

Abstract
Reliable in vitro expansion protocols of regulatory T cells (Tregs) are needed for clinical use. We studied the biology of Mauritian Cynomolgus macaque (MCM) Tregs and developed four in vitro Treg expansion protocols for translational studies. Tregs expanded three-thousand-fold when artificial APCs (aAPCs) expressing human CD80, CD58 and CD32 were used throughout the culture. When donor PBMCs were used as the single source of APCs followed by aAPCs, Tregs expanded 2,000-fold. Tregs from all protocols suppressed the proliferation of anti-CD2CD3CD28 bead-stimulated autologous PBMCs albeit with different potencies, varying from 1:2–1:4 Treg:PBMC ratios, up to >1:32. Re-culture of cryopreserved Tregs permitted re-expansion with improved suppressive activity. Occasionally, CD8 contamination was observed and resolved by resorting. Specificity studies showed greater suppression of stimulation by anti-CD2CD3CD28 beads of PBMCs from the same donor used for stimulation during the Treg cultures and of autologous cells than of third party PBMC responders. Similar to humans, the Treg-specific demethylated region (TSDR) within the Foxp3 locus correlated with suppressive activity and expression of Foxp3. Contrary to humans, FoxP3 expression did not correlate with CD45RA or CD127 expression. In summary, we have characterized MCM Tregs and developed four Treg expansion protocols that can be used for pre-clinical applications.
1. Introduction
Regulatory T cells (Tregs) are a subpopulation of CD4+ T cells that play a crucial role in self-tolerance and prevention of autoimmunity1. Tregs are currently in clinical trials for the treatment of graft-versus-host disease (GVHD)2–4, autoimmune diseases5–8 and transplant tolerance9. Limitations of these treatments include the difficulty in collecting Tregs, which are present in the circulation in very low numbers, and the requirement for substantial numbers of Tregs for infusion.
Old world monkeys are extensively used in transplantation research. Baboons are more heavily used for xenotransplantation studies while Cynomolgus or Rhesus macaques are more common in allotransplantation studies. The Mauritius Cynomolgus macaque (Macaca fascicularis) (MCM) is a widely used model in biomedical research and is increasingly being utilized for translational studies10–13 especially as many human reagents cross-react with them. Our group14 and others15–17work with MCM for immunological tolerance studies. The geographical isolation of this colony in the Mauritius Island has naturally limited their genetic diversity and maintained a limited number of MHC haplotypes,18,19 ideal for transplant studies. We aimed to characterize MCM Tregs and develop four Treg expansion protocols to produce an “off-the-shelf” Tregs for pre-clinical applications.
2. Materials and Methods
2.1. PBMC Isolation
Heparinized blood was drawn from male MCM (30–40mL). Diluted (1:2) blood with phosphate buffered saline (PBS) was overlaid on 60% Percoll (Sigma-Aldrich) and centrifuged. The buffy coat was collected, and contaminating red blood cells were lysed.
2.2. Fluorescence Activated Cell Sorting (FACS)
FACS was performed on the Influx Cell Sorter (BD Biosciences). Tregs were stained with CD4 (L200, BD), CD25 (BC96, BioLegend), CD127 (HIL-7R-M21, BD Pharmingen) and CD8 (RPA-T8, BD or BW135/80, Miltenyi). The top 1% of CD25+CD127- population within the CD4+CD8- gate was sorted and collected in sterile heat inactivated fetal bovine serum (FBS) (Gemini).
2.3. Treg Culture
Regulatory T cells (Tregs) will be referred to as CD4+ lymphocytes that express high levels of CD25 and FoxP3 with suppressive capacity. Treg growth medium consisted of 85% RPMI (Life Technologies), 10% heat inactivated FBS (Life Technologies), 1% Penicillin/Streptomycin (Life Technologies), 2% L-glutamine (Life Technologies), 1% MEM non-essential amino acids (Life Technologies) and 1% sodium pyruvate (Corning Cellgro). Interleukin-2 (IL-2) (NIH/PeproTech), anti-CD3 (SP34–2, BD Pharmingen) and rapamycin (Sigma-Aldrich) were added to the culture as per protocol.
Mouse fibroblast cells (L929) transfected with human CD32 (FcR) (to secure the Fc of the soluble anti-CD3 monoclonal antibody for T cell receptor activation), CD58 (LFA3) (used to bind to CD2 for stability) and CD80 (to provide costimulation)20,21 were used as artificial APCs (aAPCs)22. aAPCs were irradiated with 50 Gy and plated 3–24 hours before the addition of Tregs. PBMCs were irradiated with 35 Gy prior to plating. MCM PBMC stimulators were MHC mismatched to the Tregs in culture. Four protocols were developed (Tables 1):
Table 1.
