

Abstract
Vascular smooth muscle cells (vSMCs) play a crucial role in both the pathogenesis of Aneurysms and Dissections of the ascending thoracic aorta (TAAD) in humans and in the associated adaptive compensatory responses, since thrombosis and inflammatory processes are absent in the majority of cases. Aneurysms and dissections share numerous characteristics, including aetiologies and histopathological alterations: vSMC disappearance, medial areas of mucoid degeneration, and extracellular matrix (ECM) breakdown. Three aetiologies predominate in TAAD in humans: (i) genetic causes in heritable familial forms, (ii) an association with bicuspid aortic valves, and (iii) a sporadic degenerative form linked to the aortic aging process. Genetic forms include mutations in vSMC genes encoding for molecules of the ECM or the TGF-β pathways, or participating in vSMC tone. On the other hand, aneurysms and dissections, whatever their aetiologies, are characterized by an increase in wall permeability leading to transmural advection of plasma proteins which could interact with vSMCs and ECM components. In this context, blood-borne plasminogen appears to play an important role, because its outward convection through the wall is increased in TAAD, and it could be converted to active plasmin at the vSMC membrane. Active plasmin can induce vSMC disappearance, proteolysis of adhesive proteins, activation of MMPs and release of TGF-β from its ECM storage sites. Conversely, vSMCs could respond to aneurysmal biomechanical and proteolytic injury by an epigenetic phenotypic switch, including constitutional overexpression and nuclear translocation of Smad2 and an increase in antiprotease and ECM protein synthesis. In contrast, such an epigenetic phenomenon is not observed in dissections. In this context, dysfunction of proteins involved in vSMC tone are interesting to study, particularly in interaction with plasma protein transport through the wall and TGF-β activation, to establish the relationship between these dysfunctions and ECM proteolysis.
Keywords: Extracellular matrix, TGF-β, Contractile proteins, SMC tensegrity, Plasminogen activation, Epigenetics, Protease nexin-1, Endocytosis, Haemodynamics
1. Definition of aneurysms
Aneurysms are morphologically defined as localized dilations of the arterial wall with a focal loss of wall parallelism and functionally defined as a progressive loss of the arterial wall’s ability to withstand the wall tension generated by high intraluminal pressure, leading to intramural (dissection, AAD) or complete acute rupture. Aortic aneurysms, whatever their localization, ascending aorta TAA or abdominal aorta (AAA) share common pathophysiological features but differ by aetiologies and specific haemodynamics.1 Since withstanding wall tension is mainly the function of the insoluble fibrillary extracellular matrix (ECM) synthesized and matured by vascular smooth muscle cells (vSMCs) in the wall, ECM degradation by proteolytic enzymes is a common mechanism in aneurysmal pathogenesis. In this micro-environment, intrinsic frailty of the ECM, loss of vSMCs, and increased permeability to plasma zymogens, directly or indirectly potentiate the proteolytic injury. In parallel, vSMCs also respond to these injuries using their functional and epigenetic plasticity (nuclear reprogramming, phenotypic shift).2 AAAs are mainly of atherothrombotic origin,3 whereas the ascending aorta is resistant to atheroma and devoid of intraluminal thrombus formation, most likely due to the high shear and its washing effect by systolic ejection (Figure 1). Therefore, with the exception of specific rare auto-immune aetiologies such as Takayasu’s or Horton’s diseases, TAADs are characterized by the absence of myeloid cell diapedesis within the aortic tissue. Thereby TAA & D offer a unique opportunity to study the predominant roles of vSMCs in aneurysmal pathologies (i) as initiators of aneurysm formation by expression of specific genetic molecular defects; (ii) by their ability to promote blood-borne proteolytic injuries in the absence of inflammation; and (iii) to limit the risk of acute rupture, by an adaptive response of the canonical TGF-β/Smad2 pathway in vSMCs, which may help to prevent dissections through synthesizing more fibrillar ECM, along with locally secreting antiproteases,4 and clearance of protease/antiprotease complexes5 (Figure 2). The wall histopathology, involving ECM breakdown, vSMC loss co-localized with areas of mucoid degeneration, is common to TAA and dissection (AAD) whatever their aetiologies. Areas of specific mucoid degeneration are characterized by accumulation of highly hydrophilic glycosaminoglycans (GAGs). Mucoid degeneration is also observed during the aging process in human aorta.6 However, the description of the involvement of vSMCs in TAA needs to assimilate first the evolutive role of SMCs in phylogenesis, and therefore the physiology, of the arterial part of the circulation.
Figure 1.

Common features and specificities of TAA vs. AAA.
Figure 2.

vSMC involvement in TAA pathogenesis and subsequent responses.
2. Role of vSMCs in structure/function of the arterial system in mammals
Phylogenetically, the circulatory system evolved from a low-pressure closed circulating system animated by an archaic heart in fish, to a more recent, highly pressurized (potential energy) arterial system with organ-regulated directional blood flow (vasomotricity) propelled through the conductance arterial tree by the pumping action of the left ventricle. The teleonomy of arterial vSMC evolution is to structure the wall of conductance arteries and to assist the metabolic autonomisation of organ function through resistance arteries. As a main determinant, vSMC tone defines peripheral resistances to blood flow, generating arterial blood pressure.
In parallel, wall topophysiology and structure also evolved in conductance arteries in order to respond to the acquired pressure load, from a thin cellular structure to a thick matrix-rich, layered structure. In the aorta, vSMCs assume the functional roles of both producing aortic tone in response to sympathetic stimuli, and synthesizing and modelling the ECM. Biomechanically, the tensile stress supported by the wall is proportional to pressure and radius and inversely proportional to wall thickness (Laplace’s low, T = P.r/2 h). Since progressive physiological dilation of the aorta is observed with age in animals7 and humans,8 tensional stress increases with aging, independently of pressure. To respond to tensile stress, the aortic wall is structured in three spatially organized layers, from inside to outside: the intima, the media and the adventitia. The medial layer displays spatial and functional connectivity between vSMCs and ECM, assuming the function of supporting the phasic haemodynamic load (the content) within the arterial system (the container).9 vSMC differentiation and survival is dependent on cell adhesion to matrix,10 creating tensegrity11 within the cell, via ECM, intracellular cyto- and nucleoskeletal interactions,12 largely dependent on local haemodynamic parameters, the cardiac cycle and the impedance to phasic flow. In the ECM, the insoluble macro-fibrils, collagens, mainly provide the resistance to rupture whereas elastin provides the resistance to dilation.13
Outward adventitial intersititial pressure is low: 10 mmHg. Therefore, an important transmural pressure gradient (100 mmHg) exists between the intraluminal arterial blood pressure (130/80 mmHg) generated by the peripheral resistance, and adventitial interstitial pressure, creating a unidirectional outward hydraulic conductance across the arterial wall. This hydraulic conductance is responsible for advective/convective radial mass transport of soluble plasma molecules and macromolecules through the arterial wall. This biomechanical phenomenon is termed «outward convection». Outward convection is dependent, on the one hand, on haemodynamic factors, including pressure and shear14,15 and, on the other hand, on the porosity of the arterial wall, partly determined by the integrity of the elastic network16 and by vSMC tone17,18 which limit advection across the wall. In this paradigm, pressurized water and blood-borne components percolate through the wall components, creating potential interactions with vSMCs and/or the ECM and leading to retention, proteolysis, clearance and metabolism of plasma proteins or blood particles, or to their exfiltration towards the adventitia for recycling.9,19 For example, free water molecules will be retained more in hydrophilic areas of the aortic wall than in hydrophobic ones (elastin), and could contribute to the swelling of the GAG-rich mucoid degenerative areas, potentially inducing intramural delamination.20
3. Nosology and aetiologies of TAADs
TAAs are non-atheromatous aneurysmal diseases related to three main aetiologies: monogenic diseases, associated with bicuspid aortic valves (BAV) and sporadic (also termed degenerative), associated with aging.21 These different aetiologies are characterized by the age of clinical expression: younger in genetic forms, middle age in association with BAV, older in sporadic forms. Up to 30% of cases of TAAs are associated with underlying mutations in single genes. These mutations are predominantly inherited in an autosomal dominant manner and may (syndromic forms) or may not be associated with other systemic manifestations.
When compared with TAAs, characterized by a progressive dilation of the aorta, dissections are acute events, defined by intramural rupture, with or without a subjacent aneurysm, usually of small dilation. The initial intimal tear, causing blood leaks within the external part of the media, can take place in the ascending aorta (Type I or A dissections) or just below the ostia of the left subclabvicular artery developing in the descending thoracic aorta (Type III or B dissections). Type II is the extension of Type I to the thoracic descending aorta, with possible re-entry in the abdominal aorta. Types II and III dissections could generate circulating, partially circulating or thrombosed false channels which may impact the evolution of the disease.22,23 When compared with dilated aortic tissue, the dissected tissue is also characterized by areas of GAG-rich mucoid degeneration, which pave the way for initial tears and haemorrhagic suffusion and diffusion towards the external part of wall, the site of developing vasa vasorum. In this context the specific role of GAG accumulation, particularly in the tissue environment of vasa vasorum, has been recently highlighted in AAD.24 Tobacco, hypertension and intensive tonic physical effort (weightlifting) are risk factors for acute dissections.
