

Abstract
Recent work has shown that subsurface microbial communities assemble by selective survival of surface community members during sediment burial, but it remains unclear to what extent the compositions of the subsurface communities are a product of their founding population at the sediment surface or of the changing geochemical conditions during burial. Here we investigate this question for communities of sulfate-reducing microorganisms (SRMs). We collected marine sediment samples from the upper 3–5 m at four geochemically contrasting sites in the Skagerrak and Baltic Sea and measured SRM abundance (quantitative PCR of dsrB), metabolic activity (radiotracer rate measurements), and community composition (Illumina sequencing of dsrB amplicons). These data showed that SRM abundance, richness, and phylogenetic clustering as determined by the nearest taxon index peaked below the bioturbation zone and above the depth of sulfate depletion. Minimum cell-specific rates of sulfate reduction did not vary substantially between sites. SRM communities at different sites were best distinguished based on their composition of amplicon sequence variants (ASVs), while communities in different geochemical zones were best distinguished based on their composition of SRM families. This demonstrates environmental filtering of SRM communities in sediment while a site-specific fingerprint of the founding community is retained.
Subject terms: Microbial ecology, Biogeochemistry, Microbial communities
Introduction
Subsurface sediment microbial communities are descended from microorganisms from the water column and surface sediment that have undergone a selective filtering as they are buried, forming a subset of the original founding community [1–3]. Coastal marine sediments and porewater undergo changes following burial that define microbial habitats. Bioturbation, the process by which the activity of sediment fauna mixes and irrigates the sediment, allowing molecular oxygen and other water column solutes to penetrate deeper into the sediment than possible by diffusion, is generally restricted to the uppermost sediment layer [4]. Deeper sediment is characterized by changes in organic matter content and composition (increasing fraction of refractory organic compounds), decreasing sulfate concentrations, reduction of metal oxide minerals, and increasing methane concentrations [5]. Sulfate-reducing microorganisms (SRMs) carry out the terminal oxidative step in the remineralization of a substantial proportion of the detrital organic matter deposited in shelf and coastal marine sediments [6]. Like all microbial communities in marine sediments characterized by diffusive transport of solutes, SRM communities are potentially shaped by the founding community at the sediment–water interface and by the changing geochemical environment as they are buried under layers of sediment over time [2, 7]. The question of how SRM communities in the deep subsurface are formed is not only relevant due to their important geochemical role, but SRMs are also amenable to study in a way that provides broader understanding of sediment microbial ecology: they metabolically transform a compound (sulfate) that is easily measured in porewater, they perform a geochemical transformation (sulfate reduction) that is measurable using a radiotracer method [8], and they possess a universal diagnostic molecular marker gene (dsrB) to determine their abundance and community composition [9, 10].
SRMs are a diverse set of bacteria and archaea [9], with the majority of SRMs identified in marine sediment belonging to uncultivated taxa [11]. All SRMs reduce sulfate using the dissimilatory sulfite reductase (DSR) enzyme complex, including the subunit DsrB [12]. High-throughput sequencing of the gene encoding this subunit, dsrB, has been identified as an effective way of observing SRM community composition in sediment [10]. The question we address in this study is to what extent founding SRM communities in the surface sediment determine deep sediment SRM communities, and to what extent these communities are determined by changing geochemical conditions during burial? In other words, how do historical contingency and niche-based selection affect the formation of SRM communities in subsurface sediments during burial? For example, aerotolerance is a trait expected to promote SRM fitness in the uppermost bioturbated sediment layer as a consequence of bioturbation-mediated O2 penetration. In spite of the status of SRMs as anaerobic microbes, aerotolerant SRM communities have been previously described [13, 14], but different sulfate-reducing species appear to differ in their tolerance to molecular oxygen [15–17]. While one would expect all SRMs deposited on the seafloor to be aerotolerant to some extent, the degree of aerotolerance should shape the SRM community composition as O2 disappears below the bioturbation zone. Whether such bioturbation and other depth-dependent environmental factors makes the deep sediment SRM communities unrecognizable compared to the founding surface communities is unknown. Understanding the founding effect of surface sediment microbial communities on subsurface communities will help us understand the long-term impact of surface sediment perturbations and changing water column conditions on organic matter degradation in subsurface sediment.
Attention has been given to the effect of founding microbial communities on later community composition, or “historical contingency”, based on models where species colonize a new environment, and continue to arrive with time [18, 19]. The marine subsurface deviates from this model in that the environment becomes isolated, preventing the arrival of new species after a given sediment layer is buried below the upper zone mixed by faunal activities [1]. For this reason historical contingency in the deep subsurface seems almost inevitable. Indeed, previous studies have identified the effect of conditions at the time of sedimentation on the subsurface microbial community [20–22]. The question becomes not of whether historical contingency occurs, but whether it can be mitigated by environmental filtering, where selection in different subsurface sediments with similar biogeochemical conditions will produce similar SRM community structures even if surface sediment seed communities differ. One would expect that environmental filtering would result in common functional properties across similar geochemical zones of different sites, but functionally similar communities are not necessarily taxonomically similar [23].
Selection can act in different ways, leading to either phylogenetic clustering (selection of taxa in a given community that are more closely taxonomically related than expected by chance) or phylogenetic overdispersion (where niche exclusion leads to taxa being less related than expected by chance) [24]. This distinction can be made using the nearest taxon index (NTI) metric, which compares the distribution of organisms in a phylogenetic tree with a randomly simulated distribution in the same tree [25]. An NTI of >2.0 indicates selection through phylogenetic clustering to a greater degree than expected at random, an NTI between 2.0 and −2.0 indicates stochastic community formation similar to the random simulation, and NTI of <−2.0 indicates phylogenetic overdispersion with a more disperse distribution than random [26, 27]. Studies of microbial communities that have calculated the NTI have both found evidence for phylogenetic clustering and stochastic processes, with published work from a range of environments including in groundwater [26], soil [28–30], geothermal springs [31], permafrost [32], and heterotrophic lab microcosms [33]. This study is, to our knowledge, the first to calculate NTI values for bacteria in marine sediment. This study uses NTI calculations to identify which geochemical zones apply the strongest environmental filtering, and therefore which geochemical properties are driving SRM community assembly.
