

Abstract
Cysteine dioxygenase (CDO) is a non-heme iron enzyme that adds two oxygen atoms from dioxygen to the sulfur atom of L-cysteine. Adjacent to the iron site of mammalian CDO, a post-translationally generated Cys-Tyr cofactor is present, whose presence substantially enhances the oxygenase activity. The formation of the Cys-Tyr cofactor in CDO is an autocatalytic process, and it is challenging to study by traditional techniques because the crosslinking reaction is a side, uncoupled, single-turnover oxidation buried among multiple turnovers of L-cysteine oxygenation. Here, we take advantage of our recent success in obtaining a purely uncrosslinked human CDO due to site-specific incorporation 3,5-difluoro-L-tyrosine (F2-Tyr) at the crosslinking site through the genetic code expansion strategy. Using EPR spectroscopy, we show that nitric oxide (•NO), an oxygen surrogate, similarly binds to uncrosslinked F2-Tyr157 CDO as in wild-type human CDO. We determined X-ray crystal structures of uncrosslinked F2-Tyr157 CDO and mature wild-type CDO in complex with both L-cysteine and •NO. These structural data reveal that the active site cysteine (Cys93 in the human enzyme), rather than the generally expected tyrosine (i.e., Tyr157), is well aligned to be oxidized should the normal oxidation reaction uncouple. This structural based understanding is further supported by a computational study with models built on the uncrosslinked ternary complex structure. Together, these results strongly suggest that the first target to oxidize during the iron-assisted Cys-Tyr cofactor biogenesis is Cys93. Based on these data, a plausible reaction mechanism implementing a cysteine radical involved in the crosslink formation is proposed.
Graphical Abstract
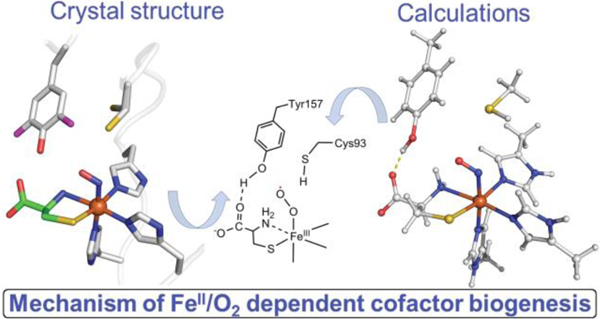
Introduction
A protein-derived cofactor is often a catalytic center or an amplifier for enhancing the catalytic activity of an enzyme. The thioether crosslinked cysteine–tyrosine (Cys-Tyr) through a carbon–sulfur bond is a protein cofactor which is found in diverse metalloproteins including non-heme iron-dependent cysteine dioxygenase (CDO) and cysteamine dioxygenase (ADO), copper-dependent galactose oxidase (GAO) and glyoxal oxidase (GLOX), heme iron-dependent cytochrome c nitrite reductase (TvNiR), heme- and [Fe4S4]-dependent sulfite reductase (NirA),1–8 and possibly in an orphan protein BF4112.9 The posttranslational modification in these proteins is accompanied by a change of the tyrosine residue for its redox potential, pKa, and also restricting its free rotation. The Cys-Tyr cofactor in the iron proteins enhances the catalytic activity8, 10, 11 while in the copper-dependent oxidases the cofactor is a copper ligand, and it is in a one-electron oxidized free radical form.6, 12 Such a Cys-Tyr radical cofactor is essential for the catalytic activity of the oxidases.13
The biogenesis mechanism of Cys-Tyr is significantly better understood for the copper-dependent oxidases,3, 14, 15 and much less is known for the iron-dependent proteins.16 The Cys-Tyr cofactor in mammalian cysteine dioxygenase (EC 1.13.11.20) is a catalytic amplifier17 located adjacent, but not directly coordinated, to the catalytic non-heme iron center.1, 18 CDO catalyzes the first and committed step of oxidative cysteine catabolism, that is, the conversion of L-cysteine to cysteinesulfinate (Scheme 1A). Cysteinesulfinate is ultimately catabolized to taurine and sulfate19 as terminal products. The activity of CDO is significant for maintaining the thiol levels and a proper balance among cysteine, taurine, and sulfate.19 The production of the Cys-Tyr cofactor in CDO is a self-processing oxidation reaction due to uncoupled oxygen activation at the non-heme iron center of the enzyme (Scheme 1B), where its active site residues are oxidized by O2 rather than the substrate, L-cysteine. It takes likely hundreds of turnovers to form the Cys-Tyr cofactor.17 Once the cofactor is composed, the mature form of CDO can metabolize high levels of cysteine and additionally increased its catalytic lifetime by ca. 3-fold.16, 20
Scheme 1.
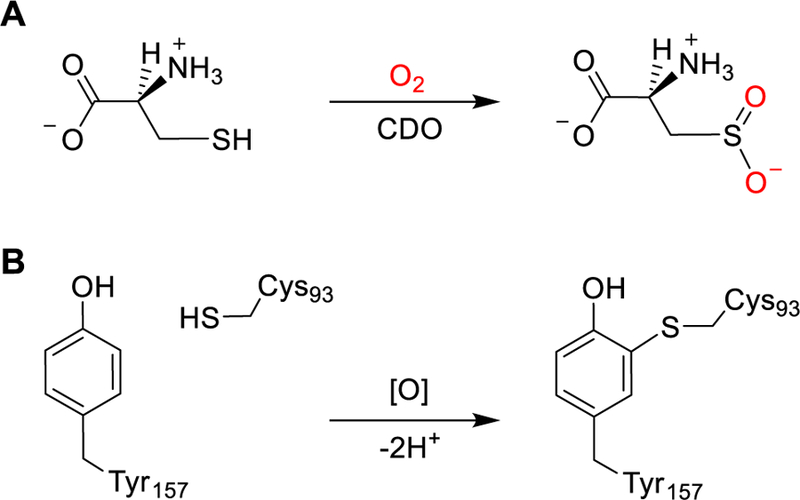
The chemical reactions catalyzed by CDO. (A) CDO catalyzes an iron dependent oxygenation reaction that activates O2 and inserts the two oxygen atoms into the sulfur group of L-cysteine to produce cysteinesulfinate. (B) An uncoupled autocatalytic oxidation leads to the formation of a Cys-Tyr cofactor. This uncoupled reaction is triggered by the presence of L-cysteine and O2.
One mechanism for the CDO cofactor biogenesis has been proposed, with which Tyr157 is oxidized to a tyrosyl radical by a metal-bound oxidant to initiate the crosslinking reaction.2, 17, 21 However, this proposal was published before the uncrosslinked starting structure was determined, and was entirely based on the mature CDO structure in which Tyr157 is closer to the iron ion than Cys93. It has been suggested that Cys93 could be oxidized before tyrosine oxidation,16, 22 however, there is no proposed mechanistic model described until our work.4 The cofactor biogenesis mechanism appears to be inaccessible in CDO by traditional experimental approaches,4, 5 and thus there is little evidence to explore the proposed mechanisms. In our studies, we employed the strategy of genetic code expansion to probe the cofactor in ADO and CDO.4, 5 Recently, we reported an uncrosslinked structure of human CDO (hCDO, GenBank ID: AAH24241) with with a 3,5-difluorotyrosine at 157, i.e., F2-Tyr157.4 Unlike those uncrosslinked CDO structures from Cys93 or Tyr157 mutants, which permanently disable the cofactor biogenesis, the uncrosslinked hCDO with unnatural amino acid F2-Tyr157 is fully capable of cofactor biogenesis for generation of a Cys-Tyr cofactor-bearing enzyme.4 The F2-Tyr157 hCDO is also catalytically active for L-cysteine dioxygenation. A notable finding from the uncrosslinked structure is that Cys93 has two distinct conformations, one of which is closer to the iron ion than Tyr157. This finding casts a serious question on which residue, Cys93 or Tyr157, is the first oxidation target during uncoupled oxidation reaction. In the present work, we took advantage of our success in obtaining a purely uncrosslinked form of hCDO and obtained ternary complex crystal structures of hCDO in both uncrosslinked and the mature forms. These new data revealed critical information about the cofactor biogenesis mechanism and also provided insights into the contribution of the Cys-Tyr cofactor to the dioxygenation mechanism. A distinct mechanistic proposal that starts with a Cys93 radical is discussed.