4. Experimental models
Experimental animal models of aortic aneurysms and dissections have been recently reviewed.25 Experimental models of progressive TAA are rare in rats and mice and limited to some pharmacological models and to mice genetically engineered to harbour gene mutations observed in humans with thoracic aortic disease, such as Marfan mouse models26 (Table 1). The Blotchy syndrome in mice remains one of the very few available models of naturally occurring, genetically determined aneurysms.27 The blotchy syndrome is due to a mutation in a copper transporter leading to copper insufficiency. Copper is the transition metal necessary for Lysyl-oxydase (LOX) enzymatic activity. As a translational example in humans, genetic copper deficiency is observed only in the rare condition, Menkes disease. Dissections have been modelled in animals for many years.28 Spontaneous dissecting aneurysms of the aorta were initially described in turkeys with stress-dependent acute hypertension.29 The frequency of these acute dissections were increased when the turkeys were fed β-aminopropionitrile (BAPN).30 BAPN is a toxic chemical isolated from a variety of sweet pea and responsible for clinical angiolathyrism. BAPN is a powerful inhibitor of LOX, impairing the maturation of elastin and collagen and thus sensitizing the ECM to proteolysis. Administration of BAPN in rats and mice also increases the risk of dissecting aneurysm.31 The effect of BAPN is dependent on the dose but also on the age of administration, with younger animals (immediate post-weaning) more likely to have dissections.
Table 1.
Experimental models of TAADs
| Spontaneous TAADs in animals |
| Turkey (aortic dissection)29 |
| Blotchy mice (mutation in ATPase copper transporter)104 |
| Genetic models K.O. and K.I. |
| Fibrillin-1 (Marfan)105 |
| Coll3A1 (Ehlers-Danlos)36 |
| FOXE3−/− (transcription factor) |
| LOX106 |
| ADAMTS151 |
| Biglycan K.O.107 |
| Filamin A108 |
| Pharmacological models |
| Angiotensin II infusion in sensitized mice (dissections)36 |
| β-aminopropionitryl (BAPN, LOX inhibitor)109 |
More recently, Daugherty and colleagues32 described that angiotensin II (AngII) infusion induces dissecting aneurysms in Apoe−/−mice.33–35 This model, easy to do, is not limited to models of atherosclerosis in mice. When angiotensin II is administered to mice sensitized by genetic mutations on ECM genes,36 BAPN exposure,37 or TGF-β antibody administration,38 a rate of dissection of 100% can be observed. Similar results were obtained in rats (JBM personal data). In these pharmacological models, the level of circulating plasma AngII is high and the mice are moderately hypertensive, suggesting that AngII acts more by causing mechanical intimal damage, rather than targeting the medial vSMCs. Physiologically, the renin/angiotensin system is compartmented and the plasma AngII level is very low, since renin acts more at a tissue level than in plasma. Thereby, it would be interesting to compare the AngII model with a similar model of renin infusion. Nevertheless these models, associating an ECM defect or vSMC relaxation39 with endovascular injury by AngII are particularly interesting for testing new therapeutic approaches in dissections. The current tendancy is to associate two models, a genetic defect with angiotensin II infusion in mice, or two pharmacological approaches (BAPN + AngII) in rats.
5. Pathogenic roles of vSMCs in TAA & D
5.1 vSMC genes predisposing to TAAs weaken ECM, disrupt vSMC tone or limit canonical TGF-β signalling
The list of genes associated with syndromic and non-syndromic ascending aortic aneurysms and dissections was recently updated.40 It has been known for many decades that one gene in the human genome can be mutated and lead to a strong familial predisposition for TAAD. Marfan syndrome (MFS) is a condition inherited in an autosomal dominant manner with skeletal (long limbs and fingers, scoliosis, pectus deformities), connective tissue (joint laxity, striae, flat feet) and ocular (ectopia lentis) complications. Affected individuals can have cardiac features (mitral insufficiency and prolapse) but the major cardiovascular complication is progressive enlargement of thoracic aortic root aneurysms and acute ascending aortic dissections, related or not to dilation. MFS is mainly due to mutations in FBN1, which encodes an ECM protein called fibrillin-1 that localizes in microfibrils. Microfibrils play an important role in the aorta; they link the elastic fibres to focal adhesions on the cell surface of vSMCs. Mutations in FBN1 lead to either less fibrillin synthesis (mutations leading to haploinsufficiency) or quantitatively decreased fibrillin-1 being incorporated into microfibrils (missense mutations).4 Other ECM genes predisposing to TAAs include other proteins in microfibrils (MFAP5)41 and loss-of-function mutations in LOX,42 which is an enzyme involved in cross-linking and maturation of the ECM. In parallel, the nosology and management of vascular Ehler-Danlos syndrome, in relation to a Col3A1 mutation,43 and the structure–dysfunction relationship created by the mutation44 have recently progressed (Table 2).
Table 2.
Genomic mutations in heritable forms of TAADs in human
| Genes encoding for ECM defects |
| Fibrillin -1 and -2 (MFS)4 |
| Microfibril-associated Protein5 (MFAP5)41 |
| Filamin A110 |
| LOX42,111 |
| Menkes disease (ATP7A copper transporter)112 |
| Elastin (ELN)113 and Fibulin (FBLN),114 (Cutis Laxa), Emilin1 (elastin microfibril interfacer 1)115 |
| Collagen 1 α2, 3 α1, 5 α1, 5 α2 chains (Elhers-Danlos) |
| ATP7A (ATPase copper transporter, Menkes disease)116 |
| Glucuronyl transferase-1 (GAG synthesis)117 |
| Dermatan-sulphate proteoglycans118, Biglycan119 |
| NOTCH1120,121 |
| TGF-b signalling pathways (loss of function) |
| TGFBR1 and TGFBR254,122 |
| SMAD355and SMAD456 |
| TGFβ-252and -β-353 |
| TGFβ-repressor SKI (Shprintzen-Goldberg syndrome)123 |
| Contractile proteins (loss of function) |
| MYH-11 (myosin heavy chain)46 |
| ACTA2 (SM Actin)45 |
| MYLCK (myosin LC kinase)47 |
| PKG (protein kinase G, gain of function)48 |
| vSMC metabolism |
| Methionine adenyl transferase124 |
| Glucose transporter (SLC2A10)125 |
| FOXE3 (Transcription factor)126 |
Another group of altered genes that predispose to TAAs and aortic dissections are genes encoding either the major structural components of the contractile unit in vSMCs or the enzymes that control vSMC contractile tone. Smooth muscle α-actin (SM α-actin), a major protein in vSMCs, is synthesized as a monomer that polymerizes to form the thin filament in the contractile unit. Heterozygous mutations, which are overwhelmingly missense mutations predicted to lead to production of a mutant monomer, predispose to TAAs and dissections.45 The mutant monomers alter the function of the thin filament, including decreasing the tensegrity pathways, the stability of the contractile filaments and decreasing the movement of the filament by the myosin motor, and thus are predicted to decrease force generation by the vSMCs. The thick filaments are composed of a smooth muscle-specific isoform of myosin heavy chain dimer (SM MHC; encoded by MYH11), and four light chains (LCs), two regulatory LCs and two essential LCs. Heterozygous MYH11 mutations also lead to an inherited predisposition for TAAs and aortic dissections.46 These exonic mutations, localized in the rod domain of MYH11, perturb the quaternary structure of the thick filament. Phosphorylation of the LC on the myosin thick filament is necessary to activate the force generating cycle of SM myosin motor heads with the actin filaments. Heterozygous loss-of-function mutations in the gene (MYLK) encoding the dedicated kinase phosphorylating the LC, myosin LC kinase, are a cause of heritable thoracic aortic disease.47 A single heterozygous gain-of-function PRKG1 mutation, p.Arg177Gln (designated R177Q), also causes a familial form of heritable thoracic aortic disease.48 PRKG1 encodes a Type I cGMP-dependent protein kinase (PKG-1), which is activated upon binding of cGMP and controls vSMC relaxation, in part through activation of the phosphatase that de-phosphorylates the LC. In summary, mutations in ACTA2, MYH11, MYLK, and PRKG1 are all predicted to disrupt force generation, especially in vSMCs, promoting TAAs and acute aortic dissections. In contrast, loss of function mutations in the NO signalling molecular pathway in vSMCs is associated with a familial form of coronary artery disease49 and Moyamoya neurovascular disease.50
This role of vSMC tone has been also recently highlighted by two experimental studies in mice. In the first one, Doyle and colleagues explored the effects of calcium channel blockers in a genetic model of Marfan mice.39 In this model, amlodipine and verapamil exacerbated the progression of the aortic dilation (monitored by echography) and decreased the survival rate, due to an increased frequency of acute ruptures (dissections) during the 3 months of calcium blocker administration. These clinical data were confirmed by the histology of the aortic wall, showing a more important fragmentation of the elastic fibres under calcium blocker treatment. They retrospectively confirmed these data in Marfan patients receiving calcium blockers (more surgery and more rapid progression of aortic dilation) as compared with other anti-hypertensive agents.
