

Mutations in the Plasmodium falciparum k13 (Pfk13) gene are linked to delayed parasite clearance in response to artemisinin-based combination therapies (ACTs) in Southeast Asia. To explore the evolutionary rate and constraints acting on this gene, k13 orthologs from species sharing a recent common ancestor with P. falciparum and Plasmodium vivax were analyzed.
KEYWORDS: Plasmodium falciparum, Plasmodium vivax, artemisinin resistance, k13 gene, kelch propeller domain, rates of evolution
ABSTRACT
Mutations in the Plasmodium falciparum k13 (Pfk13) gene are linked to delayed parasite clearance in response to artemisinin-based combination therapies (ACTs) in Southeast Asia. To explore the evolutionary rate and constraints acting on this gene, k13 orthologs from species sharing a recent common ancestor with P. falciparum and Plasmodium vivax were analyzed. These comparative studies were followed by genetic polymorphism analyses within P. falciparum using 982 complete Pfk13 sequences from public databases and new data obtained by next-generation sequencing from African and Haitian isolates. Although k13 orthologs evolve at heterogeneous rates, the gene was conserved across the genus, with only synonymous substitutions being found at residues where mutations linked to the delayed parasite clearance phenotype have been reported. This suggests that those residues were under constraint from undergoing nonsynonymous changes during evolution of the genus. No fixed nonsynonymous differences were found between Pfk13 and its orthologs in closely related species found in African apes. This indicates that all nonsynonymous substitutions currently found in Pfk13 are younger than the time of divergence between P. falciparum and its closely related species. At the population level, no mutations linked to delayed parasite clearance were found in our samples from Africa and Haiti. However, there is a high number of single Pfk13 mutations segregating in P. falciparum populations, and two predominant alleles are distributed worldwide. This pattern is discussed in terms of how changes in the efficacy of natural selection, affected by population expansion, may have allowed for the emergence of mutations tolerant to ACTs.
INTRODUCTION
Antimalarial resistance is one of the greatest threats to malaria control and its eventual elimination (1, 2). The emergence of a delayed parasite clearance phenotype following artemisinin-based therapies in Plasmodium falciparum was first reported in 2007 and 2008 in western Cambodia and was later reported in Myanmar, Thailand, Vietnam, Lao People’s Democratic Republic (Lao PDR), and South China (2–7). The occurrence of this phenotype has raised concerns that it may spread to Africa or lead to resistance, following the previous path of chloroquine (CQ) and sulfadoxine-pyrimethamine (SP) resistance (8–9). However, there is currently no evidence that this phenotype has spread outside of Southeast Asia (SEA), nor is there evidence of the emergence of resistant parasites (2).
Artemisinin delayed parasite clearance was first defined and confirmed using in vitro ring-stage survival assays (RSAs) (1, 10, 11). A series of genome-wide association studies have shown that this phenotype is associated with nonsynonymous mutations in the propeller domain of a P. falciparum kelch gene (codons 441 to 726) found on chromosome 13 (Pfk13) (12, 13). To date, 14 independent Pfk13 mutations have been associated with clinical delayed parasite clearance (2), and of these, only 5 have been validated by in vivo and in vitro experiments (2): N458Y, Y493H, R539T, I543T, and C580Y. Furthermore, there is evidence of the independent emergence of the C580Y lineage across SEA (14, 15). The most frequent and widely detected Pfk13 mutations found in Cambodia, Vietnam, and Lao PDR are Y493H, R539T, I543T, P553L, and C580Y (5, 7). The mutations F446L, P553L, N458Y, R561H, P574L, and C580Y are the most frequently observed in western Thailand, Myanmar, and China (7, 16–18). Only two mutations, P553L and C580Y, are found across SEA, with the latter mutation being more widespread in eastern Thailand (16–18).
In contrast, a high number of single nonsynonymous mutations, including some associated with delayed parasite clearance, have been found at a low frequency in Africa (19). However, many of the mutations reported in Africa have not expanded to the local parasite populations (2, 19–23). In the case of South America, only Pfk13 C580Y mutant parasites have been found in Guyana, with the flanking microsatellite profiles being different from those observed in SEA, suggesting an independent origin (24). Studies in other countries in South America have searched for polymorphisms in the Pfk13 gene, but no evidence of these mutations has been found to date outside Guyana (25, 26).
Although our understanding of which specific mutations within the PfK13 domain are associated with the delayed parasite clearance phenotype is still incomplete (especially in the context of the genetic background of parasites outside of SEA), the identification of polymorphisms in the Pfk13 gene has been used as a valuable genetic marker for the rapid detection, tracking, and monitoring of this phenotype. Here, to better understand the evolution of the k13 gene, its divergence across the genus Plasmodium and its genetic variation in P. falciparum populations were studied. In particular, we explored the mode and rate of evolution of the k13 gene by analyzing its orthologs across the Plasmodium genus and 982 Pfk13 sequences, including those in public databases, along with new data obtained by next-generation sequencing of African and Haitian isolates.
RESULTS
Phylogenetic-based analyses of the k13 gene.
This investigation reports 10 new k13 orthologous gene sequences, obtained by nested PCR, from macaque and gibbon parasite strains and chimpanzees infected with Plasmodium reichenowi, Plasmodium billcollinsi, and Plasmodium gaboni. The sequences obtained here were aligned with those available in public databases (see Materials and Methods and Data Set S1 in the supplemental material) (27). The alignment constructed with the Plasmodium species orthologous k13 genes showed that the encoded protein is remarkably conserved across the genus. Estimates of the evolutionary divergence among orthologous k13 gene sequences were low at both the nucleotide and amino acid levels compared to that of other genes (28). The k13 gene was almost identical among species belonging to the subgenus Laverania. For example, the genetic distance between P. falciparum and Plasmodium praefalciparum from gorillas was 0.00, and that between P. falciparum and P. reichenowi from chimpanzees was only 0.01 ± 0.00 (Data Sets S2 and S3). Furthermore, all k13 orthologous genes displayed a high degree of conservation in the kelch propeller domain among Plasmodium spp. (Fig. 1). In particular, the number of base substitutions per site between the two major malarial parasite k13 orthologs (Pfk13 and Plasmodium vivax k13 [Pvk13]) was low (0.09 ± 0.01) (Data Set S2) and showed synonymous substitutions between P. falciparum and P. vivax, with the exception of V568I versus V568G in Pfk13 and Pvk13, respectively (Fig. 1; Data Sets S1 and S4).
FIG 1.

Alignment of the Plasmodium K13 kelch propeller domain. The Pfk13 mutations that have been associated with clinical delayed parasite clearance (2) are shown. The five mutations validated in vivo and in vitro are indicated with an asterisk. The Haemoproteus tartakovskyi (an avian haemosporidian parasite) k13 gene was included as an outgroup.
To compare all Plasmodium species k13 orthologous genes considered in this study (including different strains of some parasites), a Bayesian phylogeny was estimated using the nucleotide sequence alignment, given the high conservation at the protein level (Fig. 2). As expected, the phylogeny was consistent with earlier reports using putatively neutral genes, genes encoding merozoite antigens, and/or mitochondrial genomes (27–29). Overall, the Laverania subgenus, which includes P. falciparum, formed a monophyletic group, as did P. vivax and its closely related nonhuman primate malaria parasites from SEA and all rodent malaria parasites. All mammal Plasmodium species shared a common ancestor with avian Plasmodium species (Fig. 2).
