

Abstract
The human ether-a-go-go-related gene (HERG) K+ channel is of great medical and pharmaceutical relevance. Inherited mutations in hERG result in congenital long QT syndrome, which is associated with a marked increased risk of cardiac arrhythmias and sudden death. hERG K+ channels are also remarkably susceptible to block by a wide range of drugs, which in turn can cause drug-induced long QT syndrome and an increased risk of sudden death. The recent determination of the near atomic resolution structure of the hERG K+ channel, using single particle cryo-EM, provides tremendous insights into the how these channels work. It also suggests the way forward in our quest to understand why they are so promiscuous with respect to drug binding.
hERG K+ channels and cardiac arrhythmias
Cardiac arrhythmias are a significant cause of morbidity and mortality [1]. The vast majority of arrhythmias occur in patients with underlying heart disease. However, just over 20 years ago, it was realised that many prescription drugs on the market, including some antibiotics, anti-histamines and anti-psychotics, could prolong the QT interval on the surface electrocardiogram and increase the risk of arrhythmias in patients with otherwise healthy hearts [2]. It had been appreciated that the rapid component of the delayed rectifier K+ channel, IKr, was the target for class III anti-arrhythmic drugs that also caused prolongation of the QT interval [3]. However, it was not until the discovery in 1995 of the human ether-a-go-go related gene (hERG) [4, 5], which encodes the pore forming subunit of IKr, that it was possible to investigate this problem in molecular detail. HERG K+ channels are voltage-gated K+ channels but they have very unusual kinetics, viz., they have slow activation and deactivation kinetics but much faster inactivation gating kinetics. As a consequence, during a cardiac action potential hERG K+ channels spend most of the time in an inactivated state but poised to quickly reactivate and terminate cardiac repolarization at just the right time [6]. The corollary of this is that drugs that block hERG K+ channels result in delayed cardiac repolarization and a markedly increased risk of cardiac arrhythmias (see Figure 1).
Figure 1: hERG K+ channels: gating and physiology.
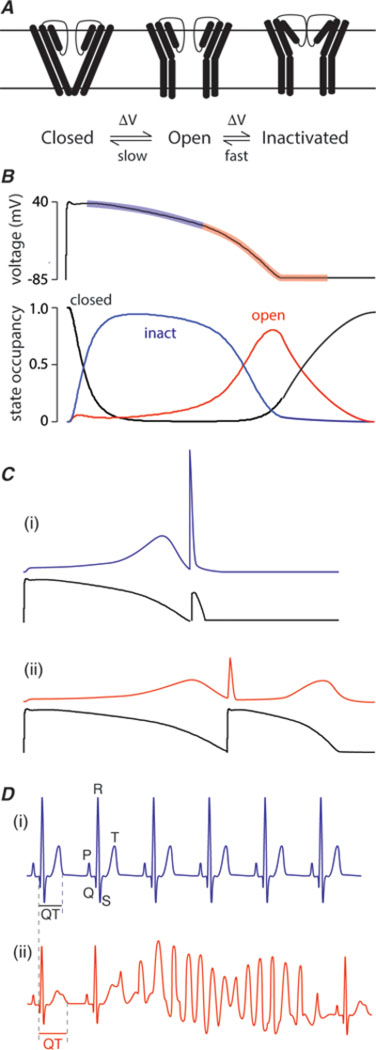
A. Simplified gating scheme for hERG K+ channels. Channels may exist in one of three main groups of states: closed states which are non-conducting, open states, which are conducting, or inactivated states, which are another non-conducting state that channels enter during a prolonged activating stimulus. The gating of hERG K+ channels is unusual in that (i) the kinetics of activation and deactivation are much slower than the kinetics of inactivation and recovery from inactivation. For example, at 0 mV, the time constant for activation is ~100 ms whereas the time constant for inactivation is ~2ms [47]. This is in marked contrast to the vast majority of voltage-gated ion channels where activation/deactivation are much more rapid than inactivation [48]. The second important feature of hERG K+ channel gating is that transitions between the open and inactivated states are voltage dependent.