MATERIALS AND METHODS
Chemicals
All primers were synthesized at the Integrated DNA Technologies. Reagents were purchased from Sigma-Aldrich, New England Biolabs (NEB), and Thermo Fisher Scientific with the highest purity grade available and used as received. DNA manipulations in Escherichia coli were carried out according to standard procedures. Ampicillin (100 μg/mL), and chloramphenicol (30 μg/mL) were used as antibiotics for selection of recombinant strains. F2-Tyr was used in the genetically modified hCDO cell culture. The synthesis of F2-Tyr from 2,6-difluorophenol by using Citrobacter Freundii (ATCC8090) tyrosine phenol-lyase (TPL) was carried out following established methods as described previously.4, 23
Preparation of human CDO proteins with a genetically incorporated unnatural tyrosine157
The construction of pVP16-hCDO plasmid for wild-type (WT) hCDO was described previously.6 The cell culture was prepared at 37 °C in Luria Bertani (LB) medium in a baffled flask at 200 rpm with the appropriate antibiotic to an optical density of 0.8 AU at 600 nm. After overnight induction with 0.5 mM isopropyl-β-thiogalactoside (IPTG) at 28 °C, the cells were harvested and resuspended in the lysis buffer, i.e., 50 mM Tris-HCl at pH 8.0 containing 300 mM NaCl, 1 mM ferrous ammonium sulfate, and then disrupted by a Microfluidizer LM20 cell disruptor. The supernatant was recovered after centrifugation (13,000×g for 30 min) at 4 °C. The His-MBP-tagged protein was separated using Ni-NTA agarose beads. After buffer exchanged with a washing buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, pH 8.0), the isolated protein was eluted with elution buffer (50 mM Tris-HCl, 300 mM NaCl, 250 mM imidazole, pH 8.0). The hCDO-containing fractions were dialyzed into the storage buffer (50 mM Tris-HCl, 100 mM NaCl, 5% glycerol, pH 8.0) and stored at −80 °C. The protein concentration was determined based on the extinction coefficient of ε280 nm = 25,440 cm−1M−1. For expression of F2-Tyr157 hCDO protein, pEVOL-F2-TyrRS was co-transformed with pVP16-hCDO157TAG into BL21(DE3).6 The transformed cells were induced with 0.5 mM IPTG and 0.02% L-arabinose at OD600 nm of 0.8 in the presence of 0.5 mM F2-Tyr. After growing for 12 h at 30 °C, the F2-Tyr157 hCDO protein was purified using the protocol described above for WT hCDO. The His-MBP tag was removed from WT and F2-Tyr157 hCDO by using a TEV protease during the last phase of the purification. The liberated native and F2-Tyr157 proteins were further purified by Superdex 75 gel-filtration column in 20 mM Tris-HCl, 50 mM NaCl (pH 8.0) buffer and were ultrafiltrated to the required concentration for subsequent experiments.
Electron paramagnetic resonance (EPR) spectroscopy
The protein samples were treated with EDTA to remove trace metals. They were anaerobically reconstituted and dialyzed with ferrous ammonium sulfate to ensure full iron occupancy. Then, they were incubated with L-cysteine and nitric oxide (•NO), a spin probe of the FeII-center and a structural analogue of the molecular oxygen. The •NO-releasing agent DEA-NONOate (Cayman Chemical Co.) was dissolved into water in the glove box, and the •NO-bound CDO samples were formed by anaerobically soaking the ES-complex with certain volume of DEA-NONOate in the presence of 20 mM L-ascorbic acid for 1 h. The samples were transferred to quartz EPR tubes and slowly frozen in liquid nitrogen. EPR spectra were recorded on a Bruker E560 X-band spectrometer equipped with a cryogen-free 4 K temperature system with an SHQE high-Q resonator at 100 kHz modulation frequency, 0.8 mW microwave power, 0.6 mT modulation amplitude at 20 – 50 K, and an average of four scans for each spectrum.
Crystallization, data collection, model building, and refinement
Crystals of the hCDO were grown at 22 °C by using the hanging-drop vapor-diffusion technique against a mother liquor composed of 0.1 M MES (pH 6.5), 2 M ammonium sulfate, 2% PEG 400, as previously described.4 After soaking in a cryoprotectant containing reservoir solution plus 20% glycerol for 30 s, the crystals were flash-frozen and stored by liquid nitrogen for data collection using synchrotron radiation. The substrate-bound structures were obtained by soaking 100 mM L-cysteine to the hCDO crystals. The anaerobic crystallization for obtaining pure uncrosslinked hCDO was conducted in an O2-free anaerobic chamber from Coy Laboratory Products. The •NO-bound crystals were formed by soaking the ES-complex crystals with the •NO-releasing agent DEA-NONOate in the presence of 0.5 mM TCEP-HCl for 15 min anaerobically before flash cooling in liquid nitrogen.
All X-ray diffraction intensity data were integrated, scaled, and merged using HKL2000.25 Molecular replacement was performed with Phenix using the crystal structure of WT hCDO as a starting model (Protein Data Bank entry 2IC1)26 except the F2-Tyr hCDO-CYS-•NO using the crystal structure of uncrosslinked F2-Tyr hCDO (6BPT)4 as the starting model. The Coot and Phenix software packages were used for the model building (main chain tracing), ligand and water finding, and real space refinement of side chains and zones (see data and refinement statistics in Table 1).27
Table 1.