FIG 2.

Bayesian phylogenetic hypothesis of Plasmodium parasites based on the k13 gene (42 sequences; 1,985 bp, excluding gaps). The values at the nodes are posterior probabilities (see Materials and Methods). Haemoproteus tartakovskyi (an avian haemosporidian parasite) k13 gene was included as an outgroup.
Then, using the Bayesian tree topology, the relative evolutionary rates of the k13 gene were estimated in a maximum likelihood framework with the RelTime method (30). This revealed a pattern in which there was a faster k13 evolutionary rate in malaria parasites infecting Southeast Asian macaques and their relatives, including the human malaria parasite P. vivax (Fig. 3). The k13 relative evolutionary rates were approximately 12 times higher in malaria parasites infecting Southeast Asian macaques and their relatives than in parasites from other mammalian hosts and African ape parasites. Differences in rates have also been observed in other genes in this clade, which includes the human parasite P. vivax (31, 32). Interestingly, Pfk13 also showed a k13 relative rate of evolution faster than and similar to that in SEA parasites (Fig. 3). The acceleration in the P. falciparum k13 evolutionary rate, however, did not translate into the gene, being under diversifying natural selection when P. falciparum diverged from its closest related species (see below).
FIG 3.
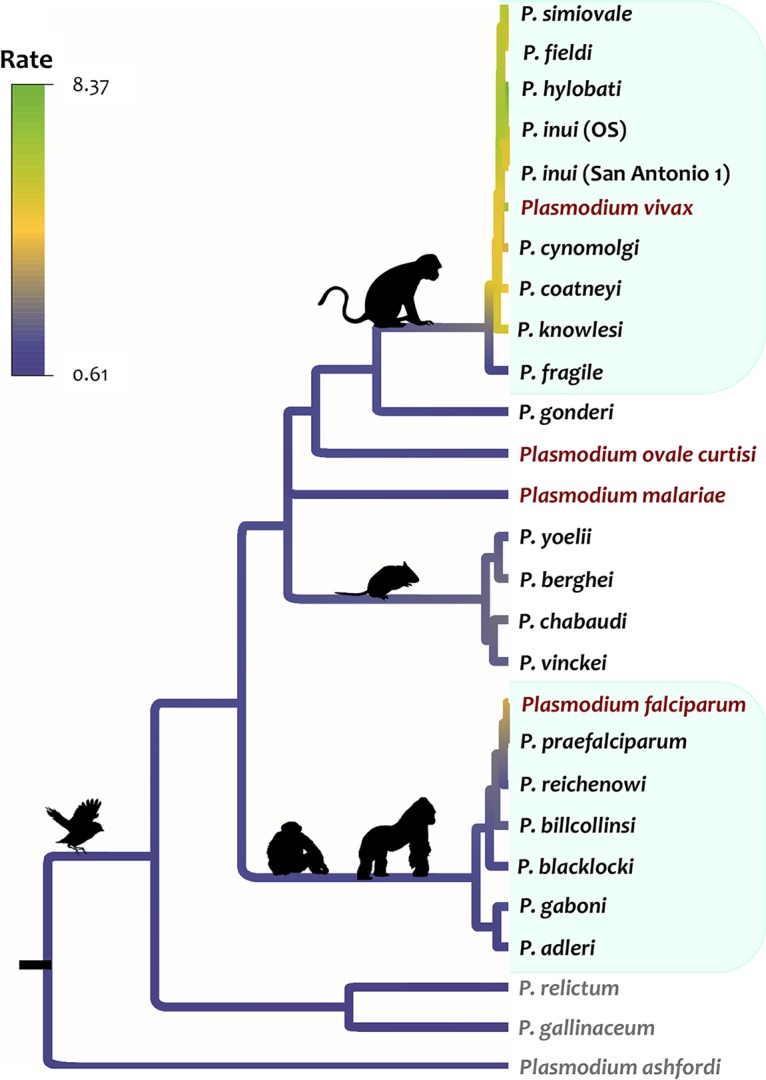
Relative evolutionary rates of the Plasmodium k13 gene. Branches are colored from high (green) to low (purple) according to their relative rates with respect to the rate at the root, which was set to 1, as estimated by RelTime without calibration constraints.
The relative evolutionary rate differences in the k13 gene between clades were further examined by estimating synonymous and nonsynonymous substitution rates per codon per million years (My) using a codon model implemented in the CodonRates (v1.0) program (33). In this analysis, the k13 nonsynonymous substitution rate (0.022 ± 0.012 per codon per My) was slightly higher (on average) than the synonymous substitution rate (0.012 ± 0.006 per codon per My) across the genus. The concordance statistic (S) showed that synonymous and nonsynonymous substitution rate changes over time were correlated (P < 0.5), suggesting that there was no pattern indicative of positive selection acting on this gene. The branch leading to P. falciparum (27, 28) from the common ancestor with its closest sister taxon (Plasmodium praefalciparum) showed a synonymous substitution rate of 0.005 per codon per My, while the nonsynonymous substitution rate was 0.001 per codon per My.
To further assess the role that selection may have had during the divergence of the k13 gene, we utilized methods implemented in the HyPhy package (34) (see Materials and Methods). A test that examines each individual site independently for evidence of episodic diversification (MEME [35]) found no evidence of episodic positive/diversifying selection acting on any site of the gene. In addition, a branch site-unrestricted statistical test for episodic diversification (BUSTED) (36), found no evidence (likelihood ratio test, P = 0.997, which is ≥0.05) of gene-wide episodic diversifying selection in any of the branches of the k13 phylogeny. Finally, RELAX (37), a test for identifying whether the strength of natural selection (positive or negative) has been relaxed or intensified along a specified set of test branches (e.g., P. falciparum), was not significant (K = 0.74, P = 0.665, likelihood ratio = 0.19). These results showed no evidence of positive diversifying selection acting on the k13 gene either during the evolution of the sampled Plasmodium species or in the lineages of P. falciparum or P. vivax.
Plasmodium falciparum population analyses.
Using an alignment with 982 complete k13 gene sequences from around the globe (519 from public databases and 463 reported in this study; see Materials and Methods and Data Set S5), low genetic diversity was found in the Pfk13 gene, as measured by π (0.0005 ± 0.0; Table 1). In contrast, the polymorphism measured using the number of polymorphic (segregating) sites had an S value of 146. The number of singleton variable sites, considering two variants, was 108. It is worth noting that no mutations associated with the delayed parasite clearance phenotype were found in our samples from Africa and Haiti. In the case of P. vivax, all 56 Pvk13 sequences available from the databases were identical.
TABLE 1.