Summary protocol A, B, C and D.
| Protocol A | Day 0 | Day 7 | Day 14 | Day 21 | ||||||
| IL-2 | x | x | x | Freeze | ||||||
| Anti-CD3 | x | x | x | |||||||
| Donor PBMCs | - | x | x | |||||||
| Artificial APCs | x | - | - | |||||||
| Rapamycin | x | - | - | |||||||
| Protocol B | Day 0 | Day 7 | Day 14 | Day 21 | Day 28 | Day 35 | Day 42 | Day 49 | Day 56 | |
| IL-2 | x | x | x | x | x | x | x | x | Freeze | |
| Anti-CD3 | x | x | x | x | x | x | x | x | ||
| Donor PBMCs | - | x | x | - | - | - | - | - | ||
| Artificial APCs | x | - | - | x | x | x | x | x | ||
| Rapamycin | x | - | - | - | - | - | - | - | ||
| Protocol C | Day 0 | Day 7 | Day 14 | Day 21 | Day 26 | |||||
| IL-2 | x | x | x | x | Freeze | |||||
| Anti-CD3 | x | x | x | x | ||||||
| Donor PBMCs | - | - | - | - | ||||||
| Artificial APCs | x | x | x | x | ||||||
| Rapamycin | x | - | - | x | ||||||
| Protocol D | Day 0 | Day 7 | Day 14 | Day 21 | Day 26 | |||||
| IL-2 | x | x | x | x | Freeze | |||||
| Anti-CD3 | x | x | x | x | ||||||
| Donor PBMCs | - | x | x | x | ||||||
| Artificial APCs | x | x | x | x | ||||||
| Rapamycin | x | - | - | x |
Protocol A: Tregs were expanded for 21 days. 200 IU/mL of IL-2 were added to the culture on days 0, 7 and 14. 100 ng/mL of anti-CD3 was added on day 0, followed by 1000 ng/mL on days 7 and 14 and 100 ng/mL of rapamycin on day 0. aAPCs were used as stimulators on day 0, but were subsequently replaced with allogeneic PBMCs (1:1 ratio PBMCs:Tregs) on days 7 and 14. Cells were harvested and cryopreserved on day 21.
Protocol B: The first 21 days mirrored Protocol A. After day 21, Tregs were continued in culture with aAPCs (1:10 ratio aAPCs:Tregs) (no PBMCs) for up to 56 days (depending on cell growth). 200 IU/mL of IL-2 were added at the time of restimulation. Anti-CD3 at a dose of 100 ng/mL was used on day 21 and thereafter.
Protocol C: Cells were cultured with aAPCs (1:10 ratio aAPCs:Tregs). 200 IU/mL of IL-2 and 100 ng/mL of anti-CD3 were added on days 0, 7, 14 and 21 and 100 ng/mL of rapamycin was added to the culture on days 0 and 21. Cells were cryopreserved on day 26. No PBMCs were added to this protocol.
Protocol D: Cells were stimulated with both, allogeneic PBMCs (1:10 ratio PBMCs:Tregs) and aAPCs (1:10 ratio aAPCs:Tregs). IL-2, anti-CD3 and rapamycin were added as in Protocol C.
2.4. Suppression Assays
PBMC responders were plated in triplicate. Tregs were plated with PBMCs starting at 1:1 Treg:PBMC ratio and serially diluted up to 1:32 Tregs:PBMCs. PBMCs were stimulated with anti-CD2CD3CD28 beads (Miltenyi Biotec). 1μCi of thymidine (Perkin, Elmer) was added to each well four days after plating. Cells were harvested (Tomtec) 24 hours after. A Perkin Elmer Plate Reader (1450 MicroBeta Life Sciences) was used to measure the thymidine uptake.
2.5. Cryopreservation
Cells were cryopreserved in 95% FBS (Gemini) and 5% dimethyl sulfoxide (DMSO) (Sigma-Aldrich). Cells were frozen at 10 million cells/mL in a step down cell freezer (CryoMed Freezer by Thermo Scientist) at 1°C/minute and stored in a liquid nitrogen freezer.
2.6. Re-expansion post-cryopreservation
Cells were thawed in a 37°C water bath. Group 1 was plated with media; Group 2 received media and 200 U/mL of IL-2; Group 3 was cultured with media, 200 U/mL of IL-2 and 100 ng/mL of anti-CD3; and Group 4 received aAPCs in addition to the same reagents as Group 3. Cells were harvested 48 hours after plating, and the absolute number, phenotype and function were assessed. Group 4 was also further expanded for 10 days and assessed at different time points.
2.7. Flow cytometry staining
Cells were stained with CD3 (SP34–2, BD Biosciences), CD4 (L200, BD Biosciences), CD8 (BW135/80, Miltenyi Biotec), CD25 (BC96, BioLegend), CD45RA (T6D11, Miltenyi Biotec), CD127 (HIL-7R-M21, BD Pharmingen and MB15–18C9, Miltenyi Biotec) and FoxP3 (236A/E7, eBioscience and 3G3, Miltenyi Biotec) antibodies, with gates drawn based on isotype controls (IS11–3B2.2.3, Miltenyi Biotec). Cells were permeabilized with the BioLegend FoxP3 Fixation/Permeabilization Buffer Set. Data was collected on a FACSCanto II (BD Bioscience) and analyzed using Flowjo VIX and VX (Tree Star) and FCS Express (De Novo Software).
2.8. Treg-Specific Demethylated Region (TSDR) studies of MCM Tregs
qPCR for the Foxp3 region was performed by Epiontis. Using a standard dilution curve, the number of demethylated and methylated genomic regions was determined, referred as TpG and CpG plasmid units, respectively. The Treg content was calculated by dividing the number of TpG plasmid units by the sum of all Foxp3 regions in the sample; TpG/(TpG+CpG).
2.9. Statistics
Data was analyzed using paired Student’s test. p values of ≤0.05 were considered statistically significant. *= p < 0.05, **= p < 0.01, ***= p < 0.001.
