
- [84].Stray SJ; Zlotnick A J. Mol. Recognit, 2006, 19, 542–8. [DOI] [PubMed] [Google Scholar]
- [85].King RW; Ladner SK; Miller TJ; Zaifert K; Perni RB; Conway SC; Otto MJ Antimicrob. Agents Chemother, 1998, 42, 3179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Delaney W. E. t.; Edwards R; Colledge D; Shaw T; Furman P; Painter G; Locarnini S Antimicrob. Agents Chemother, 2002, 46, 3057–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Li Y; Fu L; Yeo H; Zhu JL; Chou CK; Kou YH; Yeh SF; Gullen E; Austin D; Cheng YC Antivir. Chem. Chemother, 2005, 16, 193–201. [DOI] [PubMed] [Google Scholar]
- [88].Tseng YP; Kuo YH; Hu CP; Jeng KS; Janmanchi D; Lin CH; Chou CK; Yeh SF Antiviral Res, 2008, 77, 206–14. [DOI] [PubMed] [Google Scholar]
- [89].Ying C; Li Y; Leung CH; Robek MD; Cheng YC Proc. Natl. Acad. Sci. USA, 2007, 104, 8526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Durantel D; Alotte C; Zoulim F Curr. Opin. Invest. Drugs, 2007, 8, 125–9. [PubMed] [Google Scholar]
- [91].Tang KF; Abdullah MP; Yusoff K; Tan WS J. Med. Chem, 2007, 50, 5620–6. [DOI] [PubMed] [Google Scholar]
- [92].Tomai E; Butz K; Lohrey C; von Weizsacker F; Zentgraf H; Hoppe-Seyler F J. Biol. Chem, 2006, 281, 21345–52. [DOI] [PubMed] [Google Scholar]
- [93].Petersen J; Dandri M; Mier W; Lutgehetmann M; Volz T; von Weizsacker F; Haberkorn U; Fischer L; Pollok JM; Erbes B; Seitz S; Urban S Nat. Biotechnol, 2008, 26, 335–41. [DOI] [PubMed] [Google Scholar]
- [94].Heil F; Hemmi H; Hochrein H; Ampenberger F; Kirschning C; Akira S; Lipford G; Wagner H; Bauer S Science, 2004, 303, 1526–9. [DOI] [PubMed] [Google Scholar]
- [95].Heil F; Ahmad-Nejad P; Hemmi H; Hochrein H; Ampenberger F; Gellert T; Dietrich H; Lipford G; Takeda K; Akira S; Wagner H; Bauer S Eur. J. Immunol, 2003, 33, 2987–97. [DOI] [PubMed] [Google Scholar]
- [96].Cech TR Science, 2000, 289, 878–9. [DOI] [PubMed] [Google Scholar]
- [97].Herschlag D; Cech TR Nature, 1990, 344, 405–9. [DOI] [PubMed] [Google Scholar]
- [98].Long MB; Jones JP 3rd; Sullenger BA; Byun J J. Clin. Invest, 2003, 112, 312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Morrissey DV; Blanchard K; Shaw L; Jensen K; Lockridge JA; Dickinson B; McSwiggen JA; Vargeese C; Bowman K; Shaffer CS; Polisky BA; Zinnen S Hepatology, 2005, 41, 1349–56. [DOI] [PubMed] [Google Scholar]
- [100].McCaffrey AP; Nakai H; Pandey K; Huang Z; Salazar FH; Xu H; Wieland SF; Marion PL; Kay MA Nat. Biotechnol, 2003, 21, 639–44. [DOI] [PubMed] [Google Scholar]
- [101].Ren XR; Zhou LJ; Luo GB; Lin B; Xu A J. Viral Hepat, 2005, 12, 236–42. [DOI] [PubMed] [Google Scholar]
- [102].Ryther RC; Flynt AS; Phillips JA 3rd; Patton JG Gene Ther, 2005, 12, 5–11. [DOI] [PubMed] [Google Scholar]
- [103].Kakimi K; Isogawa M; Chung J; Sette A; Chisari FV J. Virol, 2002, 76, 8609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zavaglia C; Severini R; Tinelli C; Franzone JS; Airoldi A; Tempini S; Bettale G; Ideo G Dig. Dis. Sci, 2000, 45, 690–6. [DOI] [PubMed] [Google Scholar]