Polymorphism found in k13 gene of Plasmodium falciparum
| Continent | π (SE) | dS | dN | dN − dS (SEa ) | P value (Z-statistic),b hypothesis testing result | S | Tajima’s D (P value) | No. of haplotypes (Hd) |
|---|---|---|---|---|---|---|---|---|
| Asia (n = 321) | 0.0004 (0.0002) | 5.77E−05 | 0.0005 | 0.0004 (0.0002) | 0.021 (2.338), dN > dS | 32 | −2.268 (<0.01) | 30 (0.663) |
| Africa (n = 411) | 0.0006 (0.0003) | 0.0004 | 0.0005 | 0.0002 (0.0003) | 0.552 (0.597), dN = dS | 85 | −2.654 (<0.001) | 86 (0.747) |
| Americas (n = 128) | 0.0003 (0.0002) | 0.0006 | 0.0002 | −0.0004 (0.0003) | 0.271 (−1.107), dN = dS | 19 | −2.306 (<0.01) | 16 (0.469) |
| Total (n = 982) | 0.0005 (0.0002) | 0.0003 | 0.0006 | −0.0003 (0.0003) | 0.317 (1.006), dN = dS | 146 | −2.676 (<0.001) | 147 (0.742) |
Standard error (SE) estimates are from 1,000 bootstrap replicates.
P values from the codon-based Z-test are shown. Significant values (P < 0.05) are in bold.
Evidence of natural selection acting on the Pfk13 polymorphism was explored by estimating the average pairwise number of synonymous substitutions per synonymous site (dS) and the average pairwise number of nonsynonymous substitutions per nonsynonymous site (dN). This revealed a significant (P < 0.05) excess of nonsynonymous over synonymous substitutions only in Asia, the region where mutations associated with the artemisinin delayed parasite clearance phenotype were first reported (Table 1).
A total of 147 Pfk13 haplotypes were found with a haplotype (allele) diversity (Hd) of 0.742 (standard deviation [SD] = 0.012). The uneven sampling effort did not allow us to estimate the haplotype relative frequencies per country. Overall, the median-joining network for the Pfk13 gene, estimated using Network software (Fluxus Technologies, 2011), showed two star-like shapes with at least two most predominant and common haplotypes (haplotype 1 [H1] and H2; Fig. 4A) shared by the three continents (Asia, Africa, and South America) with a broad distribution. Interestingly, one of the haplotypes (H1, n = 442, 45%) is mostly distributed in Asia, and the other (H2, n = 223, 22.7%) is mostly distributed in Africa. A third haplotype (H3, n = 50, 5%), in terms of frequency, is only found in Thailand (Fig. 4A). A noteworthy finding was that haplotype H3 contains the C580Y mutation found in the SEA region. In addition, a less frequent haplotype was found only on the China-Myanmar border (H4, n = 28, 2.9%) and had the F446I mutation, one of the most common mutations found in China and on the China-Myanmar border (Fig. 4B). However, most of the haplotypes were restricted to single countries (Fig. 4A).
FIG 4.

Median-joining network of the P. falciparum k13 haplotypes. (A) Median-joining network of the P. falciparum k13 haplotypes using 982 cosmopolitan complete sequences. All the k13 gene sequences obtained here for P. falciparum population samples (n = 463), as well as those available in the PlasmoDB and NCBI databases (n = 519), were included. Branch lengths are proportional to divergence; node sizes are proportional to the total haplotype frequencies. The network shows 147 haplotypes found in 982 sequences with a haplotype diversity (Hd) of 0.742 (SD = 0.012). Every color corresponds to a different geographic origin. Lines separating haplotypes represent mutational steps. The four most frequent haplotypes are indicated. (B) Frequencies of candidate/validated mutations in the propeller domain of the Pfk13 gene that have been associated with clinical delayed parasite clearance by country (Table 2). Frequencies were estimated using an alignment constructed with 3,916 partial (n = 2,934) and complete (n = 982) k13 gene sequences. C-M, China-Myanmar.
Finally, using 3,912 partial and complete Pfk13 sequences (Data Set S5), we estimated the frequencies (by country) of candidate/validated mutations associated with a clinical delay in parasite clearance (2). Those mutations, found in the propeller domain of a Pfk13 gene, are shown in Fig. 4B (see also Table 2). As previously reported, mutations F446I and C580Y were the most frequent mutations in this data set and were mostly found in Asia (Fig. 4B; Table 2; Data Set S6).
TABLE 2.
Number of Pfk13 sequences with mutations (candidates/validated) associated with clinical delayed parasite clearance (per codon and per country)a
| Country | No. of CDS |
No. of sequences inside propeller domain (codons 440 to 680) with: |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compl. | Partial | Total | P441L | F446I | G449A | N458Y | Y493H | N537D | G538V | R539T | I543T | P553L | R561H | V568G | P574L | C580Y | A675V | |
| Kenya | 14 | 405 | 419 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | 0 | 0 |
| Mali | 23 | 213 | 236 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nigeria | 29 | 27 | 56 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Cambodia | 2 | 5 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| China | 0 | 329 | 329 | 0 | 119 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 5 | 1 | 0 | 16 | 2 | 0 |
| China-Myanmar border | 57 | 0 | 57 | 0 | 28 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| India | 0 | 140 | 140 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Myanmar | 0 | 43 | 43 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 2 | 1 | 0 | 1 | 3 | 1 |
| Thailand | 260 | 973 | 1,233 | 8 | 12 | 10 | 11 | 9 | 0 | 6 | 14 | 0 | 0 | 21 | 0 | 63 | 298 | 0 |
| Papua New Guinea | 0 | 7 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Total | 385 | 2,142 | 2,527 | 9 | 161 | 11 | 14 | 9 | 0 | 7 | 16 | 0 | 7 | 25 | 4 | 83 | 306 | 2 |
The total number of complete (Compl.) and partial sequences obtained from the databases (PlasmoDB and NCBI) are shown. CDS, coding sequences.
DISCUSSION
Unlike other genes with mutations associated with antimalarial drug resistance, such as P. falciparum dhfr (Pfdhfr) (38, 39), the Pfk13 gene shows several single mutations at a low frequency, and many of these have no clear associations with the delayed parasite clearance phenotype (2). However, similar to genes such as Pfdhfr (28, 38), the Pfk13 mutations associated with this phenotype appeared solely in the P. falciparum lineage, indicating that they are all of recent origin (40). Although they evolve at heterogeneous rates, as indicated by the relative rate analysis, it is evident that orthologous k13 genes are highly conserved across Plasmodium species (in several species of human, nonhuman primate, other mammalian, and avian parasites). Evidence specific to the P. falciparum lineage can be found in the low nonsynonymous substitution rate estimated in this study. This finding suggests that the k13 gene was under a strong constraint (negative selection) from undergoing natural variation during its evolution in the genus Plasmodium.