3. Results
3.1. Characterization of Treg phenotype in MCM
Similar to what has been published in Indian origin Cynomolgus macaque and human Tregs, MCM CD4+ Tregs co-express CD25 (the alpha chain of the IL-2 receptor) and the transcription factor FoxP323. The higher the expression of CD25, the higher the level of FoxP3 (Figure 1A). Freshly sorted Tregs (top 1% of CD4+CD25+ cells) consistently suppressed the proliferation of anti-CD2CD3CD28 bead-stimulated autologous PBMCs by at least 50% (of maximum) at a 1:2 Treg:PBMC ratio (Figure 1B). CD25hiFoxP3+ Tregs comprised 2.4±0.4% of CD4+CD3+ T cells, ranging from 2–5% (n=30) (Figure 1A, representative plot).
Figure 1. Phenotype and function of Cynomolgus macaque Tregs.

(A) FoxP3 levels based on CD25 expression in CD4+ T cells. (B) In vitro suppression of anti-CD2CD3CD28 bead-stimulated autologous-PBMCs by freshly isolated CD4+CD25hi Tregs. (C) FoxP3 expression of CD3+CD4+CD25low/negative sorted T cells after seven days of incubation with anti-CD2CD3CD28 beads. (D) FoxP3 expression in relation to CD25 and CD127 in unstimulated PBMCs (representative figure of n=4). (E) Analysis of FoxP3 expression in naïve/resting and activated Tregs in whole blood. (F) Average number of Tregs within the blood based on the expression of CD25 and CD45RA (n=16). (G) Sorting schema for the MCM Tregs.
Human activated T cells can up-regulate both CD25 and FoxP3 (albeit at lower levels than Tregs)24. We cultured CD3+CD4+CD25low/negative sorted MCM T cells with anti-CD2CD3CD28 beads for seven days and observed an upregulation of FoxP3 (Figure 1C). In humans, CD127 is expressed on effector CD4+ T cells and not on Tregs25,26. We assessed the expression of CD127 in unstimulated MCM PBMCs (n=4). Contrary to human studies, FoxP3 in the MCM was not expressed at higher levels in the CD127- cells compared to CD127+ T cells that had similar CD25 expression (Figure 1D). Therefore, CD127 may not be a reliable marker to differentiate Tregs from effector T cells in MCM.
We also assessed the expression of CD45RA (a marker of naïve T cells in humans27) within the MCM Treg populations. Based on their CD45RA expression, human Tregs have been divided into naïve/resting (rTregs; CD45RA+CD25+FoxP3lo) and activated subsets (aTregs; CD45RA-CD25hiFoxP3+)28. In the MCM peripheral blood we observed 2.0 ± 0.38% and 2.4 ±0.39% FoxP3 expression within CD45RA+CD25hi and CD45RA-CD25hi T cells respectively (n=16). Unlike human cells, these MCM populations expressed comparable levels of FoxP3 regardless of CD45RA expression (Figure 1E). CD45RA positive and negative cells and CD4CD25hi MCM cells were equally distributed (Figure 1F).
3.2. Expansion of polyclonal Tregs in vitro
We developed four protocols for the expansion of MCM Tregs (Table 1). Our gating strategy selected the top 1% of CD25+ and CD127- cells within CD4+ T cells (Figure 1G). Expansion of Tregs varied between protocols (Figure 2A). Protocol D achieved the highest Treg expansion with an average of 3,338±1,143 folds, followed by protocol B, with 2,242±400.8-fold expansion (Figure 2B). These two protocols (B and D) received a combined stimulation of aAPCs and allogeneic PBMCs. In contrast, when allogeneic PBMCs alone were used as stimulators from days 7–21 (protocol A) or aAPCs were used alone as the only stimulation source (protocol C), fewer fold expansions were obtained (687.9±149.5 for protocol A and 716.2±220.6 for protocol C) (Figure 2B). When we compared the final cell number obtained at the end of the culture prior to cryopreservation among the different protocols, we observed significant differences between protocols A and D (p=0.0037), protocols B and D (p=0.002) and protocols A and B (p=0.0436), with protocol D obtaining the highest yields (Figure 2C).
Figure 2. Expansion, suppression and phenotype of Tregs grown under four different conditions.

(A) Growth curves of Tregs cultured under four different protocols over time. Each color represents a different animal. (B) Average fold-expansion in each restimulation. Protocols A and B are identical during the first 21 days, and are therefore combined until day 21. “R” is indicative of the number of restimulations. (C) Average yield of Tregs obtained at the end of each protocol. (D) Expression of FoxP3 among CD3+CD4+CD25hi cells for each protocol. “R” is indicative of the number of restimulations. Each dot represents a different Treg line for each restimulation. (E) Suppressive function of Tregs for each expansion protocol. Suppression was measured by the ability to prevent the proliferation of anti-CD2CD3CD28 bead-stimulated-PBMCs. The dashed line denotes 50% suppression. Each dot at every dilution represents a different Treg line. Phenotype and function were tested in most (but not all) restimulations. With the exception of R4 in protocols C and D on day 26, restimulations occurred every seven days.
We then compared the FoxP3 expression prior to cryopreservation. Significant differences were found between protocols B and C (p=0.0294) and B and D (p=0.0158), with protocol B having the highest percentage of FoxP3+ cells at the end of culture. The remaining protocols showed no significant differences when compared to each other (Figure 2D).