In their recent study, Oller et al.,51 explored the impact of ADAMTS1 heterozygous K.O. in mice. They describe that the development of TAADs in these mice is dependent on inducible Nitric Oxide Synthase (iNOS or NOS2) overexpression by aortic vSMCs. iNOS induction is associated with a powerful inhibition of vSMC contractile tone. This experimental pathology is rescued, including angiotensin II-induced aortic dissection in this model, by inhibition of iNOS by L-NAME, a general inhibitor of NOSs, and by 1400 W (GW 274150) a specific inhibitor of iNOS. These experimental results (ADAMTS1 decreased expression and iNOS overexpression in vSMCs) were extended to a small series of TAA samples in MFS patients as compared with healthy human aortas. It is important to note that vSMCs cannot generate effective tensegrity unless the cells are anchored to the ECM. As mentioned previously, in the aorta, vSMCs are anchored to elastin by microfibrils. FBN1 mutations promote disruption of these connections and Fbn1 mutations in mice have been shown to decrease tensegrity of vSMCs in the aorta. The relationship between contraction/relaxation of vSMCs and outward hydraulic conductance through the wall has been recently explored ex vivo in rat aorta18 showing that induced SMC tone by addition of catecholamine, decreased mass transport through the wall. The relationship between a chronic defect in vSMC tone generation and changes in arterial wall permeability to water and plasma proteins remains to be established in vivo in human TAAs.
Heritable alterations of the genes that encode proteins in the canonical TGF-β signalling pathway can also predispose to heritable thoracic aortic disease, including TGF-β ligands (TGFB2, TGFB3),52,53 TGF-β receptors Types I and II (TGFBR1, TGFBR2)54and regulatory SMADs (SMAD3, SMAD4).55,56 Although initial studies suggested that increased TGF-β signalling was the primary driver for thoracic aortic disease, the mutations disrupting the TGF-β pathway are predicted or have been shown to decrease TGF-β signalling, limiting the ability of the vSMCs to repair the wall in response to proteolytic injury.38,57,58
5.2 vSMCs activate blood-borne proteases
In relation to the principle of advective mass transport of plasma proteins, the difference between results of proteomic analysis of the aortic wall, in which plasma proteins represent 30% of the arterial wall proteome, and results of transcriptomics, where 100% of the mRNA represent wall cell genomics, provides evidence of plasma protein enrichment of the wall. In particular, the convection of plasma proteins is largely enhanced by the increase in wall permeability in TAA (Figure 3). For example, the albumin (a neutral protein synthesized by the liver and secreted into the plasma) concentration is increased by 60% within the TAA wall as compared with healthy aorta (Figure 3), demonstrating the change in wall permeability. This increase in permeability is determined by the wall structure and function, including endothelial integrity/desintegrity, elastic network degradation, and potentially, a decrease in wall tensegrity via a decrease in vSMC tone.9 Conversely, percolation of blood components through the arterial wall may not be neutral, but could also impact permeability by modifying the connections between cells and matrix within the wall. In this paradigm, blood-borne components could injure the arterial wall. Thus, outward hydraulic conductance of blood-borne components is the largest common denominator of all TAAD, whatever their aetiology.
Figure 3.
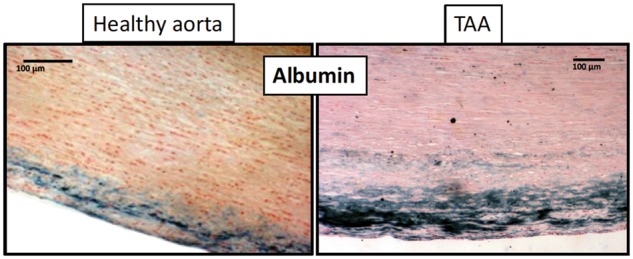
Neutral albumin staining (in blue) in the luminal media of healthy aorta vs. TAA.
Since the hydrophobic, fibrillar ECM is insoluble, the convection and interactions of plama zymogens with the wall components are of particular pathophysiological interest. We observed in human TAA tissues, whatever the underlying aetiology, that prothrombin21 and plasminogen59,60 were present in greater concentrations when compared with healthy aortic wall and could be activated in the TAA wall. Plasminogen is of particular interest because it can be activated by vSMC membranes, via an S100A4/annexin A2 heterotetramer, exposing terminal lysine-binding sites and forming ternary complexes with t-PA. A role for urokinase (u-PA) cannot be excluded.61 Plasmin is able to induce vSMC detachment from ECM (anoikis) by degrading fibronectin, one of the main adhesion proteins for vSMC integrins.62 Plasmin is the main enzyme able to mobilize TGF-β from its storage site associated with latent TGF-β-binding proteins in the ECM. Furthermore, plasmin is able to convert inactive pro-MMPs into active MMPs. In our studies, we observed that all components of the fibrinolytic system were upregulated in the TAA media, including S100A4 (mRNA and protein, providing evidence of vSMC phenotypic switching), t-PA (mRNA and protein), u-PA (mRNA and protein), and plasminogen (plasmin activity and plasmin–antiplasmin complexes).59 In contrast, plasminogen mRNA is not detected in the aortic wall, whereas the protein is, further supporting the hypothesis that it is outwardly convected from the blood.
5.3 vSMCs extracellularly accumulate GAGs
One of the common characteristics of TAA, dissections and aging in the aorta is the presence of mucoid ECM accumulation (MEMA),63 composed of alcianophilic GAGS. This osmotically active GAG accumulation evolves towards extracellular watery vacuoles, originally improperly named ‘cysts’ (but there are no ‘cysts’ because there are no capsule membranes). This mucoid degeneration is not specific of aortopathy and can also be observed in tendinous and cartilaginous, hydrophilic, GAG-rich tissues. Aortic GAG chain synthesis by vSMCs is under the control of the canonical TGF-β pathway, involving smad2 linker region phosphorylation by ERK.64 This signalling pathway activates the expression of GAG chain-synthesizing enzymes: xylosyltransferase, chondroitin sulphate synthase, and chondroitin sulfotransferase-1. GAGs are highly hydrophilic and their accumulation in specific areas leads to localized interstitial fluid retention potentially favouring the risk of dissection. Moreover areas of MEMA are able to retain specific MMPs, particularly MMP-3 (stromelysin) and MMP-7 (matrilysin), which possess a domain which interacts with the anionic structure of GAGs.65
5.4 vSMC death
Cell adhesion and tensegrity66 are requisites for cell survival within tissues. This concept is particularly relevant for vSMC differentiation, including acquisition of actomyosin complexes, allowing contractility in response to sympathetic and other stimuli, and survival within the aortic wall tissue. There is a direct relationship between tensegrity and the ability of vSMCs to resist and survive in a microenvironment of high mechanical stress. Conversely, any injury to, or defects of, the molecular tensegrity cascade67 from fibrillar ECM, adhesive intermediate proteins such as fibronectin, integrins, and focal adhesion kinases (FAK), cytoskeleton and actomyosin can induce partial or complete loss of tensegrity causing anoikis/apoptosis10 of vSMCs. In this context the activity of focal adhesion complexes, involving FAK, Src, talin, vinculin, paxillin, stimulates MEK and ERK signalling pathways and therefore promotes cell survival and growth by inhibiting anoikis signalling.68 Conversely defects in the tensegrity molecular cascade, endocellular (decrease in vSMC tone of either genetic or pharmacological origin) or extracellular (ECM defect of either genetic or pharmacological origin, pericellular proteolysis), promote cell apoptosis/anoikis by PI3K/AKT and NFkB pathways. In this context, a polycationic micro-environment is highly cytotoxic, promoting anoikis/apoptosis,69 whereas poly-anions, including negatively charged polysaccharides, are protective.70 As described earlier, activation of plasminogen on vSMC membranes by t-PA or u-PA/u-PAR, may promote cell detachment and therefore apoptosis/anoikis, mainly by degrading fibonectin which is the principal pericellular adhesive protein for vSMCs.62 Conversely, the secretion and pericellular retention of tissue anti-proteases, including Protease Nexin-1 (PN-1)71or plasminogen activator inhibitor (PAI-1),72 protect against apoptosis/anoikis.
6. vSMC responses to proteolytic injury
6.1 Transforming growth factor-β
TGF-β is a ubiquitous protein with a molecular mass of 25 kD which plays a role in tissue repair. In the aortic wall, TGF-β is secreted from vSMCs as a latent complex of TGF-β, the latency-associated peptide (LAP, interfering with αv integrin), and a molecule of latent-binding protein (LTBP).73 This inactive complex is stored within the ECM (fibrillin, fibronectin, and elastin) and is released by proteolytic activity, mainly involving plasmin and MMPs, but is also released when vSMC contractile forces stretch the ECM via integrins.74,75 Activation of TGF-β allowing receptor binding requires its dissociation from LAP. SMCs also possess TGFReceptors which are coupled to the canonical SMAD2 intracellular pathway, involving SMAD2 phosphorylation and nuclear translocation, inducing expression of numerous genes (genes coding for ECM, antiproteases, LTBP, etc). The majority of these cascade proteins present several isoforms. Canonical TGF-β extra- and intracellular pathways are physiologically involved in the protection and/or restoration of the mechano-protective function of ECM during proteolytic injury and degradation. Because of the cyclic haemodynamic stretching of the aortic ECM during the cardiac cycle, and the contractile tone of the aortic vSMCs, TGF-β release by the aortic ECM is physiologically stimulated, as a guardian of aortic ECM integrity. When ECM disruption and/or proteolytic injury, and/or specific defects in ECM interactions occur, the balance between latent, ECM-retained forms of TGF-β and active form is altered in favour of the latter, as in TAA (Figure 4).