Selection and subsequent environmental filtering of specific microbes during burial are controlled by their functional traits, but the phylogenetic level at which this selection occurs varies greatly depending on the trait [34]. Selection of more complex traits involving many genes, such as oxygenic photosynthesis or methanogenesis, takes place at the phylum level, while simpler traits such as the utilization of specific carbon compounds are determined at the genus level [35]. This study aims to determine the phylogenetic level at which selection occurs as SRM are buried in sediment, thus informing our understanding of the functional traits necessary for survival in the deep subsurface.
This study is the first to apply an amplicon sequence variant (ASV) approach to analyzing dsrB sequence libraries from environmental samples, as opposed to analyzing reads clustered into operational taxonomic units (OTUs). The ASV approach distinguishes between machine-generated errors and true microbial diversity better than previous tools [36], facilitating analysis of community structures with more accurate strain-level resolution. The ASV approach thus allows us to determine the phylogenetic level at which selection acts with higher accuracy than an OTU approach that would be more likely to view related strains as a single SRM variant [36]. This also allows us to more accurately distinguish between selection and stochastic processes, as phylogenetic clustering could be masked by an OTU approach (for example, species-level dsrB OTUs based on 90% sequence identity [11]).
The present study addresses four sites in the Skagerrak and Baltic Sea. The sediment samples were collected during an accompanying study on bioturbation and biogeochemistry [37]. That study revealed that the sites have very different near-surface sediment properties in terms of bioturbation, manganese and iron reduction, and bottom water O2 content, making it possible to disentangle the effects of founding SRM communities and selection in deeper geochemical zones.
Materials and methods
Sampling and DNA extraction
Sediment samples were collected by gravity corer, haps corer, and Rumohr corer from four sites in the Baltic Sea, Skagerrak, and Lillebælt and preserved for DNA extraction and radiotracer-based measurement of sulfate reduction rates (SRRs). DNA was extracted using bead-beating followed by phenol–chloroform–isoamylalcohol extraction. For complete sampling and DNA extraction methods, please consult the supplemental methods section.
Porewater geochemistry and sulfate reduction rates
Porewater collection with rhizon samplers and sulfate concentrations were measured using ion chromatography, ammonium concentrations were measured using colorimetric absorbance, dissolved inorganic carbon (DIC) was measured using gas chromatography, and SRRs were determined using a radiometric 35S tracer assay. For details see ref. [38] and the supplemental methods section.
Quantitative PCR and dsrB amplicon sequencing
dsrB and the gene encoding bacterial 16S ribosomal RNA (rRNA) were quantified using SYBR-green based quantitative PCR (qPCR) as previously described [39]. PCR amplification and Illumina MiSeq sequencing of an approximately 350 bp fragment of dsrB was carried out using previously designed primer set DSR1762Fmix 1–10 and DSR2107Rmix 1–13 [10]. For full details see the supplemental methods section.
Cellular sulfate reduction rate calculation
SRRs in nmol per cubic centimeter (cc) of sediment per day were converted to mean cellular SRRs (nmol per cell per day) using measured density data (grams per cc) and qPCR dsrB abundance (gene copies per gram sediment). SRR measurements from depths without density and/or qPCR data were converted to cellular measurements using the nearest available depth where a qPCR/density measurement was taken.
Bioinformatics
dsrB sequencing reads were clustered to amplicon sequence variants using dada2 [40] and further analyzed using phyloseq [41]. Classification was carried out using an existing dsrAB database and classification scheme [9]. For full details see the supplemental methods section.
Accessions
Amplicon sequencing data for dsrB generated in this study have been submitted to the NCBI (National Center for Biotechnology Information) Sequence Read Archive (SRA) and is available under BioProject accession number PRJNA485261.
Results
Geochemical zonation
Measurements of sulfate, ammonium, and DIC concentrations (Fig. 1b, c, d), together with a previously published bioturbation study carried out on cores collected from the same sites and expedition as the cores in this study [37], show key differences between the four sites. SKA4, the exposed and shallow Kattegat site, had the highest faunal abundance and greatest degree of bioturbation determined from porewater geochemistry modeling and bromide tracer incubation. The greatest observed depth of bioirrigation at SKA4 was ~30 cm below seafloor (cmbsf) according to reverse modeling of porewater geochemistry. SKA5, the Little Belt site, had no fauna and no detected bioirrigation, as a consequence of anoxic bottom water at this site. The deeper Skagerrak sites SKA1 and SKA2 show intermediate degrees of bioturbation, in between the two extreme cases of SKA4 and SKA5. Bioirrigation was evident to a depth of ~15 cmbsf at SKA1 and to ~10 cmbsf at SKA2. Kristensen et al. [37] also estimated Fe(III) and Mn(IV) reduction rates for the uppermost 20 cm of the sediment, with evidence for Fe(III) reduction at all sites, albeit highest at site SKA4. Mn(IV) reduction rates were highest at SKA2 which had very high concentrations of solid-phase Mn(IV) [42, 43]. Ammonium and DIC concentration profiles show the most organic matter mineralized at SKA5, the lowest at SKA2, with SKA1 and SKA4 showing moderate organic matter mineralization (Fig. 1).
Fig. 1.

Map of sampling sites in this study (a) and porewater geochemistry concentrations of sulfate (b), ammonium (c), and dissolved inorganic carbon (DIC - d)
The depth of the sulfate reduction zone, and of the sulfate-methane transition zone (SMTZ) at its base, is a function of the amount of organic matter electron donor available for SRMs and the presence of other competing electron acceptors [44]. Site SKA5 had the shallowest SMTZ (20 cmbsf, Fig. 1b), a consequence of relatively high organic matter deposition and degradation rates (Fig. 1c, d) and no O2 penetration into the seafloor due to intense oxygen consumption, anoxic bottom water, and absence of bioturbating fauna [37]. The deepest SMTZ was at site SKA2, where it was below the deepest sampling depth (385 cmbsf). Sites SKA1 and SKA4 had similar intermediate SMTZ depths, at 91 and 98 cmbsf, respectively (Fig. 1b).
