

Abstract
Aim: Endothelin-1 (ET-1) and angiotensin II (Ang II) are multifunctional peptide hormones that regulate the function of the cardiovascular and renal systems. Both hormones increase the intracellular production of inositol-1,4,5-trisphosphate (IP3) by activating their membrane-bound receptors. We have previously demonstrated that IP3-mediated sarcoplasmic reticulum (SR) Ca2+ release results in mitochondrial Ca2+ uptake and activation of ATP production. In this study, we tested the hypothesis that intact SR/mitochondria microdomains are required for metabolic IP3-mediated SR/mitochondrial feedback in ventricular myocytes.
Methods: As a model for disrupted mitochondrial/SR microdomains, cardio-specific tamoxifen-inducible mitofusin 2 (Mfn2) knock out (KO) mice were used. Mitochondrial Ca2+ uptake, membrane potential, redox state, and ATP generation were monitored in freshly isolated ventricular myocytes from Mfn2 KO mice and their control wild-type (WT) littermates.
Results: Stimulation of ET-1 receptors in healthy control myocytes increases mitochondrial Ca2+ uptake, maintains mitochondrial membrane potential and redox balance leading to the enhanced ATP generation. Mitochondrial Ca2+ uptake upon ET-1 stimulation was significantly higher in interfibrillar (IFM) and perinuclear (PNM) mitochondria compared to subsarcolemmal mitochondria (SSM) in WT myocytes. Mfn2 KO completely abolished mitochondrial Ca2+ uptake in IFM and PNM mitochondria but not in SSM. However, mitochondrial Ca2+ uptake induced by beta-adrenergic receptors activation with isoproterenol (ISO) was highest in SSM, intermediate in IFM, and smallest in PNM regions. Furthermore, Mfn2 KO did not affect ISO-induced mitochondrial Ca2+ uptake in SSM and IFM mitochondria; however, enhanced mitochondrial Ca2+ uptake in PNM. In contrast to ET-1, ISO induced a decrease in ATP levels in WT myocytes. Mfn2 KO abolished ATP generation upon ET-1 stimulation but increased ATP levels upon ISO application with highest levels observed in PNM regions.
Conclusion: When the physical link between SR and mitochondria by Mfn2 was disrupted, the SR/mitochondrial metabolic feedback mechanism was impaired resulting in the inability of the IP3-mediated SR Ca2+ release to induce ATP production in ventricular myocytes from Mfn2 KO mice. Furthermore, we revealed the difference in Mfn2-mediated SR-mitochondrial communication depending on mitochondrial location and type of communication (IP3R-mRyR1 vs. ryanodine receptor type 2-mitochondrial calcium uniporter).
Keywords: mitofusin 2, IP3 signaling, SR/mitochondria metabolic feedback, mitochondrial mRyR1, ATP generation, endothelin-1, Mfn2 KO mice
Introduction
Heart failure (HF) is one of the most common chronic conditions associated with aging. Despite advances in HF treatment, it has a poor prognosis with a steady increase in mortality and incidence in the aging population (Lloyd-Jones et al., 2002; Nieminen et al., 2006). Current treatments of HF rely almost entirely on altering the neurohormonal milieu, using agents such as angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers, and beta-adrenergic blockers to improve cardiac function and reduce mortality. Importantly, there are no proven interventions that are directly targeted toward maintaining cardiac function and myocyte survival. The pathogenesis of HF is the result of numerous changes in the whole organism with striking subcellular modifications especially at the level of mitochondria: the number of cristae of the inner mitochondrial membrane (IMM) is reduced, and due to subcellular changes, the contact sites of mitochondria and the sarcoplasmic reticulum (SR) are significantly diminished. Depending on the type of cardiomyopathy, both decreased respiratory chain activity due to impaired mitochondrial complexes I and III (Ventura-Clapier et al., 2011; Thai et al., 2018) or increased respiratory chain activity due to increased expression of uncoupling proteins (Sebastiani et al., 2007), could lead to increased proton leak and decline in ATP generation. The heart has the highest energy demand by weight of any organ in the body, and its function fails within minutes if mitochondrial ATP production is interrupted. Therefore, addressing mitochondrial dysfunction and decrease in mitochondrial ATP generation may represent an effective treatment for HF directly targeting the key features in its pathogenesis.
An increase in mitochondrial calcium (Ca2+) concentration ([Ca2+]m) is thought to be one of the major factors responsible for the activation of Ca2+-dependent dehydrogenases eventually leading to stimulation of the oxidative phosphorylation and ATP generation (Balaban, 2009). Recent studies utilizing mitochondrial Ca2+ uniporter (MCU) knockout (KO) mice (Pan et al., 2013; Kwong et al., 2015) questioned this accepted dogma since mitochondrial bioenergetics and ATP generation were not significantly compromised in those animals. Only when cardiac myocytes were challenged with the beta-adrenergic agonist isoproterenol (ISO), MCU KO myocytes were not able to match ATP production and respiratory rates in response to an increased work load (Kwong et al., 2015). These data indicate that although MCU is considered the major pathway for mitochondrial Ca2+ uptake in cardiac myocytes, other pathways located in the inner mitochondrial membrane (IMM) may compensate for the lack of MCU and play a role in the regulation of mitochondrial Ca2+ uptake and ATP generation. The mitochondrial ryanodine receptor (mRyR1) was proposed to be an alternative mitochondrial Ca2+ uptake pathway in cardiac myocytes (Beutner et al., 2005; Jakob et al., 2014) while the physiological function and relevance of this pathway are still controversial. Recently, we were able to demonstrate that Ca2+ released from the sarcoplasmic reticulum (SR) via the inositol 1,4,5-trisphosphate receptor (IP3R) is taken up into the mitochondrial matrix via the mRyR1 (Seidlmayer et al., 2016), where it activates ATP production. This IP3-mediated mitochondria/SR feedback is essential for the adaptation of mitochondrial metabolism to chronic stress as induced by endothelin-1 (ET-1) or angiotensin (Ang II).
Ca2+ release from intracellular stores is the essential signal generated by electrical activation in cardiomyocytes, which is required to initiate cell contraction in the process known as excitation-contraction coupling (ECC). To release Ca2+ from the SR, cardiac myocytes express two different Ca2+ release channels: (1) the ryanodine receptor type 2 (RyR2), which is Ca2+ sensitive and releases Ca2+ from the SR during ECC upon Ca2+ entrance through the voltage-gated L-type Ca2+ channels and (2) the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R), which releases Ca2+ after binding of IP3 independently from ECC. IP3 is cleaved from phosphatidylinositol-4,5-bisphosphate (PIP2) by phospholipase C when hormones like ET-1 or Ang II bind to their membrane bound G-protein-coupled receptors. Although IP3Rs are less abundant in ventricular myocytes compared to RyR2s, their activation results in measurable Ca2+ increase during diastole and systole creating areas with highly, localized concentrations of Ca2+, known as microdomains. These microdomains are typically formed at the junction between the SR and mitochondria resulting in elevation of the cytosolic Ca2+ from 0.1 to ~30 μM, which leads to the activation of mitochondrial Ca2+ uptake (Szalai et al., 2000; Dedkova and Blatter, 2008; Dedkova et al., 2013; Dorn and Maack, 2013).
Cardiac myocytes display a highly organized structure: mitochondria, SR, and the contractile apparatus are tightly packed. Mitochondria occupy about 40% of the myocytes volume (Zhou et al., 1998). Based on the proximity of mitochondria to the Ca2+ release sites, mitochondria are divided into subsarcolemmal (SSM), interfibrillar (IFM) and perinuclear mitochondria (PNM). All three subgroups differ in enzymatic activity (Palmer et al., 1977; Matlib et al., 1978; McMillin-Wood et al., 1980) and shape (Fawcett and McNutt, 1969; Lukyanenko et al., 2009). SSM are located underneath the sarcolemma, PNM are located around nuclei, while IFM are localized between the myofilaments where they are in close contact to the Ca2+ release sites in the junctional SR (Chikando et al., 2011).
In addition to this spatial relationship, mitochondria and the SR are linked physically by electron dense structures called tethers (Csordás et al., 2006). In mammalian cells, these tethers are formed by protein complexes containing IP3R (Szabadkai et al., 2006) and mitofusins, mainly containing mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2). Despite both molecules are homologues, Mfn1 mainly mediates mitochondrial fusion (Cipolat et al., 2004), whereas Mfn2 plays an important role in mitophagy (Schrepfer and Scorrano, 2016), controls the shape of the endoplasmic reticulum, and tethers mitochondria and the SR mechanically to each other to ensure the proper communication between the two organelles (de Brito and Scorrano, 2008). This latter function of Mfn2 has been challenged by Filadi et al. (2015) who suggested that Mfn2-tethers actually keep mitochondria far away from the SR to prevent mitochondrial Ca2+ overload. In a series of experiments, they demonstrated that KO of Mfn2 in cultured cells improved the communication between the two organelles, suggesting that Mfn2 is a negative regulator of tethering. Critical re-evaluation of Mfn2 function, however, confirmed the original discovery from Scorrano’s group that Mfn2 tethers serve to mediate the proper transfer of Ca2+ from the SR to mitochondria (Naon et al., 2016). In non-excitable cells, it has been shown that IP3Rs-mediated Ca2+ tunneling to mitochondria is an essential modulator of cell bioenergetics and ATP production (Cárdenas et al., 2010). However, the role of Mfn2 in IP3Rs-mediated Ca2+ tunneling to mitochondria and energetics in cardiac myocytes has not been evaluated yet.