Figure 4.

Different impacts of vSMC tensegrity on the main molecular components of TAA and AAD pathophysiology (ECM, TGF-β canonical pathway, SMC cytoskeletton and nucleoskeletton: (A) physiological tensegrity involving intracellular components and interactions within SMC, and ECM submitted to cyclic hemodynamic stretching; (B) impact of SMC relaxation on tensegrity and consequences. The behaviour of TGF-β in this context remains to be defined (?); (C) Defect in ECM (enzymatic, genetic, and pharmacologic) released active TGF-β; (D) Progressive dilation increases stretching and induces chromatin remodelling via tensegrity-induced cyto/nucleoskeletton more interactions.
For instance, recent studies76,77 have explored this molecular diversity in the context of Marfan models. A first study of the New York Marfan group, using a fibrillin-1- deficient model,78 explored the specificity of LTBP isoform binding to microfibrils, and demonstrated that LTBP-1 mainly binds to fibronectin, whereas LTBP-2 and -3 mainly bind to fibrillin.76 In a second study, the same group demonstrated, in the same model, that FBN mutations disrupt the interaction of LTBP-3 with fibrillin, causing aneurysm formation, elastin degradation, and lethal dissections, whereas genetic deletion of LTBP-3 rescues this morbid phenotype. Of note, in this model, fibrillin deficiency is associated with experimental TAAD but not with a defect in elastin maturation.78 These important studies, limited to Marfan mice, but not extended to TAAD, illustrate the complexity introduced by molecular isoforms in the genetics as well as pathophysiology, and the potential functional overlap of different molecules, and the necessary condition of proteolysis to produce elastin breakdown.
In our first study on the tissue TGF-β pathway in human healthy aorta and in TAA, we observed an increased TGF-β1 storage in TAA within the ECM without changes in TGF-β1synthesis, but an increase in LTBP synthesis.79 In healthy ascending aorta as in TAA, TGF-β accumulates in the external third of the media, with a gradient similar to that of vSMC myosin where the concentration is greatest in proximity to the adventitial sympathetic contractile stimuli.
In this context, it has been demonstrated that TGF-β overexpression is protective, limiting aneurysmal progression in rats.57 More recently, injections of TGF-β-neutralizing antibodies promoted aortic dissections and death in response to AngioII infusion in mice.38 These early results have now been confirmed by different groups.80–82 These repeated experimental observations, associated with clinical observations and therapeutic assays, lead to the conclusion that strategies aimed at inhibiting canonical TGF-β-dependent signalling are unlikely to provide any benefit to patients with TAADs58.
6.2 Epigenetic adaptation of vSMCs in TAA
6.2.1 Epigenetic modifications
Epigenetic modifications can be defined as the introduction of new stable heritable traits independently of changes in the DNA sequence. In many cardiovascular disorders, significant epigenetic modifications have been shown to affect disease development or progression. Epigenetic modifications encompass different mechanisms: modifications of DNA-associated histones, DNA methylation and non-coding RNA-mediated modifications. These mechanisms target DNA molecules, transcriptional machinery or transcription products, resulting in modulation of gene expression and consequent protein synthesis.
6.2.2 Histones
An epigenetically-determined constitutive overexpression of active Smad2 in vSMCs was identified in TAAs.83 By CHIP (CHromatin Immuno Precipitation) assays on human tissues and cells, it was shown that this constitutive overexpression involved the use a Smad2 alternative promoter that was related to the acetylation of histone, the recruitment of histone acetyl transferase, a shift of Myc repressor transcription factor to P53 activator, where the TRRAP co-factor facilitated the formation of this new molecular complex.84 The epigenetic nature of this phenotypic change was confirmed by the demonstration that this phenotype (constitutive overexpression of Smad2 as compared with vSMCs derived from healthy aorta) is conserved, in primary culture of vSMCs derived from TAA tissue, throughout successive passages. Moreover, vSMCs derived from TAA no longer responded to exogenous TGF-β1, whereas those derived from healthy aorta did. In parallel, TAA-derived vSMCs are more resistant to plasminogen-induced anoikis than healthy aorta-derived cells. This chromatin remodelling is potentially related to the modifications in mechanical strain which occur in TAAs. Indeed, the chronic dilation characteristic of TAAs is responsible for an increase in the wall tension according to Laplace’s law. The nuclear envelope is mechanically coupled to other vectors of mechanotransduction (tensegrity) via vSMC adhesion to the ECM, coupling of integrins to intracellular actin and linkers of nucleoskeleton to cytoskeleton.85,86 It has been proposed that the mechanical environment impacts the chromatin state of vSMCs thereby controlling vascular gene expression and function.87 It is likely that the increased wall tension in TAAs modifying mechanotransduction between matrix and nuclei will have an impact on the chromatin remodelling in vSMCs, although this issue has never been directly addressed. Interestingly, this chromatin remodelling is observed only during progressive chronic dilation, i.e. a progressive increase in wall tension, but not during acute intramural rupture.88 Thereby chromatin remodelling in vSMCs may be a hallmark of TAAs as compared with aortic dissection.
6.2.3 DNA methylation
This is carried out by DNA methyl transferase which covalently, but reversibly, binds a methyl group to the DNA base cytosine, which limits the transcription factor accessibility to the methylated sequence. Therefore, DNA methylation usually leads to repression of gene transcription. One study reports identification of abnormal DNA methylation in vSMCs from TAAs associated with BAV,89 but not in those from TAAs associated with TAV. No vSMCs from healthy aorta were studied here.
6.2.4 Non-coding RNAs
Non-coding RNAs are defined as RNA molecules lacking protein-coding potential. They are generally classified according to their size: small non-coding RNAs contain <200 nucleotides (miRNAs) while long non-coding RNAs contain at least 200 base pairs. These epigenetic aspects are more well-documented than DNA methylation. Different studies underline the impact of miRNA-29a and b on aneurysms in general, and more particularly on non-syndromic and Marfan TAA and dissections. The non-coding RNAs and their impact on vSMCs are reviewed in this issue (Leeper N.J. and Maegdefessel L.).
6.3 Increase in antiprotease secretion by vSMCs
The canonical TGF-β/Smad 2 pathway controls the expression of numerous genes, including genes responsible for ECM synthesis and maturation. It can be triggered either by TGF-β1 release from its ECM storage site and/or by the epigenetic overexpression and nuclear translocation of Smad2. Connective Tissue Growth Factor gene is the archetypal gene controlled by TGF/Smad2 pathway directly involved in ECM synthesis and modelling.90 We observed an increase in fibronectin turn-over and LTBP accumulation in TAA as compared with healthy human aorta. Because we were interested in plasminogen activation by vSMCs in TAA, we studied the impact of the constitutive overexpression of Smad2 on antiprotease synthesis and accumulation. We previously showed that PN-1 a tissue inhibitor of thrombin, plasmin, t-PA and u-PA, is highly expressed by vSMCs in response to TGF-β1.91 We also observed a high concentration of PN-1 in TAA media as compared with healthy aorta. We demonstrated that this PN-1 enrichment is under the control of Smad2 both in aortic tissue and in cultured vSMCs. By CHIPS assay we observed a highly significant increase in Smad2 binding to the PN-1 promoter, leading to overexpression and accumulation of the protein. Similar results, but less specific of Smad2, were reported for the PAI-1 promotor. These epigenetic modifications make TAA-derived cultured vSMCs more resistant to plasminogen-induced anoikis62 than healthy aorta-derived cells.