There was no evidence of fixed nonsynonymous differences between P. falciparum and its closest related Laverania species (P. praefalciparum and P. reichenowi). Together, this evidence indicates that all Pfk13 mutations, including those associated with delayed parasite clearance, are of recent occurrence. The presence of several Pfk13 mutations found at a low frequency translates into the extraordinary haplotype diversity found in this study. A naive interpretation of this pattern could find it to be contradictory to the strong conservation observed among Plasmodium species at the protein level (indicative of negative selection). However, this finding is consistent with the proposed historical population expansion of P. falciparum following expansion of the human population (41). Population expansions can increase the number of segregating mutations (even semideleterious mutations) by making negative selection less effective (42, 43). Nonetheless, as parasite populations expand, negative selection becomes more efficient in eliminating those semideleterious mutations and maintaining them at a low frequency. The spatial differential distribution of those Pfk13 mutations at a low frequency is also expected because P. falciparum is not a single population, so there are local population expansions where different mutations occur. This pattern of population expansions is summarized in Tajima’s D results, where the low value of π and a high number of segregating sites (mostly singletons) lead to a significant negative estimate for the statistic D (−2.683, P < 0.001). Thus, considering the parasite demography, the Pfk13 gene is expected to have several recent geographically distinct mutations at a low frequency, with each individual parasite lineage carrying one or very few of those segregating sites (42, 43). Although in P. vivax there is also evidence of a recent population expansion (44, 45), no segregating mutations were found in Pvk13. Given the limitation of our sample (n = 53), we cannot discern whether this is a real pattern or a sampling artifact.
The data are consistent with both selection and demography driving the dynamic of Pfk13 mutations conferring artemisinin-based combination therapy (ACT) tolerance (46). The parasite population expansion (demography) relaxes purifying (negative) selection, allowing for semideleterious mutations to segregate (41, 43, 46, 47), and the introduction of ACTs selects for mutations conferring delayed parasite clearance/tolerance out of those several variants present at a low frequency, even if such variants have a fitness cost in the absence of drug pressure (8, 46–48). Under such dynamics, as the data in Africa seem to indicate, an asymptomatic untreated reservoir will favor ACT-sensitive parasites, keeping a negative selective pressure on those semideleterious mutations conferring tolerance to the drugs (47–50). Consequently, the Pfk13 mutations present today are unlikely to increase in frequency in regions such as Africa, where their relative advantage is still overcome by their fitness cost in the absence of treatment (e.g., subclinical or asymptomatic infections at a high frequency [49, 50]), and parasites with those mutatiosn are still susceptible to aggressive/monitored drug treatment (8, 46). The interplay between the putative fitness cost of these mutations and the absence of drug pressure in asymptomatic individuals could be facilitated by the higher population sizes of P. falciparum in Africa, making differences in selection regimens prevail over genetic drift in the dynamics of these mutations (46, 48). Under the current scenario, interventions directed to reduce the asymptomatic reservoir, such as targeted mass drug administration (TMDA) with ACTs, will remain effective because there are not yet Pfk13 mutations conferring complete resistance (46, 51).
Although the observed patterns are robust, it is worth noting that many studies in P. falciparum directly report the frequency of mutations related to ACT-delayed parasite clearance. This practice not only may lead to underreporting of mutations occurring at a low frequency but also limits our capacity to track geographic changes in haplotype frequencies or infer other patterns. As an example, the frequency of the C580Y mutation is high in western Cambodia (52, 53), but the deposited sequence data are scarce. Whether there is an expansion of a particular haplotype in the parasite population cannot be addressed across studies by using mutation frequency data.
The proposed dynamics explain why the actions deployed thus far have limited the expansion of these mutations from Asia, and there has been only a single haplotype of C580Y identified in Guyana. Whether the spread will remain limited (17, 24) is a matter that requires continued monitoring. Importantly, reporting novel mutations is critical since they may change the parasite fitness under drug pressure. Thus, considering that all observed mutations in Pfk13 are of recent origin, understanding their evolutionary dynamics at the early stages of drug resistance could further inform containment strategies wherever putative resistance mutations start to increase in frequency.
MATERIALS AND METHODS
Samples/sequences from the genus Plasmodium.
Orthologous k13 genes were sequenced from macaque and gibbon parasite laboratory strains provided by the Centers for Disease Control and Prevention (CDC): Plasmodium inui (OS strain), Plasmodium hylobati, Plasmodium fieldi (N-3 strain), Plasmodium simiovale, and Plasmodium gonderi. Information on these species and strains can be found elsewhere (54). Archived remainders of blood samples from chimpanzees infected with Plasmodium reichenowi (Shegue and 20 isolates [28]), Plasmodium billcollinsi (the Ithaito isolate [28]), and Plasmodium gaboni (Shegue and 39 isolates [28]) were also used in this study (see Data Set S1 in the supplementary material). The chimpanzees were housed at the Jane Goodall Institute (JGI) Tchimpounga Chimpanzee Rehabilitation Center in the Democratic Republic of Congo (DRC), and their blood samples were collected throughout 2009 and 2010 by the veterinary staff as part of the chimpanzees’ routine health examinations, following standards approved by the Pan Africa Sanctuary Alliance. More information on these species and ethical guidelines can be found in reference 28.
In addition, all orthologous k13 gene sequences from different nonhuman Plasmodium species and Plasmodium vivax with publicly available genomes were retrieved from the PlasmoDB (v38) (55) and MalAvi (56) databases (Data Sets S1 and S4).
Plasmodium falciparum samples/sequences.
Plasmodium falciparum samples from Africa (n = 244) and Haiti (n = 103) and samples from U.S. malaria cases without travel histories (n = 116) reported to the CDC malaria surveillance network from 2014 to 2017 (n = 463) were used (Data Set S5). Genomic DNA isolated from P. falciparum strains 7G8 and HB3 was used as a positive control, and DNA isolated from whole blood from individuals without malaria was used as a negative control. The use of U.S. surveillance P. falciparum samples was approved by the Office of the Associate Director of Science, Center for Global Health, Centers for Disease Control and Prevention, as research not involving human subjects (protocol 2017-192). In addition, the methodology employed here for the amplification of the k13 gene was also approved by the same office as a surveillance activity (protocol 2017-309).
The data were expanded by retrieving complete and partial k13 gene sequences for P. falciparum from publicly available genomes/sequences found in the PlasmoDB (55) and NCBI (57) databases. All sequences were analyzed together with the new data obtained here from African and Haitian isolates (Data Set S5).
DNA extraction and Sanger sequencing for nonhuman primate parasites.