Tregs cultured with protocols A and B suppressed bead-stimulated autologous PBMC proliferation more efficiently than Tregs cultured under protocols C and D (Figures 2E). When we compared suppression across the protocols at the 1:2 Treg:PBMC ratio, we observed significant differences between protocols A and D (p=0.0834) and B and D (p=0.0041) (Figure 2E).
In summary, protocol A had the best suppressive capacity followed by protocol B, with moderate/high expansion of Tregs. FoxP3 expression was the highest in protocol B, though meaningful cell numbers for infusions took longer to attain. Protocol D yielded higher cell numbers over a shorter period of time (Figure 2C), but those cells were less suppressive and in some cases showed substantial loss of FoxP3 expression (Figure 2D and 2E). Protocol C was abandoned early because cells exhibited poor suppression and low yield.
3.3. Management of outgrowth of CD8+ T cells in Treg cultures
CD8+ T cells have been reported to contaminate cultured Tregs29. In agreement with these reports, we occasionally observed the outgrowth of CD8+ T cells in our cultures. Because host effector CD8+ T cells are undesirable in transplant studies because of their potential to induce rejection, we assessed approaches to prevent them from contaminating our cultures. Our first approach attempted to exclude CD8+ cells by using anti-CD8 antibodies at the initial FACS (Figure 1G). This approach was not always successful, as CD8+ T cells were still detected in the cultured cell lines (Figure 3A and 3B).
Figure 3. Removal of CD8 from Treg cultures. (A) and (B) CD8, FoxP3 expression and in vitro suppressive capacity of two different Treg lines with comparable CD8 contamination and different FoxP3 expression.

Treg lines from Figure 3A and 3B were expanded under protocol B. (C) CD8 single positive (SP) contamination on day 21 of culture between a Treg line that underwent MACS purification (left panel) and one that did not (right panel). (D) FoxP3 expression and suppressive capacity at the end of the culture period of Tregs that underwent CD8 MACS depletion prior to FACS sorting on day 0. (E) CD8 depletion by FACS early in culture to eliminate CD8 contamination. (F) Cell growth of a Treg line that underwent FACS on day 15 of culture to eliminate CD8 T cells. (G) FoxP3 and (H) suppressive capacity of a Treg line at the end of the culture period (day 56) after undergoing FACS on day 15 due to CD8 T cell contamination.
Pre-enrichment of CD4+ T cells from PBMCs by magnetic-activated cell sorting (MACS) prior to FACS was shown to efficiently remove CD8+ T cells (Figure 3C), but Treg lines grown following this technique consistently failed to maintain FoxP3 expression and suppressive activity (Figure 3D). Hence, in our hands, MACS may have removed or damaged natural Tregs.
We subsequently attempted to re-sort Tregs mid-way through the culture period. Lines were re-sorted when >4% of CD8+ T cells were detected. This approach proved to be successful in eliminating CD8+ T cells (Figure 3E), but was associated with significant loss of Tregs, thereby prolonging the culture period (Figure 3F). However, despite culturing Tregs for >50 days (protocol B), expression of FoxP3 and suppressive function was maintained at high levels (Figure 3G and 3H).
Finally, we assessed the suppression by Treg cultures that had comparable CD8 contaminations. Differences in suppression were directly related to the levels of FoxP3 expression of the CD4+ T cells (Figure 3A and 3B), indicating that the Treg stability/purity could override the proliferation of CD8+ T cells (Figures 3A and 3B).
3.4. FoxP3 expression is maintained in CD45RA+ and CD45RA- cells in long-term cultures
Human Treg studies suggest that CD45RA+ and not CD45RA- Tregs maintain FoxP3 expression during prolonged cultures30. We thus examined the correlation between CD45RA and FoxP3 in our in vitro expanded Tregs cultured under protocols B and D (Figure 4A and 4B). Regardless of the protocol, the expression of CD45RA in the cultured cells did not correlate with FoxP3 expression. CD45RA intermediate/low cells also expressed high levels of FoxP3 in some cases. Therefore, and contrary to human studies, reduced FoxP3 was not associated with loss of CD45RA expression in MCM.
Figure 4. CD45RA expression in cultured Tregs and cryopreservation and recovery of Tregs under different conditions.

(A) Correlation between FoxP3 and CD45RA expression in protocol B. (B) Correlation between FoxP3 and CD45RA expression in protocol D. (C) Average cell survival after 48 hours of culture post-thaw (n=3). Cell death was assessed by trypan blue and propidium iodide (PI). (D) FoxP3 expression after two days of culture under various conditions. (E) Average suppressive activity of freshly thawed Tregs and (F) 48 hours after culture (n=3).
3.5. Recovery of Tregs after cryopreservation
In order to be useful as an “off-the-shelf” product, Tregs must be functional after thawing. Cryopreservation has been reported to impact Treg phenotype and suppressive activity31. We thus studied the effects of cryopreservation on MCM Tregs. Cryopreserved and thawed Treg lines were plated under four culture conditions (see Materials and Methods). Regardless of condition, the viability was low after 48 hours, ranging from 5.1 to 24.8% (Figure 4C). A distinct FoxP3 negative population was observed in all the conditions except the media alone (Figure 4D). Freshly thawed cells had 50% suppression at 1:4–1:8 Treg:PBMC ratio (Figure 4E), whereas suppression 48 hours post-reculture varied from 1:2 to 1:8 (Figure 4F). Thus, reculturing Tregs for 48 hours (regardless of condition) did not improve FoxP3 expression or suppression.