B. As a consequence of their unusual gating kinetics, during the plateau phase of the cardiac myocyte action potential hERG K+ channels reside predominantly in the inactivated state (blue transparent region on action potential trace highlights the period when hERG K+ channels are predominantly inactivated). As the channels passing inward currents start to inactivate the membrane potential slowly starts to repolarize and this allows hERG K+ channels to recover from inactivation. The more the hERG K+ channels recover from inactivation the more outward current they pass and the more rapidly the membrane potential repolarizes (red transparent region on action potential trace). After the membrane potential has recovered to resting levels it still takes 200–300 ms for all the hERG K+ channels to return to the closed state.
C. (i) As a consequence of the slow deactivation of hERG K+ channels after the membrane potential has returned to the resting level, a premature stimulus (such as can occur with an ectopic beat) will result in a large “spike” of outward current through the still open hERG K+ channels, which then rapidly inactivate[26]. (ii) In patients with reduced hERG K+ channel activity, e.g. due to drug block, the reduced hERG K+ current results in a longer action potential as well as lower current response to premature beats [25].
D. (i) The surface electrocardiogram represents the summed activity of all the cells in the heart with the major deflections being the P-wave (represents atrial depolarization), QRS complex (represents ventricular depolarization) and the T-wave (represents ventricular repolarization). The duration of the interval from the start of the QRS complex to the end of the T-wave (QT interval) is usually ~400ms (at a heart rate of 60 beats per minute). (ii) Patients with reduced hERG K+ channel activity have prolonged QT intervals on their surface electrocardiogram and an increased risk of developing ventricular arrhythmias initiated by ectopic beats. In particular, they are prone to develop a particular arrhythmia called “torsades-de-pointes”.
The discovery of the potentially lethal consequences of inadvertent hERG drug block led to a major shakeup in the regulation of the drug approval process [7]. Twelve drugs (out of 1453 drugs that have ever been brought to market) were withdrawn from the market or had their use severely curtailed due to unacceptably high risk of sudden death [2]. Another 4% of drugs still on the market have been associated with documented torsades-de-pointes arrhythmias and 15% of drugs still on the market can cause QT prolongation (data taken from www.crediblemeds.org). Furthermore, an estimated 60% of drugs in development (in all areas of clinical application) show hERG liability [8]. Whilst regulators and the pharmaceutical industry have focused on reducing the impact of hERG drug block [9], the fundamental question as to why hERG K+ channels are so much more problematic than any other ion channel has puzzled researchers for the last two decades. The recent determination of the near atomic resolution structure of the hERG K+ channel [10], using single particle cryo-EM, represents a major breakthrough in our quest to answer this question.
The first near atomic resolution structure of the hERG K+ channel
Over the past few years, determination of the structure of membrane proteins has been greatly enhanced by spectacular developments in cryo-electron microscopy. Improvements in sample freezing, electron guns, camera technology and computer algorithms running on much faster computers have all contributed to the resolution revolution [11, 12]. In April this year, the Mackinnon lab published the first single particle cryo-EM structure of the hERG K+ channel [10]. To achieve this, they deleted most of the cytoplasmic regions predicted to be unstructured (Δ141–350 in the N-terminus and Δ 871–1005 in the C-terminus, see Figure 2A) but all of the features critical for drug binding were preserved. Furthermore, the baseline construct (which they called hERGT) retained gating characteristics very similar to full-length channels. Therefore one would expect that when the channels were extracted from the cell membrane (and held at 0 mV) that the channels would be in the inactivated state. This baseline structure was determined to a resolution of 3.8Å with the highest resolution regions being in the central region, which fortuitously is where the drug binding cavity is located. In addition to hERGT they determined the structure of another construct that had a larger deletion in the N-terminus, Δ141–380 (which they called hERGTs) to a resolution of 3.7A. Finally, they determined the structure of the S631A mutant in the hERGTs construct. The S631A mutation results in channels that will be mostly open, rather than inactivated, at 0 mV [13].