6.4 vSMCs clear protease/antiprotease complexes
It is well accepted that vSMCs in the arterial wall are able to ingest macromolecules by endocytosis,92,93 via scavenger receptors, and to engulf particles94 and cells by phagocytosis.95 LRP-1 (LDL receptor-related protein-1) is a major scavenger receptor for modified LDL in vSMCs,92 but also an endocytic receptor for protease/antiprotease complexes.96 We recently observed that circulating plasminogen, which had entered the aortic wall and had been converted into plasmin on vSMC contact, can bind to tissue PN-1 present in the pericellular GAG environment. Plasmin/PN-1 complexes can then be endocytosed by vSMCs in an LRP-1 dependent manner, whereas plasmin alone cannot.5 LRP-1 is also able to engulf MMP/TIMP complexes,97 so that this system may be involved the in situ clearance of protease/antiprotease complexes by vSMCs. Therefore, this clearance function of vSMCs, which is dependent on the in situ presence of antiproteases, may limit proteolytic injury within the aortic wall. Also, in keeping with this hypothesis, a decrease in LRP-1 expressed by vSMCs in genetically modified mice leads to aortic aneurysm formation.98,99 In parallel, a mutation on the LRP-1 gene has been recently published in a chinese Marfan-like family100 and an LRP-1 variant has been associated with AAA in humans.101
6.5 vSMCs induce inward neo-angiogenesis
Neo-angiogenesis from the adventitia to the medial layer is another consequence of both outward convection of mediators through the aortic wall, and the avascular nature of the aortic tissue. In the ascending aorta, only the external quarter of the media is vascularized, i.e. contains arterioles, capillaries and venules. However, there are no lymphatics in the medial layer. This external physiological vascularisation arises from the adventitia and is relatively scarce in normal aorta, but can extend across the full medial thickness in TAA in humans. As we have demonstrated in initial human aortic atheroma,102 this inward neo-angiogenesis, generated by sprouting of endothelial cells from the adventitia or external media, is related to the outward convection of angiogenic mediators synthesized and secreted by vSMCs. In contrast to atheroma,102 the inward neo-angiogenesis present in TAA is not related to lipid mediators or VEGF overexpression and secretion by vSMCs. Similarly, the markers of hypoxia, specifically HIF and sirtuine-1, are not altered in TAA when compared with control aorta. In contrast other angiogenic mediators are increased in the media of TAA when compared with healthy aorta60 (angiopetin-1 and -2 (proteins and mRNA), thrombospodin-1 (TSP-1) and -2, platelet-derived endothelial growth factor and IGFBP-1. Although TSP-1 and angiopoietin-1 overexpression is clear, the stimuli have not yet been determined. Neo-angiogenesis seems to be more pronounced in degenerative and Marfan TAA, whereas, in degenerative forms, a patchy intimal proliferation of SMCs is observed.
7. Aneurysms vs. dissections
Similar activation of plasminogen and the presence of areas of mucoid degeneration are observed in both chronic dilation (TAA) and acute dissection. However, in the aorta after dissection no epigenetic overexpression of Smad2 is observed, which limits the activation of TGF-β pathway and the ability of vSMCs to prevent acute intramural rupture (PN1 secretion…) is thus limited88 (Figure 5).
Figure 5.

Schematic representation of the pathophysiological determinants of TAA and AAD and the central role of vSMCs.
8. Conclusions
As described earlier, TAA in humans is a model for the interaction between vSMCs and outwardly-convected plasma components. Syndromic or non-syndromic monogenic diseases secondary to mutations in a gene altering the contractile apparatus of the vSMC, an ECM protein, or a protein of the canonical TGF-β signalling pathway have the potential to sensitize the aortic wall to plasma-borne, proteolytic injury.4 Disruption of the ECM or TGF-β signalling or an excess of vSMC relaxation in the aorta may lead to aneurysm formation involving the aortic root. As first illustrated by Leonardo da Vinci, the sinuses of Valsalva are the site of physiological vortexing of blood during diastole, in relation to the closing of the aortic valve, blood stagnation in the aorta, and coronary inflow. This blood stagnation and vortexes could potentially enhance outward hydraulic conductance of plasma components through the aortic wall.
In contrast, in TAA associated with BAVs, haemodynamic modifications may promote aneurysmal development on the outer curvature of the ascending aorta. Morphological ovalization of the aortic ring associated with BAVs alters the blood flow patterns in the ascending aorta, creating a hot spot of velocity vector dispersion (Tranverse Wall Shear Stress103) and mechanical impedance on the convex side of the ascending aorta.
vSMCs, as the main mesenchymal cells of the aortic wall, play a central role in the genesis and evolution of TAA and D, involving different vSMC functions and different extracellular and intracellular signalling pathways within the aortic wall. vSMC defects may be pathogenic in TAA and D, but TGF-β activation and vSMC responses to aneurysmal injury may also represent repair mechanisms. Despite important progress in understanding TAA & D pathophysiology, some interactions between vSMC physiology and pathology, remain to be further explored. This may be crucial for the development of new therapeutic approaches, potentially involving other antihypertensive compounds associated with higher sympathetic activity, blockade of plasmin generation, and specific inhibition of iNOS signalling, for preventing the development of aneurysms of the ascending aorta.
Acknowledgements
We aknowledge Mary Osborne-Pellegrin for editing English of this review.
Conflict of interest: none declared.
Funding
The following sources provided funding for these studies: RO1 HL62594, P01HL110869-01, John Ritter Foundation, and Richard T. Pisani Funds (D.M.M.), Fighting Aneurysmal Disease (FAD, EU FP-7 200647), ANR (French National Research Agency) NONAGE and Inserm (J.B.M and G.J.).
References
- 1. Michel J-B, Biology of Vascular Wall Dilation and Rupture. Oxford, UK: Oxford University Press; 2017. [Google Scholar]
- 2. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB.. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 2012;95:194–204. [DOI] [PubMed] [Google Scholar]
- 3. Michel JB, Martin-Ventura JL, Egido J, Sakalihasan N, Treska V, Lindholt J, Allaire E, Thorsteinsdottir U, Cockerill G, Swedenborg J, Consortium FE.. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc Res 2011;90:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jondeau G, Michel JB, Boileau C.. The translational science of Marfan syndrome. Heart 2011;97:1206–1214. [DOI] [PubMed] [Google Scholar]
- 5. Boukais K, Borges LF, Venisse L, Touat Z, François D, Arocas V, Jondeau G, Declerk P, Bouton MC, Jb M.. Clearance of plasmin-PN-1 complexes by vascular smooth muscle cells in human aneurysm of the ascending aorta. Cardiovascular Pathology 2017;(in press). [DOI] [PubMed] [Google Scholar]
- 6. Schlatmann TJ, Becker AE.. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol 1977;39:13–20. [DOI] [PubMed] [Google Scholar]
- 7. Michel JB, Heudes D, Michel O, Poitevin P, Philippe M, Scalbert E, Corman B, Levy BI.. Effect of chronic ANG I-converting enzyme inhibition on aging processes. II. Large arteries. Am J Physiol 1994;267:R124–R135. [DOI] [PubMed] [Google Scholar]
- 8. Virmani R, Avolio AP, Mergner WJ, Robinowitz M, Herderick EE, Cornhill JF, Guo SY, Liu TH, Ou DY, O'Rourke M.. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am J Pathol 1991;139:1119–1129. [PMC free article] [PubMed] [Google Scholar]
- 9. Michel JB, Thaunat O, Houard X, Meilhac O, Caligiuri G, Nicoletti A.. Topological determinants and consequences of adventitial responses to arterial wall injury. Arterioscler Thromb Vasc Biol 2007;27:1259–1268. [DOI] [PubMed] [Google Scholar]
- 10. Michel JB. Anoikis in the cardiovascular system: known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol 2003;23:2146–2154. [DOI] [PubMed] [Google Scholar]
- 11. Ingber DE. Tensegrity-based mechanosensing from macro to micro. Prog Biophys Mol Biol 2008;97:163–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Milewicz DM, Trybus KM, Guo DC, Sweeney HL, Regalado E, Kamm K, Stull JT.. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol 2017;37:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobrin PB, Baker WH, Gley WC.. Elastolytic and collagenolytic studies of arteries. Implications for the mechanical properties of aneurysms. Arch Surg 1984;119:405–409. [DOI] [PubMed] [Google Scholar]
- 14. Caro CG, Lever MJ.. Factors influencing arterial wall mass transport. Biorheology 1984;21:197–205. [DOI] [PubMed] [Google Scholar]
- 15. Caro CG. Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol 2009;29:158–161. [DOI] [PubMed] [Google Scholar]
- 16. Tada S, Tarbell JM.. Internal elastic lamina affects the distribution of macromolecules in the arterial wall: a computational study. Am J Physiol Heart Circ Physiol 2004;287:H905–H913. [DOI] [PubMed] [Google Scholar]