We then studied whether longer periods of culture post-thaw would enhance the purity and number of Tregs beyond that observed after 48 hours. Cells were cultured with IL-2, anti-CD3 and aAPCs. During the first 24–48 hours of culture the majority of cells again died (Figure 5A). However, maintaining the cells in vitro for a longer period allowed them to expand to the original number by day +3, by a log by day +5 and 30±7.1 fold by the end of the culture on day 10 (Figure 5A). FoxP3 expression was maintained throughout the culture period post-cryopreservation (Figure 5B). In addition, Treg function was improved compared to the thawing day (Figure 5C). These results suggest that a minimum of 3-day re-culture period can improve function and re-expand Treg numbers under the described conditions.
Figure 5. Cryopreservation and recovery of Tregs after 10 days of culture post-thaw.

(A) Cell growth of thawed Tregs plated with IL-2, anti-CD3 and aAPCs for 10 days (n=4). (B) FoxP3 expression of freshly thawed Tregs (Day 0) and over the course of the culture (n=4). (C) Suppressive capacity of freshly thawed Tregs (Day 0) and over the course of the culture (n=4).
3.6. Superior suppression of stimulator versus third-party PBMCs by polyclonal Tregs
In order to assess the specificity of our cultured cells, Tregs from protocol B (which provided the best outcomes) were used. We previously assessed the suppressive abilities of these Tregs in in vitro MLRs and observed a comparable suppression of recipient and donor responders stimulated by donor and third-party stimulators14. Because a maximum of 10–20% of the T cells in an MLR proliferate, we asked whether these Treg suppressive qualities were maintained under maximal stimulation where responders were stimulated with anti-CD3CD28 beads. We challenged the cultured Tregs to suppress the response of T cells from either self, “stimulator-type” (which are MHC-mismatched PBMCs used in the Treg cultures) or third-party cells (cells that are MHC-mismatched to both Tregs and stimulator PBMCs and never exposed to the Tregs in culture) (Figure 6A). The assays were performed using Tregs that had been exposed to stimulator-type PBMCs for two weeks. Stronger suppression of responses by autologous or “stimulator- type” PBMCs (PBMCs from the animal used for the expansion of the Tregs) than of third-party cells was observed. However even under these challenging conditions, a 50% suppression at a 1:4 Treg:Teffector ratio was uniformly achieved (Figure 6A).
Figure 6. Specificity study of in vitro expanded MCM Tregs and demethylation status of their TSDR.

(A) Host, donor and third party PBMCs stimulated with anti-CD2CD3CD28 beads (n=3). Tregs were cultured following protocol B. Donor and third party PBMCs are full-MHC mismatched to host. Donor and third party PBMCs do not share any MHC. (B) Demethylation status of the TSDR of in vitro cultured Tregs grown under protocol A, B and D was analyzed in correlation with the FoxP3 expression and suppressive capacity of the same cell lines. (C) Study of the correlation between the TSDR demethylation status, FoxP3 expression and suppressive capacity of two Treg lines grown under protocol B over time.
3.7. Demethylation of the TSDR correlates with high FoxP3 expression and good suppressive capacity in MCM
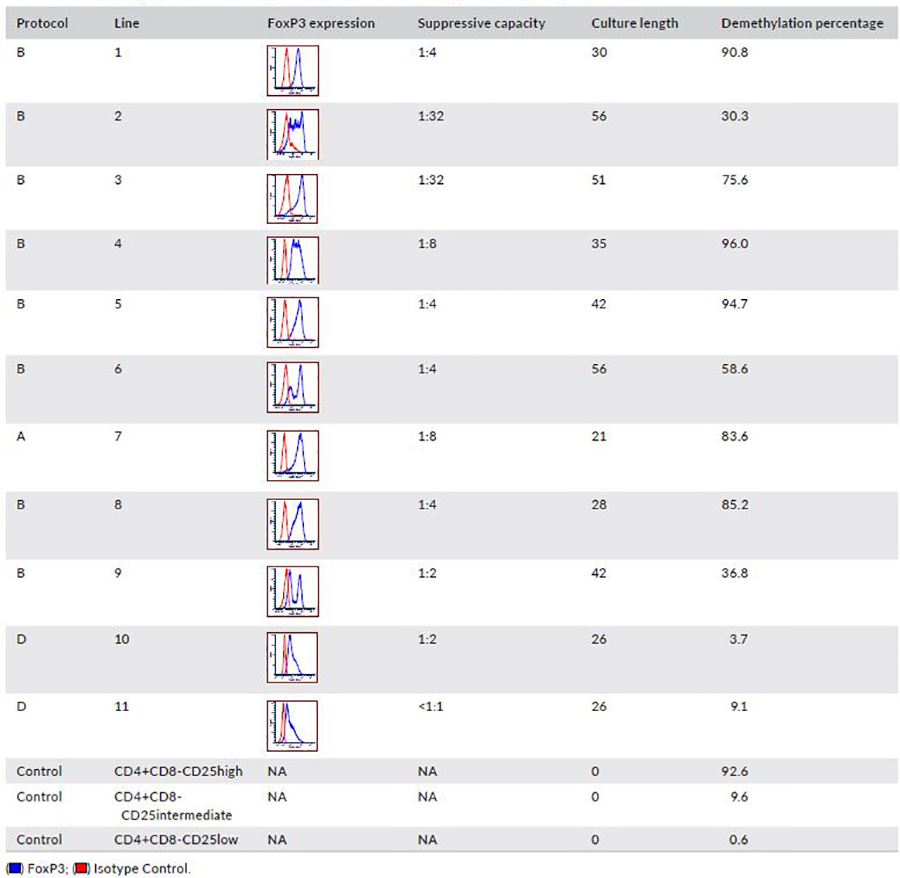
Demethylation of the TSDR has been shown to identify stable Tregs32. We therefore analyzed the TSDR of MCM Tregs that were grown under protocols A, B and D (Figure 6B and Table 2). Treg lines that concurrently had ≥50% suppression at a 1:4 or lower Treg:PBMC ratio and FoxP3 expression >80% (as a single peak) all had TSDR demethylation >75%. Tregs from cultures which lost FoxP3 expression and were less suppressive in some cases had greater TSDR methylation (Figure 6B and Table 2). One Treg line with high FoxP3 expression (80%) showed a low percentage of TSDR demethylation, possibly reflecting two peaks of FoxP3 expression (Table 2; Line 2). A second outlier had good suppressive capacity with low TSDR demethylation percentage, possibly due to a high percentage of CD8+ T cells in this line (9.6%). In addition, in two cell lines that were studied over time, we observed a decrease in the TSDR demethylation in association with a decrease in FoxP3 expression and suppressive capacity (Figure 6C). These results mirror what has been found in humans33 and further support the MCM as a good model for Treg studies.