Figure 2: Overall structure of the hERG K+ channel.
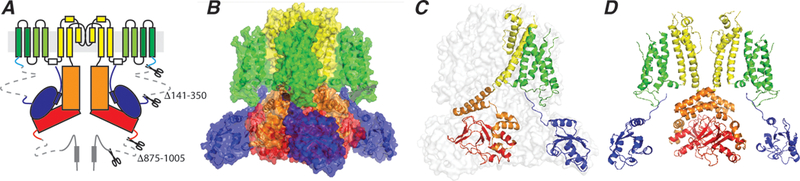
A. Topology of the hERG K+ channel showing two of the four subunits that constitute the tetrameric channel. The full-length monomer is 1159 residues. To generate a biochemically stable channel, residues 141–350 in the N-terminus and residues 875–1005 in the C-terminus were deleted.
B. The channel structure shown in surface representation, colour coded according to the scheme shown in panel A.
C. hERG structure shown in transparent surface representation with one subunit shown in cartoon representation colour coded by domains as shown in panel A. Note that the voltage sensor of each subunit interacts with the pore domain within its own subunit (in contrast to the domain swapped structure in classical voltage-gated ion channels).
D. Two opposing subunits are shown to highlight the open intracellular activation gate and the “domain swapped” architecture within the cytoplasmic C-terminal domains.
Overall the architecture of the hERG K+ channel tetramer looks similar to that of other voltage-gated ion channels (Figure 2B). The pore domain is open at the intracellular end (Figure 2D) and the voltage sensor domains are in the activated or “up” state with the three principal charge-carrying residues (K525, R528 and R531, [14]) all located extracellular to the charge transfer centre [15]. There are however some notable differences to the classical voltage-gated K+ channels. First, the voltage sensors are not domain swapped and thus interact with the pore domain of the same subunit (Figure 2C). Conversely, there is extensive domain swapping between subunits within the cytoplasmic regions of the hERG K+ channel (Figure 2B,D). Second, the structure of the upper region of the pore cavity of hERG contains 4 hydrophobic pouches, which could be important for drug binding.
Structural Insights into hERG K+ channel function
Activation gating
The structure of the hERG K+ channel is very similar to the structure of the EAG1 channel [16]. The major difference in the structure of these two closely related channels is that the EAG1 channel was captured with the intracellular activation gate in a closed conformation (due to the presence of calmodulin, [16]) whereas in hERG the activation gate is in an open conformation. An overlap of the pore structures of hERG and EAG1 shows that the two structures start to deviate at a glycine-gating hinge (G648 in hERG, G460 in EAG1), which is in a very similar location to the gating hinge observed in other K+ channels [17]. In this respect it looks as though activation gating in the ether-a-go-go family of channels is similar to that seen in other K+ channels [17].
The non-domain swapped architecture of the voltage sensor domains seen in the hERG and EAG1 channel suggests that the mechanism by which movements in the voltage sensor in these channels are transduced to opening of the activation gate must be quite different to what is seen in the classical voltage-gated K+ channels [18]. The recent finding that cutting the S4S5 linker in hERG or EAG1 (i.e., co-expressing separate N-terminal and C-terminal halves of the protein) does not significantly perturb activation gating kinetics [19] also supports the idea that electromechanical coupling in these channels must be different to that in classical voltage-gated K+ channels. However, before we can start to understand how electromechanical coupling is achieved in the hERG K+ channel, it will be necessary to determine structures of the hERG channel with the voltage sensors in both the “up” and “down” conformations.
In contrast to the lack of domain swapping in the VSD region of hERG, the subunits undergo domain swapping within the cytoplasmic regions. This likely contributes to fine-tuning the gating of the channels and stabilising the tetrameric assembly [20, 21] (see Figure 2D). An important feature of the hERG channel structure is the location of the N-terminal tail, an important contributor to the slow deactivation kinetics [22–24]. The tip of the tail is located close to the bottom of the VSD. This suggests that there may be an interaction between the N-tail and the VSD in its activated state, which would help to stabilise the activated conformation and so contribute to the slow deactivation kinetics that are so critical to how hERG K+ channels respond to premature beats [25, 26].