In the present study, we aimed to examine the role of Mfn2 in the IP3Rs-mediated SR/mitochondria metabolic feedback mechanism described above. The hypothesis is that an intact linkage of SR and mitochondria is required for basal metabolic control of the IP3-mediated Ca2+ release in cardiac myocytes. Our hypothesis was tested using the cardiac-specific, tamoxifen-induced knockout of Mfn2 (Mfn2 KO). We found that when the physical link between SR and mitochondria by Mfn2 was disrupted, the SR/mitochondrial metabolic feedback mechanism was impaired resulting in the inability of the IP3-mediated SR Ca2+ release to induce ATP production in ventricular myocytes from Mfn2 KO mice. We further revealed the difference in Mfn-2 mediated SR-mitochondrial communication depending on mitochondrial location and type of receptor (IP3R vs. Ryanodine receptor), which can explain the existing controversy in the field.
Materials and Methods
Animal Model
Mfn2loxp/loxp/cre (Mfn2 KO) mice were obtained from the laboratory of Prof. Gerald W. Dorn, II in St. Louis, Missouri, USA. For induction of Cre-mediated deletion of Mfn2, mice were fed 20 mg tamoxifen/kg body weight/day for 7 days. Littermates of Mfn2 KO mice (heterozygous for Mfn2) were used as control animals and designated as wild-type (WT) group. All experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th Edition, 2011) and approved by the University of Würzburg Institutional Animal Care and Use Committee.
Cell Isolation
Adult left ventricular myocytes were isolated from 10- to 16-week-old Mfn2 KO mice and their healthy littermates. Mice were anesthetized with isoflurane and sacrificed by cervical dislocation. The excised heart was mounted to a Langendorff perfusion system and retrogradely perfused with Ca2+-free washing solution followed by enzyme solution containing Liberase TH (Research Grade, Roche, Basel, Switzerland) as described previously (Seidlmayer et al., 2016). Isolated myocytes were kept in Tyrode solution containing 1 mM Ca2+.
Immunoblot Analysis
Left ventricular tissue was homogenized in ice-cold lysis buffer (50 mM Tris pH 7.5, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% triton X-100, 50 mM NaF, 5 mM Na4P2O7, 1 mM Na3VO4, 1 mM DTT, protease inhibitor cocktail), and protein content was determined using the BCA assay. About 30 μg of protein was separated by 10% SDS-PAGE and transferred onto 0.2 μm PVDF membrane and blocked with 5% nonfat dry milk in phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBS-T). Blocked membranes were exposed to primary antibodies toward total Mfn2 (Abcam mouse monoclonal anti-Mfn2, ab56889, 1:2000 dilution) and GAPDH (Cell Signaling antibodies, 36,835, 1:2000 dilution). Sheep anti-mouse immunoglobulin G (IgG) antibodies (GE Healthcare NA9310V) in 1:4000 dilution were used as secondary antibodies. Protein bands were visualized by enhanced chemiluminescence (GE Healthcare Life Sciences). Mfn2 bands were normalized to GAPDH bands using the software ScanPack 3.0.
Real-Time PCR
After extraction of total RNA using a RNA fibrous tissue mini kit (Qiagen, Venlo, Netherlands), cDNA was synthesized from 1 μg of RNA using the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed with commercial and customized TaqMan probes (Life Technologies™, Waltham, Massachusetts, USA). Target gene mRNA levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which served as housekeeping gene for comparison.
Confocal Microscopy
Laser scanning confocal microscopy (Leica SP5 and a Zeiss LSM 780) was used to follow the changes in cytosolic ([Ca2+]i) and mitochondrial ([Ca2+]m) Ca2+ concentration, mitochondrial membrane potential (ΔΨm), reactive oxygen species (ROS) generation, and mitochondrial ATP generation using specific fluorescent indicators. All fluorescent signals were recorded in electrically stimulated (0.5 Hz) single cardiac myocytes. For measurements, the myocytes were plated on laminin-covered coverslips and incubated in Tyrode solution containing 1 mM Ca2+ at room temperature. The fluorescence image was recorded every 10 s. Fluorescence levels were corrected for background fluorescence and normalized to untreated control (F0) (F/F0).
[Ca2+]m Measurements
Changes in mitochondrial Ca2+ concentration ([Ca2+]m) were measured in intact cells using the Ca2+-sensitive fluorescent probe X-Rhod-1/AM (molecular probes, λex = 543 nm and λem = 552–617 nm). Myocytes were loaded with the dye for 30 min at 37°C. This loading procedure favors mitochondrial localization of X-Rhod-1. To quench the remaining cytosolic fluorescence, the myocytes were incubated in 1 mM CoCl2-containing Tyrode during washout period as published before (Seidlmayer et al., 2015, 2016).
Analysis of Mitochondrial Subfractions
For the analysis of [Ca2+]m levels in SSM, a longitudinal region of interest (ROI) was placed directly underneath the sarcolemma and for IFM in the center of the cell excluding the nuclei. Changes in [Ca2+]m levels in perinuclear regions (PNM) were detected in the ROIs located from each side of the nucleus. Fluorescence changes over time were analyzed.
ATP Measurement
ATP concentration was measured indirectly via the free magnesium (Mg2+) concentration using the fluorescent probe mag-fluo-4 AM (Invitrogen) and directly using a commercially available ATP luciferase assay (Invitrogen, Waltham, Massachusetts, USA, #A22066) (Seidlmayer et al., 2016). This indirect method is based on the fact that ATP forms complexes with Mg2+. Since free [Mg2+]i is kept constant within a rather narrow range, ATP hydrolysis leads to concomitant increase in free [Mg2+]i as measured with fluorescent Mg2+ indicators such as mag-fluo-4. Therefore, an increase in mag-fluo-4 fluorescence indicates a decrease in ATP concentration. For indirect ATP measurements, myocytes were loaded with 10 μM mag-fluo-4 (λex = 488 nm and λem = 565–605 nm) for 30 min at 37°C. All data are expressed as an inverted ratio R = 1 − F/F0.
Mitochondrial Membrane Potential Measurements
Mitochondrial membrane potential (ΔΨm) was measured in intact myocytes using the potentiometric probe tetramethylrhodamine methyl ester (TMRM) (λex = 543 nm and λem = 565–605 nm) (Dedkova and Blatter, 2012; Seidlmayer et al., 2015, 2016). The myocytes were loaded with 10 nM TMRM for 15 min at 37°C. 10 nM TMRM was present in all solutions during the experiments. In the end of each experiment, 5 μM carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) and 1 μM oligomycin were added to calibrate the signal. All data were background corrected.
Redox Measurements
Isolated adult ventricular myocytes from WT mice were field-stimulated at 37°C with a stimulation rate of 0.5 Hz and superfused with normal Tyrode’s (NT) solution containing (in mM) NaCl 130, KCl 5, MgCl2 1, CaCl2 1, Na-HEPES 10, glucose 10, sodium pyruvate 2, and ascorbic acid 0.3, pH 7.4. After 120 s, cardiac myocytes were superfused with NT solution containing either 10 nM endothelin or 500 nM isoproterenol. We monitored changes in redox status of cardiomyocytes by measuring autofluorescence of two endogenous tissue fluorophores: reduced form of nicotinamide adenine dinucleotide (NADH, transfers electrons to molecular oxygen) and flavin adenine dinucleotide (FAD+, which is an electron acceptor) (Dedkova and Blatter, 2012). NADH has fluorescence excitation and emission maxima at 350 and 460 nm, respectively. FAD+ has fluorescence excitation and emission maxima at 450 and 535 nm, respectively. During this protocol, NADH (λex = 340 nm; λem = 450 nm) and FAD+ (λex = 480 nm; λem = 520 nm) autofluorescence were recorded using an IonOptix set-up as described previously in (Kohlhaas et al., 2017). Calibration was performed with FCCP (5 μmol/L) and cyanide (4 mmol/L). “Redox ratio,” which represents approximation of the reduction-to-oxidation status of the mitochondrial matrix space, was calculated by dividing the fluorescence intensity of NADH to the fluorescence intensity of FAD+.