Genomic DNA was extracted from whole blood of the nonhuman primate Plasmodium species using a DNeasy blood and tissue kit (Qiagen, GmbH, Hilden, Germany). Orthologous k13 genes from macaque and gibbon parasites were amplified by nested PCR using external primers forward 5′-C ATT TCC AAC TTC TCC GTC-3′ (58) and reverse 5′-TAT ATT TGC TAT TAR NAC NGA RTG-3′ and internal primers forward 5′-A TGA CGT ATG ADA GRG ART CNG-3′ and reverse 5′-AA TCT GGG AAC TAA TAR DGR DGG-3′. The primary PCR amplifications were carried out in a 50-μl reaction volume using 5 μl of total genomic DNA, 2.5 mM MgCl2, 1× PCR buffer, 1.25 mM each deoxynucleoside triphosphate, 0.4 mM each primer, and 0.03 U/μl AmpliTaq polymerase (Applied Biosystems, Roche, USA). The primary PCR conditions were a partial denaturation at 94°C for 4 min and 36 cycles with 1 min at 94°C, 1 min at 54°C, and a 2-min extension at 72°C, and a final extension of 10 min at 72°C was added in the last cycle. Then, the nested PCR mixtures were also made in 50-μl reaction volume using 1 μl of the primary PCR mixture, 2.5 mM MgCl2, 1× PCR buffer, 1.25 mM each deoxynucleoside triphosphate, 0.4 mM each primer, and 0.03 U/μl AmpliTaq polymerase. The nested PCR conditions were a partial denaturation at 94°C for 4 min and 25 cycles with 1 min at 94°C, 1 min at 56°C, and a 2-min extension at 72°C, and a final extension of 10 min at 72°C was added in the last cycle. Then, orthologous k13 genes from chimpanzee parasites, which share a recent common ancestor with P. falciparum, were amplified by using the same protocol described above, but with external primers forward 5′-GAA TTT TTC TAT NAC RTA YGA DAG-3′ and reverse 5′-ATT TGC TAT TAR NAC NGA RTG NCC-3′ and internal primers forward 5′-A TGA CGT ATG ADA GRG ART CNG-3′ and reverse 5′-AA TCT GGG AAC TAA TAR DGR DGG-3′. Then, two independent PCR products (50 μl) were excised from the gel (bands of approximately 2 kbp), purified using a QIAquick gel extraction kit (Qiagen, GmbH, Hilden, Germany), and cloned into the pGEM-T Easy vector system (Promega, USA). To detect mixed infections, at least two independent PCR products were cloned, and four clones were sequenced from each sample. Both strands for all the k13 orthologous genes were sequenced using an Applied Biosystems 3730 capillary sequencer. All the sequences obtained were identified as the k13 gene using analysis with the BLAST program (59). The sequences were submitted to GenBank and may be found under accession numbers MK103884 to MK103893.
DNA extraction from human samples and next-generation sequencing.
DNA from human samples was extracted using the whole-blood protocol for the QIAamp DNA blood kit (Qiagen, GmbH, Hilden, Germany). Then, an end-to-end Illumina targeted amplicon deep sequencing (TADS) protocol and bioinformatics pipeline for molecular surveillance of drug resistance in P. falciparum, called malaria resistance surveillance (MaRS), was used to amplify the Pfk13 gene (60). First, the gene was amplified using a New England BioLabs (NEB) High Fidelity PCR kit (catalog no. 51104; New England BioLabs, USA) according to the manufacturer’s instructions with a 50.0-μl master mix preparation using 5× GC buffer. Next, 10.0 μl of each PCR product for each sample was used to generate the next-generation sequencing library. Pooled library fragment sizes were checked using an Agilent D5000 ScreenTape station (catalog numbers G2940C, 5067-4626, and G2940CA; Agilent Technologies), and the pooled library concentration was measured using a Qubit (v3.0) fluorometer (catalog numbers Q33216 and Q32853; Life Technologies Corp.). Lastly, a MiSeq desktop sequencer (Illumina Inc.) was used to obtain the sequences, and a consensus sequence for each sample was assembled using a pipeline. Additional details about the MaRS protocol are provided in Text S1 in the supplemental material of reference 60, including primer sequences, thermocycling conditions, and library amplification, purification, quantification, and normalization. The Pfk13 sequences obtained here were submitted to the GenBank database and may be found under accession numbers MK103421 to MK103883.
Phylogenetic-based method analyses of the k13 gene.
First, a nucleotide sequence alignment was performed by using 42 sequences (1,985 bp, excluding gaps), including the entire orthologous k13 gene sequences obtained here, the sequences of the different haplotypes for some well-identified parasite species (e.g., P. reichenowi and P. gaboni), as well as sequences available in the PlasmoDB and MalAvi databases (Fig. 1 and 2; Data Set S1). Then, a second alignment based on 28 orthologous k13 gene sequences selected from different species of the first alignment was performed (duplicate haplotypes were not included; Fig. 3). All the alignments were generated using the ClustalX (v2.0.12) and Muscle programs implemented in the SeaView (v4.3.5) program (61) with manual editing. In both alignments, the Haemoproteus tartakovskyi (an avian haemosporidian parasite) k13 gene was included as an outgroup (56).
Using both alignments, phylogenetic relationships were inferred using the Bayesian method implemented in MrBayes (v3.2.6) software with the default priors (62). A general time-reversible model with gamma-distributed substitution rates and a proportion of invariant sites (GTR + Γ + I), which was the model with the lowest Bayesian information criterion (BIC) scores, as estimated by MEGA (v7.0.26) software, was used (63). Bayesian support for the nodes was inferred in MrBayes by sampling every 1,000 generations from two independent chains lasting 4 × 106 Markov chain Monte Carlo (MCMC) steps. The chains were assumed to have converged once the average standard deviation of the posterior probability was below 0.01 and the value of the potential scale reduction factor (PSRF) was between 1.00 and 1.02 (62). As a burn-in, 25% of the sample was then discarded once convergence was reached. The phylogenetic trees were visualized and edited using the FigTree (v1.4.3) program (64).
To detect changes in the rate of evolution, relative evolutionary rates of the k13 gene were estimated on the second alignment using a maximum likelihood framework with the RelTime method (30), as implemented in the command line version implemented in MEGA (vX) software (65). The substitution model was the same as that used for Bayesian analyses (GTR + Γ + I). Then, the tree was visualized in the FigTree (v1.4.3) program and colored using the relative evolutionary rate estimates of the k13 gene (Fig. 3). To quantitate the rate of evolution of this gene, absolute rates of synonymous and nonsynonymous substitutions per codon per My for the k13 gene were obtained using the CodonRates (v1.0) program in a Bayesian framework (33). In addition, by disentangling synonymous from nonsynonymous substitution rates, this analysis provides a first assessment of how natural selection may have affected the evolution of the gene. Because this method requires a tree topology and time calibration priors, including one for the in-group root node (the Plasmodium clade in this case), the Bayesian methods program BEAST (v2.4.1) (66) was used to estimate the origin of the Plasmodium species included in this investigation.
In BEAST, the relaxed clock (67) was applied with the GTR + Γ + I evolutionary model with a Yule model as the tree prior. Two independent chains lasting 108 MCMC steps were run until convergence. One scenario, using a combination of two calibration constraints that consider information on vertebrate hosts’ fossils (see the Paleobiology Database at http://www.paleodb.org/; last accessed 13 November 2017) was explored, and the results were compared with those of earlier studies based on the mitochondrial genomes (29). In all cases, uniform priors were used to calibrate divergences. Therefore, no particular time point within a given time interval was favored, and no punctual calibrations were used. The first calibration constraint considers that African parasites found in Mandrillus spp. (Plasmodium gonderi) diverged from those Plasmodium species found in Southeast Asia macaques (a monophyletic group) when Macaca branched from Papio (29, 68, 69). This event assumed that the divergence at that node took place at any time between 6 and 14.2 My ago (Ma) using a uniform prior. This time interval includes molecular estimates for the same Papio-Macaca divergence event (29, 69), and it is also consistent with the age of older fossils reported for Macaca spp. (see the Paleobiology Database at http://www.paleodb.org/; last accessed 13 November 2017). This calibration constraint considers the fact that P. gonderi has also been seen in Chlorocebus (a member of the Cercopithecini). The second fossil-based calibration constraint is the minimum of 23.5 Ma for the human-Macaca split (70), which is assumed to be the minimum time when the monophyletic group that includes P. malariae (a parasite found in humans) and all the Asian parasites originated. This event is used to inform a calibration prior with a maximum of 65 Ma that allows this parasite clade to be as old as the origin of primates (71). More details about this method can be found elsewhere (29, 69).