dsrB abundance and sulfate reduction rates
At the sediment surface, dsrB abundances measured by qPCR ranged from ~107 to 109 copies per gram sediment, with the highest abundance (109 copies g−1) at site SKA5 (shallowest SMTZ of all sites, no bioturbation) and the lowest abundance (107 copies g−1) at site SKA2 (deepest SMTZ, moderate bioturbation, dominated by Mn(IV) reduction). At sites SKA1, SKA2, and SKA4, dsrB abundance decreased with depth by about one order of magnitude from the shallowest to the deepest samples. At site SKA5, this decrease was about 3 orders of magnitude. The ratio of dsrB to 16S ribosomal RNA (rRNA) gene abundance was consistently between 0.1 and 0.4 in the sulfate reduction zone at all sites, dropping below 0.1 in the bioturbation zone and below the SMTZ. At sites with a bioturbation zone (SKA1, SKA2, and SKA4), fractional and absolute dsrB abundances peaked just below the bioturbation zone, and then dropped deeper in the sediment (Fig. 2). At SKA5, where there was no bioturbation in the surface layer, dsrB abundance was highest at the shallowest sampled depth. Trends in SRRs typically followed trends in dsrB abundance, making cellular sulfate respiration rates differ less from site to site than overall SRRs (Fig. 2). A lower rate boundary of approximately 0.1 fmol cell−1 day−1 is evident at deep Skagerrak sites SKA1 and SKA2 in spite of differences in dsrB abundance and SRRs of about an order of magnitude between the two sites. Sites SKA4 and SKA5 have more scattered dsrB abundance and SRR, but still centered around 0.1 fmol cell−1 day−1 (Fig. 2).
Fig. 2.

Quantitative PCR (qPCR) measurements of dsrB abundance in sediment, dsrB abundance as a fraction of 16S rRNA gene abundance, radiotracer measurements of sulfate reduction rates, and mean cellular sulfate reduction rates based on bulk radiotracer measurements divided by dsrB qPCR abundance. Lower limits of bioturbation are taken from the bioturbation study from the same expedition [37]
SRM community composition
Only a minor fraction (<0.1%) of dsrB genes represent the oxidative type of DSR, and the dsrB abundance patterns thus, as expected, indicate the presence of SRM (or sulfite-reducing microorganisms) rather than sulfur-oxidizing microorganisms. SRM communities were dominated by three bacterial families: Desulfobacteraceae, Syntrophobacteraceae, and the Desulfatiglans-anilini-family-level lineage (Fig. 3). The general trend at all sites was a decrease of Syntrophobacteraceae fractional abundance with depth and an increase in the fractional abundance of the Desulfatiglans-anilini-family-level lineage. Desulfobacteraceae made up ~10–75% of the community at all sites and all depths. Only a small fraction of the community could be assigned to cultivated genera of SRMs, with at most 4% of the community belonging to the cultivated genus Desulfosarcina and at most 0.4% of the community belonging to Azospirillum (Supplemental Figure SF1).
Fig. 3.

Community composition of sulfate-reducing microorganisms based on amplicon sequencing of dsrB including calculated richness and nearest taxon index (NTI). NTI >2 indicates selective pressure. Lower limits of bioturbation are taken from the bioturbation study from the same expedition [37]
SRM community richness was in the range of ~200–1200 amplicon sequence variants (ASVs). Rarefaction curves showing the number of ASVs obtained with increasing sequencing depth become flat, suggesting that extra sequencing would not have revealed further ASVs and sequencing depth was appropriate for the diversity of these samples (Supplemental Figure SF2). At sites with a bioturbation zone (SKA1, SKA2, and SKA4), richness gradually increased from the surface down into the bioturbation zone, peaking in the sulfate-reduction zone and falling again with depth (Fig. 3). At SKA5, with no bioturbation, SRM richness was highest at the surface and dropped steadily with depth. These richness trends match the dsrB gene abundance trends described above (Supplemental Figure SF3).
The NTI is a metric of the strength of selection, with NTI >2.0 indicating coexisting taxa more closely related than by chance, i.e,. environmental filtering resulting in phylogenetic clustering [26]. The site with the densest fauna and strongest bioirrigation, SKA4, has NTI <2 from the surface to 53 cmbsf, suggesting reduced selective pressure, i.e., a greater degree of stochastic community assembly, in the upper 53 cmbsf. Bioirrigation was measured at this site down to ~30 cmbsf. SKA5, with no bioturbation, starts from an NTI of ~5 at the surface and then gradually drops with depth. SKA1 and SKA2, the sites with moderate bioturbation, have NTI >2 at all depths apart from the uppermost depth (1 cmbsf).
Ordination plots (Fig. 4) of the SRM communities in each sample show different patterns depending on the ordination method used. Methods based on the fractional abundance of the different ASVs, with phylogenetic distance between the ASVs not taken into account, show a clear separation between sites, and a clear separation between different geochemical zones within each site (Bray–Curtis/principal coordinates analysis (PCoA) and Bray–Curtis/non-metric multidimensional scaling (NMDS)). Methods that take phylogenetic distances into account (weighted Unifrac and double principal coordinates analysis (DPCoA)) show a clearer separation between different geochemical zones than between different sites, with samples from the same geochemical zone at different sites clustering together (Fig. 4). This shows that specific SRM taxa are selected for at each geochemical zone. To determine what phylogenetic level this selection acts on, we calculated ANOSIM R for clusters of ASVs at 1% increments from 50 to 100% nucleotide sequence identities (Fig. 5). ANOSIM (analysis of similarity) is a method of comparing groups of microbial communities. If R = 0.0, then the microbial communities in different groups are indistinguishable from one another, and if R = 1.0, then the groups are completely separate [45]. For the different sites in this study, increasing identity percentages led to increasing ANOSIM R. This means that an increasingly more granular phylogenetic resolution of SRMs allows SRM communities at different sites to be better differentiated from one another, right up to 100% identity (unclustered ASVs). In contrast, ANOSIM R distinguishing SRM communities in the different geochemical zones of the 4 investigated sites peaked for 75% sequence identity clusters before dropping again at higher identities (Fig. 5).
Fig. 4.