Statistics
The data are normalized to untreated control and background corrected. All data are presented as mean ± standard error of the mean for the indicated number (n) of experiments. For comparisons of multiple groups in Figure 1, ANOVA was used. A significant overall F-test multi-group comparison was done via Tukey’s test (including correction for multiple testing) for treatment vs. control. For comparisons of only two groups, Student’s t-test was used. Data were considered significant at p < 0.05.
Figure 1.

Expression of Mfn2 was effectively abolished by 7 days of tamoxifen treatment in Mfn2 KO mice. (A) Original Western blots of Mfn2 and GAPDH in WT and Mfn2 KO mice exposed to tamoxifen for 0, 1, and 7 days. (B) Mfn2 normalized to GAPDH showing a significant decrease of Mfn2 in tamoxifen-treated mice. (C) Fold change expression normalized to GAPDH of Mfn2 after 7 days of tamoxifen treatment compared to untreated WT measured by real-time PCR. (D) Expression level of RyR1, RyR2, MCU, and VDAC1 and VDAC 2 in WT and Mfn2 KO mice measured by real-time PCR. *p ≤ 0.05 compared to WT, **p ≤ 0.01 compared to WT.
Results
Expression of Mfn2 was Effectively Abolished by Tamoxifen Treatment in Mfn2 KO Mice
To test our hypothesis that disturbed SR/mitochondrial contacts due to Mfn2 knockout affect the SR/mitochondrial metabolic feedback mechanism, experiments were performed in tamoxifen-inducible cardiac-specific Mfn2 knockout (Mfn2 KO) mice (Chen et al., 2011). To verify that Mfn2 protein was effectively reduced by tamoxifen, Western blot analysis was performed in heart tissue lysates collected 1 and 7 days after beginning of tamoxifen treatment. As shown in representative Western blot images in Figure 1A, Mfn2 expression was significantly reduced after 1 day of tamoxifen treatment and almost completely eliminated (90% decrease compared to WT myocytes) after 7 days of tamoxifen (Figure 1B). These results were confirmed by quantifying RNA levels of Mfn2 using real-time PCR. As shown in Figure 1C, the relative RNA in Mfn2 KO was 0.09 ± 0.07 (n = 3) compared to 1.0 ± 0.37 (n = 4), in WT mice. To verify that knockout of Mfn2 has no effect on the proteins involved in cardiac ECC, we tested the expression levels of RyR types 1 and 2, MCU, and voltage-dependent anion channel 1 (VDAC1) and 2 (VDAC2) by real-time PCR. As demonstrated in Figure 1D, expression levels of both RyR types 1 and 2, MCU, VDAC1, and VDAC2 were not altered by Mfn2 knockout.
Ca2+ Released via the IP3R Cannot Be Taken Up Into Mitochondria in Mfn2 KO mice
We have recently demonstrated (Seidlmayer et al., 2016) that Ca2+ released from the SR upon IP3R activation is taken up into the mitochondrial matrix in WT cardiomyocytes. 10 nM ET-1 induced a significant increase in mitochondrial Ca2+, which could be abolished by cell treatment with 3 μM of the IP3R antagonist 2-APB (Seidlmayer et al., 2016). Since IP3-mediated global cytosolic Ca2+ changes are very small, experiments were designed to test the importance of the subcellular contacts between SR and mitochondria in Ca2+ tunneling directly from SR to mitochondria in Mfn2 KO mice in which the physical tethering of SR and mitochondria is disrupted. Consistent with our previous reports (Seidlmayer et al., 2016), addition of 10 nM ET-1 to electrically field-stimulated (0.5 Hz) ventricular myocytes induced a significant increase in mitochondrial Ca2+ uptake in WT myocytes (+35 ± 6%, n = 7, Figures 2A,B, solid green trace). This ET-1-induced mitochondrial Ca2+ uptake was significantly blunted in Mfn2 KO myocytes (+17 ± 4%, n = 9) (Figures 2A,B, dashed green trace) confirming the importance of Mfn2 in ET-1 mediated Ca2+ transfer to mitochondria. Furthermore, we confirmed that blocking mitochondrial Ca2+ uptake with the mRyR1 inhibitor dantrolene (1 μM), led to a corresponding increase in systolic Ca2+ level in the cytosol (Supplementary Figure S1). This indicates that when mitochondrial Ca2+ uptake via mRyR1 is not possible, Ca2+ released from SR is leaking out into cytosol. This Ca2+ leak from the SR in cytosol could contribute to pro-arrhythmic events described before (Proven et al., 2006).
Figure 2.

In Mfn2 KO mice Ca2+ released via the IP3R cannot be taken up into mitochondria. (A) The representative confocal traces recorded from adult ventricular myocytes loaded with the Ca2+-sensitive fluorescent dye X-Rhod-1 upon electrical field stimulation (FS, 0.5 Hz) and subsequent exposure to 10 nM ET-1 (WT: solid green, Mfn2 KO: dashed green) or 500 nM ISO (WT: solid blue, Mfn2 KO: dashed blue), respectively. Changes in fluorescence reflect changes in mitochondrial Ca2+. (B) Mean values of the mitochondrial Ca2+ levels change during 20 min of observation. ET-1 increased mitochondrial Ca2+ concentration ([Ca2+]m) by +35 ± 6% (n = 7) in healthy WT myocytes, whereas ISO-induced elevation of [Ca2+]m was significantly smaller (+20 ± 2%, n = 7). In Mfn2 KO myocytes, the mitochondrial Ca2+ uptake induced by ET-1 was significantly smaller compared to WT myocytes (ET-1: +17 ± 4%, n = 9). However, the effect of ISO on mitochondrial Ca2+ uptake was not altered by Mfn2 KO (ISO: +19 ± 4%, n = 7). No significant changes in [Ca2+]m were observed in untreated WT (solid black) and Mfn2 KO (dashed black) myocytes during 20 min of observation. *p < 0.05 compared to untreated WT, **p ≤ 0.05 compared to untreated WT, #p ≤ 0.05 compared to WT treated with ET-1.
Control time recordings performed in untreated electrically field-stimulated (0.5 Hz) myocytes from WT (solid black trace, −1 ± 4%, n = 6) and Mfn2 KO mice (dashed black trace, +3 ± 5%, n = 4) confirmed that there was no measurable increase in mitochondrial Ca2+ levels in the absence of ET-1 stimulation (Figure 2B), confirming that the observed increase in mitochondrial Ca2+ was due to ET-1 activation of IP3 receptors.
Mfn2 KO Does Not Block Global ISO-Induced Mitochondrial Ca2+ Uptake
We have previously shown that when cytosolic Ca2+ was increased by the non-specific beta-adrenergic receptor agonist isoproterenol (ISO) in WT myocytes, which induces a global increase in cytosolic Ca2+ concentration, only a small increase in mitochondrial Ca2+ uptake was observed (Seidlmayer et al., 2016). In this study, we performed experiments in cardiomyocytes from Mfn2 KO mice, and their control littermates (WT) to examine whether intact Mfn-2-mediated contact sites are required for ISO-induced changes in [Ca2+]m. As shown in Figure 2A, we found that mitochondrial Ca2+ increase induced by ISO (solid blue line) in electrically field-stimulated (0.5 Hz) ventricular myocytes was significantly smaller (+20 ± 2%, n = 7) compared to the ET-1-induced response (solid green line). Furthermore, in contrast to the data obtained with ET-1, Mfn2 KO had no effect on average ISO-induced mitochondrial Ca2+ uptake (+19 ± 4%, n = 7, dashed blue line) (Figures 2A,B), indicating that intact spatial arrangement of SR and mitochondria was not critical for ISO-induced mitochondrial Ca2+ uptake.