X-ray crystallographic data collection and refinement statistics1
| Description (PDB ID) (crosslink status) |
F2-Tyr hCDO- CYS-•NO (6BPR) (Uncrosslinked) |
WT hCDO (6E87) (Crosslinked) |
WT hCDO-CYS (6N42) (Crosslinked) |
WT hCDO- CYS-•NO (6N43) (Crosslinked) |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | SBC-19-BM | SSRL-BL9–2 | SBC-19-BM | SSRL-BL9–2 |
| Space group | P65 | |||
| Wavelength (Å) | 50.00 – 1.96 (1.99 – 1.96) |
50.00 – 1.95 (1.98 – 1.95) |
50.00 – 2.20 (2.25 – 2.20) |
50.00 – 2.29 (2.34 – 2.29) |
| a, b, c (Å) | 131.4, 131.4, 34.2 | 131.7, 131.7, 34.3 | 131.2, 131.2, 34.3 | 131.2, 131.2, 34.2 |
| α, β, γ (°) | 90, 90, 120 | |||
| Completeness (%) | 100.0 (100.0) | 99.5 (94.1) | 100.0 (100.0) | 92.5 (99.2) |
| No. of unique reflections | 24681 (1224) | 25141 (1169) | 17616 (1173) | 15449 (954) |
| I / σ | 19.9 (2.6) | 13.5 (1.4) | 15.1 (2.6) | 9.0 (1.5) |
| CC1/2 last shell | 0.851 | 0.746 | 0.829 | 0.757 |
| Redundancy | 11.1 (11.0) | 6.3 (4.1) | 12.5 (12.4) | 5.2 (3.3) |
| Rmerge | 14.3 (99.9) | 13.6 (67.7) | 17.4 (98.9) | 14.5 (46.7) |
| Wilson B factor (Å2) | 25.1 | 26.4 | 26.1 | 32.4 |
| Refinement | ||||
| Resolution (Å) | 32.83 – 1.96 | 43.12 – 1.95 | 32.84 – 2.20 | 32.73 – 2.29 |
| No. of reflections | 24659 | 25129 | 17598 | 15431 |
| Rwork / Rfree | 16.0 / 18.8 | 17.1 / 19.7 | 17.2 / 21.0 | 17.2 / 21.0 |
| RMSD for bond lengths (Å) | 0.008 | 0.006 | 0.008 | 0.008 |
| RMSD for bond angles (°) | 0.9 | 0.9 | 0.9 | 1.1 |
| Ramachandran statistics2 | ||||
| Preferred (%) | 99.4 | 98.4 | 99.5 | 98.4 |
| Allowed (%) | 0.6 | 1.1 | 0.5 | 1.6 |
| Outliers (%) | 0 | 0.5 | 0 | 0 |
| No. of atoms | ||||
| Protein | 1565 | 1540 | 1523 | 1502 |
| L-cysteine | 7 | NA3 | 7 | 7 |
| FeII | 1 | 1 | 1 | 1 |
| •NO | 2 | NA | NA | 2 |
| Water | 191 | 155 | 121 | 98 |
| Average B-factors (Å2) | ||||
| Protein | 29.2 | 33.7 | 31.2 | 37.9 |
| L-Cysteine | 31.9 | N/A | 43.0 | 41.9 |
| FeII | 21.4 | 28.4 | 41.7 | 34.5 |
| NO | 30.8 | NA | NA | 44.0 |
| Water | 40.8 | 42.7 | 38.6 | 42.1 |
Values in parentheses are for the highest-resolution shell.
Ramachandran statistics were analyzed using MolProbity.24
NA, no applicable.
Computational methods
We utilized Cartesian coordinates determined experimentally in the crystal structure of F2-Tyr157 hCDO ternary complex (PDB entry 6BPR) as the starting structure for the DFT calculations. The iron complex in the model with the following ligands: three histidine residues (His66, His88 and His140), the bound substrate L-cysteine, and •NO. Other than the precursor residues of the cofactor, i.e., the uncrosslinked form of Cys93 and F2-Tyr157, we also considered Tyr58, Arg60, Glu104, and His155, as part of the active site of F2-Tyr157 hCDO. The histidine, cysteine, tyrosine, arginine, and glutamate residues were truncated to methylimidazole, methylsulfide, p-methylphenol, propylguanidinium and propanoate groups, respectively. Hydrogen atoms were modeled in with standard bond metrics, and their positions were optimized while freezing all heavy atoms. Attempts to optimize with fewer constraints lead to the sulfur of Cys93 moving away from the iron center and are not reported. The constrained geometry optimizations and single point calculation were performed using the B3LYP28, 29 hybrid functional and RIJCOSX approximation30, 31, together with the def2-TZVP32, 33 basis set with auxiliary basis sets def2/J34 for Coulomb fitting were used for all atoms in the gas phase. The EPR parameter were calculated on the geometry based on crystal structure, with three functionals B3LYP, PBE0 and PWP1 and a combination of basis sets: CP(PPP) basis set35 for Fe, Kutzalnigg’s basis set for NMR and EPR parameters (IGLO-III)36 for the NO atom of the NO ligand and def2-TZVP basis set with auxiliary basis sets def2/J for the remaining atoms. A higher-resolution radical grid with an integration accuracy of 7.0 was used for Fe atom and N atoms. In these calculations, the tight self-consistent field (SCF) convergence criteria were used. Scalar relativistic effects were corrected using the all-electron zero-order-relativistic-approximation (ZORA).37, 38 DFT calculations were carried out with the ORCA (4.0.1) quantum chemistry program package39 using the high-performance computational clusters of UTSA and Texas Advanced Computing Center (TACC).
RESULTS
X-band EPR spectra of substrate-bound iron-nitrosyl F2-Tyr157 CDO
The diatomic molecule •NO has previously been employed as a spin probe and an O2 surrogate for the ferrous ion in CDO.22, 40, 41 An EPR study was performed to verify that the electronic structure and geometry of the hCDO active site in the substrate-bound ternary complex are minimally perturbed in F2-Tyr157 hCDO. When •NO binds to the WT hCDO and F2-Tyr157 hCDO in the presence of L-cysteine, three sets of EPR signals were observed in both proteins (Figure 1). The major resonances arise from a characteristic, broad low-spin (S = 1/2) non-heme, L-cysteine-bound, ferrous-nitrosyl {Fe(NO)}7 complex as was well-documented in a previous study,22 and a more narrow signal from an axial dinitrosyl {Fe(NO)2}9 complex. The S = 1/2 total ground spin state of the dinitrosyl complex is due to an antiferromagnetic coupling between the high-spin ferrous iron (SFe = 2) and the overall (NO)2− ligand (S(NO)2 = 3/2), which has been found in synthetic and protein-based non-heme iron centers.42–49 An additional minor axial signal was also observed which was present in controls that did not contain L-cysteine and thus attributed to the substrate-free form of the ferrous-nitrosyl complex (Figure S1). These EPR data illustrate that the ternary complex, which mimics the substrate-bound FeII-oxygen species in the uncrosslinked form of hCDO, are nearly electronically identical in WT hCDO and F2-Tyr157 hCDO. Given the high sensitivity of EPR spectroscopy in detecting the geometry and electronic structures of the iron center, these results suggest that the ternary complex of hCDO with an unnatural tyrosine F2-Tyr157 could faithfully represent the corresponding ternary complex chemical structure of the wild-type protein.
Figure 1.
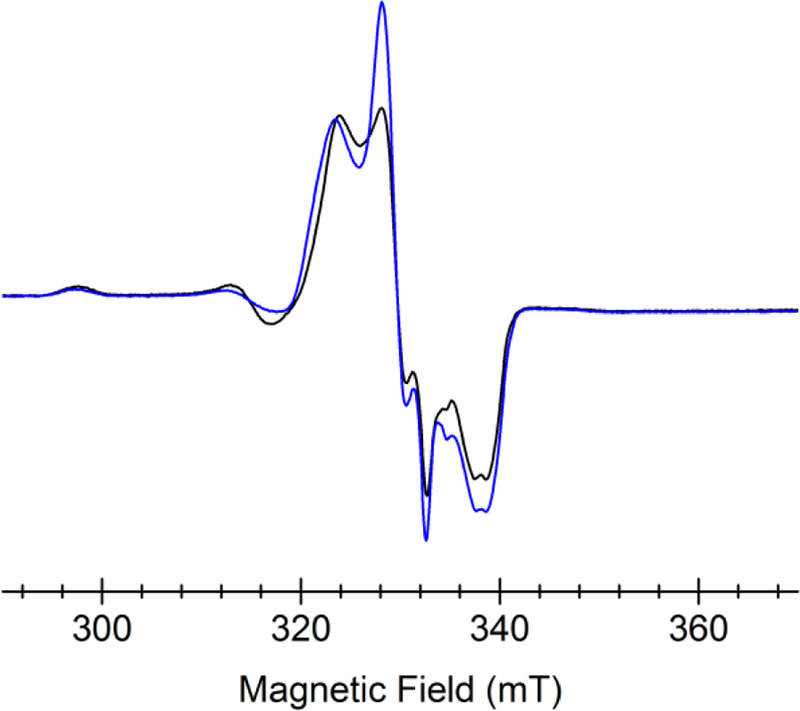
EPR spectra of CDO (FeII)-L-cysteine-•NO complex (black trace, wild-type enzyme; blue trace, F2-Tyr157 hCDO) These spectra were collected at 50 K, 0.8 mW microwave power, 0.6 mT modulation amplitude, and 100 kHz modulation frequency.