Table 2.
Summary of the TSDR methylation status, FoxP3 and suppressive capacity study.
 |
4. Discussion
Pre-clinical and clinical Treg studies support their safe and efficacious use for the treatment of autoimmune diseases, prevention of rejection and GVHD in allotransplantation2,5–8. Their low number in the circulation is a major limitation for their widespread therapeutic use. Development of efficient and consistent Treg expansion protocols has been a major focus of many groups34–37. Here, we provide our experience with MCM Tregs and document four Treg expansion protocols.
MCM Tregs show a direct correlation between FoxP3 and CD25 expression in CD4+ T cells, comprising 2.4±0.4% of the CD4+ T cells, similar to humans (2–4%)28. In MCM, the fidelity of the expression of CD127 and FoxP3 was less clear as to what has been demonstrated in published human studies38 and further studies will likely be needed to understand whether the IL-7R and FoxP3 expression have similar biology as in humans. In addition, CD45RA has been utilized to describe different subsets of Tregs in humans. In our hands, CD45RA and FoxP3 expression did not correlate with in vitro expanded MCM Tregs. Hence, this suggests that though CD4+CD25hi Tregs function similarly as human Tregs, in the MCM (and in our culture conditions) the expression of CD45RA may have different biological significance. Further studies will be required to better understand the relevance of such differences.
The expansion of CD8+ cells in MCM Treg cultures is a difficulty that has also been encountered in human Treg cultures29. Inclusion of the CD8 marker in our sorting strategy did not completely prevent outgrowth of this population. Pre-selection of CD4+ cells through MACS prior to sorting prevented CD8 contamination, but these Tregs were poor suppressors, suggesting a potential loss of or damage to highly suppressive natural Tregs. We succeeded in controlling CD8 growth by re-sorting cultures early by FACS. The impact of re-sorting forced us to prolong the Treg cultures in order to expand sufficient cell numbers for transplantation14. The effects observed in MCM Tregs after MACS might not be encountered with human cells as cliniMACS is being used safely in the clinic. However, we recommend to thoroughly assess human Tregs after MACS selection based on our studies.
Clinical-grade beads may be favored over cellular reagents. However, exploiting the use of clinical-grade irradiated aAPCs may be less costly. There is precedence for the use of aAPCs clinically such as K562 cells in vaccine studies (such as the GVAX trial)39.
In humans, it has been reported that extended Treg cultures can lead to loss of FoxP3 expression and suppressive function. MCM Tregs grown under protocol B maintained potent suppression and high FoxP3 levels after prolonged culture periods (up to 2 months). However, when aAPCs were used throughout the 26-day culture period, we observed a loss of FoxP3. These Tregs may have been too aggressively stimulated or overtaken by contaminating conventional T cells. In summary, protocols A and B provided the best Tregs but required 4–8 weeks of culture and use of aAPCs alone did not generate highly suppressive MCM Tregs.
We aimed to develop a protocol that creates “off-the-shelf” polyclonal Tregs potent enough to prevent rejection of organs from any deceased donor. We have previously shown that Treg suppression of the response of autologous naïve T cells was similar when the T cells were stimulated by PBMCs from either the same donor used during the Treg expansion or by a random third-party (mismatched to both, the Treg recipient and PBMC used in Treg expansion at MHC class I and II)14. Here, we tested the suppression by Tregs of the T cell response from different sources (autologous, the same donor used during Treg expansion and a random third-party) when all T cell responders were robustly stimulated via their TCR using anti-CD2CD3CD28 beads. Suppression of polyclonally stimulated T cells from the same donor that was used during the Treg expansion was superior to the suppression of third-party responders. The degree of enrichment of antigen-specific Tregs will require TCR repertoire studies. However, overall, polyclonally expanded Tregs generated in protocol B were able to suppress all responders robustly, suggesting that this Treg expansion method may be clinically relevant as a product for any donor-recipient pair.