Interfibrillar and Perinuclear Mitochondria Take Up More Ca2+ Following IP3-Mediated SR Ca2+ Release Compared to Subsarcolemmal Mitochondria
Since mitochondrial morphology and function vary between different mitochondrial subpopulations in the cell (Fawcett and McNutt, 1969; Palmer et al., 1977; Matlib et al., 1978; McMillin-Wood et al., 1980; Lukyanenko et al., 2009), we tested the importance of physical tethering of SR and mitochondria in different mitochondrial regions. As we mentioned before, subsarcolemmal mitochondria (SSM) exist below the cell membrane, interfibrillar mitochondria (IFM) reside in rows between the myofibrils, and perinuclear mitochondria (PNM) are located around the nuclei. Electron microscopy studies revealed that the SR/mitochondrial cleft is only 10–25 nm (Chen et al., 2012). Since, due to their intracellular localization, the interorganellar contact differs between the different mitochondrial subpopulations, we analyzed differences in IP3-mediated mitochondrial Ca2+ uptake in these mitochondrial subfractions. For the analysis of [Ca2+]m levels in SSM, a longitudinal region of interest (ROI) was placed directly underneath the sarcolemma. For [Ca2+]m levels in IFM, ROIs were placed in the center of the cell excluding the nuclei. Changes in [Ca2+]m levels in perinuclear regions (PNM) were detected in the ROIs located from each side of the nucleus. As shown in Figure 3, subsarcolemmal mitochondria of WT myocytes take up significantly less Ca2+ than interfibrillar and perinuclear mitochondria following ET-1 stimulation (IFM: +47 ± 8% after 20 min, n = 6; PNM: +47 ± 6% after 20 min, n = 6 vs. SSM: +30 ± 8% after 20 min, n = 6). In Mfn2 KO mice, mitochondrial Ca2+ uptake was significantly inhibited in IFM (+24 ± 4%, n = 6, p < 0.05 compared to WT) and PNM (+19 ± 4%, n = 6, p < 0.05) mitochondria following ET-1 stimulation. There was no difference in [Ca2+]m levels upon ET-1 treatment in Mfn2 KO mitochondria from SSM regions (+20 ± 2%, n = 8, p = 0.23) (Figure 3). These data indicate that disruption of the SR-mitochondrial tethering in specific regions abolishes the differences in mitochondrial Ca2+ uptake induced by IP3-mediated Ca2+ release from SR in ventricular myocytes.
Figure 3.
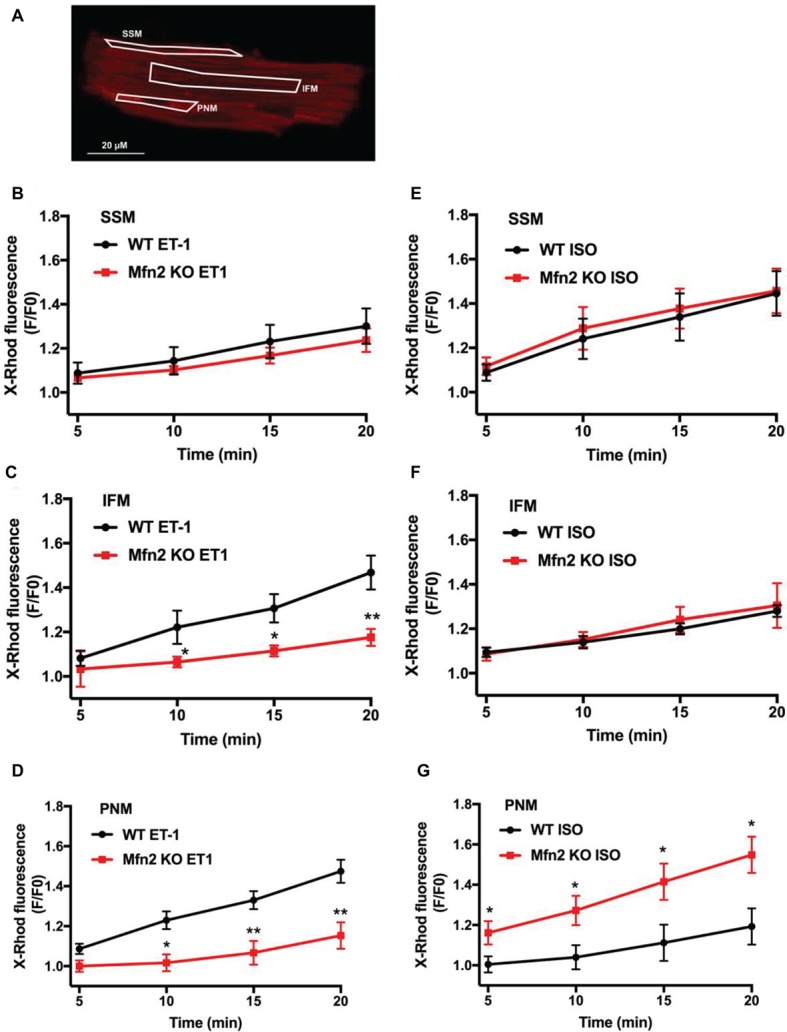
IFM and PNM take up more Ca2+ than SSM following IP3-mediated SR Ca2+ release. (A) Original confocal image of an adult murine ventricular myocyte loaded with X-Rhod-1. Shown in white are regions of interest (ROIs) placed over SSM, IFM, and PNM for the analysis of Ca2+ changes in different mitochondrial regions. (B) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in SSM normalized to untreated controls (black: WT myocytes+ET-1; red: Mfn2 KO myocytes + ET-1). (C) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in IFM normalized to untreated controls (black: WT myocytes+ET-1; red: Mfn2 KO myocytes + ET-1). (D) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in PNM normalized to untreated controls (black: WT myocytes+ET-1; red: Mfn2 KO myocytes + ET-1). (E) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in SSM normalized to untreated controls (black: WT myocytes+ISO; red: Mfn2 KO myocytes + ISO). (F) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in IFM normalized to untreated controls (black: WT myocytes+ISO; red: Mfn2 KO myocytes + ISO). (G) Mean values for mitochondrial Ca2+ changes measured as changes in X-Rhod-1 fluorescence in PNM normalized to untreated controls (black: WT myocytes+ISO; red: Mfn2 KO myocytes + ISO). *p ≤ 0.05 compared to WT ET-1 or ISO vs. Mfn2 KO, **p ≤ 0.001 compared to WT ET-1 or ISO vs. Mfn2 KO.
Interestingly, the effect of ISO on [Ca2+]m levels in the three mitochondrial subgroups was different from ET-1. The highest mitochondrial Ca2+ uptake was detected in subsarcolemmal mitochondria (+44 ± 12% after 20 min, n = 7), which gradually declined in IFM (+20 ± 9% after 20 min, n = 7) and was the smallest in perinuclear (+11 ± 9% after 20 min, n = 7) mitochondria. Furthermore, while Mfn-2 KO had no effect on ISO-induced [Ca2+]m levels in SSM and IFM mitochondrial regions (Figures 3E,F), mitochondrial Ca2+ uptake was actually increased in perinuclear mitochondria (Figure 3G) in cardiomyocytes from Mfn2 KO animals.
These data suggest that tethering between SR and mitochondria is heterogeneous within ventricular myocytes to support the privileged SR-mitochondrial communication between IP3 receptors and mRyR1. When this communication was lost in Mfn-2 KO mice, perinuclear mitochondria were able to pick more Ca2+ upon ISO stimulation.
IP3-Mediated Increase in Mitochondrial Membrane Potential was Dependent on Mfn2
The Ca2+ taken up by mitochondria following ET-1 stimulation is activating mitochondrial oxidative phosphorylation, which results in a significant increase in ATP levels (Seidlmayer et al., 2016). Here, we measured changes in mitochondrial membrane potential (ΔΨm) following ET-1 stimulation using the potentiometric probe TMRM in cardiac myocytes from WT and Mfn2 KO mice (Figure 4A). In WT myocytes, ET-1 induced a significant hyperpolarization over 20 min (+37 ± 3%, n = 13, p < 0.01 compared to untreated controls), whereas in Mfn-2 KO ΔΨm decreased upon exposure to ET-1 (−11 ± 6%, n = 4, p < 0.01 compared to WT + ET-1) myocytes, indicating the importance of intact SR-mitochondrial contacts for mitochondrial ΔΨm maintenance.
Figure 4.

IP3R stimulation with ET-1 increases mitochondrial membrane potential in WT myocytes but not in Mfn2 KO myocytes. (A) Normalized mean values of TMRM fluorescence changes in WT (black line) and Mfn2 KO (red line) cardiac myocytes after exposure to 10 nM ET-1 for 20 min. TMRM signal was normalized to the level measured in untreated WT and Mfn2 KO myocytes. (B) Mean values of basal TMRM fluorescence in untreated myocytes from WT (black) and Mfn2 KO (red) mice. (C) Mean values of NADH in % of reduced NADH following ISO (red) and ET-1 (blue) treatment as well as untreated control (black) over 20 min. (D) Mean values of FAD+ in % of oxidized FAD+ following ISO (red) and ET-1 (blue) treatment as well as untreated control (black) over 20 min. (E) The ratio of NADH/FADH following the treatment with ET-1 (blue) and ISO (red). For comparison, untreated control is shown in black. (F) The normalized ratio of NADH/FADH following treatment with ET-1 (blue) and ISO (red) is shown. Black is the untreated control with electrical field stimulation at frequency 0.5 Hz. (A) *p ≤ 0.05 compared to untreated control, **p ≤ 0.001 compared to untreated control, #p ≤ 0.05 compared to WT treated with ET-1, ##p ≤ 0.001 compared to WT treated with ET-1. (C–F) #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001; black – ISO vs. ctrl, gray – ET-1 vs. ctrl.