Determination of the ternary complex structures of uncrosslinked and crosslinked hCDO
The crystal structure of CDO with bound L-cysteine substrate and an O2 surrogate is crucial for understanding the mechanism of the Cys-Tyr cofactor formation as it mimics the first reactive complex structure of the enzyme. Thus, we determined a crosslink-free complex structure of F2-Tyr157 hCDO by soaking the crystals with both L-cysteine and •NO under anaerobic conditions. The ternary complex structure of uncrosslinked F2-Tyr157 hCDO, refined to 1.96 Å resolution (Table 1), shows additional density for the substrates bound to the iron ion and no crosslink between Cys93 and F2-Tyr157 (Figure 2A). The final refined structure was obtained by fitting the excess density with •NO at 100% occupancy. The Fe-N-O angle is 116 degrees, consistent with the angle found in the crystal structure of isopenicillin N synthase (IPNS) complexed with its substrate and •NO (Figure S2), an enzyme that also has substrate-based thiolate ligation to a catalytic non-heme iron center.50 The Fe-N bond for the •NO ligand is 1.94 Å. The distance between the oxygen of the •NO ligand and the Nδ of His155 is 3.1 Å, allowing for a weak H-bond interaction between them (Figure 2B). The occupancy of the water ligand in the ternary complex structure is lower than that in the crystal structure of uncrosslinked F2-Tyr157 hCDO in the substrate-free form, and the B-factor changes from 15.6 to 37.8. The lower occupancy and higher B-factor indicate that the water ligand has a low affinity to the iron ion and is replaced by •NO (Figure S3, A–C). Of note, the hydroxyl group of F2-Tyr157 forms a strong H-bond with the carboxyl group of the iron-bound L-cysteine (2.6 Å), as was observed in a previous computational study.51 Interestingly, the •NO ligand is in close contact with one of the conformations of Cys93 (3.1 Å) (Figure 2B). The potential interaction between •NO with Cys93 suggests a possible reaction between the iron-bound dioxygen, mostly in the FeIII-superoxide form and Cys93 during cofactor biogenesis should the uncoupled oxidation reaction occur. The oxygen of the •NO ligand is 3.1 Å from the F2-Tyr157 hydroxyl group (Figure 2B), however, the strong H-bond between the tyrosyl hydroxyl group and the carboxyl group of the substrate should decrease the probability of the tyrosine residue oxidation via hydrogen atom transfer.
Figure 2.
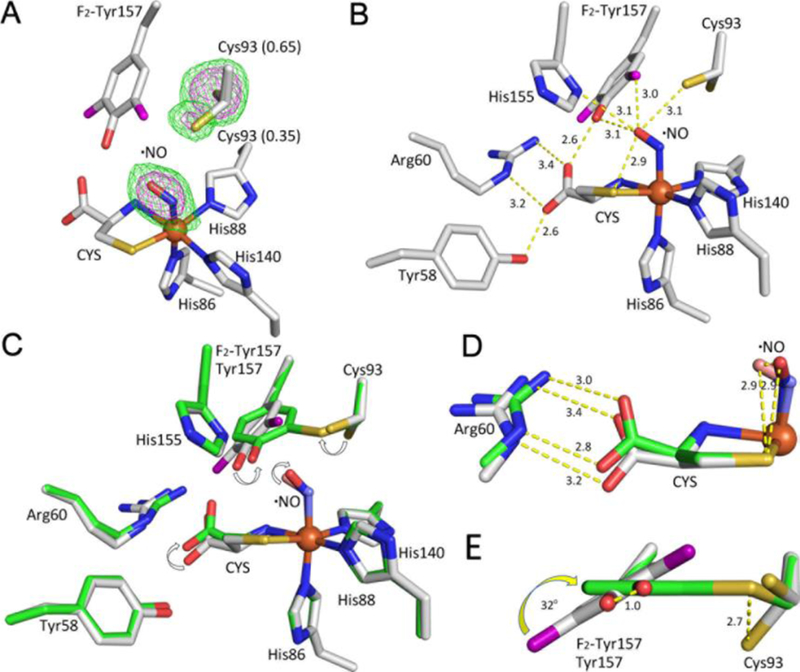
A ternary complex of the 100% uncrosslinked F2-Tyr157 hCDO bound with L-cysteine and •NO. The L-cysteine substrate is labeled as CYS. The fluorine atoms are shown in purple color. (A) The omit Fo−Fc electron densities of the •NO ligand and Cys93 (which has two conformations) contoured at 3 σ (green) and 6 σ (purple), respectively. (B) The details of the key interactions. The distances are in angstrom. (C) Alignment of the F2-Tyr157 CDO (grey) and the matured WT CDO (green). (D) Both the CYS and •NO rotate during the cofactor formation. (E) Tyr157 rotates during the cofactor biogenesis.
We also obtained a crosslinked ternary complex structure of WT hCDO with the same method. The crystal structure of the ternary complex of the mature protein shows additional density bound to the iron ion and the crosslinked Cys-Tyr cofactor. Fitting with •NO at 100% occupancy resulted in a ternary complex structure. This structure represents an analogous complex for oxygenation but not for cofactor biogenesis, as the Cys-Tyr cofactor is already fully installed. Obtaining such a structure would allow identification of any cofactor formation-induced structural rearrangement at the enzyme active site. The water ligand in this structure was not found in the iron center after soaking with L-cysteine and •NO (Table 1 and Figure S3, D–F). The hydroxyl group of the Cys-Tyr cofactor forms a strong H-bond with both the •NO ligand and the carboxyl group of the substrate L-cysteine (Figure S4). Tyr157 rotates during cofactor formation, and the position of •NO moves together with Tyr157 possibly because of their interactions (Figure 2C). The structural data indicate that the orientation of the carboxyl group of the iron-bound L-cysteine substrate is sensitive to the movement of Tyr157. The autocatalytic formation of the cofactor moves F2-Tyr157 (1 Å difference) away from the guanidyl group of Arg60 and rotates it by ca. 32 degrees, pulling the carboxyl group of the substrate toward Arg60 (Figure 2 D–E).
The Fe-N-O angle in the crosslinked complex structure is 139 degrees, significantly large than in the uncrosslinked structure (Figure 2D). Notably, the dihedral angles of L-Cys (S)-Fe-N-O are −4.5 and −66.2 degrees in the crosslinked and uncrosslinked structures, respectively. These differences may be important for understanding the contribution of the Cys-Tyr cofactor in tuning the reactivity of the iron-bound oxygen towards the bound L-cysteine.
The Fe-N-O angle in the crosslinked complex structure is 139 degrees, significantly large than in the uncrosslinked structure (Figure 2D). Notably, the dihedral angles of L-Cys (S)-Fe-N-O are −4.5 and −66.2 degrees in the crosslinked and uncrosslinked structures, respectively. Since the distance from the distal oxygen to the substrate sulfur shows little change upon cofactor formation, these changes in ligand orientation should be significant for understanding the contribution of the Cys-Tyr cofactor in tuning the reactivity of the iron-bound oxygen towards the bound L-cysteine.