- 17. Chooi KY, Comerford A, Sherwin SJ, Weinberg PD.. Intimal and medial contributions to the hydraulic resistance of the arterial wall at different pressures: a combined computational and experimental study. J R Soc Interface 2016;13:20160234.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chooi KY, Comerford A, Sherwin SJ, Weinberg PD.. Noradrenaline has opposing effects on the hydraulic conductance of arterial intima and media. J Biomech 2017;54:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, Dai J, Gervais-Taurel M, Eberl G, Michel JB, Caligiuri G, Nicoletti A.. M1 macrophages act as LTbetaR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res 2014;101:434–443. [DOI] [PubMed] [Google Scholar]
- 20. Humphrey JD. Possible mechanical roles of glycosaminoglycans in thoracic aortic dissection and associations with dysregulated transforming growth factor-beta. J Vasc Res 2013;50:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Touat Z, Lepage L, Ollivier V, Nataf P, Hvass U, Labreuche J, Jandrot-Perrus M, Michel JB, Jondeau G.. Dilation-dependent activation of platelets and prothrombin in human thoracic ascending aortic aneurysm. Arterioscler Thromb Vasc Biol 2008;28:940–946. [DOI] [PubMed] [Google Scholar]
- 22. Tsai TT, Fattori R, Trimarchi S, Isselbacher E, Myrmel T, Evangelista A, Hutchison S, Sechtem U, Cooper JV, Smith DE, Pape L, Froehlich J, Raghupathy A, Januzzi JL, Eagle KA, Nienaber CA.. International Registry of Acute Aortic D. Long-term survival in patients presenting with type B acute aortic dissection: insights from the International Registry of Acute Aortic Dissection. Circulation 2006;114:2226–2231. [DOI] [PubMed] [Google Scholar]
- 23. Sakalihasan N, Nienaber CA, Hustinx R, Lovinfosse P, El Hachemi M, Cheramy-Bien JP, Seidel L, Lavigne JP, Quaniers J, Kerstenne MA, Courtois A, Ooms A, Albert A, Defraigne JO, Michel JB.. (Tissue PET) Vascular metabolic imaging and peripheral plasma biomarkers in the evolution of chronic aortic dissections. Eur Heart J Cardiovasc Imaging 2015;16:626–633. [DOI] [PubMed] [Google Scholar]
- 24. Osada H, Kyogoku M, Ishidou M, Morishima M, Nakajima H.. Aortic dissection in the outer third of the media: what is the role of the vasa vasorum in the triggering process? Eur J Cardiothorac Surg 2013;43:e82–e88. [DOI] [PubMed] [Google Scholar]
- 25. Senemaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R.. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2017;37:401–410. [DOI] [PubMed] [Google Scholar]
- 26. Milewicz DM, Prakash SK, Ramirez F.. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from predisposing genes and mouse models. Annu Rev Med 2017;68:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coutard M. Experimental cerebral aneurysms in the female heterozygous Blotchy mouse. Int J Exp Pathol 1999;80:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang DS, Yi X, Zhu XH, Wei X.. Experimental in vivo and ex vivo models for the study of human aortic dissection: promises and challenges. Am J Transl Res 2016;8:5125–5140. [PMC free article] [PubMed] [Google Scholar]
- 29. Krista LM, Waibel PE, Shoffner RN, Sautter JH.. Natural dissecting aneurysm (aortic rupture) and blood pressure in the turkey. Nature 1967;214:1162–1163. [DOI] [PubMed] [Google Scholar]
- 30. Simpson CF, Boucek RJ.. The B-aminopropionitrile-fed turkey: a model for detecting potential drug action on arterial tissue. Cardiovasc Res 1983;17:26–32. [DOI] [PubMed] [Google Scholar]
- 31. Hosoda Y, Iri H.. Angiolathyrism. 2. Elastin, collagen, and hexosamine content of the lathyritic rat aorta. Angiology 1967;18:616–627. [DOI] [PubMed] [Google Scholar]
- 32. Daugherty A, Manning MW, Cassis LA.. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000;105:1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saraff K, Babamusta F, Cassis LA, Daugherty A.. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2003;23:1621–1626. [DOI] [PubMed] [Google Scholar]
- 34. Trachet B, Fraga-Silva RA, Piersigilli A, Tedgui A, Sordet-Dessimoz J, Astolfo A, Van der Donckt C, Modregger P, Stampanoni MF, Segers P, Stergiopulos N.. Dissecting abdominal aortic aneurysm in Ang II-infused mice: suprarenal branch ruptures and apparent luminal dilatation. Cardiovasc Res 2015;105:213–222. [DOI] [PubMed] [Google Scholar]
- 35. Trachet B, Aslanidou L, Piersigilli A, Fraga-Silva RA, Sordet-Dessimoz J, Villanueva-Perez P, Stampanoni MFM, Stergiopulos N, Segers P.. Angiotensin II infusion into ApoE-/- mice: a model for aortic dissection rather than abdominal aortic aneurysm? Cardiovasc Res 2017;113:1230–1242. [DOI] [PubMed] [Google Scholar]
- 36. Faugeroux J, Nematalla H, Li W, Clement M, Robidel E, Frank M, Curis E, Ait-Oufella H, Caligiuri G, Nicoletti A, Hagege A, Messas E, Bruneval P, Jeunemaitre X, Bergaya S.. Angiotensin II promotes thoracic aortic dissections and ruptures in Col3a1 haploinsufficient mice. Hypertension 2013;62:203–208. [DOI] [PubMed] [Google Scholar]
- 37. Ren W, Liu Y, Wang X, Jia L, Piao C, Lan F, Du J.. beta-Aminopropionitrile monofumarate induces thoracic aortic dissection in C57BL/6 mice. Sci Rep 2016;6:28149.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z.. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest 2010;120:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doyle JJ, Doyle AJ, Wilson NK, Habashi JP, Bedja D, Whitworth RE, Lindsay ME, Schoenhoff F, Myers L, Huso N, Bachir S, Squires O, Rusholme B, Ehsan H, Huso D, Thomas CJ, Caulfield MJ, Van Eyk JE, Judge DP, Dietz HC; Gen TACRC Consortium ML. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brownstein AJ, Ziganshin BA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA.. Genes associated with thoracic aortic aneurysm and dissection: an update and clinical implications. Aorta 2016;5:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barbier M, Gross MS, Aubart M, Hanna N, Kessler K, Guo DC, Tosolini L, Ho-Tin-Noe B, Regalado E, Varret M, Abifadel M, Milleron O, Odent S, Dupuis-Girod S, Faivre L, Edouard T, Dulac Y, Busa T, Gouya L, Milewicz DM, Jondeau G, Boileau C.. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet 2014;95:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo D, Regalado ES, Gong L, Duan X, Santos-Cortez RL, Arnaud P, Ren Z, Cai B, Hostetler EM, Moran R, Liang D, Estrera AL, Safi HJ, Leal SM, Bamshad MJ, Shendure J, Nickerson DA, Jondeau G, Boileau C, Milewicz DM.. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ Res 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, Wheeldon N.. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Genet 2017;175:40–47. [DOI] [PubMed] [Google Scholar]
- 44. Parkin J. D, San Antonio J D, Persikov A V, Dagher H, Dalgleish R, Jensen S T, Jeunemaitre X, Savige J, Zheng J.. The collalphagen III fibril has a “flexi-rod” structure of flexible sequences interspersed with rigid bioactive domains including two with hemostatic roles. PLoS One 2017;12:e0175582.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM.. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007;39:1488–1493. [DOI] [PubMed] [Google Scholar]
- 46. Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X.. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 2006;38:343–349. [DOI] [PubMed] [Google Scholar]
- 47. Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM.. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 2010;87:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA, Gen TACRC, National Heart L Blood Institute Grand Opportunity Exome Sequencing P Kim C, Milewicz DM.. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet 2013;93:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, de Wit C, Kaiser F J, Braunholz D, Medack A, Fischer M, Zimmermann ME, Tennstedt S, Graf E, Eck S, Aherrahrou Z, Nahrstaedt J, Willenborg C, Bruse P, Brænne I, Nöthen MM, Hofmann P, Braund PS, Mergia E, Reinhard W, Burgdorf C, Schreiber S, Balmforth AJ, Hall AS, Bertram L, Steinhagen-Thiessen E, Li S-C, März W, Reilly M, Kathiresan S, McPherson R, Walter U, Ott J, Samani NJ, Strom TM, Meitinger T, Hengstenberg C, Schunkert H.. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 2013;504:432–436. [DOI] [PubMed] [Google Scholar]
- 50. Herve D, Philippi A, Belbouab R, Zerah M, Chabrier S, Collardeau-Frachon S, Bergametti F, Essongue A, Berrou E, Krivosic V, Sainte-Rose C, Houdart E, Adam F, Billiemaz K, Lebret M, Roman S, Passemard S, Boulday G, Delaforge A, Guey S, Dray X, Chabriat H, Brouckaert P, Bryckaert M, Tournier-Lasserve E.. Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet 2014;94:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oller J, Méndez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, Briones AM, Alberca R, Lozano-Vidal N, Hurlé MA, Milewicz D, Evangelista A, Salaices M, Nistal JF, Jiménez-Borreguero LJ, De Backer J, Campanero MR, Redondo JM.. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med 2017;23:200–212. [DOI] [PubMed] [Google Scholar]