Treg cryopreservation will be necessary for clinical application using “off-the-shelf” Tregs. Because Tregs can suppress third-party PBMCs reliably, self or third-party Tregs could be used as an “off-the-shelf” product to mitigate post-transplant alloresponses. Testing third-party Tregs (exposed during expansion to several PBMC stimulators) in an NHP model will help in predicting an effective therapeutic dose. Furthermore, we demonstrated that Tregs can improve their suppressive function and numbers after cryopreservation if allowed to expand for a minimum of 3 days post-thaw, despite some contradictory reports31. In our in vivo studies, MCM underwent a protocol for the induction of tolerance through the mixed chimerism approach with a non-myeloablative conditioning regimen, BMT and recipient expanded Tregs (grown under protocol A)14. Tregs were infused after thawing without letting the cells rest prior to infusion. Based on our in vitro results, this direct infusion of our Tregs could have limited the efficacy of the study, suggesting that the infusion of freshly expanded or thawed and recultured Tregs may be beneficial. Further studies in vivo are necessary to assess this possibility.
The methylation status of MCM Tregs paralleled what has been observed in humans. We observed a correlation between high FoxP3 expression, suppressive capacity and demethylation of the TSDR. In addition, longer culture periods decreased the demethylation percentage in correlation with FoxP3 expression and suppressive activity. These observations raise questions about the utility of culturing the lines longer for the purpose of achieving higher cell yields.
Despite the beneficial outcomes that Tregs have shown in transplant tolerance protocols in different reported models, Thomson et al. reported in their heart transplant model that ATG-lymphodepleted MCM that received Tregs during the early post-transplant period had increased effector memory T cells and anti-donor antibodies40. These were associated with deficient graft survival and function compared to non-Treg recipients. Although Tregs in vitro were shown to be potent suppressors, this report argued against their use in the context of severe lymphodepletion in heart transplantation. In contrast to that study, the infusion of high doses of expanded recipient Tregs to animals receiving MHC-mismatched BM following a non-myeloablative conditioning regimen (consisting of low dose total body irradiation, thymic irradiation, horse ATG, anti-CD40L and cyclosporine or rapamycin) achieved more persistent chimerism and more robust tolerance than control recipients14. Significant differences in the preparative regimens may have influenced the outcome of both studies (by impacting the survival of the Tregs). Though so far infusion of Tregs has been shown to be safe, further work is needed to define the quality, potency and dosing of Tregs in pre-clinical large animal models to better design successful clinical applications.
In summary, MCM Tregs operate similarly to human Tregs. We demonstrate the feasibility of expanding large number of MCM Tregs in vitro for pre-clinical studies. We have generated four protocols, all of which expanded MCM Tregs; however, protocol B yielded the best outcomes with potent suppressive capacity, FoxP3 expression, and acceptable cell growth. Protocol-B Tregs could expand (or re-expand cryopreserved) Tregs for two months while maintaining robust suppressive function. However, they are greatly impacted by the culture method and careful quality assessment is necessary prior to infusion. Our most successful Treg expansion protocol allows Tregs to suppress robustly host or donor bead-stimulated effector T cells (if donor PBMCs are used during the expansion). Furthermore, MHC-mismatched third-party effector cells could be suppressed, supporting their potential use in patients receiving brain-dead donor grafts. Our approach is safe and allows Tregs to maintain their phenotype and function over long-culture periods demonstrated by the TSDR stability confirming their “natural” thymic origin. Finally, though MCM Tregs did not share all phenotypic characteristics with human Tregs, overall they very closely resemble their biology, which argues in favor of their utility in key proof of concept, large animal pre-clinical studies.
Acknowledgments
Artificial antigen presenting cells (aAPC) that express CD80, CD58 and CD32 were kindly provided by Dr. Megan Levings, University of British Columbia, Vancouver, Canada.