Structural models of the ternary complex of F2-Tyr157 hCDO bound with L-cysteine and •NO
To probe the feasibility of Cys93 as the initial target for oxidation by the putative FeIII-superoxide intermediate, a computational study was executed to predict hydrogen atom positions in the ternary complex. Figure 3 shows models of the ternary complex mimic of the active sites of F2-Tyr157 hCDO and WT hCDO. Heavy atom positions were obtained from the •NO-bound crystal structure (6BPR), and hydrogen atoms positions were calculated by a constrained geometry optimization using the density functional theory (DFT) method (see the experimental procedures).
Figure 3.
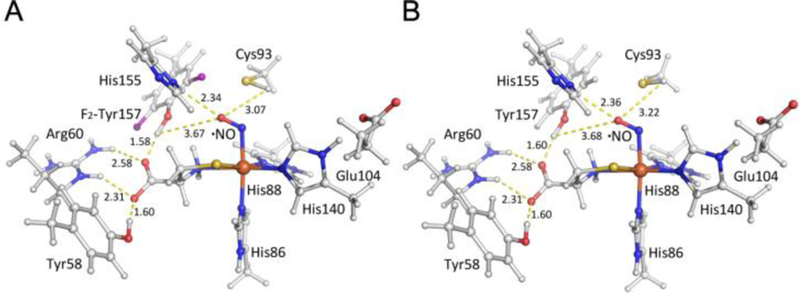
Structural models of the ternary complex of F2-Tyr157 hCDO bound with L-cysteine and •NO (A) along with the ternary complex models of WT hCDO bound with L-cysteine and •NO (B). Hydrogen atoms were determined by constrained geometry optimizations with other atoms fixed in the coordinates determined in the crystal structure as described in the text.
In order to verify that the models represent reasonable structures, EPR parameters, principal values of g-tensor and 14N hyperfine tensor (A-tensor), were calculated using DFT calculation with three different functionals for comparison with experimental data and other published DFT structures. Table S1 shows the calculated principal values as well as reported values listed from the literature.22 The calculated g values are nearly consistent with the reported values for CDO (S = ½) complex within ±0.01 difference. The principal axis of largest component of 14N hyperfine tensor (denoted as Ay in Table S1) is predicted to be nearly collinear to the axis of middle component (gy) of g-tensor from which it deviates slightly by 15° or less in case of using PBE0 and PW91. Negative components of 14N hyperfine tensor, which come from slightly dominant nuclear spin-electron spin dipolar interaction in Ax and Az, are predicted by the DFT calculation, but not clearly recognized in the experimental spectra due to line broadening in gz and gx. The same constrained geometry optimization was carried out also for the uncrosslinked WT hCDO by substituting fluorine atoms with hydrogen atoms in the model before energy minimizations (Figure 3B), and similar results were observed.
To further examine the oxidation target during the initial steps of cofactor biogenesis, we implemented single point energy calculations for different positions of the hydrogen atom in the hydroxyl group of F2-Tyr157 or the thiol group of Cys93 by changing the dihedral angles (θ, ϕ) defined at every 20 degrees. The relative single point energies for different dihedral angles are shown in Figure S5. In this set of the calculations, F2-Tyr157 hCDO and WT hCDO gave the minimum energy at the dihedral angle θ of 11.8 and 10.2 (Figure 3 A–B and Figure S5A), indicating that the O-H bond of F2-Tyr157 or Tyr157 prefers an orientation pointing towards the carboxyl group of substrate L-cysteine to form a H-bond, rather than •NO. According to the energy profile, the structure with θ of 100 degrees gives the highest energy. In this case, the tyrosine O-H group approaches the amino group of the substrate L-cysteine. There is a local minimum point at around θ = 160 degrees with an energy higher than lowest point by 29 kcal/mol at around θ = 20 degrees, in which O-H bond comes close to the •NO ligand. We observed that F2-Tyr157 hCDO and WT hCDO yield minimum energies at the dihedral angle ϕ of 166.8 and 189.8 degrees, respectively (Figure 3 A–B and Figure S5B). In this case, the S-H bond points further away from F2-Tyr157 or Tyr157. According to the energy profile for the different hydrogen positions of thiol group of Cys93, two local minima were predicted at the dihedral angle ϕ of 60 and 280 degrees, in which hydrogen of S-H bond is positioned away from closer fluorine atom by 2.2 to 2.3 Å. Together, these computational results suggest that the first target of the oxidation by the iron-bound oxygen during cofactor biogenesis is Cys93, not Tyr157, and that His155 may play a role in the O2 binding.
DISCUSSION
This work addresses the most critical chemical question in the cofactor formation mechanism in CDO, i.e., which residue, Cys93 or Tyr157, is first oxidized by an iron-bound oxygen intermediate. It has been speculated that an iron-bound oxygen species oxidizes Tyr157 as the first step of the cofactor biogenesis.2, 17, 21 This is nearly entirely based on the fact that protein-based tyrosyl radical is more frequently observed, and that in the mature enzyme structure, Tyr157 is closer to the Fe ion than Cys93. However, Siakkou et al.16 and Pierce et al.22 suggest the possibility that the iron-bound oxygen could oxidize Cys93 to form a thiyl radical. In our newly obtained uncrosslinked structure and ternary complex structure, one conformer of Cys93 is a similar distance to the Fe center as F2-Tyr157, the hydroxyl group of which is stabilized by the carboxyl group of the substrate. In addition to the structural data, our working model is consistent with the fact that cysteine is more easily oxidized than tyrosine.
Since fluorine is a relatively small atom and its introduction to Tyr157 does not abrogate cofactor biogenesis, the success for obtaining the first uncrosslinked ternary complex hCDO structure provides a structural platform for the mechanistic considerations of the autocatalytic cofactor formation. We also determined the equivalent ternary complex crystal structure with a mature cofactor. These successes allowed us to compare the starting and the ending (mature) structures to determine the changes that further shed light on the cofactor biogenesis mechanism with otherwise inaccessible information. It should be noted that our findings are consistent with a proactive description of the substrate binding and activation provided by Driggers et al. based on the structures of Y157F and C93A.11 We also noted a substantial rotation and ca. 1.0 Å shift in the side chain of residue F2-Tyr157 or Tyr157 between the uncrosslinked and mature forms of the protein structure (Figure 2E), and this rotation could be detected in crystallo. To assess the degree to which the observed rotation may be influenced by the fluorine substitution, we compare the ternary complex of uncrosslinked F2-Tyr157 hCDO, Y157F rCDO (Figure S6A), and C93A rCDO (Figure S6B)11. The overlays show that the aromatic rings are in similar positions, but the ring of F2-Tyr157 has significant rotation as compared to Tyr157 and Phe157. Thus the rotation we observed during cofactor formation may be largely due to the presence of fluorine, however the translocation of the phenol ring should be a general phenomenon for formation of the thioether bond of the cofactor.
It is very likely that such a movement enhances the catalytic activity by positioning the hydroxyl group of Tyr157 to enable its intended catalytic role in the mature form of the enzyme for optimal chemistry. In the uncrosslinked form, the hydroxyl group of Tyr157 is either incapable or in a less optimal position to fulfill such a role. The less efficient stabilization of the ternary complex not only slows down the chemistry, but may also cause uncoupled oxygen activation to provide the driving force for Tyr157 and Cys93 crosslinking. Moreover, the carboxyl group of the substrate L-cysteine moves toward Arg60 after the crosslink formation due to a strong H-bond between the carboxyl group of the substrate and the hydroxyl group of Tyr157 (Figure 2D). If there is no crosslink to stabilize the position of Tyr157, the hydroxyl group of Tyr157 is easily disturbed by the incoming substrate, and the substrate L-cysteine may bind less tightly. The movements of these residues and substrate are consistent with the reported computational work of uncrosslinked and crosslinked mouse CDO.51 Based on the two ternary complex structures and the movements of the residues and substrates, we conclude that there is a strong H-bond between the carboxyl group of substrate L-cysteine and hydroxyl group of Tyr157. Cys93 adopted two conformations in the structure of Y157F rCDO (Figure S6).11 In our uncrosslinked structures, we also found Cys93 with two conformations, and there is an interaction between nitric oxide and one conformation of Cys93.