Inactivation gating
Inactivation gating in hERG K+ channels involves rearrangements of the extracellular entrance to the pore domain [13, 26–28]. It is also worth noting that this region, which contains the selectivity filter, has a number of subtle sequence differences compared to that seen in voltage-gated K+ channels. For example, in KCNA2, the selectivity filter sequence is 373-TTVGYGDMV-381 whereas in hERG it is 623-TSVGFGNVS-631 (sequence differences are underlined). Most notably for the purposes of this review, the aromatic residue in the middle of the selectivity filter is a phenylalanine in hERG rather than a tyrosine in most other K+ channels. Mutation of the Ser residue at the end of selectivity filter sequence in hERG to alanine results in a channel that is largely open at 0 mV whereas wild type hERG channels are largely inactivated at 0 mV [10, 13]. In addition to the structure of a WT-like channel, Wang and Mackinnon also determined the structure of a S631A-hERG construct. They noted a small but clear difference in the orientation of the phenylalanine side chain in the selectivity filter between the baseline and S631A channels. Whilst such a subtle change would be consistent with the rapidity of the kinetics of inactivation in hERG channels, it is not consistent with the more marked structural changes seen in the selectivity filter of other channels that have been captured in conducting and non-conducting states [29]. Furthermore, there are no clear changes in the extracellular S5P domains between the baseline and S631A constructs, which is surprising given the importance of this domain for channel inactivation [30, 31]. Perhaps inactivation in hERG channels involves much more subtle rearrangements than previously imagined [28]. An alternative explanation for why the structures of the baseline and S631A structures are so similar is that the S631A mutant and baseline constructs, when they are resuspended in lipid-detergent micelles, adopt subtle variations of the same conformational state. Given the significance of the open and inactivated states for drug binding it is going to be important to clarify the structures of the open and inactivated states, which we suggest will require analysis of additional mutants that stabilize the open or inactivated state of hERG and ideally structures determined at higher resolution.
Structural insights into the pharmacology of hERG K+ channels
Over the last 20 years considerable progress has been made in understanding the basic features of drug binding to hERG K+ channels. (i) Drugs bind in the central cavity of the pore region of hERG [32] and the channels need to open before this can occur [33] (ii) Two pore lining aromatic residues (Tyr652 and Phe656) are the most critical residues for drug binding [32]. It is important to remember that hERG channels are tetrameric and so there are a total of 8 aromatic residues (2 from each subunit) lining the pore. There are also three residues at the bottom of the pore helix, Thr623, Ser624 and Val625, which contribute to binding (at least for some drugs [32]) and mutations to Phe557 on the S5 helix can also affect the binding of some drugs [34]. (iii) The pore cavity of the hERG K+ channel is large enough to trap drugs at least as large as MK499, which is ~20 Å by ~7Å [35]. (iv) The majority of high affinity drugs, which includes all the most problematic drugs, preferentially bind to the inactivated state rather than the open state, with the affinity of drugs for the inactivated versus open state varying by between 2–3 fold up to 100 fold [36].
Drug binding pocket
There are several notable findings in the hERG structure that relate to the presumed drug-binding pocket. Note we say presumed, simply because we have not yet seen a drug bound. First, the sidechains of the residues most critical for drug binding, F656 and Y652, as well as V625, S624 and T623, all point towards a putative site located just below the selectivity filter (see Figure 3). Furthermore, the sidechain of F557 sits in between Y652 and F656 of the same subunit. Second, the central cavity of the channel in the region just below the selectivity filter is slightly narrower than that seen in the shaker-like voltage-gated K+ channel structure. As a consequence, there is a greater negative electrostatic potential in this region of the cavity, i.e., the fields originating from the pore helix dipoles [37] are less shielded by water and thus “concentrated” in this smaller volume. The most unexpected finding in the study is the presence of cylindrical hydrophobic pouches extending out from the central pore cavity (see Figure 3). The authors suggest that the size of these pouches (diameter ~8Å, length ~11Å, based on atom centre-centre distances) may be large enough, if one also allows for protein flexibility, to accommodate a substituted aromatic ring, such as those found on most high affinity drugs. It is worth noting that many previous studies that have attempted to simulate drug docking in hERG K+ channels (see e.g. [38–40]). Unsurprisingly, none of these included the side cavities, simply because the hERG homology models were based on other K+ channels that do not possess them. One should, however, bear in mind that it is possible that not all drugs will bind in the hydrophobic pockets, but rather some may still be accommodated within the central cavity.