- 52. Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, National Heart L, Blood Institute Go Exome Sequencing P, Jondeau G, Milewicz DM.. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 2012;44:916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JM, de Graaf BM, van de Beek G, Gallo E, Kruithof BP, Venselaar H, Myers LA, Laga S, Doyle AJ, Oswald G, van Cappellen GW, Yamanaka I, van der Helm RM, Beverloo B, de Klein A, Pardo L, Lammens M, Evers C, Devriendt K, Dumoulein M, Timmermans J, Bruggenwirth HT, Verheijen F, Rodrigus I, Baynam G, Kempers M, Saenen J, Van Craenenbroeck EM, Minatoya K, Matsukawa R, Tsukube T, Kubo N, Hofstra R, Goumans MJ, Bekkers JA, Roos-Hesselink JW, van de Laar IM, Dietz HC, Van Laer L, Morisaki T, Wessels MW, Loeys BL.. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol 2015;65:1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC.. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006;355:788–798. [DOI] [PubMed] [Google Scholar]
- 55. van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collee M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM.. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011;43:121–126. [DOI] [PubMed] [Google Scholar]
- 56. Teekakirikul P, Milewicz DM, Miller DT, Lacro RV, Regalado ES, Rosales AM, Ryan DP, Toler TL, Lin AE.. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet A 2013;161A:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dai J, Losy F, Guinault AM, Pages C, Anegon I, Desgranges P, Becquemin JP, Allaire E.. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation 2005;112:1008–1015. [DOI] [PubMed] [Google Scholar]
- 58. Mallat Z, Ait-Oufella H, Tedgui A.. The pathogenic transforming growth factor-beta overdrive hypothesis in aortic aneurysms and dissections: a mirage? Circ Res 2017;120:1718–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Borges LF, Gomez D, Quintana M, Touat Z, Jondeau G, Leclercq A, Meilhac O, Jandrot-Perrus M, Gutierrez PS, Freymuller E, Vranckx R, Michel JB.. Fibrinolytic activity is associated with presence of cystic medial degeneration in aneurysms of the ascending aorta. Histopathology 2010;57:917–932. [DOI] [PubMed] [Google Scholar]
- 60. Kessler K, Borges LF, Ho-Tin-Noé B, Jondeau G, Michel J-B, Vranckx R.. Angiogenesis and remodelling in human thoracic aortic aneurysms. Cardiovasc Res 2014;104:147–159. [DOI] [PubMed] [Google Scholar]
- 61. Carmeliet P, Moons L, Lijnen R, Baes M, Lemaitre V, Tipping P, Drew A, Eeckhout Y, Shapiro S, Lupu F, Collen D.. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet 1997;17:439–444. [DOI] [PubMed] [Google Scholar]
- 62. Meilhac O, Ho-Tin-Noé B, Houard X, Philippe M, Michel J-B, Anglés-Cano E.. Pericellular plasmin induces smooth muscle cell anoikis. Faseb J 2003;17:1301–1303. [DOI] [PubMed] [Google Scholar]
- 63. Halushka MK, Angelini A, Bartoloni G, Basso C, Batoroeva L, Bruneval P, Buja LM, Butany J, d'Amati G, Fallon JT, Gallagher PJ, Gittenberger-de Groot AC, Gouveia RH, Kholova I, Kelly KL, Leone O, Litovsky SH, Maleszewski JJ, Miller DV, Mitchell RN, Preston SD, Pucci A, Radio SJ, Rodriguez ER, Sheppard MN, Stone JR, Suvarna SK, Tan CD, Thiene G, Veinot JP, van der Wal AC.. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: iI. Noninflammatory degenerative diseases - nomenclature and diagnostic criteria. Cardiovasc Pathol 2016;25:247–257. [DOI] [PubMed] [Google Scholar]
- 64. Rostam MA, Kamato D, Piva TJ, Zheng W, Little PJ, Osman N.. The role of specific Smad linker region phosphorylation in TGF-beta mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell Signal 2016;28:956–966. [DOI] [PubMed] [Google Scholar]
- 65. Borges LF, Touat Z, Leclercq A, Zen AA, Jondeau G, Franc B, Philippe M, Meilhac O, Gutierrez PS, Michel JB.. Tissue diffusion and retention of metalloproteinases in ascending aortic aneurysms and dissections. Hum Pathol 2009;40:306–313. [DOI] [PubMed] [Google Scholar]
- 66. Ingber DE, Wang N, Stamenovic D.. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep Prog Phys 2014;77:046603.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ingber DE. Mechanical control of tissue morphogenesis during embryological development. Int J Dev Biol 2006;50:255–266. [DOI] [PubMed] [Google Scholar]
- 68. Lee S, Choi E, Cha MJ, Hwang KC.. Cell adhesion and long-term survival of transplanted mesenchymal stem cells: a prerequisite for cell therapy. Oxid Med Cell Longev 2015;2015:632902.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parhamifar L, Andersen H, Wu L, Hall A, Hudzech D, Moghimi SM.. Polycation-mediated integrated cell death processes. Adv Genet 2014;88:353–398. [DOI] [PubMed] [Google Scholar]
- 70. Cado G, Kerdjoudj H, Chassepot A, Lefort M, Benmlih K, Hemmerle J, Voegel JC, Jierry L, Schaaf P, Frere Y, Boulmedais F.. Polysaccharide films built by simultaneous or alternate spray: a rapid way to engineer biomaterial surfaces. Langmuir 2012;28:8470–8478. [DOI] [PubMed] [Google Scholar]
- 71. Rossignol P, Ho-Tin-Noé B, Vranckx R, Bouton M-C, Meilhac O, Lijnen H. R, Guillin M-C, Michel J-B, Anglés-Cano E.. Protease nexin-1 inhibits plasminogen activation-induced apoptosis of adherent cells. J Biol Chem 2004;279:10346–10356. [DOI] [PubMed] [Google Scholar]
- 72. Schneider DJ, Chen Y, Sobel BE.. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb Haemost 2008;100:1037–1040. [PubMed] [Google Scholar]
- 73. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB.. Latent TGF-beta-binding proteins. Matrix Biol 2015;47:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hinz B. The extracellular matrix and transforming growth factor-beta1: tale of a strained relationship. Matrix Biol 2015;47:54–65. [DOI] [PubMed] [Google Scholar]
- 75. Bochaton-Piallat ML, Gabbiani G, Hinz B.. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res 2016;5:752.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY, Rifkin DB.. Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol 2012;227:3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB.. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci U S A 2015;112:14012–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F.. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A 1999;96:3819–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gomez D, Al Haj Zen A, Borges LF, Philippe M, Gutierrez PS, Jondeau G, Michel JB, Vranckx R.. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 2009;218:131–142. [DOI] [PubMed] [Google Scholar]
- 80. Chen X, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, Daugherty A, Aikawa E.. TGF-beta Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS One 2016;11:e0153811.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Angelov SN, Hu JH, Wei H, Airhart N, Shi M, Dichek DA.. TGF-beta (transforming growth factor-beta) signaling protects the thoracic and abdominal aorta from angiotensin II-induced pathology by distinct mechanisms. Arterioscler Thromb Vasc Biol 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA.. Aortopathy in a mouse model of marfan syndrome is not mediated by altered transforming growth factor beta signaling. J Am Heart Assoc 2017;6:e004968.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB, Vranckx R.. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res 2011;89:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gomez D, Kessler K, Michel JB, Vranckx R.. Modifications of chromatin dynamics control smad2 pathway activation in aneurysmal smooth muscle cells. Circ Res 2013;113:881–890. [DOI] [PubMed] [Google Scholar]
- 85. Wang N, Tytell JD, Ingber DE.. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol 2009;10:75–82. [DOI] [PubMed] [Google Scholar]
- 86. Isermann P, Lammerding J.. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol 2013;23:R1113–R1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen LJ, Wei SY, Chiu JJ.. Mechanical regulation of epigenetics in vascular biology and pathobiology. J Cell Mol Med 2013;17:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gomez D, Kessler K, Borges LF, Richard B, Touat Z, Ollivier V, Mansilla S, Bouton MC, Alkoder S, Nataf P, Jandrot-Perrus M, Jondeau G, Vranckx R, Michel JB.. Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler Thromb Vasc Biol 2013;33:2222–2232. [DOI] [PubMed] [Google Scholar]
- 89. Shah AA, Gregory SG, Krupp D, Feng S, Dorogi A, Haynes C, Grass E, Lin SS, Hauser ER, Kraus WE, Shah SH, Hughes GC.. Epigenetic profiling identifies novel genes for ascending aortic aneurysm formation with bicuspid aortic valves. Heart Surg Forum 2015;18:E134–E139. [DOI] [PubMed] [Google Scholar]
- 90. Ungvari Z, Valcarcel-Ares MN, Tarantini S, Yabluchanskiy A, Fulop GA, Kiss T, Csiszar A.. Connective tissue growth factor (CTGF) in age-related vascular pathologies. Geroscience 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bouton MC, Boulaftali Y, Richard B, Arocas V, Michel JB, Jandrot-Perrus M.. Emerging role of serpinE2/protease nexin-1 in hemostasis and vascular biology. Blood 2012;119:2452–2457. [DOI] [PubMed] [Google Scholar]