The oxidation of Cys93 by the non-heme iron-bound oxidant is further supported by a computational study with models built from the uncrosslinked ternary complex structure. This understanding is consistent with the fact that cysteine is more reactive than tyrosine and that this enzyme is designed to oxidize cysteine, and it is not wholly unsurprising as the iron-bond oxidant in this enzyme is intended to oxidize a cysteine substrate rather than a tyrosine.
Together, these findings led us to propose a new mechanistic model for the formation of the Cys-Tyr cofactor found in CDO (Figure 4A). Dioxygen binds to the FeII ion of the enzyme-substrate complex, producing an iron-bound superoxide anion radical. Subsequent oxidation of Cys93 though hydrogen atom abstraction by the FeIII-superoxide generates a thiyl radical at Cys93 and an iron-bound hydroperoxide species. A similar example is that a CuII-superoxide is active toward H atom abstraction from a cysteine residue.3, 6 The oxidative attack on Tyr157 by the thiyl radical creates the crosslink with the formation of a thioether bond and a tyrosyl-like radical. The generation of a transient tyrosyl-like radical at Tyr157 induces deprotonation of the hydroxyl group and promotes the formation of a hydrogen peroxide side product with the iron-bound hydroperoxide. In the case of F2-Tyr157 hCDO (Figure 4B), the proposed mechanism is analogous to the native enzyme yet distinct in that F− is the anticipated side product rather than hydrogen peroxide.4 The transient fluorotyrosyl-like radical may abstract one hydrogen from the iron-bound hydroperoxide, producing the iron-bound superoxide anion radical that may react with the substrate. Overall, the formation of the Cys-Tyr cofactor in WT CDO consumes one molecule of O2, whereas O2 is expected to be a spectator in the case of F2-Tyr157 hCDO.
Figure 4.

Working models for cofactor biogenesis in the (A) wild-type and (B) F2-Tyr157 hCDO. In the proposed pathway, substrate binding to the ferrous iron center generates an iron-bound superoxide radical, which subsequently oxidizes Cys93 and produces an iron-bound hydroperoxide and a thiol radical. Oxidation of Tyr157 by the thiol radical leads to the crosslink between Cys93 and Tyr157, concomitant with the production of a transient-state radical species in Tyr157. Deprotonation of the hydroxyl group to the iron-bound hydroperoxide results in a ketone species, which drives the C-H bond cleavage. There are two less electrons in the transient-state radical species in F2-Tyr157 (indicated by brackets) compared with the radical species in Tyr157.
The ternary complex structure obtained from the wild-type human CDO (Fig. 5A) is not merely a control for understanding the cofactor biogenesis, it may also be useful for understanding the role of the Cys-Tyr cofactor in catalysis. A similar ternary complex is not previously available, but a cysteine-derived persulfenate species was previously characterized (Figure 5B).52, 53 It is still under debate whether this persulfenate species is an intermediate of the catalytic cycle, and if so, it would strongly favor a concerted dioxygenation mechanism rather than a stepwise the O-atom transfer model. The comparison of the two structures shows that the Cys-Tyr cofactor and the active site residues are all nearly aligned, as well as the distal oxygen of the dioxygen in the persulfenate species and the •NO ligand. The most notable difference is the proximal oxygen and the nitrogen in the •NO ligand (Figure 5C). It has been a general understanding that •NO binds to the nonheme Fe(II) center in enzymes at the same location where O2 binds based on a large amount of the data from other Fe(II)-dependent proteins. Thus, the ternary nitrosyl complex structure we reported here is a reasonable structural analog of the reactive ternary complex of the catalytic cycle. It is important to note both the distal oxygen of the ligand in the nitrosyl complex and the persulfenate interacts with multiple active site residues. It is 2.9 Å from the phenolic oxygen of the cofactor, 3.1 Å from His155, and 3.2 Å to the sulfur atom of the cofactor (Figure 5A). Similar interactions are present in the putative the persulfenate intermediate (Figure 5B) and the uncrosslinked enzyme (Figure 5D). In contrast, the proximal oxygen and the nitrogen of the ligands only interact with the phenolic oxygen of the cofactor at 2.9 and 3.2 Å distances, respectively. This proximal atom forms a stronger interaction with the target sulfur atom of the bound substrate in the persulfenate structure as compared to the nitrosyl complex, 1.7 and 2.8 Å, respectively. One potential interpretation is that the Cys-Tyr cofactor and His155 moieties ‘hold’ the distal oxygen, allowing the proximal oxygen to oxidize the target sulfur atom of the substrate, so that a concerted oxygenation mechanism may be preferred.52, 53 In the cofactor-free structure, the nitrogen atom of the ligand is 4.1 Å from the cofactor and less likely to be pushed to the substrate. Although the new ternary complex structure cannot prove or disapprove the validity of the putative persulfenate species as a catalytic intermediate, this new structural data are more supportive of the concerted mechanistic model illustrated in Figure 6. Considering the changes of the Fe-N-O angle and the dihedral angle of L-Cys (S)-Fe-N-O in the crosslinked and uncrosslinked structures described earlier (Figure 2 and associated text), the presence of a Cys-Tyr cofactor appears to facilitate a more efficient dioxygenation pathway whereas its absence renders the iron center in between the stepwise oxygen insertion pathway or a borderline concerted dioxygen transfer. Future spectroscopic and computational studies are needed to further understandings about the oxygenation mechanism of this thiol dioxygenase.
Figure 5.
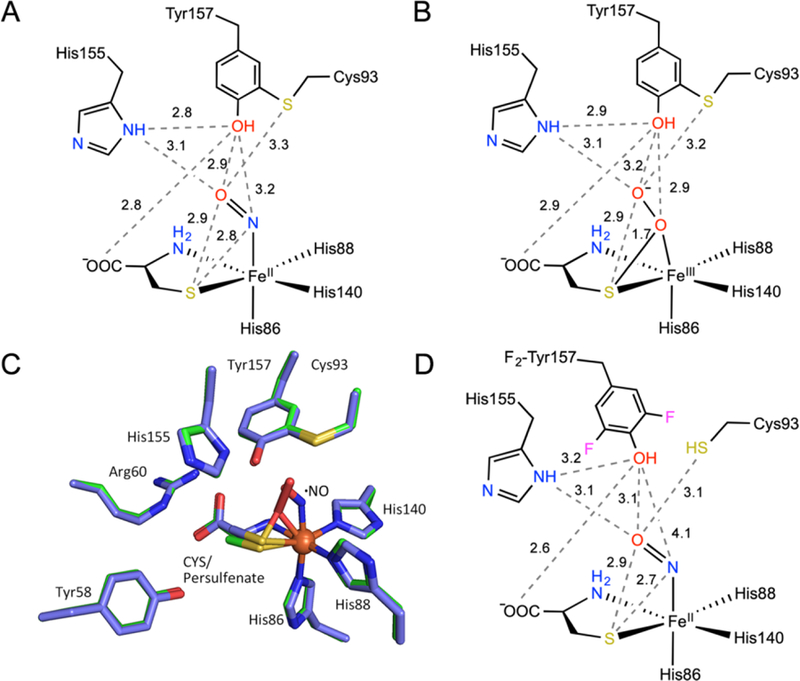
Comparison of the active site of CDO in the ternary nitrosyl complex and a previously characterized putative intermediate of the enzyme. (A) 2D interaction diagram of the active site of the ternary complex of crosslinked wild-type human CDO and (B) the putative cysteine-derived persulfenate intermediate trapped in rat CDO. (C) Overlay of the ternary complex of human CDO (green) and the persulfenate species (light blue). (D) For comparison, a 2D diagram of the active site of the ternary nitrosyl complex of uncrosslinked F2-Tyr157 CDO is included.