To test if basal ΔΨm was different in WT and Mfn2 KO myocytes, we measured TMRM fluorescence at the beginning of the experiment before cell treatment with ET-1. No significant difference in basal ΔΨm was detected (Figure 4B), suggesting that there is no difference in basal respiratory activity in WT and Mfn2 KO mice.
In support of these data, we also found that while both ET-1 and ISO induced a decrease in reduced NADH (Figure 4C), changes in both reduced NADH and oxidized FAD+ levels (Figure 4D) were less pronounced upon ET-1 stimulation resulting in minimal net redox ratio NADH/FAD+ changes (Figures 4E,F) normalized to untreated conditions whereas, in cells treated with ISO, the ratio declined significantly. Altogether, these data indicate that ET-1 maintains mitochondrial membrane potential and respiratory chain activity.
IP3-Mediated ATP Production Following ET-1 Stimulation is Impaired in Mfn2 KO Mice
Next, we monitored changes in [ATP]i indirectly using the [Mg2+]-sensitive dye mag-fluo-4 in WT and Mfn-2 KO cardiac myocytes treated with either 10 nM ET-1 or 100 nM ISO. This method takes advantage of the fact that ATP is a major Mg2+ buffer, such that an increase in [ATP]i level reduces free [Mg2+]i (Dedkova and Blatter, 2012). Therefore, increased ATP production causes mag-fluo-4 fluorescence to decline. In Figure 5, the mag-fluo-4 signal was inverted so that an increase in the signal reflects an increase in [ATP]i. Similar to previous experiments, cardiac myocytes were electrically stimulated at the frequency of 0.5 Hz to maintain Ca2+ fluxes during ECC, and then ET-1 (10 nM) and ISO (500 nM) were applied to activate the correspondent receptors. As shown in Figure 5A, ET-1 but not ISO induced an increase in ATP generation in WT ventricular myocytes, which was abolished in Mfn2 KO myocytes emphasizing the importance of Mfn2 in Ca2+ transfer to mitochondria to mediate ATP production. As summarized in Figure 5B, ET-1 induced a significant increase in ATP levels over 20 min (+19.18 ± 3.7%, n = 6), whereas in Mfn2 KO myocytes the effect of ET-1 on ATP generation was completely abolished (+0.74 ± 1.5%, n = 5). To the contrary, ISO-treatment caused a significant decrease in ATP levels in WT (−8.7 ± 1.2%, n = 4, p < 0.01), while a significant increase in ATP (+13.1 ± 3%, n = 7, p < 0.01) was observed in Mfn-2 KO myocytes upon ISO stimulation.
Figure 5.

ATP production following IP3-mediated SR Ca2+ release is impaired in Mfn2 KO mice. (A) Original confocal traces of ATP levels recorded in WT myocytes stimulated with 10 nM ET-1 (black) or 500 nM ISO (blue) and Mfn2 KO cardiac myocytes stimulated with ET-1 (red) or ISO (green), respectively. Mg2+-sensitive fluorescent dye mag-fluo-4 was used to monitor ATP levels inside cardiac myocytes. As free Mg2+ and ATP form complexes, increase in ATP concentration reduces free Mg2+ and thus mag-fluo-4 fluorescence. Traces are inverted for better understanding and presented as 1-F/F0. (B) Mean values of the experiment described in (A) in %. *p ≤ 0.05 compared to untreated WT; **p ≤ 0.01 compared to untreated WT; #p ≤ 0.05 compared to WT treated with ET-1 or ISO, respectively; ##p ≤ 0.01 compared to WT treated with ET-1 or ISO, respectively. (C–H) Subgroup analysis of subsarcolemmal (SSM, C,D), interfibrillar (IFM, E,F), and perinuclear (PNM, G,H) mitochondria in myocytes from WT (black) and Mfn2 KO (red) mice loaded with mag-fluo-4 as described in (A). The myocytes were treated with either ET-1 (left) or ISO (right). *p ≤ 0.05 compared to WT, **p ≤ 0.01 compared to WT.
We further analyzed the effect of ET-1 and ISO on ATP content in mitochondrial subgroups as we did for mitochondrial Ca2+ uptake. The results are summarized in Figure 5C. As expected, in Mfn2 KO myocytes, ET-1 was not able to induce a significant increase in ATP levels in all subgroups. However, there was no significant difference in ET-1-induced ATP increase between IFM, SSM, and PNM in myocytes from WT or Mfn2 KO mice.
A decrease in ATP levels upon ISO exposure was more pronounced in IFM and SSM mitochondrial subgroups but remained unchanged in PNM (Figure 5C). In agreement, with the data presented in Figure 5B, ISO addition to Mfn2 KO myocytes increased ATP levels in all groups significantly compared to the corresponding groups in WT myocytes. Furthermore, PNM in Mfn 2 KO myocytes, which took more Ca2+ into the mitochondria, also increased ATP levels significantly higher than IFM and SSM from Mfn2 KO myocytes.
Discussion
In this study, we examined the role of SR-mitochondrial tethering by Mfn2 in activation of mitochondrial Ca2+ uptake and metabolism using mice with a cardiac-specific deletion of Mfn2 (Figure 1). We tested the hypothesis that physical linkage of SR and mitochondria via Mfn2 is essential for IP3R-mediated signal transduction to mitochondria to maintain ATP levels in cardiac myocytes under physiological conditions (i.e., normal levels of IP3R expression and circulating ET-1). In our previous study (Seidlmayer et al., 2016), we found that the stimulation of cardiac myocytes with IP3R agonists induced mitochondrial Ca2+ uptake via mRyR1 and increased ATP content. In contrast, under basal conditions, beta-adrenergic stimulation with ISO was not able to induce measurable ATP production. Since the global changes in cytosolic Ca2+ concentration produced by IP3-mediated SR Ca2+ release are very small compared to RyR-mediated Ca2+ changes, here, we examined the significance of intact SR-mitochondrial tethering by Mfn2 for IP3-mediated stimulation of mitochondrial Ca2+ uptake and ATP generation.
We found that the ability of ET-1 to induce a significant IP3-dependent mitochondrial Ca2+ uptake was lost in myocytes from Mfn2 KO mice (Figure 2). Interestingly, when myocytes from Mfn2 KO mice were stimulated with the beta-adrenergic receptor agonist ISO, mitochondrial Ca2+ uptake was not affected in SSM and IFM but significantly increased in PNM regions. When the influence of beta-adrenergic and IP3R stimulation on ATP content was examined (Figure 5), ET-1 did not induce an increase in ATP in myocytes from Mfn2 KO mice, indicating the importance of physical tethering of mitochondria and the SR for metabolic control by IP3 under physiological conditions. To the contrary, in Mfn2 KO myocytes, ISO significantly increased ATP content under conditions of electrical field stimulation, whereas in WT myocytes ISO led to a decrease in mitochondrial ATP content. The latter finding agrees with previous studies showing that sudden increase in work load causes a decrease in mitochondrial NADH (and presumably ATP levels), which recovered slowly over the time in a Ca2+-dependent manner (Brandes and Bers, 1997). Furthermore, another study demonstrated that beta-adrenergic stimulation with ISO induced only a transient MCU-dependent increase in oxygen consumption, suggesting that MCU only plays a role in acute matching of the mitochondrial energy output with increase in cardiac metabolic demand (Kwong et al., 2015). However, over the time, oxygen consumption was not different between WT and MCU KO cardiac myocytes, indicating that another mitochondrial Ca2+ influx pathway contributes to the sustained energy generation in cardiac myocytes. Our data demonstrate that IP3R-mRyR1-mediated mitochondrial Ca2+ uptake contributes to the sustained ATP generation (Figure 6) and that this Ca2+ transfer from SR is regulated by Mfn2 under physiological conditions. When this regulation is lost in Mfn2 KO mice, IP3-mediated Ca2+ release is not capable to stimulate ATP generation anymore, as observed in pathological conditions. Altogether, our data suggest that Mfn2 can provide a fine tuning to regulate Ca2+-dependent energy production under physiological conditions upon ET-1 stimulation, but when Mfn2-mediated tethering is disrupted, mitochondria can adapt to respond to global Ca2+ elevation induced by ISO with increase in ATP generation.
Figure 6.