Figure 3: Key features of the drug-binding cavity in the hERG K+ channel pore.
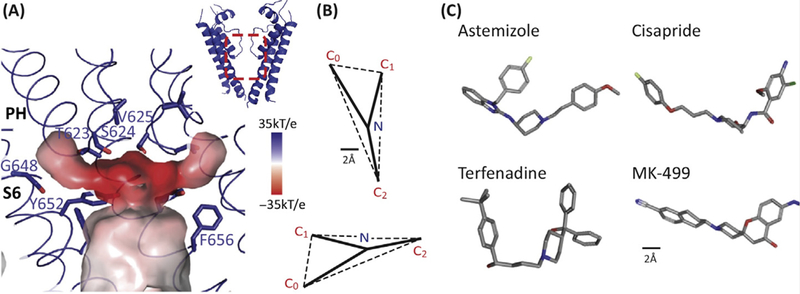
A. The central cavity of the pore domain was generated using Hollow (http://hollow.sourceforge.net). The colour scheme represents the electrostatic potential with the darker the red the more negative the electrostatic potential. There are four hydrophobic pouches (one per subunit) extending from the central cavity. Whilst the central cavity has a smaller diameter than that seen for the open conformation of other voltage-gated K+ channels, the hydrophobic pouches would create a much larger drug-binding cavity. Figure reproduced with permission from Figure 5 of [10].
B. Pharmacophore for hERG drug binding adapted with permission from [41]. The size bar is 2Å with the pharmacophore scaled to match the dimensions of the structure shown in panel A.
C. 3D conformers for 4 potent hERG K+ channel blockers taken from PubChem (https://pubchem.ncbi.nlm.nih.gov/): Astemizole (CID: 2247), Cisapride (CID: 2769), Terfenadine (CID: 5405) and MK-499 (CID: 9934294).
Quantitative structure-activity relationship (QSAR) analysis of drugs that inhibit hERG led to the identification of a pharmacophore for drugs that bind to hERG [41]. The pharmacophore consists of three centers of mass (usually aromatic rings) and an amino group (usually charged at physiological pH) that together form a flattened tetrahedron (Figure 3B,C). One or more of the aromatic side chains could fit into one or more of the hydrophobic pouches. The negative electrostatic potential in the centre of the pore cavity could explain why so many drugs that bind to hERG contain a positive charge. However, one needs to bear in mind that the smaller cavity would also disfavour charged drug binding due to the greater Born dehydration energy cost [37]. It has also been suggested that the charged moiety can undergo cation-pi interactions and the aromatic rings can be involved in pi-pi stacking or hydrophobic interactions with Tyr652 and Phe656 on hERG [42, 43]. Both of these observations are plausible given the close proximity of the Tyr652 and Phe656 side-chains in the recently determined structure of the hERG channel.