Figure 6.
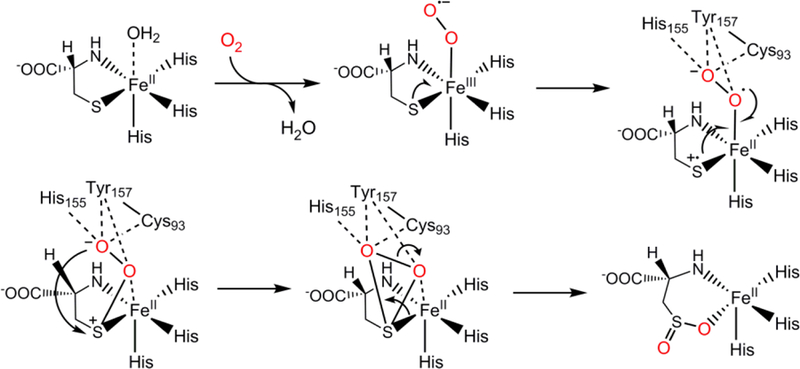
Proposed dioxygenase pathway with the assistance of the Cys-Tyr cofactor
In conclusion, by using •NO as an oxygen surrogate and the strategy of an expanded genetic code, we obtained fully uncrosslinked and mature human CDO structures bound with L-cysteine and nitric oxide, which shed light on the cofactor biogenesis mechanism. Based on the geometric details and the conformational changes in both ternary structures, we found that the cysteine (Cys93), rather than the generally expected tyrosine (Tyr157), is likely the first target for oxidation during iron-assisted Cys-Tyr cofactor biogenesis. This conclusion is further supported by a computational study with models built from the crystal structure. These findings will facilitate the structure and mechanism studies of CDO and other relevant metalloenzymes.
Supplementary Material
Acknowledgments
Funding
The work is supported by the National Science Foundation grant CHE-1808637, and in part by the National Institutes of Health Grants GM107529 and the Lutcher Brown Distinguished Chair Endowment fund (to A.L.). X-ray synchrotron data were collected at the beamlines of the Advanced Photon Source Section 19, Structural Biology Center user program GUP-48198, Argonne National Laboratory and the beamline BL9–2 of the Stanford Synchrotron Radiation Lightsource (SSRL) under the user program #5B14, SLAC National Accelerator Laboratory. The beamline staff scientists are acknowledged for the assistance of the remote data collections. The Advanced Photon Source is a U.S. Department of Energy, Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. SSRL is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515 and by the National Institutes of Health (P41GM103393). We are also indebted to the supercomputer clusters of The University of Texas at San Antonio and the Texas Advanced Computing Center (TACC).
Footnotes
Protein accession ID
Human CDO: GenBank ID: AAH24241
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00006.
Six supporting figures and a table include more spectroscopic data, structural refinement details, a reference of complex for comparison, and more computational details.
The authors declare no competing financial interest.
References
- (1).McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, and Phillips GN (2006) Structure and mechanism of mouse cysteine dioxygenase, Proc. Natl. Acad. Sci. U. S. A 103, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA, and Stipanuk MH (2006) Crystal structure of mammalian cysteine dioxygenase - A novel mononuclear iron center for cysteine thiol oxidation, J. Biol. Chem 281, 18723–18733. [DOI] [PubMed] [Google Scholar]
- (3).Cowley RE, Cirera J, Qayyum MF, Rokhsana D, Hedman B, Hodgson KO, Dooley DM, and Solomon EI (2016) Structure of the reduced copper active site in preprocessed galactose oxidase: Ligand tuning for one-electron O2 activation in cofactor biogenesis, J. Am. Chem. Soc 138, 13219–13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Li J, Griffith WP, Davis I, Shin I, Wang J, Li F, Wang Y, Wherritt DJ, and Liu A (2018) Cleavage of a carbon–fluorine bond by an engineered cysteine dioxygenase, Nat. Chem. Biol 14, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang Y, Griffith WPWP, Li J, Koto T, Wherritt DJDJJ, Fritz E, and Liu A (2018) Cofactor biogenesis in cysteamine dioxygenase: C−F bond cleavage with genetically incorporated unnatural tyrosine, Angew. Chem. Int. Ed 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Whittaker JW (2003) Free radical catalysis by galactose oxidase, Chem. Rev 103, 2347–2363. [DOI] [PubMed] [Google Scholar]
- (7).Polyakov KM, Boyko KM, Tikhonova TV, Slutsky A, Antipov AN, Zvyagilskaya RA, Popov AN, Bourenkov GP, Lamzin VS, and Popov VO (2009) High-resolution structural analysis of a novel octaheme cytochrome c nitrite reductase from the haloalkaliphilic bacterium Thioalkalivibrio nitratireducens, J. Mol. Biol 389, 846–862. [DOI] [PubMed] [Google Scholar]
- (8).Schnell R, Sandalova T, Hellman U, Lindqvist Y, and Schneider G (2005) Siroheme- and [Fe4S4]-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site, J. Biol. Chem 280, 27319–27328. [DOI] [PubMed] [Google Scholar]
- (9).Hromada SE, Hilbrands AM, Wolf EM, Ross JL, Hegg TR, Roth AG, Hollowell MT, Anderson CE, and Benson DE (2017) Protein oxidation involved in Cys-Tyr post-translational modification, J. Inorg. Biochem 176, 168–174. [DOI] [PubMed] [Google Scholar]
- (10).Trofimov AA, Polyakov KM, Tikhonova TV, Tikhonov AV, Safonova TN, Boyko KM, Dorovatovskii PV, and Popov VO (2012) Covalent modifications of the catalytic tyrosine in octahaem cytochrome c nitrite reductase and their effect on the enzyme activity, Acta Crystallogr. Sect. D Biol. Crystallogr 68, 144–153. [DOI] [PubMed] [Google Scholar]
- (11).Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, and Karplus PA (2016) Structure-based insights into the role of the Cys-Tyr crosslink and inhibitor recognition by mammalian cysteine dioxygenase, J. Mol. Biol 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Whittaker MM, Kersten PJ, Nakamura N, Sanders-Loehr J, Schweizer ES, and Whittaker JW (1996) Glyoxal oxidase from Phanerochaete chrysosporium is a new radical-copper oxidase, J. Biol. Chem 271, 681–687. [DOI] [PubMed] [Google Scholar]
- (13).Baron AJ, Stevens C, Wilmot C, Seneviratne KD, Blakeley V, Dooley DM, Phillips SE, Knowles PF, and McPherson MJ (1994) Structure and mechanism of galactose oxidase. The free radical site, J. Biol. Chem 269, 25095–25105. [PubMed] [Google Scholar]
- (14).Whittaker MM, and Whittaker JW (2003) Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor, J. Biol. Chem 278, 22090–22101. [DOI] [PubMed] [Google Scholar]
- (15).Rogers MS, Hurtado-Guerrero R, Firbank SJ, Halcrow MA, Dooley DM, Phillips SE, Knowles PF, and McPherson MJ (2008) Cross-link formation of the cysteine 228-tyrosine 272 catalytic cofactor of galactose oxidase does not require dioxygen, Biochemistry 47, 10428–10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Siakkou E, Rutledge MT, Wilbanks SM, and Jameson GN (2011) Correlating crosslink formation with enzymatic activity in cysteine dioxygenase, Biochim. Biophys. Acta 1814, 2003–2009. [DOI] [PubMed] [Google Scholar]