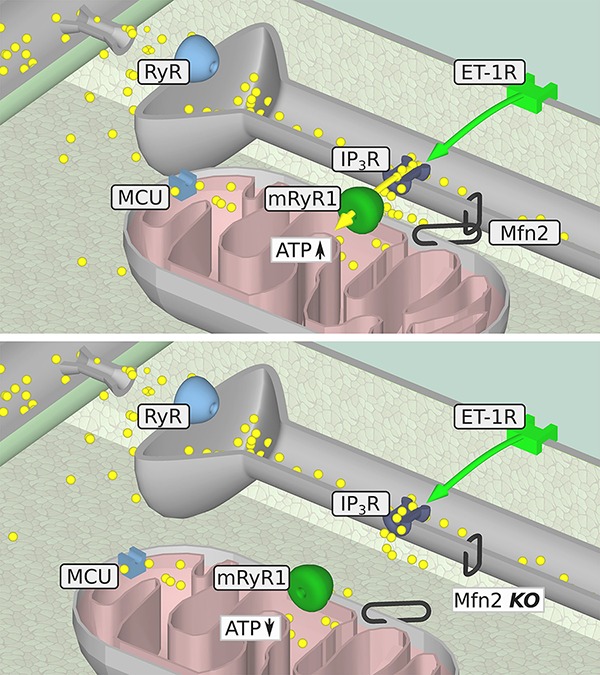
Summary of the proposed metabolic IP3-mediated SR/mitochondrial signaling pathway in the presence (upper panel) or absence (lower panel) of intact Mfn2-tethers. IP3-mediated mitochondrial Ca2+ uptake in WT myocytes (upper panel), which is followed by an increase in ATP generation. When the physical contact of mitochondria and the SR is disturbed by knockout of Mfn2 (lower panel), Ca2+ released from the SR by IP3 cannot be taken up into mitochondria anymore leading to the decrease in ATP level. Yellow circles: Ca2+, yellow arrow: Ca2+ movement, green arrow: IP3 movement.
The disruption of Mfn2-mediated intact ER-mitochondrial contacts exists in conditions of heart failure induced by transverse aortic constriction (TAC), which is associated with a decreased excitability and force generation in cardiac myocytes (Goh et al., 2016). ET-1 levels are typically increased in chronic stress as in the setting of congestive heart failure (CHF) (Wei et al., 1994) along with increased myocardial expression of ETA receptors (Picard et al., 1998; Pieske et al., 1999). To determine if this increase in the ET-1 axis is pathologic, a mouse model was developed to conditionally overexpress ET-1 in the myocardium that led to nearly 10-fold increase in ET-1 concentration in the heart, without any significant increase in circulating ET-1 levels (Yang et al., 2004). Mice with elevated cardiac ET-1 expression presented with severe heart inflammation, hypertrophy leading to dilated cardiomyopathy, CHF, and death within 5 weeks of induction. However, a whole body ET-1 KO (ET-1−/−) homozygous mice developed by disrupting exon 2 of the ET-1 gene were neonatally lethal (Kurihara et al., 1994). ET-1−/− mice delivered by caesarean section at day 18.5 postcoitum, all displayed significant craniofacial and cardiac abnormalities, highlighting the critical role of ET-1 in the development (Kurihara et al., 1994, 1995b). Moreover, ET-1−/− mice had reduced neonatal weight, impaired thyroid and thymus development (Kurihara et al., 1995a), and reduced cardiac sympathetic innervation (Ieda et al., 2004). Mice heterozygous for ET-1 deletion, ET-1+/−, appeared normal, were fertile, and had reduced lung and plasma ET-1 concentration; however, they had elevated systolic, diastolic, and mean arterial blood pressure (Kurihara et al., 1994). These observations suggest that while 10-fold elevation in ET-1 levels in cardiac myocytes is clearly pathological, basal levels of ET-1 are required for maintaining normal physiological function in cardiac myocytes.
Indeed, myocardial ET-1 depicts beneficial effects depending on the physiological situation. In the myocardium, ET-1 prevents excessive apoptosis after cardiac overload induced by aortic banding (Zhao et al., 2006). Signaling of the anti-apoptotic effects of ET-1 involves calcineurin, the mitochondrial function, and the classical MEK1/2-ERK1/2 and PI3 kinase pathways (Iwai-Kanai and Hasegawa, 2004). It has been reported that ET-1 prevents the early phase of doxorubicin-induced cytotoxicity via the upregulation of the antioxidant manganese superoxide dismutase through the ETA receptor in cultured cardiomyocytes (Suzuki and Miyauchi, 2001). Preconditioning infusion of ET-1 can reduce infarct size in rats subjected to ischemia-reperfusion (Gourine et al., 2005). The transgenic expression of ET-1 in mice lacking a functional gene for eNOS restores diastolic function presumably through modulation of oxidative stress and a change of metabolic substrate from fatty acid oxidation toward enhanced glycolysis (Vignon-Zellweger et al., 2011). In line with this, the enhanced glycolysis in the failing heart, which is a beneficial compensatory mechanism, is accompanied by a hypoxia-inducible factor-1α-dependent elevation of ET-1 expression (Kakinuma et al., 2001).
Our study indicates that stimulation of ET-1 receptors in healthy control myocytes leads to mitochondrial Ca2+ uptake, maintains mitochondrial membrane potential, and activates ATP generation. Furthermore, we determined that intact Mfn2 tethers were required to mediate Ca2+ uptake into mitochondria and stimulate ATP production. The significance of a physical linkage of SR and mitochondria in cardiac myocytes is still debated. Our data add new aspects to this discussion revealing the importance of Mfn2 tethers in IP3-mediated SR/mitochondrial metabolic feedback in ventricular myocytes. As shown by our group before (Seidlmayer et al., 2016), Ca2+ released from the SR via the IP3R is taken up into mitochondria where it increases ATP content. Here, we were able to demonstrate that this metabolic feedback is severely disturbed in Mfn2-deficient mice. When cardiac myocytes from these mice were stimulated with the IP3R agonist ET-1, no mitochondrial Ca2+ uptake could be detected. In agreement with this finding, IP3-mediated SR Ca2+ release in Mfn2-deficient myocytes was not able to increase cellular ATP content. These results clearly show that intact tethering of SR and mitochondria via Mfn2 is crucial for the channeling of intracellular Ca2+ signals from IP3R to mRyR1 located in mitochondria. In support of this, data presented in Supplementary Figure S1 demonstrate that when cells were treated with the mRyR1 inhibitor, 1 μM Dantrolene, the systolic Ca2+ level in cytosol was significantly elevated.
It is generally accepted that Ca2+ crosses the outer mitochondrial membrane via non-specific high-conductance voltage-dependent anion channels (VDAC) (see Shoshan-Barmatz et al., 2008, for review). Specifically, it has been demonstrated that VDAC1 plays a major role in ER/mitochondria-Ca2+ cross-talk (Shoshan-Barmatz et al., 2017) by forming a supra-molecular complex with the IP3 receptor in the SR and VDAC1 in the OMM, linked by a chaperone called GRP75 (Csordás et al., 2006), together with Mfn2 (de Brito and Scorrano, 2008). It is plausible that Mfn2-mediated tethering creates a microenvironment with highly elevated Ca2+ levels to enhance Ca2+ transfer via VDAC1; however, the direct experimental evidence of the enhanced Ca2+ channeling via VDAC1 by Mfn2 is lacking. Several studies, however, demonstrated that VDAC1 is highly Ca2+-permeable and modulates the accessibility of Ca2+ to the intermembrane space (IMS) (Gincel et al., 2001; Rapizzi et al., 2002; De Stefani et al., 2012). The importance of the 18-kDa outer mitochondrial membrane transporter protein (TSPO) in mitochondrial Ca2+ transport and its role in heart failure have been recently revealed by Thai et al. (2018) ringing attention to the outer mitochondrial membrane proteins in regulation of the mitochondrial Ca2+ uptake and bioenergetics.
Interestingly, not all mitochondria within the myocyte take up Ca2+ into their matrix following IP3R activation to the same extent. Detailed analysis of mitochondrial subpopulations revealed that in WT cardiac myocytes IFM and PNM take up almost two times more Ca2+ following IP3-mediated SR Ca2+ release compared to the SSM regions (Figure 3). In Mfn2-deficient myocytes, mitochondrial Ca2+ uptake following IP3R activation was not different between all three mitochondrial subpopulations. This finding is supported by several studies providing evidence for not only structural differences of IFM (Fawcett and McNutt, 1969; Shimada et al., 1984; Lukyanenko et al., 2009), PNM, and SSM but also functional differences between these mitochondrial populations. It has been suggested that IFM produce ATP primarily for contraction, PNM for nuclear processes, and SSM provide ATP for active transport processes across the sarcolemma (Müller, 1976; Palmer et al., 1977; Shimada et al., 1984; Rosca and Hoppel, 2013). Since ET-1 has a positive inotropic effect and activates pathological gene transcription in the nucleus, a preferential effect of ET-1 on IFM and PNM is in agreement with these findings.