Ordination plots of sulfate-reducing microorganism (SRM) communities at different sites and geochemical zones based on dsrB sequencing. The four plots display the same data with four different distance/ordination methods - Bray-Curtis/PCoA (a), Bray-Curtis/NMDS (b), Unifrac/PCoA (c), and DPCoA (d). For Bray–Curtis principal coordinates analysis (PCoA) and non-metric multidimensional scaling (NMDS) plots, samples from each site are surrounded by a black border line (two samples appear to have been mistakenly mislabeled between SKA4 and SKA2)
Fig. 5.
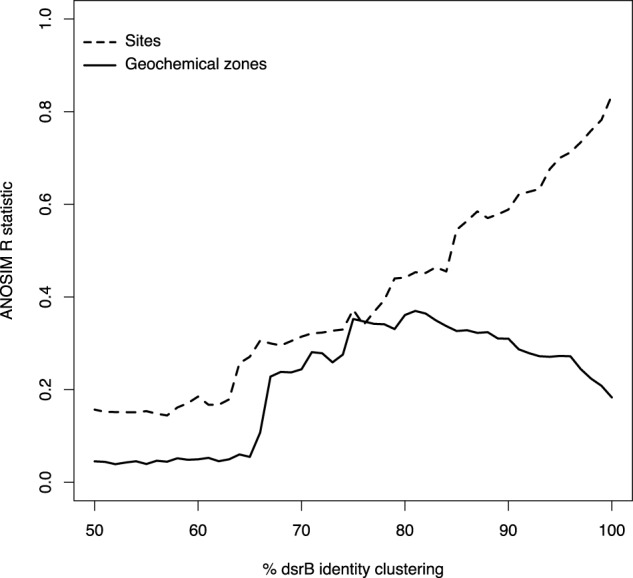
The analysis of similarity (ANOSIM) R statistics to distinguish sites or geochemical zones (bioturbation zone, sulfate zone, sub-sulfate zone). An R value (y axis) of 1.0 means completely distinct groups of communities, while R = 0.0 means completely indistinguishable groups of communities
Discussion
Observations of sediment SRM communities, abundance, and activity at these four contrasting sites provide insights into the factors that shape SRM community assembly in marine sediments.
Microorganisms capable of using a more favorable electron acceptor will typically outcompete those that use a less favorable electron acceptor. This means that in the presence of more favorable electron acceptors than SO42−, like O2, NO3−, Mn(IV) or Fe(III), SRMs are likely to be outcompeted [43]. Such more favorable electron acceptors are available in bioturbated sediment where the burrowing and irrigation activity of sediment-dwelling animals delivers O2 and NO3− from the water column to the sediment and thereby also reoxidizes reduced iron and manganese. Moreover, O2 damages the fraction of the SRM community that is not aerotolerant, further reducing SRM activity and growth. Our data support this model, as at stations SKA1, 2, and 4, the relative and absolute abundance and richness of SRMs and SRRs peaked below the lower limit of bioturbation (Figs. 2 and 3). In contrast, in the absence of bioturbation at station SKA5, these parameters peak at the sediment surface. At SKA2, SRM abundances and activity were about an order of magnitude lower than at the other sites, which could be explained by being the deepest site (at 586 m water depth, compared to the next deepest, SKA1, at 318 m) (Fig. 2). Organic matter is therefore more likely to be degraded by the time it reaches the seafloor, decreasing the energy available to SRMs. Moreover, the higher abundance of Mn(IV)-containing minerals at SKA2 will lead to the increased activity of metal-reducing microorganisms that compete with the SRMs [37, 42, 43]. As expected, SRM abundance and SRR tend to decrease with increasing depth in the sediment, consistent with the decreasing degradability of the remaining organic matter as the deeper and older sediment has been subjected to microbial degradation over longer time. Interestingly, SRM presence and sulfate reduction continue deeper than the main sulfate penetration (Fig. 2). This is consistent with previous observations of marine sediments, and is likely either a result of sulfate reducers switching to a sulfate-free metabolism, most likely a syntrophic metabolism where they produce H2 consumed by a methanogenic partner [46, 47], or a result of the cryptic sulfur cycle whereby SRMs reduce sulfate that is concurrently produced through sulfide oxidation coupled to iron(III) reduction [48]. In spite of the significant variability in SRM abundance and SRR between different sites and depths, it is striking that the mean cell-specific SRR is much less variable, converging at around 0.1 fmol cell−1 day−1. This suggests a narrow physiological range of minimum cellular rates for sulfate reduction, with variability in SRRs controlled by the degradability of the organic matter and subsequent changes in SRM abundance rather than by acclimation or by the observed differences in SRM community composition. Similar results were obtained from measurements in a Greenlandic fjord, where mean cell-specific SRRs converged to a lower limit of around 0.01 fmol cell−1 day−1, albeit using microscopic cell counts to correct for DNA extraction efficiency [49]. If we apply a DNA extraction efficiency of 7% (as calculated in another study comparing qPCR results to cell counts [50]) to this study, then we see a similar lower limit of mean cell-specific rates in both datasets. On the other hand, the peak in cellular SRR in the SMTZ at SKA5 suggests that cellular rates can increase dramatically (by two orders of magnitude) where there is likely anaerobic oxidation of methane.
The SRM community richness reflected SRR through the sediment, with both the highest SRM richness and the highest SRR below the deepest extent of bioturbation but above the deepest extent of porewater sulfate at all sites. Typically, marine sediment acts as a “filter”, with the maximum richness in the shallowest sediment decreasing with depth, as taxa poorly adapted to the deep subsurface are being filtered out as they are buried more deeply in the sediment [1]. In our study, this “typical” richness pattern is only present at SKA5, where there is no bioturbation zone (Fig. 3). At the remaining three sites, richness is highest at the base of the bioturbation zone or between the bioturbation zone and the lowest extent of sulfate. Richness therefore follows the same pattern as SRM abundance and SRR. This can also be seen by the degree of selection at each depth as indicated by the NTI (Fig. 3). The two end-member sites with respect to bioturbation identified by Kristensen et al. [37], SKA4 (highest bioturbation) and SKA5 (no bioturbation), show distinctly different patterns in their NTI with respect to depth, with SKA4 showing less selection of SRM from the sediment surface to 53 cmbsf, while SKA5 shows highest selection levels at the surface, steadily decreasing with depth. SKA1 and SKA2 show a scattered NTI pattern that matches neither of the two extreme scenarios in this study. The collective picture is that SRM activity, abundance, and selection are all highest below the bioturbation zone. By comparing the situation at bioturbation-free SKA5 to bioturbated sites SKA1, SKA2, and SKA4, it appears that bioturbation makes the seafloor at SKA5 resemble the base of the bioturbation zone at the other sites, with (a) competition and inhibition from O2 and other electron acceptors delivered from the water column, and (b) mixing of the sediment allowing new SRM to seed the sediment at the base of the bioturbation zone rather than at the sediment surface. This dramatic effect of the bioturbation zone on microbial community composition has been observed before in Baltic sediments [51], but here the direct impact of bioturbation on SRM community composition, abundance, activity, and selection is directly evident by comparison with a bioturbation-free site.