- (17).Dominy JE Jr., Hwang J, Guo S, Hirschberger LL, Zhang S, and Stipanuk MH (2008) Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity, J. Biol. Chem 283, 12188–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stipanuk MH, Ueki I, Dominy JE Jr., Simmons CR, and Hirschberger LL (2009) Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels, Amino Acids 37, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stipanuk MH, Simmons CR, Karplus PA, and Dominy JE Jr. (2011) Thiol dioxygenases: unique families of cupin proteins, Amino Acids 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fellner M, Siakkou E, Faponle AS, Tchesnokov EP, de Visser SP, Wilbanks SM, and Jameson GNL (2016) Influence of cysteine 164 on active site structure in rat cysteine dioxygenase, J. Biol. Inorg. Chem 21, 501–510. [DOI] [PubMed] [Google Scholar]
- (21).Njeri CW, and Ellis HR (2014) Shifting redox states of the iron center partitions CDO between crosslink formation or cysteine oxidation, Arch. Biochem. Biophys 558, 61–69. [DOI] [PubMed] [Google Scholar]
- (22).Pierce BS, Gardner JD, Bailey LJ, Brunold TC, and Fox BG (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications, Biochemistry 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- (23).Seyedsayamdost MR, Yee CS, and Stubbe J (2007) Site-specific incorporation of fluorotyrosines into the R2 subunit of E. coli ribonucleotide reductase by expressed protein ligation, Nature Protocols 2, 1225–1235. [DOI] [PubMed] [Google Scholar]
- (24).Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (26).Ye S, Wu X. a., Wei L, Tang D, Sun P, Bartlam M, and Rao Z (2007) An insight into the mechanism of human cysteine dioxygenase - Key roles of the thioether-bonded tyrosine-cysteine cofactor, J. Biol. Chem 282, 3391–3402. [DOI] [PubMed] [Google Scholar]
- (27).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (28).Lee CT, Yang WT, and Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron-density, Phys. Rev. B 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (29).Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys 98, 5648–5652. [Google Scholar]
- (30).Neese F, Wennmohs F, Hansen A, and Becker U (2009) Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange, Chem. Phys 356, 98–109. [Google Scholar]
- (31).Neese F (2003) An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comp. Chem 24, 1740–1747. [DOI] [PubMed] [Google Scholar]
- (32).Schafer A, Horn H, and Ahlrichs R (1992) Fully optimized contracted Gaussian-basis sets for atoms Li to Kr, J. Chem. Phys 97, 2571–2577. [Google Scholar]
- (33).Weigend F, and Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (34).Weigend F (2006) Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (35).Neese F (2002) Prediction and interpretation of the 57Fe isomer shift in Mossbauer spectra by density functional theory, Inorg. Chim. Acta 337, 181–192. [Google Scholar]
- (36).Kutzelnigg W, Fleischer U, and Schindler M (1991) The IGLO-Method: Ab-initio calculation and interpretation of NMR chemical shifts and magnetic susceptibilities, In Deuterium and Shift Calculation, pp 165–262, Springer Berlin Heidelberg, Berlin, Heidelberg. [Google Scholar]
- (37).Vanlenthe E, Baerends EJ, and Snijders JG (1993) Relativistic regular 2-component Hamiltonians, J. Chem. Phys 99, 4597–4610. [Google Scholar]
- (38).van Wullen C (1998) Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations, J. Chem. Phys 109, 392–399. [Google Scholar]
- (39).Neese F (2012) The ORCA program system, WIREs Comput. Mol. Sci 2, 73–78. [Google Scholar]
- (40).Blaesi EJ, Gardner JD, Fox BG, and Brunold TC (2013) Spectroscopic and computational characterization of the NO adduct of substrate-bound Fe(II) cysteine dioxygenase: Insights into the mechanism of O2 activation, Biochemistry 52, 6040–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Blaesi EJ, Fox BG, and Brunold TC (2015) Spectroscopic and computational investigation of the H155A variant of cysteine dioxygenase: geometric and electronic consequences of a third-sphere amino acid substitution, Biochemistry 54, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Orville AM, Chen VJ, Kriauciunas A, Harpel MR, Fox BG, Munck E, and Lipscomb JD (1992) Thiolate ligation of the active site Fe2+ of isopenicillin N synthase derives from substrate rather than endogenous cysteine: spectroscopic studies of site-specific Cys-Ser mutated enzymes, Biochemistry 31, 4602–4612. [DOI] [PubMed] [Google Scholar]
- (43).Rogers PA, and Ding H (2001) L-cysteine-mediated destabilization of dinitrosyl iron complexes in proteins, J. Biol. Chem 276, 30980–30986. [DOI] [PubMed] [Google Scholar]
- (44).Tinberg CE, Tonzetich ZJ, Wang H, Do LH, Yoda Y, Cramer SP, and Lippard SJ (2010) Characterization of iron dinitrosyl species formed in the reaction of nitric oxide with a biological Rieske center, J. Am. Chem. Soc 132, 18168–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).D’Autreaux B, Horner O, Oddou JL, Jeandey C, Gambarelli S, Berthomieu C, Latour JM, and Michaud-Soret I (2004) Spectroscopic description of the two nitrosyl-iron complexes responsible for fur inhibition by nitric oxide, J. Am. Chem. Soc 126, 6005–6016. [DOI] [PubMed] [Google Scholar]
- (46).Vanin AF (2016) Dinitrosyl iron complexes with thiol-containing ligands as a “working form” of endogenous nitric oxide, Nitric Oxide 54, 15–29. [DOI] [PubMed] [Google Scholar]
- (47).Tonzetich ZJ, Do LH, and Lippard SJ (2009) Dinitrosyl iron complexes relevant to Rieske cluster nitrosylation, J. Am. Chem. Soc 131, 7964–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vanin AF, Poltorakov AP, Mikoyan VD, Kubrina LN, and Burbaev DS (2010) Polynuclear water-soluble dinitrosyl iron complexes with cysteine or glutathione ligands: electron paramagnetic resonance and optical studies, Nitric Oxide 23, 136–149. [DOI] [PubMed] [Google Scholar]
- (49).Ye S, and Neese F (2010) The unusual electronic structure of dinitrosyl iron complexes, J. Am. Chem. Soc 132, 3646–3647. [DOI] [PubMed] [Google Scholar]
- (50).Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Schofield CJ, Hajdu J, and Baldwin JE (1997) Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation, Nature 387, 827–830. [DOI] [PubMed] [Google Scholar]
- (51).Li W, Blaesi EJ, Pecore MD, Crowell JK, and Pierce BS (2013) Second-sphere interactions between the C93-Y157 cross-link and the substrate-bound Fe site influence the O2 coupling efficiency in mouse cysteine dioxygenase, Biochemistry 52, 9104–9119. [DOI] [PubMed] [Google Scholar]
- (52).Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, and Karplus PA (2013) Cysteine dioxygenase structures from pH 4 to 9: Consistent Cys-persulfenate formation at intermediate pH and a Cys-Bound enzyme at higher pH, J. Mol. Biol 425, 3121–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Begley TP, Stipanuk MH, and Karplus PA (2008) A putative Fe2+-bound persulfenate intermediate in cysteine cioxygenase, Biochemistry 47, 11390–11392 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.