Given that the time estimated here for the avian Plasmodium-primate/rodent Plasmodium split (45.36 Ma; 95% credibility intervals, 33.23 to 61.10 Ma) was comparable to previous estimates also obtained using BEAST (45.31 Ma; credibility intervals, 38.95 to 51.96 [29]), the absolute rates of synonymous and nonsynonymous substitutions per codon per My were then estimated by using CodonRates (33) with a calibration prior of 45.4 ± 10 Ma for the in-group root (genus Plasmodium), as estimated from BEAST. This analysis was performed on a tree topology estimated with MrBayes that used Haemoproteus tartakovskyi as an outgroup.
Finally, to elucidate selective pressures during the evolution of the k13 gene, several methods were performed with the HyPhy package (34) implemented in the DataMonkey web server (http://www.datamonkey.org/; last accessed August 2018). Evidence of both episodic and pervasive positive selection at the level of an individual site was identified using a mixed-effects model of evolution, MEME (35), which detects sites evolving under positive selection under a proportion of branches. To investigate whether the k13 gene has experienced positive selection at least at one site on at least one branch, a branch site-unrestricted statistical test for episodic diversification (BUSTED) was used. This provides a gene-wide (not site-specific) test for positive selection (36). Sites positively selected were identified at a significance level (P value) of <0.05. In addition, to test whether the strength of natural selection has been relaxed or intensified along a specified set of test branches, the RELAX method (37) was performed. RELAX is most useful for identifying trends and/or shifts in the stringency of natural selection on a given gene.
Plasmodium falciparum population analyses.
To study the genetic relationships among worldwide haplotypes of the k13 gene, a third alignment was performed using 982 cosmopolitan complete sequences, including all the k13 gene sequences obtained here for P. falciparum population samples (n = 463), as well as those available in the PlasmoDB and NCBI databases (n = 519). Evidence of natural selection was explored by estimating the average number of synonymous substitutions per synonymous site (dS) and the average number of nonsynonymous substitutions per nonsynonymous site (dN) between a pair of sequences under the Nei-Gojobori method with the Jukes and Cantor corrections, as implemented in the MEGA (v7) program (63). The difference between dS and dN with its standard error (estimated using bootstrap analysis with 1,000 pseudoreplications) was calculated, and significance was tested by a two-tailed codon-based Z-test on the difference between dS and dN, as described by Nei and Kumar (72). The null hypothesis was that the observed polymorphism is not under selection (dS = dN).
The alignments were used to estimate median-joining networks using Network (v4.6.1.0) software (Fluxus Technologies, 2011). Transversions were set equal to transitions, and the epsilon parameter was set equal to 0 with only one round of star contraction, which collapses star-like structures in the network into single nodes. The total number of sites included in this analysis, excluding gaps or missing data, was 2,137 bp out of 2,181 bp using the P. falciparum 3D7 strain as a reference. Estimates of nucleotide diversity, haplotype diversity, and Tajima’s D test parameter were obtained using DnaSP (v5.50) software (73). Lastly, the frequency of candidate/validated mutations located in the propeller domain of a P. falciparum kelch (Pfk13) gene that have been associated with clinical delayed parasite clearance was estimated by country, as well as by continent, using a fourth alignment constructed with 3,916 partial (n = 2,934) and complete (n = 982) k13 gene sequences (Table 2; Data Sets S5 and S6).
Accession number(s).
The Pfk13 sequences obtained here were deposited in GenBank under accession numbers MK103421 to MK103893.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Li for the silhouettes design and the DNA laboratory at the School of Life Sciences (Arizona State University) for technical support.
This study was supported in part by grants from the U.S. National Institutes of Health (2U19 AI089681-08 and R01AI130473). Computer resources were supported by a grant from the Pennsylvania Department of Health (TU-420721).
We declare that no conflicts of interest exist.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02550-18.
REFERENCES
- 1.Dondorp AM, Smithuis FM, Woodrow C, Seidlein L. 2017. How to contain artemisinin- and multidrug-resistant falciparum malaria. Trends Parasitol 33:353–363. doi: 10.1016/j.pt.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2017. Artemisinin and artemisinin-based combination therapy resistance: status report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium. 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 4.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders DL, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program, National Center for Parasitology Entomology and Malaria Control. Royal Cambodian Armed Forces. 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 7.Imwong M, Suwannasin K, Kunasol C, Sutawong K, Mayxay M, Rekol H, Smithuis FM, Hlaing TM, Tun KM, van der Pluijm RW, Tripura R, Miotto O, Menard D, Dhorda M, Day NPJ, White NJ, Dondorp AM. 2017. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis 17:491–497. doi: 10.1016/S1473-3099(17)30048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escalante AA, Smith DL, Kim Y. 2009. The dynamics of mutations associated with anti-malarial drug resistance in Plasmodium falciparum. Trends Parasitol 25:557–563. doi: 10.1016/j.pt.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prosser C, Meyer W, Ellis J, Lee R. 2018. Evolutionary ARMS race: antimalarial resistance molecular surveillance. Trends Parasitol 34:322–334. doi: 10.1016/j.pt.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WR, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale JC, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Ménard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Ménard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Ménard D, Fidock DA. 2015. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takala-Harrison S, Jacob CG, Arze C, Cummings MP, Silva JC, Dondorp AM, Fukuda MM, Hien TT, Mayxay M, Noedl H, Nosten F, Kyaw MP, Nhien NT, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Ariey F, Mercereau-Puijalon O, Menard D, Newton PN, Khanthavong M, Hongvanthong B, Starzengruber P, Fuehrer HP, Swoboda P, Khan WA, Phyo AP, Nyunt MM, Nyunt MH, Brown TS, Adams M, Pepin CS, Bailey J, Tan JC, Ferdig MT, Clark TG, Miotto O, MacInnis B, Kwiatkowski DP, White NJ, Ringwald P, Plowe CV. 2015. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 211:670–679. doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talundzic E, Okoth SA, Congpuong K, Plucinski MM, Morton L, Goldman IF, Kachur PS, Wongsrichanalai C, Satimai W, Barnwell JW, Udhayakumar V. 2015. Selection and spread of artemisinin-resistant alleles in Thailand prior to the global artemisinin resistance containment campaign. PLoS Pathog 11:e1004789. doi: 10.1371/journal.ppat.1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ménard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, Rahim-Awab G, Barnadas C, Berry A, Boum Y, Bustos MD, Cao J, Chen JH, Collet L, Cui L, Thakur GD, Dieye A, Djallé D, Dorkenoo MA, Eboumbou-Moukoko CE, Espino FE, Fandeur T, Ferreira-da-Cruz MF, Fola AA, Fuehrer HP, Hassan AM, Herrera S, Hongvanthong B, Houzé S, Ibrahim ML, Jahirul-Karim M, Jiang L, Kano S, Ali-Khan W, Khanthavong M, Kremsner PG, Lacerda M, Leang R, Leelawong M, Li M, Lin K, Mazarati JB, Ménard S, Morlais I, Muhindo-Mavoko H, Musset L, Na-Bangchang K, Nambozi M, Niaré K, Noedl H, et al. 2016. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med 374:2453–2464. doi: 10.1056/NEJMoa1513137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobasa T, Talundzic E, Sug-Aram R, Boondat P, Goldman IF, Lucchi NW, Dharmarak P, Sintasath D, Fukuda M, Whistler T, MacArthur J, Udhayakumar V, Prempree P, Chinanonwait N. 2018. Emergence and spread of kelch13 mutations associated with artemisinin resistance in Plasmodium falciparum parasites in 12 Thai provinces from 2007 to 2016. Antimicrob Agents Chemother 62:e02141-17. doi: 10.1128/AAC.02141-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WWARN K13 Genotype-Phenotype Study Group. 2019. Association of mutations in the Plasmodium falciparum Kelch13 gene (Pf3D7_1343700) with parasite clearance rates after artemisinin-based treatments-a WWARN individual patient data meta-analysis. BMC Med 17:1. doi: 10.1186/s12916-018-1207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plucinski MM, Dimbu PR, Macaia AP, Ferreira CM, Samutondo C, Quivinja J, Afonso M, Kiniffo R, Mbounga E, Kelley JS, Patel DS, He Y, Talundzic E, Garrett DO, Halsey ES, Udhayakumar V, Ringwald P, Fortes F. 2017. Efficacy of artemether-lumefantrine, artesunate-amodiaquine, and dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum malaria in Angola, 2015. Malar J 16:62. doi: 10.1186/s12936-017-1712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madamet M, Kounta MB, Wade KA, Lo G, Diawara S, Fall M, Bercion R, Nakoulima A, Fall KB, Benoit N, Gueye MW, Fall B, Diatta B, Pradines B. 2017. Absence of association between polymorphisms in the K13 gene and the presence of Plasmodium falciparum parasites at day 3 after treatment with artemisinin derivatives in Senegal. Int J Antimicrob Agents 49:754–756. doi: 10.1016/j.ijantimicag.2017.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Gupta H, Macete E, Bulo H, Salvador C, Warsame M, Carvalho E, Ménard D, Ringwald P, Bassat Q, Enosse S, Mayor A. 2018. Drug-resistant polymorphisms and copy numbers in Plasmodium falciparum, Mozambique, 2015. Emerg Infect Dis 24:40–48. doi: 10.3201/eid2401.170864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Laurent ZR, Chebon LJ, Ingasia LA, Akala HM, Andagalu B, Ochola-Oyier LI, Kamau E. 2018. Polymorphisms in the K13 gene in Plasmodium falciparum from different malaria transmission areas of Kenya. Am J Trop Med Hyg 98:1360–1366. doi: 10.4269/ajtmh.17-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chenet SM, Akinyi Okoth S, Huber CS, Chandrabose J, Lucchi NW, Talundzic E, Krishnalall K, Ceron N, Musset L, Macedo de Oliveira A, Venkatesan M, Rahman R, Barnwell JW, Udhayakumar V. 2016. Independent emergence of the Plasmodium falciparum kelch propeller domain mutant allele C580Y in Guyana. J Infect Dis 213:1472–1475. doi: 10.1093/infdis/jiv752. [DOI] [PubMed] [Google Scholar]
- 25.Mita T, Culleton R, Takahashi N, Nakamura M, Tsukahara T, Hunja CW, Win ZZ, Htike WW, Marma AS, Dysoley L, Ndounga M, Dzodzomenyo M, Akhwale WS, Kobayashi J, Uemura H, Kaneko A, Hombhanje F, Ferreira MU, Björkman A, Endo H, Ohashi J. 2016. Little polymorphism at the K13 propeller locus in worldwide Plasmodium falciparum populations prior to the introduction of artemisinin combination therapies. Antimicrob Agents Chemother 60:3340–3347. doi: 10.1128/AAC.02370-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes LR, Lavigne A, Peterka CL, Brasil P, Ménard D, Daniel-Ribeiro CT, Ferreira-da-Cruz MF. 2018. Absence of K13 polymorphism in Plasmodium falciparum from Brazilian areas where the parasite is endemic. Antimicrob Agents Chemother 62:e00354-18. doi: 10.1128/AAC.00354-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto TD, Gilabert A, Crellen T, Böhme U, Arnathau C, Sanders M, Oyola SO, Okouga AP, Boundenga L, Willaume E, Ngoubangoye B, Moukodoum ND, Paupy C, Durand P, Rougeron V, Ollomo B, Renaud F, Newbold C, Berriman M, Prugnolle F. 2018. Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nat Microbiol 3:687–697. doi: 10.1038/s41564-018-0162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacheco MA, Cranfield M, Cameron K, Escalante AA. 2013. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J 12:328. doi: 10.1186/1475-2875-12-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pacheco MA, Matta NE, Valkiunas G, Parker PG, Mello B, Stanley CE Jr, Lentino M, Garcia-Amado MA, Cranfield M, Kosakovsky Pond SL, Escalante AA. 2018. Mode and rate of evolution of haemosporidian mitochondrial genomes: timing the radiation of avian parasites. Mol Biol Evol 35:383–403. doi: 10.1093/molbev/msx285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S. 2012. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci U S A 109:19333–19338. doi: 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. 2008. Big bang in the evolution of extant malaria parasites. Mol Biol Evol 25:2233–2239. doi: 10.1093/molbev/msn171. [DOI] [PubMed] [Google Scholar]
- 32.Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, Alsisto S, Ramirez D, Escalante AA. 2015. Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol 32:422–439. doi: 10.1093/molbev/msu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo TK, Kishino H, Thorne JL. 2004. Estimating absolute rates of synonymous and non-synonymous nucleotide substitution in order to characterize natural selection and date species divergences. Mol Biol Evol 217:1201–1213. doi: 10.1093/molbev/msh088. [DOI] [PubMed] [Google Scholar]
- 34.Pond SLK, Frost SDW, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 215:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 35.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 87:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, Scheffler K, Kosakovsky Pond SL. 2015. Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371. doi: 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. 2015. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 323:820–832. doi: 10.1093/molbev/msu400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCollum AM, Poe AC, Hamel M, Huber C, Zhou Z, Shi YP, Ouma P, Vulule J, Bloland P, Slutsker L, Barnwell JW, Udhayakumar V, Escalante AA. 2006. Antifolate resistance in Plasmodium falciparum: multiple origins and identification of novel dhfr alleles. J Infect Dis 194:189–197. doi: 10.1086/504687. [DOI] [PubMed] [Google Scholar]