This selection in the sulfate-reducing zone is also shown by ordination analyses (Fig. 4). Clustering methods that do not take phylogeny into account (Bray–Curtis distances with PCoA or NMDS) showed clearly separated sites, so that while the effect of geochemical zonation on community composition was evident within each site, the SRM communities were different at each site. Changing conditions over time may change the input communities that led to the observed subsurface communities, but the fact that the samples at different depths for a given site cluster together suggests that temporal community variation at the surface of a given site is less than the between-site variation. This observation supports a community assembly model where subsurface community membership is governed by selective survival of populations in the sediment surface community during burial in to the sediment [1]. This model predicts that sites with different surface community membership will also differ in the subsurface community membership. Such differences in surface SRM community may result from differences in limitations of dispersal [52], sediment properties including metal content and bioturbation activity, or climate conditions [21]. The proximity and connectedness of the four sites suggests that dispersal limitation or climatic differences would have a limited impact, leaving the majority of differences to be driven by sediment properties. This matches the ordination plots, with the most similar sites in terms of organic matter degradation rates and bioturbation (SKA1 and SKA4) clustering together in the ordination, and the two more unique sites (SKA2 and SKA5) appearing as outliers (Fig. 4a, b).
While two of the ordination plots show that the surface sediment characteristics drive community composition in deeper sediment, ordination analyses that take into account the phylogeny of the SRMs (weighted Unifrac and DPCoA) showed clearer clustering of geochemical zones than of sites. Moreover, the phylogeny-based PCoA plots (Fig. 4c, d) explained a higher fraction of variation in the data than the Bray–Curtis PCoA (Fig. 4a). In these plots (Fig. 4), a clear clustering of SRM communities in the bioturbation layer and sub-sulfate layer is evident with clusters containing samples from all sites where the relevant layer is present. The sulfate-reducing layer clusters in between the bioturbation and sub-sulfate layers, albeit with some overlap with the neighboring geochemical zones. These ordination plots (Fig. 4c, d) show that there are some environmental factors in each geochemical zone that all sites have in common that shape the SRM community in a common way, but that this effect does not act on individual ASVs (Fig. 4a, b), but rather on higher phylogenetic groupings of ASVs. It is also interesting to note that the weighted Unifrac and DPCoA methods show a young, but anoxic, SKA5 surface sediment with high similarity to old SKA1, 2, and 4 sediments in the sulfate-reducing layer. This matches the pattern observed for SRM abundance and richness: that a bioturbating layer shifts the effective seabed layer deeper into the sediment.
To determine which phylogenetic level the geochemical-zone-based differentiation acts on, we clustered ASVs based on nucleotide sequence identity at different minimum identity cutoffs from 50 to 100%. Using these clustered ASVs, we carried out ANOSIM to see how different the SRM communities in each geochemical zone or site were at each minimum identity. Geochemical zones showed a pattern of ANOSIM R increasing up to about 75% identity, and then dropping again at higher identities (Fig. 5). This shows that geochemical zones could be best differentiated from one other if ASVs were clustered at 75% identity. A dsrB identity of 75% roughly corresponds to family-level SRM division [9]. This 75% peak for distinguishing SRM communities in different geochemical zones is in contrast to differentiation of communities at the four different sites, which peaks at 100% identity (i.e., by comparing the true ASVs). This means that sites are best differentiated at the most granular phylogenetic level possible with the methods used (ASVs), while geochemical zones are best differentiated at the family level. In other words, processes driving differentiation in SRM communities act at the ASV level for sites, but at the family level for geochemical zones. It should be emphasized that this conclusion has been reached based on the analysis of just four sites, and that the inclusion of more sites, especially using more replicates of similar types of site, may change this picture. However, the uniform patterns across the diverse sites analyzed in this study give us reasonable confidence that future studies with more sediment sites will confirm the present findings.
Different processes acting to form SRM communities at sites or geochemical zones can explain the differentiation of sites and geochemical zones at different phylogenetic levels. The processes that form microbial communities in sediment are dispersal, diversification, drift, and selection [1]. With limited motility and long generation times below the bioturbation zone [7], dispersal, diversification, and drift are limited in their influence on microbial communities as they become buried beneath the bioturbated zone, leaving selection as the main factor driving microbial community assembly in the aforementioned “filtering” [1]. Different sites, on the other hand, can be seeded by different communities from the water column and bioturbation layer, making the three aforementioned processes other than selection more likely in the surface sediment than in deeper sediment. While all processes might drive the differentiation of SRM communities at different locations in the seabed, the only process that drives the modification of SRM communities as they are buried into changing geochemical layers is selection. Our data show that this environmental filtering results in the selection of certain families in subsurface sediment. With more processes affecting SRM diversity in the bioturbation layer, there is a larger set of possible founding SRMs, giving each site a more unique signature at the ASV level. This is the same conclusion that another recent study reached [53]. Goldford et al. [53] used an experimental approach to the question of microbial community assembly, where founding communities from different sources were subjected to the same laboratory microcosm conditions. It was found that in time the communities in each microcosm converged to the same family composition, even though each microcosm remained unique in terms of its genus or ASV competition. It is essentially the same experiment that has been carried out naturally with SRMs in Baltic and Skagerrak sediment, only over a much larger scale of time and space.