As expected, since mitochondrial Ca2+ uptake following ET-1 stimulation was abolished in Mfn-2 KO mice, ATP production was not detectable in Mfn2-deficient cardiomyocytes. Interestingly, the basal mitochondrial membrane potential measured with TMRM was not different between the two genotypes, which is in accordance with the results of Chen et al. (2012); however, ET-1 stimulation led to the increase in mitochondrial membrane potential (Figure 4), which contributes to the enhanced electron transfer chain coupling and ATP generation (Figure 5). Taken together, these results indicate a crucial role of physical tethering of SR and mitochondria by Mfn2 for adaptation of energy production to chronic stress. Interestingly, the effect of ISO on [Ca2+]m levels in the three mitochondrial subgroups was different from ET-1. The highest mitochondrial Ca2+ uptake was detected in subsarcolemmal mitochondria, which gradually declined in IFM and was the smallest in perinuclear mitochondria. Furthermore, while Mfn-2 KO had no effect on ISO-induced [Ca2+]m levels in SSM and IFM mitochondrial regions (Figures 3E,F), mitochondrial Ca2+ uptake actually increased in perinuclear mitochondria (Figure 3G) in cardiomyocytes from Mfn2 KO animals.
These data suggest that tethering between SR and mitochondria is heterogeneous within ventricular myocytes to support the privileged SR-mitochondrial communication between IR3 receptors and mRyR1. When this communication was lost in Mfn-2 KO mice, perinuclear mitochondria picked up significantly more Ca2+ upon ISO stimulation. This increase in mitochondrial Ca2+ during ISO stimulation in PNM regions in Mfn-2 KO mice was also associated with the highest ATP levels in Mfn-2 deficient myocytes (see Figure 5). This difference between ET-1 and ISO responses in WT and Mfn-2 KO mice could explain the existent controversy in the field [(Filadi et al., 2015) vs. (de Brito and Scorrano, 2008) and (Naon et al., 2016)] on the role of Mfn2-mediated tethers in SR-mitochondrial Ca2+ communication.
Conclusions
Our study demonstrates that in ventricular myocytes, basal ET-1 signaling is required for maintaining normal cardiac function and bioenergetics (Figure 6). We have shown that when the physical linkage between SR and mitochondria Mfn-2 is disrupted, the SR/mitochondrial metabolic feedback mechanism is severely impaired resulting in the inability of the IP3-mediated SR Ca2+ release to induce ATP production. These findings could explain the impaired cardiac function observed in aged mice and young mice following TAC (Goh et al., 2016) where Mfn2 levels were decreased leading to an impaired metabolic IP3-mediated SR/mitochondrial feedback.
Ethics Statement
This study was carried out in accordance with the recommendations of the National Institutes of Health (NIH Publication NO. 85-23, revised 1996). The protocol was approved by the University of Würzburg Institutional Animal Care and Use Committee.
Author Contributions
LS and ED designed the study and wrote the manuscript. LS, CMag, AB, and MKa performed the experiments. LS, CMag, MKa, and ED analyzed the data. MS and GD provided Mfn 2 KO mice. PE-N and PA-L supported equipment. ED, LS, BG, CMaa, and MKo edited the manuscript. BG and SF provided financial support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00733/full#supplementary-material
Glossary
Abbreviations
- ΔΨm
Mitochondrial membrane potential
- ACE
Angiotensin converting enzyme
- Ang II
Angiotensin II
- Ca2+
Calcium
- [Ca2+]i
Cytosolic calcium concentration
- [Ca2+]m
Mitochondrial calcium concentration
- CHF
Congestive heart failure
- ECC
Excitation-contraction coupling
- ET-1
Endothelin-1
- FAD
Flavin adenine dinucleotide
- IFM
Interfibrillar mitochondria
- IMM
Inner mitochondrial membrane
- IP3
Inositol 1,4,5-trisphosphate
- IP3R
Inositol 1,4,5-trisphosphate receptor
- ISO
Isoproterenol
- KO
Knockout
- MCU
Mitochondrial Ca2+ uniporter
- Mfn2
Mitofusin 2
- mRyR1
Mitochondrial ryanodine receptor type 1
- NADH
Reduced nicotinamide adenine dinucleotide
- PNM
Perinuclear mitochondria
- RyR1
Ryanodine receptor type 1
- SR
Sarcoplasmic reticulum
- SSM
Subsarcolemmal mitochondria
- VDAC1
Voltage-dependent anion channel 1
- VDAC2
Voltage-dependent anion channel 2
- WT
Wild-type.
Footnotes
Funding. This project was supported by the German Heart Research Foundation award number F/40/15 (to LS) and by the American Heart Association Grant-In-Aid 15GRNT25090220 (to ED).
References
- Balaban R. S. (2009). The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 1787, 1334–1341. 10.1016/j.bbabio.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G., Sharma V. K., Lin L., Ryu S.-Y., Dirksen R. T., Sheu S.-S. (2005). Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim. Biophys. Acta 1717, 1–10. 10.1016/j.bbamem.2005.09.016 [DOI] [PubMed] [Google Scholar]
- Brandes R., Bers D. M. (1997). Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ. Res. 80, 82–87. 10.1161/01.RES.80.1.82, PMID: [DOI] [PubMed] [Google Scholar]
- Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. 10.1016/j.cell.2010.06.007, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Csordás G., Jowdy C., Schneider T. G., Csordás N., Wang W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 111, 863–875. 10.1161/CIRCRESAHA.112.266585, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Y., Dorn G. W. (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. 10.1161/CIRCRESAHA.111.258723, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikando A. C., Kettlewell S., Williams G. S., Smith G., Lederer W. J. (2011). Ca2+ dynamics in the mitochondria–state of the art. J. Mol. Cell. Cardiol. 51, 627–631. 10.1016/j.yjmcc.2011.08.003, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 101, 15927–15932. 10.1073/pnas.0407043101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. 10.1083/jcb.200604016, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito O. M., Scorrano L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. 10.1038/nature07534, PMID: [DOI] [PubMed] [Google Scholar]
- De Stefani D., Bononi A., Romagnoli A., Messina A., De Pinto V., Pinton P., et al. (2012). VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 19, 267–273. 10.1038/cdd.2011.92, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova E. N., Blatter L. A. (2008). Mitochondrial Ca2+ and the heart. Cell Calcium 44, 77–91. 10.1016/j.ceca.2007.11.002, PMID: [DOI] [PubMed] [Google Scholar]
- Dedkova E. N., Blatter L. A. (2012). Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 52, 48–61. 10.1016/j.yjmcc.2011.08.030, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova E. N., Seidlmayer L. K., Blatter L. A. (2013). Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J. Mol. Cell. Cardiol. 59, 41–54. 10.1016/j.yjmcc.2013.01.016, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G. W., Maack C. (2013). SR and mitochondria: calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 55, 42–49. 10.1016/j.yjmcc.2012.07.015, PMID: [DOI] [PubMed] [Google Scholar]
- Fawcett D. W., McNutt N. S. (1969). The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J. Cell Biol. 42, 1–45. 10.1083/jcb.42.1.1, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R., Greotti E., Turacchio G., Luini A., Pozzan T., Pizzo P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 112, E2174–E2181. 10.1073/pnas.1504880112, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gincel D., Zaid H., Shoshan-Barmatz V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155. 10.1042/0264-6021:3580147, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh K. Y., Qu J., Hong H., Liu T., Dell’Italia L. J., Wu Y., et al. (2016). Impaired mitochondrial network excitability in failing Guinea-pig cardiomyocytes. Cardiovasc. Res. 109, 79–89. 10.1093/cvr/cvv230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine A. V., Molosh A. I., Poputnikov D., Bulhak A., Sjöquist P.-O., Pernow J. (2005). Endothelin-1 exerts a preconditioning-like cardioprotective effect against ischaemia/reperfusion injury via the ET(A) receptor and the mitochondrial K(ATP) channel in the rat in vivo. Br. J. Pharmacol. 144, 331–337. 10.1038/sj.bjp.0706050, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M., Fukuda K., Hisaka Y., Kimura K., Kawaguchi H., Fujita J., et al. (2004). Endothelin-1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J. Clin. Invest. 113, 876–884. 10.1172/JCI200419480, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai-Kanai E., Hasegawa K. (2004). Intracellular signaling pathways for norepinephrine- and endothelin-1-mediated regulation of myocardial cell apoptosis. Mol. Cell. Biochem. 259, 163–168. 10.1023/B:MCBI.0000021368.80389.b9, PMID: [DOI] [PubMed] [Google Scholar]