- 39.McCollum AM, Schneider KA, Griffing SM, Zhou Z, Kariuki S, Ter-Kuile F, Shi YP, Slutsker L, Lal AA, Udhayakumar V, Escalante AA. 2012. Differences in selective pressure on dhps and dhfr drug resistant mutations in western Kenya. Malar J 11:77. doi: 10.1186/1475-2875-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson TJ, Nair S, McDew-White M, Cheeseman IH, Nkhoma S, Bilgic F, McGready R, Ashley E, Pyae Phyo A, White NJ, Nosten F. 2017. Population parameters underlying an ongoing soft sweep in Southeast Asian malaria parasites. Mol Biol Evol 34:131–144. doi: 10.1093/molbev/msw228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanabe K, Mita T, Jombart T, Eriksson A, Horibe S, Palacpac N, Ranford-Cartwright L, Sawai H, Sakihama N, Ohmae H, Nakamura M, Ferreira MU, Escalante AA, Prugnolle F, Björkman A, Färnert A, Kaneko A, Horii T, Manica A, Kishino H, Balloux F. 2010. Plasmodium falciparum accompanied the human expansion out of Africa. Curr Biol 20:1283–1289. doi: 10.1016/j.cub.2010.05.053. [DOI] [PubMed] [Google Scholar]
- 42.Gazave E, Chang D, Clark AG, Keinan A. 2013. Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics 195:969–978. doi: 10.1534/genetics.113.153973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gravel S. 2016. When is selection effective? Genetics 203:451–462. doi: 10.1534/genetics.115.184630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, Herrera S, Kim JY, Menard D, Póvoa MM, Villegas L, Mulyanto Snounou G, Cui L, Zeyrek FY, Escalante AA. 2013. The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol 30:2050–2064. doi: 10.1093/molbev/mst104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, Vallejo AF, Herrera S, Arévalo-Herrera M, Fan Q, Wang Y, Cui L, Lucas CM, Durand S, Sanchez JF, Baldeviano GC, Lescano AG, Laman M, Barnadas C, Barry A, Mueller I, Kazura JW, Eapen A, Kanagaraj D, Valecha N, Ferreira MU, Roobsoong W, Nguitragool W, Sattabonkot J, Gamboa D, Kosek M, Vinetz JM, González-Cerón L, Birren BW, Neafsey DE, Carlton JM. 2016. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet 48:953–958. doi: 10.1038/ng.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim Y, Escalante AA, Schneider KA. 2014. A population genetic model for the initial spread of partially resistant malaria parasites under anti-malarial combination therapy and weak intrahost competition. PLoS One 9:e101601. doi: 10.1371/journal.pone.0101601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straimer J, Gnädig NF, Stokes BH, Ehrenberger M, Crane AA, Fidock DA. 2017. Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. mBio 8:e00172-17. doi: 10.1128/mBio.00172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair S, Li X, Arya GA, McDew-White M, Ferrari M, Nosten F, Anderson TJC. 2018. Fitness costs and the rapid spread of kelch13-C580Y substitutions conferring artemisinin resistance. Antimicrob Agents Chemother 62:e00605-18. doi: 10.1128/AAC.00605-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ataide R, Ashley EA, Powell R, Chan JA, Malloy MJ, O’Flaherty K, Takashima E, Langer C, Tsuboi T, Dondorp AM, Day NP, Dhorda M, Fairhurst RM, Lim P, Amaratunga C, Pukrittayakamee S, Hien TT, Htut Y, Mayxay M, Faiz MA, Beeson JG, Nosten F, Simpson JA, White NJ, Fowkes FJ. 2017. Host immunity to Plasmodium falciparum and the assessment of emerging artemisinin resistance in a multinational cohort. Proc Natl Acad Sci U S A 114:3515–3520. doi: 10.1073/pnas.1615875114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein EY, Smith DL, Boni MF, Laxminarayan R. 2008. Clinically immune hosts as a refuge for drug-sensitive malaria parasites. Malar J 7:67. doi: 10.1186/1475-2875-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghani AC. 2018. Can improving access to care help to eliminate malaria? Lancet 391:1870–1871. doi: 10.1016/S0140-6736(18)30910-3. [DOI] [PubMed] [Google Scholar]
- 52.Dwivedi A, Khim N, Reynes C, Ravel P, Ma L, Tichit M, Bourchier C, Kim S, Dourng D, Khean C, Chim P, Siv S, Frutos R, Lek D, Mercereau-Puijalon O, Ariey F, Menard D, Cornillot E. 2016. Plasmodium falciparum parasite population structure and gene flow associated to anti-malarial drugs resistance in Cambodia. Malar J 15:319. doi: 10.1186/s12936-016-1370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kheang ST, Sovannaroth S, Ek S, Chy S, Chhun P, Mao S, Nguon S, Lek DS, Menard D, Kak N. 2017. Prevalence of K13 mutation and day-3 positive parasitaemia in artemisinin-resistant malaria endemic area of Cambodia: a cross-sectional study. Malar J 16:372. doi: 10.1186/s12936-017-2024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coatney RG, Collins WE, Warren M, Contacos PG. 1971. The primate malarias. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- 55.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ Jr, Treatman C, Wang H. 2009. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bensch S, Hellgren O, Pérez-Tris J. 2009. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour 95:1353–1358. doi: 10.1111/j.1755-0998.2009.02692.x. [DOI] [PubMed] [Google Scholar]
- 57.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW 2012. 2013. GenBank. Nucleic Acids Res 41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Talundzic E, Chenet SM, Goldman IF, Patel DS, Nelson JA, Plucinski MM, Barnwell JW, Udhayakumar V. 2015. Genetic analysis and species specific amplification of the artemisinin resistance-associated kelch propeller domain in P. falciparum and P. vivax. PLoS One 10:e0136099. doi: 10.1371/journal.pone.0136099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Talundzic E, Ravishankar S, Kelley J, Patel D, Plucinski M, Schmedes S, Ljolje D, Clemons B, Madison-Antenucci S, Arguin PM, Lucchi NW, Vannberg F, Udhayakumar V. 2018. Next-generation sequencing and bioinformatics protocol for malaria drug resistance marker surveillance. Antimicrob Agents Chemother 62:e02474-17. doi: 10.1128/AAC.02474-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 272:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 62.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 1912:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 63.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 337:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rambaut A. 2006. FigTree tree figure drawing tool version 1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, United Kingdom: http://tree.bio.ed.ac.uk/software/figtree/. Accessed 31 January 2018. [Google Scholar]
- 65.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mu J, Joy DA, Duan J, Huang Y, Carlton J, Walker J, Barnwell J, Beerli P, Charleston MA, Pybus OG, Su XZ. 2005. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol 22:1686–1693. doi: 10.1093/molbev/msi160. [DOI] [PubMed] [Google Scholar]
- 69.Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, Rabetafika L, Snounou G, Jones-Engel L, Escalante AA. 2011. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol 11:299. doi: 10.1186/1471-2148-11-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benton MJ, Donoghue PC. 2007. Paleontological evidence to date the tree of life. Mol Biol Evol 241:26–53. doi: 10.1093/molbev/msl150. [DOI] [PubMed] [Google Scholar]
- 71.Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, Schneider MP, Silva A, O’Brien SJ, Pecon-Slattery J. 2011. A molecular phylogeny of living primates. PLoS Genet 73:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nei M, Kumar S. 2000. Molecular evolution and phylogenetics. Oxford University Press, New York, NY. [Google Scholar]
- 73.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.