Funding for these studies was provided by NIH grant R01OD017949, by startup funds from Columbia University Departments of Medicine and Surgery (to MS and RDS), the Banting Foundation (to MS), the Columbia University Core award (to RDS), and the Irving Pilot Translational Science award for new investigators (to RDS). Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under the award S10OD020056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- aAPC
Artificial antigen presenting cell
- MCM
Mauritius Cynomolgus macaque
- MHC
Major histocompatibility complex
- NHP
Non-human primate
- PBMC
Peripheral blood mononuclear cell
- Treg
Regulatory T cell
- TSDR
Treg-specific demethylated region
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nature reviews Immunology. 2010;10(7): 490–500. [DOI] [PubMed] [Google Scholar]
- 2.Trzonkowski P, Bieniaszewska M, Juscinska J, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. 2009;133(1): 22–26. [DOI] [PubMed] [Google Scholar]
- 3.Brunstein CG, Miller JS, Cao Q, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3): 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Ianni M, Falzetti F, Carotti A, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14): 3921–3928. [DOI] [PubMed] [Google Scholar]
- 5.Frey O, Petrow PK, Gajda M, et al. The role of regulatory T cells in antigen-induced arthritis: aggravation of arthritis after depletion and amelioration after transfer of CD4+CD25+ T cells. Arthritis Res Ther. 2005;7(2): R291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care. 2012;35(9): 1817–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol. 2014;153(1): 23–30. [DOI] [PubMed] [Google Scholar]
- 8.Bluestone JA, Buckner JH, Fitch M, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315): 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Todo S, Yamashita K, Goto R, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64(2): 632–643. [DOI] [PubMed] [Google Scholar]
- 10.Prost S, Le Dantec M, Auge S, et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest. 2008;118(5): 1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butenhoff JL, Kennedy GL Jr., Hinderliter PM, et al. Pharmacokinetics of perfluorooctanoate in cynomolgus monkeys. Toxicol Sci. 2004;82(2): 394–406. [DOI] [PubMed] [Google Scholar]
- 12.Roitberg B, Khan N, Tuccar E, et al. Chronic ischemic stroke model in cynomolgus monkeys: behavioral, neuroimaging and anatomical study. Neurol Res. 2003;25(1): 68–78. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Cosimi AB, Wee SL, et al. Effect of mixed hematopoietic chimerism on cardiac allograft survival in cynomolgus monkeys. Transplantation. 2002;73(11): 1757–1764. [DOI] [PubMed] [Google Scholar]
- 14.Duran-Struuck R, Sondermeijer HP, Buhler L, et al. Effect of Ex Vivo-Expanded Recipient Regulatory T Cells on Hematopoietic Chimerism and Kidney Allograft Tolerance Across MHC Barriers in Cynomolgus Macaques. Transplantation. 2017;101(2): 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai T, Sachs DH, Cosimi AB. Tolerance to vascularized organ allografts in large-animal models. CurrOpinImmunol. 1999;11(5): 516–520. [DOI] [PubMed] [Google Scholar]
- 16.Nadazdin O, Boskovic S, Murakami T, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci TranslMed. 2011;3(86): 86ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada TO Yohei, Boskovic Svjetlan, Nadazdin Ognjenka, Oura Tetsu, Schoenfeld David, Cappetta Kate, Smith Rex-Neal, Madsen Joren, Sachs David H., Benichou Gilles, Cosimi A. Benedict, and Tatsuo Kawai. Use of CTLA4Ig for Induction of Mixed Chimerism and Renal Allograft Tolerance in Nonhuman Primates (IN PRESS). American Journal of Transplantation. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budde ML, Wiseman RW, Karl JA, Hanczaruk B, Simen BB, O’Connor DH. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics. 2010;62(11–12): 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Connor SL, Blasky AJ, Pendley CJ, et al. Comprehensive characterization of MHC class II haplotypes in Mauritian cynomolgus macaques. Immunogenetics. 2007;59(6): 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malefyt RD, Verma S, Bejarano MT, Ranesgoldberg M, Hill M, Spits H. Cd2/Lfa-3 or Lfa-1/Icam-1 but Not Cd28/B7 Interactions Can Augment Cytotoxicity by Virus-Specific Cd8+ Cytotoxic Lymphocytes-T. Eur J Immunol. 1993;23(2): 418–424. [DOI] [PubMed] [Google Scholar]
- 21.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129(4): 263–276. [DOI] [PubMed] [Google Scholar]
- 22.Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. JExpMed. 2001;193(11): 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zitsman JS, Alonso-Guallart P, Ovanez C, et al. Distinctive Leukocyte Subpopulations According to Organ Type in Cynomolgus Macaques. Comp Med. 2016;66(4): 308–323. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37(1): 129–138. [DOI] [PubMed] [Google Scholar]
- 25.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7): 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seddiki N, Santner-Nanan B, Martinson J, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7): 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada R, Kondo T, Matsuki F, Takata H, Takiguchi M. Phenotypic classification of human CD4+ T cell subsets and their differentiation. Int Immunol. 2008;20(9): 1189–1199. [DOI] [PubMed] [Google Scholar]
- 28.Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6): 899–911. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104(3): 895–903. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann P, Eder R, Boeld TJ, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108(13): 4260–4267. [DOI] [PubMed] [Google Scholar]
- 31.Golab K, Leveson-Gower D, Wang XJ, et al. Challenges in cryopreservation of regulatory T cells (Tregs) for clinical therapeutic applications. Int Immunopharmacol. 2013;16(3): 371–375. [DOI] [PubMed] [Google Scholar]
- 32.Polansky JK, Kretschmer K, Freyer J, et al. DNA methylation controls Foxp3 gene expression. EurJImmunol. 2008;38(6): 1654–1663. [DOI] [PubMed] [Google Scholar]
- 33.Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37(9): 2378–2389. [DOI] [PubMed] [Google Scholar]
- 34.Bluestone JA, Buckner JH, Fitch M, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science Translational Medicine. 2015;7(315). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Putnam AL, Safinia N, Medvec A, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. 2013;13(11): 3010–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald KG, Orban PC, Levings MK. T regulatory cell therapy in transplantation: stability, localization and functional specialization. Current Opinion in Organ Transplantation. 2012;17(4): 343–348. [DOI] [PubMed] [Google Scholar]
- 37.Riley JL, June CH, Blazar BR. Human T Regulatory Cell Therapy: Take a Billion or So and Call Me in the Morning. Immunity. 2009;30(5): 656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7): 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemunaitis J, Jahan T, Ross H, et al. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006;13(6): 555–562. [DOI] [PubMed] [Google Scholar]
- 40.Ezzelarab MB, Zhang H, Guo H, et al. Regulatory T Cell Infusion Can Enhance Memory T Cell and Alloantibody Responses in Lymphodepleted Nonhuman Primate Heart Allograft Recipients. American Journal of Transplantation. 2016;16(7): 1999–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]