- [105].Lebray P; Vallet-Pichard A; Michel ML; Fontaine H; Sobesky R; Brechot C; Pol S J. Hepatol, 2003, 39, (Suppl 1), S151–9. [DOI] [PubMed] [Google Scholar]
- [106].Loggi E; Gramenzi A; Margotti M; Cursaro C; Galli S; Vitale G; Grandini E; Scuteri A; Vukotic R; Andreone P; Bernardi M J. Viral Hepat, 2008, 15, 442–8. [DOI] [PubMed] [Google Scholar]
- [107].Robek MD; Boyd BS; Wieland SF; Chisari FV Proc. Natl. Acad. Sci. USA, 2004, 101, 1743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Robek MD; Boyd BS; Chisari FV J. Virol, 2005, 79, 3851–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Guilhot S; Guidotti LG; Chisari FV J. Virol, 1993, 67, 7444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kovacs JA; Lempicki RA; Sidorov IA; Adelsberger JW; Sereti I; Sachau W; Kelly G; Metcalf JA; Davey RT Jr.; Falloon J; Polis MA; Tavel J; Stevens R; Lambert L; Hosack DA; Bosche M; Issaq HJ; Fox SD; Leitman S; Baseler MW; Masur H; Di Mascio M; Dimitrov DS; Lane HC J. Clin. Invest, 2005, 115, 2139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tilg H; Vogel W; Tratkiewicz J; Aulitzky WE; Herold M; Gruber M; Geissler D; Umlauft F; Judmaier G; Schwulera U;et al. J. Hepatol, 1993, 19, 259–67. [DOI] [PubMed] [Google Scholar]
- [112].Kimura K; Kakimi K; Wieland S; Guidotti LG; Chisari FV J. Virol, 2002, 76, 10702–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Xiong SQ; Lin BL; Gao X; Tang H; Wu CY Int. Immunopharmacol, 2007, 7, 578–87. [DOI] [PubMed] [Google Scholar]
- [114].Szkaradkiewicz A; Jopek A; Wysocki J Antiviral Res, 2005, 66, 23–7. [DOI] [PubMed] [Google Scholar]
- [115].Hasebe A; Akbar SM; Furukawa S; Horiike N; Onji M Clin. Exp. Immunol, 2005, 139, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Akbar SM; Furukawa S; Hasebe A; Horiike N; Michitaka K; Onji M Int. J. Mol. Med, 2004, 14, 295–9. [PubMed] [Google Scholar]
- [117].Isogawa M; Robek MD; Furuichi Y; Chisari FV J. Virol, 2005, 79, 7269–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kimura K; Kakimi K; Wieland S; Guidotti LG; Chisari FV J. Immunol, 2002, 169, 5188–95. [DOI] [PubMed] [Google Scholar]
- [119].Mancini-Bourgine M; Fontaine H; Brechot C; Pol S; Michel ML Vaccine, 2006, 24, 4482–9. [DOI] [PubMed] [Google Scholar]
- [120].Horiike N; Fazle Akbar SM; Michitaka K; Joukou K; Yamamoto K; Kojima N; Hiasa Y; Abe M; Onji M J. Clin. Virol, 2005, 32, 156–61. [DOI] [PubMed] [Google Scholar]
- [121].Vitiello A; Ishioka G; Grey HM; Rose R; Farness P; LaFond R; Yuan L; Chisari FV; Furze J; Bartholomeuz R; Chestnut RW J. Clin. Invest, 1995, 95, 341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].BenMohamed L; Wechsler SL; Nesburn AB Lancet Infect. Dis, 2002, 2, 425–31. [DOI] [PubMed] [Google Scholar]
- [123].Heathcote J; McHutchison J; Lee S; Tong M; Benner K; Minuk G; Wright T; Fikes J; Livingston B; Sette A; Chestnut R Hepatology, 1999, 30, 531–6. [DOI] [PubMed] [Google Scholar]