- Jakob R., Beutner G., Sharma V. K., Duan Y., Gross R. A., Hurst S., et al. (2014). Molecular and functional identification of a mitochondrial ryanodine receptor in neurons. Neurosci. Lett. 575, 7–12. 10.1016/j.neulet.2014.05.026, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma Y., Miyauchi T., Yuki K., Murakoshi N., Goto K., Yamaguchi I. (2001). Novel molecular mechanism of increased myocardial endothelin-1 expression in the failing heart involving the transcriptional factor hypoxia-inducible factor-1alpha induced for impaired myocardial energy metabolism. Circulation 103, 2387–2394. 10.1161/01.CIR.103.19.2387, PMID: [DOI] [PubMed] [Google Scholar]
- Kohlhaas M., Nickel A. G., Bergem S., Casadei B., Laufs U., Maack C. (2017). Endogenous nitric oxide formation in cardiac myocytes does not control respiration during β-adrenergic stimulation. J. Physiol. 595, 3781–3798. 10.1113/JP273750, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y., Kurihara H., Maemura K., Kuwaki T., Kumada M., Yazaki Y. (1995a). Impaired development of the thyroid and thymus in endothelin-1 knockout mice. J. Cardiovasc. Pharmacol. 26(Suppl. 3), S13–S16. [PubMed] [Google Scholar]
- Kurihara Y., Kurihara H., Oda H., Maemura K., Nagai R., Ishikawa T., et al. (1995b). Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Invest. 96, 293–300. 10.1172/JCI118033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y., Kurihara H., Suzuki H., Kodama T., Maemura K., Nagai R., et al. (1994). Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature 368, 703–710. 10.1038/368703a0, PMID: [DOI] [PubMed] [Google Scholar]
- Kwong J. Q., Lu X., Correll R. N., Schwanekamp J. A., Vagnozzi R. J., Sargent M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. 10.1016/j.celrep.2015.06.002, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones D. M., Larson M. G., Leip E. P., Beiser A., D’Agostino R. B., Kannel W. B., et al. (2002). Lifetime risk for developing congestive heart failure: the Framingham heart study. Circulation 106, 3068–3072. 10.1161/01.CIR.0000039105.49749.6F, PMID: [DOI] [PubMed] [Google Scholar]
- Lukyanenko V., Chikando A., Lederer W. J. (2009). Mitochondria in cardiomyocyte Ca2+ signaling. Int. J. Biochem. Cell Biol. 41, 1957–1971. 10.1016/j.biocel.2009.03.011, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlib M. A., Rouslin W., Kraft G., Berner P., Schwartz A. (1978). On the existence of two populations of mitochondria in a single organ. Respiration, calcium transport and enzyme activities. Biochem. Biophys. Res. Commun. 84, 482–488. 10.1016/0006-291X(78)90194-8, PMID: [DOI] [PubMed] [Google Scholar]
- McMillin-Wood J., Wolkowicz P. E., Chu A., Tate C. A., Goldstein M. A., Entman M. L. (1980). Calcium uptake by two preparations of mitochondria from heart. Biochim. Biophys. Acta 591, 251–265. [DOI] [PubMed] [Google Scholar]
- Müller W. (1976). Subsarcolemmal mitochondria and capillarization of soleus muscle fibers in young rats subjected to an endurance training. A morphometric study of semithin sections. Cell Tissue Res. 174, 367–389. 10.1007/BF00220682, PMID: [DOI] [PubMed] [Google Scholar]
- Naon D., Zaninello M., Giacomello M., Varanita T., Grespi F., Lakshminaranayan S., et al. (2016). Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 113, 11249–11254. 10.1073/pnas.1606786113, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen M. S., Brutsaert D., Dickstein K., Drexler H., Follath F., Harjola V.-P., et al. (2006). EuroHeart failure survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur. Heart J. 27, 2725–2736. 10.1093/eurheartj/ehl193, PMID: [DOI] [PubMed] [Google Scholar]
- Palmer J. W., Tandler B., Hoppel C. L. (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739. PMID: [PubMed] [Google Scholar]
- Pan X., Liu J., Nguyen T., Liu C., Sun J., Teng Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. 10.1038/ncb2868, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard P., Smith P. J., Monge J. C., Rouleau J. L., Nguyen Q. T., Calderone A., et al. (1998). Coordinated upregulation of the cardiac endothelin system in a rat model of heart failure. J. Cardiovasc. Pharmacol. 31(Suppl. 1), S294–S297. [DOI] [PubMed] [Google Scholar]
- Pieske B., Beyermann B., Breu V., Löffler B. M., Schlotthauer K., Maier L. S., et al. (1999). Functional effects of endothelin and regulation of endothelin receptors in isolated human nonfailing and failing myocardium. Circulation 99, 1802–1809. 10.1161/01.CIR.99.14.1802, PMID: [DOI] [PubMed] [Google Scholar]
- Proven A., Roderick H. L., Conway S. J., Berridge M. J., Horton J. K., Capper S. J., et al. (2006). Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. J. Cell Sci. 119, 3363–3375. 10.1242/jcs.03073, PMID: [DOI] [PubMed] [Google Scholar]
- Rapizzi E., Pinton P., Szabadkai G., Wieckowski M. R., Vandecasteele G., Baird G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624. 10.1083/jcb.200205091, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca M. G., Hoppel C. L. (2013). Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 18, 607–622. 10.1007/s10741-012-9340-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrepfer E., Scorrano L. (2016). Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694. 10.1016/j.molcel.2016.02.022, PMID: [DOI] [PubMed] [Google Scholar]
- Sebastiani M., Giordano C., Nediani C., Travaglini C., Borchi E., Zani M., et al. (2007). Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 50, 1362–1369. 10.1016/j.jacc.2007.06.035 [DOI] [PubMed] [Google Scholar]
- Seidlmayer L. K., Juettner V. V., Kettlewell S., Pavlov E. V., Blatter L. A., Dedkova E. N. (2015). Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 106, 237–248. 10.1093/cvr/cvv097, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidlmayer L. K., Kuhn J., Berbner A., Arias-Loza P., Williams T., Kaspar M., et al. (2016). IP3 mediated SR-mitochondrial crosstalk influences ATP production via mitochondrial Ca2+ uptake through the mRyR1 in cardiac myocytes. Cardiovasc. Res. 112, 491–501. 10.1093/cvr/cvw185, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Horita K., Murakami M., Ogura R. (1984). Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell Tissue Res. 238, 577–582. 10.1007/BF00219874, PMID: [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V., Keinan N., Zaid H. (2008). Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191. 10.1007/s10863-008-9147-9, PMID: [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V., Maldonado E. N., Krelin Y. (2017). VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 1, 11–36. 10.15698/cst2017.10.104, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Miyauchi T. (2001). A novel pharmacological action of ET-1 to prevent the cytotoxicity of doxorubicin in cardiomyocytes. Am. J. Phys. Regul. Integr. Comp. Phys. 280, R1399–R1406. 10.1152/ajpregu.2001.280.5.R1399, PMID: [DOI] [PubMed] [Google Scholar]
- Szabadkai G., Bianchi K., Várnai P., De Stefani D., Wieckowski M. R., Cavagna D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. 10.1083/jcb.200608073, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G., Csordás G., Hantash B. M., Thomas A. P., Hajnóczky G. (2000). Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 275, 15305–15313. 10.1074/jbc.275.20.15305, PMID: [DOI] [PubMed] [Google Scholar]
- Thai P. N., Daugherty D. J., Frederich B. J., Lu X., Deng W., Bers D. M., et al. (2018). Cardiac-specific conditional knockout of the 18-kDa mitochondrial translocator protein protects from pressure overload induced heart failure. Sci. Rep. 8:16213. 10.1038/s41598-018-34451-2, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R., Garnier A., Veksler V., Joubert F. (2011). Bioenergetics of the failing heart. Biochim. Biophys. Acta 1813, 1360–1372. 10.1016/j.bbamcr.2010.09.006 [DOI] [PubMed] [Google Scholar]
- Vignon-Zellweger N., Relle K., Kienlen E., Alter M., Seider P., Sharkovska J., et al. (2011). Endothelin-1 overexpression restores diastolic function in eNOS knockout mice. J. Hypertens. 29, 961–970. 10.1097/HJH.0b013e3283450770, PMID: [DOI] [PubMed] [Google Scholar]
- Wei C. M., Lerman A., Rodeheffer R. J., McGregor C. G., Brandt R. R., Wright S., et al. (1994). Endothelin in human congestive heart failure. Circulation 89, 1580–1586. 10.1161/01.CIR.89.4.1580, PMID: [DOI] [PubMed] [Google Scholar]
- Yang L. L., Gros R., Kabir M. G., Sadi A., Gotlieb A. I., Husain M., et al. (2004). Conditional cardiac overexpression of endothelin-1 induces inflammation and dilated cardiomyopathy in mice. Circulation 109, 255–261. 10.1161/01.CIR.0000105701.98663.D4, PMID: [DOI] [PubMed] [Google Scholar]
- Zhao X.-S., Pan W., Bekeredjian R., Shohet R. V. (2006). Endogenous endothelin-1 is required for cardiomyocyte survival in vivo. Circulation 114, 830–837. 10.1161/CIRCULATIONAHA.105.577288, PMID: [DOI] [PubMed] [Google Scholar]
- Zhou Z., Matlib M. A., Bers D. M. (1998). Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J. Physiol. 507, 379–403. 10.1111/j.1469-7793.1998.379bt.x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.