- 92. Costales P, Fuentes-Prior P, Castellano J, Revuelta-Lopez E, Corral-Rodríguez MÁ, Nasarre L, Badimon L, Llorente-Cortes V.. K Domain CR9 of low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is critical for aggregated LDL-induced foam cell formation from human vascular smooth muscle cells. J Biol Chem 2015;290:14852–14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boukais K, Bayles R, Borges LDF, Louedec L, Boulaftali Y, Ho-Tin-Noé B, Arocas V, Bouton M-C, Michel J-B.. Uptake of plasmin-PN-1 complexes in early human atheroma. Front Physiol 2016;7:273.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bennett MR, Gibson DF, Schwartz SM, Tait JF.. Binding and phagocytosis of apoptotic vascular smooth muscle cells is mediated in part by exposure of phosphatidylserine. Circ Res 1995;77:1136–1142. [DOI] [PubMed] [Google Scholar]
- 95. Delbosc S, Bayles RG, Laschet J, Ollivier V, Ho-Tin-Noé B, Touat Z, Deschildre C, Morvan M, Louedec L, Gouya L, Guedj K, Nicoletti A, Michel J-B.. Erythrocyte efferocytosis by the arterial wall promotes oxidation in early-stage atheroma in humans. Front Cardiovasc Med 2017;4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gettins PG, Olson ST.. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem J 2016;473:2273–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thevenard J, Verzeaux L, Devy J, Etique N, Jeanne A, Schneider C, Hachet C, Ferracci G, David M, Martiny L, Charpentier E, Khrestchatisky M, Rivera S, Dedieu S, Emonard H, Lenting PJ.. Low-density lipoprotein receptor-related protein-1 mediates endocytic clearance of tissue inhibitor of metalloproteinases-1 and promotes its cytokine-like activities. PLoS One 2014;9:e103839.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J.. LRP: role in vascular wall integrity and protection from atherosclerosis. Science 2003;300:329–332. [DOI] [PubMed] [Google Scholar]
- 99. Boucher P, Li W-P, Matz RL, Takayama Y, Auwerx J, Anderson RGW, Herz J, Hatakeyama M.. LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One 2007;2:e448.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li G, Yu J, Wang K, Wang B, Wang M, Zhang S, Qin S, Yu Z.. Exome sequencing identified new mutations in a Marfan syndrome family. Diagn Pathol 2014;9:25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Badger SA, Bradley DT, Burnand K, Child AH, Clough RE, Cockerill G, Hafez H, Scott DJ, Futers S, Johnson A, Sohrabi S, Smith A, Thompson MM, van Bockxmeer FM, Waltham M, Matthiasson SE, Thorleifsson G, Thorsteinsdottir U, Blankensteijn JD, Teijink JA, Wijmenga C, de Graaf J, Kiemeney LA, Assimes TL, McPherson R, Consortium CA, Global BC, Consortium D, Consortium V, Folkersen L, Franco-Cereceda A, Palmen J, Smith AJ, Sylvius N, Wild JB, Refstrup M, Edkins S, Gwilliam R, Hunt SE, Potter S, Lindholt JS, Frikke-Schmidt R, Tybjaerg-Hansen A, Hughes AE, Golledge J, Norman PE, van Rij A, Powell JT, Eriksson P, Stefansson K, Thompson JR, Humphries SE, Sayers RD, Deloukas P, Samani NJ.. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet 2011;89:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ho-Tin-Noé B, Michel J-B.. Initiation of angiogenesis in atherosclerosis: smooth muscle cells as mediators of the angiogenic response to atheroma formation. Trends Cardiovasc Med 2011;21:183–187. [DOI] [PubMed] [Google Scholar]
- 103. Mohamied Y, Sherwin SJ, Weinberg PD.. Understanding the fluid mechanics behind transverse wall shear stress. J Biomech 2017;50:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Andrews EJ, White WJ, Bullock LP.. Spontaneous aortic aneurysms in blotchy mice. Am J Pathol 1975;78:199–210. [PMC free article] [PubMed] [Google Scholar]
- 105. Ramirez F, Dietz HC.. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev 2007;17:252–258. [DOI] [PubMed] [Google Scholar]
- 106. Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R.. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation 2002;106:2503–2509. [DOI] [PubMed] [Google Scholar]
- 107. Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P.. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 2007;115:2731–2738. [DOI] [PubMed] [Google Scholar]
- 108. Retailleau K, Arhatte M, Demolombe S, Jodar M, Baudrie V, Offermanns S, Feng Y, Patel A, Honore E, Duprat F.. Smooth muscle filamin A is a major determinant of conduit artery structure and function at the adult stage. Pflugers Arch 2016;468:1151–1160. [DOI] [PubMed] [Google Scholar]
- 109. Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, Itoh H, Hori S, Aikawa N, Okada Y.. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation 2012;126:3070–3080. [DOI] [PubMed] [Google Scholar]
- 110. Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA.. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci U S A 2006;103:19836–19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, Bierhals AJ, Vuzman D, Brigham Genomic M, Mecham RP, Frank NY, Stitziel NO.. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A 2016;113:8759–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kaler SG, ATP7A-related copper transport disorders In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N (eds). GeneReviews(R). Seattle, WA: University of Washington; 1993, p1993–2018. [PubMed] [Google Scholar]
- 113. Szabo Z, Crepeau MW, Mitchell AL, Stephan MJ, Puntel RA, Yin Loke K, Kirk RC, Urban Z.. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet 2006;43:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A.. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet 2002;11:2113–2118. [DOI] [PubMed] [Google Scholar]
- 115. Capuano A, Bucciotti F, Farwell KD, Tippin Davis B, Mroske C, Hulick PJ, Weissman SM, Gao Q, Spessotto P, Colombatti A, Doliana R.. Diagnostic exome sequencing identifies a novel gene, EMILIN1, associated with autosomal-dominant hereditary connective tissue disease. Hum Mutat 2016;37:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. de Figueiredo Borges L, Martelli H, Fabre M, Touat Z, Jondeau G, Michel JB.. Histopathology of an iliac aneurysm in a case of Menkes disease. Pediatr Dev Pathol 2010;13:247–251. [DOI] [PubMed] [Google Scholar]
- 117. Baasanjav S, Al-Gazali L, Hashiguchi T, Mizumoto S, Fischer B, Horn D, Seelow D, Ali BR, Aziz SA, Langer R, Saleh AA, Becker C, Nurnberg G, Cantagrel V, Gleeson JG, Gomez D, Michel JB, Stricker S, Lindner TH, Nurnberg P, Sugahara K, Mundlos S, Hoffmann K.. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet 2011;89:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mizumoto S, Kosho T, Yamada S, Sugahara K.. Pathophysiological significance of dermatan sulfate proteoglycans revealed by human genetic disorders. Pharmaceuticals (Basel) 2017;10:34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Meester JA, Vandeweyer G, Pintelon I, Lammens M, Van Hoorick L, De Belder S, Waitzman K, Young L, Markham LW, Vogt J, Richer J, Beauchesne LM, Unger S, Superti-Furga A, Prsa M, Dhillon R, Reyniers E, Dietz HC, Wuyts W, Mortier G, Verstraeten A, Van Laer L, Loeys BL.. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med 2017;19:386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM. 3rd. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg 2007;134:290–296. [DOI] [PubMed] [Google Scholar]
- 121. Proost D, Vandeweyer G, Meester JA, Salemink S, Kempers M, Ingram C, Peeters N, Saenen J, Vrints C, Lacro RV, Roden D, Wuyts W, Dietz HC, Mortier G, Loeys BL, Van Laer L.. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat 2015;36:808–814. [DOI] [PubMed] [Google Scholar]
- 122. Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, De Backer J, Muino-Mosquera L, Naudion S, Zordan C, Morisaki T, Morisaki H, Von Kodolitsch Y, Dupuis-Girod S, Morris SA, Jeremy R, Odent S, Ades LC, Bakshi M, Holman K, LeMaire S, Milleron O, Langeois M, Spentchian M, Aubart M, Boileau C, Pyeritz R, Milewicz DM, Montalcino A. C. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet 2016;9:548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D, Gillis E, Mortier G, Homfray T, Sauls K, Norris RA, Huso ND, Leahy D, Mohr DW, Caulfield MJ, Scott AF, Destree A, Hennekam RC, Arn PH, Curry CJ, Van Laer L, McCallion AS, Loeys BL, Dietz HC.. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet 2012;44:1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Guo DC, Gong L, Regalado ES, Santos-Cortez RL, Zhao R, Cai B, Veeraraghavan S, Prakash SK, Johnson RJ, Muilenburg A, Willing M, Jondeau G, Boileau C, Pannu H, Moran R, Debacker J, GenTac Investigators NHL, Blood Institute Go Exome Sequencing P, Montalcino Aortic C, Bamshad MJ, Shendure J, Nickerson DA, Leal SM, Raman CS, Swindell EC, Milewicz DM.. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet 2015;96:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Callewaert BL, Willaert A, Kerstjens-Frederikse WS, De Backer J, Devriendt K, Albrecht B, Ramos-Arroyo MA, Doco-Fenzy M, Hennekam RC, Pyeritz RE, Krogmann ON, Gillessen-Kaesbach G, Wakeling EL, Nik-Zainal S, Francannet C, Mauran P, Booth C, Barrow M, Dekens R, Loeys BL, Coucke PJ, De Paepe AM.. Arterial tortuosity syndrome: clinical and molecular findings in 12 newly identified families. Hum Mutat 2008;29:150–158. [DOI] [PubMed] [Google Scholar]
- 126. Kuang SQ, Medina-Martinez O, Guo DC, Gong L, Regalado ES, Reynolds CL, Boileau C, Jondeau G, Prakash SK, Kwartler CS, Zhu LY, Peters AM, Duan XY, Bamshad MJ, Shendure J, Nickerson DA, Santos-Cortez RL, Dong X, Leal SM, Majesky MW, Swindell EC, Jamrich M, Milewicz DM.. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest 2016;126:948–961. [DOI] [PMC free article] [PubMed] [Google Scholar]