Identification of the family level as decisive for selection by geochemical zones leaves the question of what physiological differences between families drive this selection. Succession of family composition through each geochemical zone is evident from the classification of dsrB ASVs (Fig. 3): the transition from bioturbation zone through sulfate-reducing zone to sub-sulfate zone is accompanied by a transition in the relative abundances of dominant SRM families from Syntrophobacteraceae to the Desulfatiglans anilini family-level lineage (labeled Desulfatiglans_uncultured_lineage), with Desulfobacteraceae present at all depths. The receding fraction of Syntrophobacteraceae with depth has also been observed in another study of SRM community composition in marine sediment [39]. The physiology of isolates from Syntrophobacteraceae is diverse, with no common features that could hint at selection or counter-selection with depth—the family contains isolates that are both syntrophic and non-syntrophic [54], complete oxidizers [54–57] and incomplete oxidizers [58], thermophiles [55, 58], and mesophiles [56, 57]. Oxygen tolerance in SRMs is another factor that might influence selection, but of the six Syntrophobacteraceae genomes available on the Integrated Microbial Genomes database, we could not identify any genes associated with O2 protection (cytochromes, catalase, superoxide dismutase [59]) that were common to all six. A phylogenetic tree with the Syntrophobacteraceae sequences from the 100 most abundant ASVs and all SRM genome sequences show that Syntrophobacteraceae from this study comprise a sibling cluster to other Syntrophobacteraceae, making it impossible to relate the ASVs in this study to any specific bacteria at more specific phylogenetic levels within Syntrophobacteraceae (Supplemental Figure SF4). It is questionable whether this clade should even be considered part of the Syntrophobacteraceae, but future cultivation-based or metagenomic studies of this clade will hopefully shed more light on their phylogeny and ecophysiology. For the family-level Desulfatiglans anilini lineage, there are only two isolates at this time, Desulfatiglans anilini and Desulfatiglans parachlorophenolica, both of which are capable of using aromatic electron donors, including phenol, benzoate, and related compounds [60]. A recent study on two single-cell amplified Desulfatiglans genomes from Baltic sediment suggests that they also use aromatic organic compounds as electron donors [61]. This may support survival in older, deeper sediment where more labile organic compounds have been exhausted. Among the identified cultivated genera in the dataset, Desulfosarcina (a member of the Desulfobacteraceae) showed higher abundances at SKA5 relative to the other three sites (Supplemental Figure SF1). One key characteristic of Desulfosarcina that differentiates it from other SRM is its ability to form aggregates. Aggregate forming may be selected for under planktonic conditions rather than in sediment, and SKA5 has the best conditions for the growth of SRM under planktonic conditions due to its anoxic bottom water. As such, anoxic bottom water may explain the elevated abundance of Desulfosarcina at SKA5. Apart from this, the exact nature of the functional traits that drive the selection of different SRMs is not possible to determine with the data at hand, but the fact that selection occurs at the family level suggests that there are some family-level properties that help Syntrophobacteraceae thrive in the bioturbation zone and help the Desulfatiglans anilini lineage thrive in the sulfate-reducing and sub-sulfate zone. These adaptations might be related to energy limitation in the marine subsurface [5], the utilization of more refractory organic compounds following the consumption of the most labile organic matter in the shallowest part of the sediment, the ability to switch from a sulfate-reducing lifestyle to a syntrophic metabolism beneath the SMTZ, or aerotolerance.
Conclusion
Comparing trends in sulfate-reduction rates and the abundance and community composition of SRMs at four different sites show how different sediment conditions affect SRM communities and their activity. These contrasts reveal that each site has a distinct signature of unique SRM ASVs, but that on a broader, family-based level the sites are similar, with selection of specific SRM families in each geochemical zone. We therefore conclude that both the founding surface SRM community and the selection by geochemical zonation form the SRM community in sediment, with geochemical zonation acting on a broader level to determine what SRM families are present and the founding surface community determining what specific ASVs are present within each family. It is unclear from this study what physiological characteristics form the SRM family-niche relationships that guide selection during burial in marine sediment, but the knowledge that selection acts on the family level will help guide future research into the traits of SRMs that make them successful in sediment.
Supplementary information
Acknowledgements
We thank all participants and crew in the Aurora SKA cruise, 2014, and Britta Poulsen, Jeanette Johansen, Karina Bomholt Oest and Susanne Nielsen for excellent technical assistance in the lab. The work was supported by the Danish National Research Foundation (grant no DNRF104); the ERC Advanced Grant MICROENERGY [grant no 294200], and the VILLUM Experiment project “FISHing for the ancestors of the eukaryotic cell”. The sampling cruise was supported by the Danish Center for Marine Research.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-019-0387-y) contains supplementary material, which is available to authorized users.