State dependence of drug binding
HERG and EAG1 channels have the same pore lining aromatic residues yet EAG1 is orders of magnitude less sensitive to inhibition by most of the drugs that block hERG [42]. One key difference between hERG and EAG1 is that EAG1 channels undergo minimal inactivation (at most 5% at potentials > 0 mV, [44]) whereas hERG channels undergo rapid and complete inactivation (>95% inactivated at potentials >0 mV). The contribution of inactivation to high affinity binding is further substantiated by the observation that it is possible to generate inactivating hEAG-hERG chimaeras (composed mostly of EAG1 but containing the upper half of the pore domain of hERG) that bind drugs with almost hERG like affinity [27]. In the structures reported by Wang and Mackinnon there are only very subtle differences in the structures of the baseline (presumed to be in the inactivated state) and S631A (presumed to be in the open state) and it is difficult to see how these subtle differences could explain the state-dependent changes in drug affinity. One possibility is that, if the hydrophobic pouches are important for drug binding in the inactivated state but not in the open state, then even subtle differences in the size of the entrances to these pouches could explain changes in affinity. Unfortunately, the resolution of the structures is not sufficient to address this question.
The major difference in the structures of the hERG and EAG1 channels is that the EAG1 channels have a closed intracellular gate whereas in hERG this gate is open. Whilst the positions of the sidechains of the top four residues lining the drug binding pocket (T623, S624, V625 and Y652 in hERG) are in very similar locations in the hERG and EAG1 structures, the location of the Phe656 sidechain is very different. Some but not all drugs that bind to hERG appear to be able to bind to the closed state (i.e. they are trapped behind the closed activation gate). If the closed conformation of hERG is similar to that of EAG1 then this would suggest that binding to Phe656 may be an important determinant of whether drugs dissociate from the closed state or remain bound within the cavity in the closed state.
Where exactly do drugs bind?
Based on the findings discussed above, a plausible mechanism for the promiscuity of drug binding is starting to emerge. Still, this hypothesis will ultimately need to be substantiated once a hERG structure is obtained with a drug bound within this pocket. The authors suggest two factors that might have prevented them identifying bound drugs in the structures they determined. First, even in the absence of a drug there is a density present in the hERG pore cavity (presumably a potassium ion) and at the resolution they can obtain using cryo-EM it is not possible to clearly differentiate the drug from other ion densities. This might be a consequence of the strong negative electrostatic potential at the centre of the presumed drug-binding cavity. Second, and more importantly, the tetrameric symmetry of the channel means that an asymmetric drug will be able to bind to one of four identical binding sites, with each one occupied ~1/4 of the time, and this will result in a reduced density that cannot be seen above the noise. The determination of binding of an asymmetric drug in the symmetric cavity of hERG will be a challenge. It is likely that molecular dynamics (MD) simulations will be needed to investigate how drugs bind to the channels. MD simulations performed with fully-atomistic models provide a cogent way to observe, describe and quantify drug binding (see e.g. [45]), allowing identification of the possible sites, poses, interactions and protein structural responses.
Concluding Remarks
The present hERG structures provide a much-needed reference for decades-worth of data in biophysics and molecular pharmacology and it represents a critical first step towards elucidating the mechanisms of hERG function and drug binding at atomic level. As with any significant advance, the present study generates a number of questions (see the outstanding questions box) that should encourage further research.
(i) Where do the drugs actually bind? The first priority will be to determine structures of drugs in the binding site. Do drugs insert into a single hydrophobic pouch or could they span two pouches? Further, it is likely that different drugs with different structures will have slightly different binding orientations. Ultimately, to get further detailed chemical insights into the nature of drug binding we will need not just structures at much higher resolutions, but MD simulations to add a quantitative dimension to our understanding of drug binding.
(ii) How stable are the hydrophobic pouches and do they change in size/shape between the open and inactivated states. Also, if they do change shape and/or size does this explain the higher affinity for the inactivated states for some drugs [36]. MD simulations would be particularly valuable here as they could also capture any changes in the pouches induced by drug binding in both the putative open and inactivated states.
(iii) If these pouches collapse in the closed sate, which Wang and Mackinnon suggest may be the case given that the pouches are not seen in the closed pore-conformation of the EAG1 channel [16], then how does that affect the binding of drugs that get trapped in the closed state. Again, we need to determine structures of the hERG channel in more conformational states (as for example has been done with the Slo2.2 channel [46], and especially in the closed state). Moreover, MD simulations will also help capture the drug-bound conformations and quantify the state-dependent free energies of binding of drugs responsible for allosteric modulation of the hERG channel.
(iv) To what extent are residues Y652 and F656 as well as T623, S624, V625 and F557 involved in direct drug binding versus sculpting the shape of the hydrophobic pouches that presumably allow drug binding. Again, we need more structures and MD simulations of mutant channels, ideally in the presence of drugs.
Trends Box:
Background to hERG K+ channels and drug-induced cardiac arrhythmias
The first near-atomic level view of the hERG K+ channel solved using single particle cryo-electron microscopy.
Insights into the structural basis of function and drug binding to hERG K+ channels
The presence of hydrophobic pouches sprouting from the central cavity provides a plausible explanation for the promiscuity of drug binding to hERG K+ channels
Outstanding Questions Box
Do drugs insert into the hydrophobic cavity pouches and if so do they insert into a single pouch or could they span two pouches?
How stable are the hydrophobic pouches, do they have a different shape and/or size in different conformational states of the channel?
Elucidation of the chemical basis of drug binding will require additional higher resolution structures, combined with molecular dynamics simulations, to identify exactly where and how drugs bind to hERG K+ channels and why they bind differently to different conformational states of the channel
Acknowlegements:
JIV is a National Health and Medical Research Council Principal Research Fellow (App1116948) and his work is funded by the Australian Resarch Council (ARC, DP150101929), the National Health and Medical Research Council (NHMRC, App1074386), the St Vincent’s Clinic Foundation and an infrastructure grant from the NSW Department of Health. EP is supported by the National Institutes of Health (NIH R01-GM057846) and the Membrane Protein Structural Dynamics Consortium (U54 GM087519). TWA is supported by the NHMRC (App1104259), ARC (DP170101732), NIH (U01–11567710) and the Medical Advances Without Animals Trust.
Footnotes
Conflict of Interest: None
References
- 1.Albert CM and Stevenson WG (2016) The Future of Arrhythmias and Electrophysiology. Circulation 133 (25), 2687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roden DM (2004) Drug-induced prolongation of the QT interval. N Engl J Med 350 (10), 1013–22. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC and Jurkiewicz NK (1990) Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 96 (1), 195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trudeau MC et al. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269 (5220), 92–5. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC et al. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81 (2), 299–307. [DOI] [PubMed] [Google Scholar]
- 6.Vandenberg JI et al. (2012) hERG K(+) channels: structure, function, and clinical significance. Physiol Rev 92 (3), 1393–478. [DOI] [PubMed] [Google Scholar]
- 7.Food and Drug Administration, H. (2005) International Conference on Harmonisation; guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals; availability. Notice. Fed Regist. 70 (202), 61133–4. [PubMed] [Google Scholar]
- 8.Raschi E et al. (2008) The hERG K+ channel: target and antitarget strategies in drug development. Pharmacol Res 57 (3), 181–95. [DOI] [PubMed] [Google Scholar]
- 9.Sager PT et al. (2014) Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 167 (3), 292–300. [DOI] [PubMed] [Google Scholar]
- 10.Wang W and MacKinnon R (2017) Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG. Cell 169 (3), 422–430 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y et al. (2015) A primer to single-particle cryo-electron microscopy. Cell 161 (3), 438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merk A et al. (2016) Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 165 (7), 1698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schonherr R and Heinemann SH (1996) Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol 493 ( Pt 3), 635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M et al. (2004) Gating charges in the activation and inactivation processes of the HERG channel. J Gen Physiol 124 (6), 703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao X et al. (2010) A gating charge transfer center in voltage sensors. Science 328 (5974), 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whicher JR and MacKinnon R (2016) Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 353 (6300), 664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y et al. (2002) The open pore conformation of potassium channels. Nature 417 (6888), 523–6. [DOI] [PubMed] [Google Scholar]
- 18.Toombes GE and Swartz KJ (2016) STRUCTURAL BIOLOGY. Twists and turns in gating ion channels with voltage. Science 353 (6300), 646–7. [DOI] [PubMed] [Google Scholar]