References
- 1.Petro C, Starnawski P, Schramm A, Kjeldsen KU. Microbial community assembly in marine sediments. Aquat Microb Ecol. 2017;79:177–95. doi: 10.3354/ame01826. [DOI] [Google Scholar]
- 2.Starnawski P, Bataillon T, Ettema TJG, Jochum LM, Schreiber L, Chen X, et al. Microbial community assembly and evolution in subseafloor sediment. Proc Natl Acad Sci USA. 2017;114:2940–5. doi: 10.1073/pnas.1614190114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh EA, Kirkpatrick JB, Rutherford SD, Smith DC, Sogin M, D'Hondt S. Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 2016;10:979–89. doi: 10.1038/ismej.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kristensen E, Penha-Lopes G, Delefosse M, Valdemarsen T, Quintana CO, Banta GT. What is bioturbation? The need for a precise definition for fauna in aquatic sciences. Mar Ecol Prog Ser. 2012;446:285–302. doi: 10.3354/meps09506. [DOI] [Google Scholar]
- 5.Jørgensen BB, Marshall IPG. Slow microbial life in the seabed. Annu Rev Mar Sci. 2016;8:311–32. doi: 10.1146/annurev-marine-010814-015535. [DOI] [PubMed] [Google Scholar]
- 6.Jørgensen BB. Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature. 1982;296:643–5. doi: 10.1038/296643a0. [DOI] [Google Scholar]
- 7.Hoehler TM, Jørgensen BB. Microbial life under extreme energy limitation. Nat Rev Microbiol. 2013;11:83–94. doi: 10.1038/nrmicro2939. [DOI] [PubMed] [Google Scholar]
- 8.Jørgensen BB. A comparison of methods for the quantification of bacterial sulfate reduction in coastal marine sediments. Geomicro J. 1978;1:11–27. doi: 10.1080/01490457809377721. [DOI] [Google Scholar]
- 9.Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015;9:1152–65. doi: 10.1038/ismej.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelikan C, Herbold CW, Hausmann B, Müller AL, Pester M, Loy A. Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrAand dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ Microbiol. 2016;18:2994–3009. doi: 10.1111/1462-2920.13139. [DOI] [PubMed] [Google Scholar]
- 11.Vigneron A, Cruaud P, Alsop E, Rezende JR, Head IM, Tsesmetzis N. Beyond the tip of the iceberg; a new view of the diversity of sulfite- and sulfate-reducing microorganisms. ISME J. 2018;12:2096–9. doi: 10.1038/s41396-018-0155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabus Ralf, Venceslau Sofia S., Wöhlbrand Lars, Voordouw Gerrit, Wall Judy D., Pereira Inês A.C. Advances in Microbial Physiology. 2015. A Post-Genomic View of the Ecophysiology, Catabolism and Biotechnological Relevance of Sulphate-Reducing Prokaryotes; pp. 55–321. [DOI] [PubMed] [Google Scholar]
- 13.Canfield DE, Marais Des DJ. Aerobic sulfate reduction in microbial mats. Science. 1991;251:1471–3. doi: 10.1126/science.11538266. [DOI] [PubMed] [Google Scholar]
- 14.Jørgensen BB, Bak F. Pathways and microbiology of thiosulfate transformations and sulfate reduction in a marine sediment (Kattegat, Denmark) Appl Environ Microbiol. 1991;57:847–56. doi: 10.1128/aem.57.3.847-856.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cypionka H, Widdel F, Pfennig N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol Ecol. 1985;31:39–45. doi: 10.1111/j.1574-6968.1985.tb01129.x. [DOI] [Google Scholar]
- 16.Sass H, Cypionka H, Babenzien HD. Vertical distribution of sulfate-reducing bacteria at the oxic-anoxic interface in sediments of the oligotrophic Lake Stechlin. FEMS Microbiol Ecol. 1997;22:245–55. doi: 10.1111/j.1574-6941.1997.tb00377.x. [DOI] [Google Scholar]
- 17.Minz D, Flax JL, Green SJ, Muyzer G, Cohen Y, Wagner M, et al. Diversity of sulfate-reducing bacteria in oxic and anoxic regions of a microbial mat characterized by comparative analysis of dissimilatory sulfite reductase genes. Appl Environ Microbiol. 1999;65:4666–71. doi: 10.1128/aem.65.10.4666-4671.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukami T. Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu Rev Ecol Evol S. 2015;46:1–23. doi: 10.1146/annurev-ecolsys-110411-160340. [DOI] [Google Scholar]
- 19.Vass M, Langenheder S. The legacy of the past: effects of historical processes on microbial metacommunities. Aquat Microb Ecol. 2017;79:13–9. doi: 10.3354/ame01816. [DOI] [Google Scholar]
- 20.Marshall IPG, Karst SM, Nielsen PH, Jørgensen BB. Metagenomes from deep Baltic Sea sediments reveal how past and present environmental conditions determine microbial community composition. Mar Genomics. 2018;37:58–68. doi: 10.1016/j.margen.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Orsi WD, Coolen MJL, Wuchter C, He L, More KD, Irigoien X, et al. Climate oscillations reflected within the microbiome of Arabian Sea sediments. Sci Rep. 2017;7:619. doi: 10.1038/s41598-017-05590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyra C, Sinkko H, Rantanen M, Paulin L, Kotilainen A. Sediment bacterial communities reflect the history of a sea basin. PLoS ONE. 2013;8:e54326. doi: 10.1371/journal.pone.0054326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–7. doi: 10.1126/science.aaf4507. [DOI] [PubMed] [Google Scholar]
- 24.Emerson BC, Gillespie RG. Phylogenetic analysis of community assembly and structure over space and time. Trends Ecol Evol. 2008;23:619–30. doi: 10.1016/j.tree.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Webb CO. Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am Nat. 2000;156:145–55. doi: 10.1086/303378. [DOI] [PubMed] [Google Scholar]
- 26.Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6:1653–64. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Ning D. Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev. 2017;81:e00002–17. doi: 10.1128/MMBR.00002-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen C, Ni Y, Liang W, Wang J, Chu H. Distinct soil bacterial communities along a small-scale elevational gradient in alpine tundra. Front Micro. 2015;6:582. doi: 10.3389/fmicb.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripathi BM, Kim M, Kim Y, Byun E, Yang JW, Ahn J, et al. Variations in bacterial and archaeal communities along depth profiles of Alaskan soil cores. Sci Rep. 2018;8:504. doi: 10.1038/s41598-017-18777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeAngelis KM, Firestone MK. Phylogenetic clustering of soil microbial communities by 16S rRNA but not 16S rRNA genes. Appl Environ Microbiol. 2012;78:2459–61. doi: 10.1128/AEM.07547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyd ES, Hamilton TL, Spear JR, Lavin M, Peters JW. [FeFe]-hydrogenase in Yellowstone National Park: evidence for dispersal limitation and phylogenetic niche conservatism. ISME J. 2010;4:1485–95. doi: 10.1038/ismej.2010.76. [DOI] [PubMed] [Google Scholar]