- 19.Lorinczi E et al. (2015) Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat Commun 6, 6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haitin Y et al. (2013) The structural mechanism of KCNH-channel regulation by the eag domain. Nature 501 (7467), 444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng CA et al. (2014) Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J Biol Chem 289 (37), 25822–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muskett FW et al. (2011) Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J Biol Chem 286 (8), 6184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng CA et al. (2011) The N-terminal tail of hERG contains an amphipathic alpha-helix that regulates channel deactivation. PLoS One 6 (1), e16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J et al. (1998) Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. J Gen Physiol 112 (5), 637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Y et al. (2001) Effects of premature stimulation on HERG K(+) channels. J Physiol 537 (Pt 3), 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith PL et al. (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379 (6568), 833–6. [DOI] [PubMed] [Google Scholar]
- 27.Herzberg IM et al. (1998) Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J Physiol 511 ( Pt 1), 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang DT et al. (2011) Mapping the sequence of conformational changes underlying selectivity filter gating in the K(v)11.1 potassium channel. Nat Struct Mol Biol 18 (1), 35–41. [DOI] [PubMed] [Google Scholar]
- 29.Cuello LG et al. (2010) Structural mechanism of C-type inactivation in K(+) channels. Nature 466 (7303), 203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J et al. (2002) Structural and functional role of the extracellular s5-p linker in the HERG potassium channel. J Gen Physiol 120 (5), 723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres AM et al. (2003) Structure of the HERG K+ channel S5P extracellular linker: role of an amphipathic alpha-helix in C-type inactivation. J Biol Chem 278 (43), 42136–48. [DOI] [PubMed] [Google Scholar]
- 32.Mitcheson JS et al. (2000) A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A 97 (22), 12329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiehn J et al. (1996) Molecular physiology and pharmacology of HERG. Single-channel currents and block by dofetilide. Circulation 94 (10), 2572–9. [DOI] [PubMed] [Google Scholar]
- 34.Saxena P et al. (2016) New potential binding determinant for hERG channel inhibitors. Sci Rep 6, 24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitcheson JS et al. (2000) Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol 115 (3), 229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrin MJ et al. (2008) Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels. Mol Pharmacol 74 (5), 1443–52. [DOI] [PubMed] [Google Scholar]
- 37.Roux B and MacKinnon R (1999) The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science 285 (5424), 100–2. [DOI] [PubMed] [Google Scholar]
- 38.Stansfeld PJ et al. (2007) Drug block of the hERG potassium channel: insight from modeling. Proteins 68 (2), 568–80. [DOI] [PubMed] [Google Scholar]
- 39.Masetti M et al. (2008) Modeling the hERG potassium channel in a phospholipid bilayer: Molecular dynamics and drug docking studies. J Comput Chem 29 (5), 795–808. [DOI] [PubMed] [Google Scholar]
- 40.Durdagi S et al. (2012) Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J Chem Inf Model 52 (10), 2760–74. [DOI] [PubMed] [Google Scholar]
- 41.Cavalli A et al. (2002) Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K(+) channel blockers. J Med Chem 45 (18), 3844–53. [DOI] [PubMed] [Google Scholar]
- 42.Chen J et al. (2002) Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A 99 (19), 12461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez D et al. (2004) Physicochemical features of the HERG channel drug binding site. J Biol Chem 279 (11), 10120–7. [DOI] [PubMed] [Google Scholar]
- 44.Garg V et al. (2012) Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule. J Gen Physiol 140 (3), 307–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boiteux C et al. (2014) Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U S A 111 (36), 13057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hite RK and MacKinnon R (2017) Structural Titration of Slo2.2, a Na+-Dependent K+ Channel. Cell 168 (3), 390–399 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandenberg JI et al. (2006) Temperature dependence of human ether-a-go-go-related gene K+ currents. Am J Physiol Cell Physiol 291 (1), C165–75. [DOI] [PubMed] [Google Scholar]
- 48.Yellen G (2002) The voltage-gated potassium channels and their relatives. Nature 419 (6902), 35–42. [DOI] [PubMed] [Google Scholar]