- 32.Hu W, Zhang Q, Tian T, Li D, Cheng G, Mu J, et al. Relative roles of deterministic and stochastic processes in driving the vertical distribution of bacterial communities in a permafrost core from the Qinghai-Tibet Plateau, China. PLoS ONE. 2015;10:e0145747. doi: 10.1371/journal.pone.0145747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nemergut DR, Knelman JE, Ferrenberg S, Bilinski T, Melbourne B, Jiang L, et al. Decreases in average bacterial community rRNA operon copy number during succession. ISME J. 2016;10:1147–56. doi: 10.1038/ismej.2015.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martiny JBH, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: a phylogenetic perspective. Science. 2015;350:aac9323. doi: 10.1126/science.aac9323. [DOI] [PubMed] [Google Scholar]
- 35.Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013;7:830–8. doi: 10.1038/ismej.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kristensen E, Røy H, Debrabant K, Valdemarsen T. Carbon oxidation and bioirrigation in sediments along a Skagerrak-Kattegat-Belt Sea depth transect. Mar Ecol Prog Ser. 2018;604:33–50. doi: 10.3354/meps12734. [DOI] [Google Scholar]
- 38.Flury S, Røy H, Dale AW, Fossing H, Tóth Z, Spiess V, et al. Controls on subsurface methane fluxes and shallow gas formation in Baltic Sea sediment (Aarhus Bay, Denmark) Geochim Cosmochim Act. 2016;188:297–309. doi: 10.1016/j.gca.2016.05.037. [DOI] [Google Scholar]
- 39.Jochum LM, Chen X, Lever MA, Loy A, Jørgensen BB, Schramm A, et al. Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay. Appl Environ Microbiol. 2017;83:e01547–17. doi: 10.1128/AEM.01547-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canfield DE, Jørgensen BB, Fossing H, Glud R, Gundersen J, Ramsing NB, et al. Pathways of organic carbon oxidation in three continental margin sediments. Mar Geol. 1993;113:27–40. doi: 10.1016/0025-3227(93)90147-N. [DOI] [PubMed] [Google Scholar]
- 43.Canfield DE, Thamdrup B, Hansen JW. The anaerobic degradation of organic matter in Danish coastal sediments: iron reduction, manganese reduction, and sulfate reduction. Geochim Cosmochim Act. 1993;57:3867–83. doi: 10.1016/0016-7037(93)90340-3. [DOI] [PubMed] [Google Scholar]
- 44.Canfield DE, Thamdrup B. Towards a consistent classification scheme for geochemical environments, or, why we wish the term “suboxic” would go away. Geobiology. 2009;7:385–92. doi: 10.1111/j.1472-4669.2009.00214.x. [DOI] [PubMed] [Google Scholar]
- 45.Ramette A. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 2007;62:142–60. doi: 10.1111/j.1574-6941.2007.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plugge CM, Zhang W, Scholten JCM, Stams AJM. Metabolic flexibility of sulfate-reducing bacteria. Front Micro. 2011;2:81. doi: 10.3389/fmicb.2011.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beulig F, Røy H, Glombitza C, Jørgensen BB. Control on rate and pathway of anaerobic organic carbon degradation in the seabed. Proc Natl Acad Sci USA. 2018;115:367–72. doi: 10.1073/pnas.1715789115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holmkvist L, Ferdelman T, Jørgensen B. A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark) Geochim Cosmochim Act. 2011;75:3581–99. doi: 10.1016/j.gca.2011.03.033. [DOI] [Google Scholar]
- 49.Jaussi M. Microbial metabolism in the deep biosphere. PhD dissertation. Aarhus: Aarhus University; 2017.
- 50.Petro C. Assembly and activity of microbial communities in marine sediments. PhD dissertation. Aarhus: Aarhus University; 2018.
- 51.Chen X, Andersen TJ, Morono Y, Inagaki F, Jørgensen BB, Lever MA. Bioturbation as a key driver behind the dominance of Bacteria over Archaea in near-surface sediment. Sci Rep. 2017;7:2400. doi: 10.1038/s41598-017-02295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol. 2012;10:497–1093. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- 53.Goldford JE, Lu N, Bajić D, Estrela S, Tikhonov M, Sanchez-Gorostiaga A, et al. Emergent simplicity in microbial community assembly. Science. 2018;361:469–74. doi: 10.1126/science.aat1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka K, Stackebrandt E, Tohyama S, Eguchi T. Desulfovirga adipica gen. nov., sp. nov., an adipate-degrading, gram-negative, sulfate-reducing bacterium. Int J Syst Evol Microbiol. 2000;50(Pt 2):639–44. doi: 10.1099/00207713-50-2-639. [DOI] [PubMed] [Google Scholar]
- 55.Sievert SM, Kuever J. Desulfacinum hydrothermale sp. nov., a thermophilic, sulfate-reducing bacterium from geothermally heated sediments near Milos Island (Greece) Int J Syst Evol Microbiol. 2000;50(Pt 3):1239–46. doi: 10.1099/00207713-50-3-1239. [DOI] [PubMed] [Google Scholar]
- 56.Davidova IA, Duncan KE, Choi OK, Suflita JM. Desulfoglaeba alkanexedens gen. nov., sp. nov., an n-alkane-degrading, sulfate-reducing bacterium. Int J Syst Evol Microbiol. 2006;56(Pt 12):2737–42. doi: 10.1099/ijs.0.64398-0. [DOI] [PubMed] [Google Scholar]
- 57.Oude Elferink SJ, Maas RN, Harmsen HJ, Stams AJ. Desulforhabdus amnigenus gen. nov. sp. nov., a sulfate reducer isolated from anaerobic granular sludge. Arch Microbiol. 1995;164:119–24. doi: 10.1007/BF02525317. [DOI] [PubMed] [Google Scholar]
- 58.Baena S, Perdomo N, Carvajal C, Díaz C, Patel BKC. Desulfosoma caldarium gen. nov., sp. nov., a thermophilic sulfate-reducing bacterium from a terrestrial hot spring. Int J Syst Evol Microbiol. 2011;61(Pt 4):732–6. doi: 10.1099/ijs.0.020586-0. [DOI] [PubMed] [Google Scholar]
- 59.Cypionka H. Oxygen respiration by desulfovibrio species. Annu Rev Microbiol. 2000;54:827–48. doi: 10.1146/annurev.micro.54.1.827. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki D, Li Z, Cui X, Zhang C, Katayama A. Reclassification of Desulfobacterium anilini as Desulfatiglans anilini comb. nov. within Desulfatiglans gen. nov., and description of a 4-chlorophenol-degrading sulfate-reducing bacterium, Desulfatiglans parachlorophenolica sp. nov. Int J Syst Evol Microbiol. 2014;64(Pt 9):3081–6. doi: 10.1099/ijs.0.064360-0. [DOI] [PubMed] [Google Scholar]
- 61.Jochum LM, Schreiber L, Marshall IPG, Jørgensen BB, Schramm A, Kjeldsen KU. Single-cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans-related Deltaproteobacteria widely distributed in marine sediment. Front Micro. 2018;9:2038. doi: 10.3389/fmicb.2018.02038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.