

The title compound has a single protonated psilacetin cation and one half of a fumarate dianion in the asymmetric unit. The ions are held together through N—H⋯O hydrogen bonds in infinite one-dimensional chains along [111].
Keywords: crystal structure, tryptamines, hydrogen bonding
Abstract
The title compound (systematic name: bis{2-[4-(acetyloxy)-1H-indol-3-yl]ethan-1-aminium} but-2-enedioate), 2C14H19N2O2 +·C4H2O4 2−, has a single protonated psilacetin cation and one half of a fumarate dianion in the asymmetric unit. There are N—H⋯O hydrogen bonds between the ammonium H atoms and the fumarate O atoms, as well as N—H⋯O hydrogen bonds between the indole H atoms and the fumarate O atoms. The hydrogen bonds hold the ions together in infinite one-dimensional chains along [111].
Chemical context
Psychedelic agents have received a great deal of interest lately as potential pharmaceuticals to treat mood disorders, including depression and post traumatic stress disorder (PTSD) (Carhart-Harris & Goodwin, 2017 ▸). Psilocybin, a naturally occurring tryptamine derivative found in ‘magic’ mushrooms, is a prodrug of psilocin. When consumed orally, psilocybin hydrolyzes to generate psilocin, a serotonin-2a agonist, producing mood-altering or ‘psychedelic’ effects (Dinis-Oliveira, 2017 ▸). Like psilocybin, psilacetin serves as a prodrug of psilocin. Compared to psilocybin, psilacetin is easier and less expensive to synthesize. This suggests that administering psilacetin (instead of psilocybin) represents a better means of delivery for the active psilocin. Psilacetin was first reported in 1999 by Nichols and co-workers (Nichols & Frescas, 1999 ▸), generally producing the molecule as its crystalline fumarate salt. Psilacetin was structurally characterized earlier this year (Chadeayne et al., 2019 ▸). Herein we report the structure of a new crystalline form of psilacetin, in which two psilacetin molecules are protonated, and charge-balanced by one fumarate dianion.
Structural commentary
The molecular structure of bis(4-acetoxy-N,N-dimethyltryptammonium) fumarate is shown in Fig. 1 ▸. The cation possesses a near-planar indole, with a mean deviation from planarity of 0.04 Å. The acetate on the 4-position of the indole is approximately perpendicular, with the angles between the indole and acetate planes being 100.85 (1)°. Half of a fumarate ion is present in the asymmetric unit, with the full dianion produced through inversion. The fumarate shows a near planar trans configuration with a deviation from planarity of 0.019 Å. A series of N—H⋯O hydrogen bonds hold the ions together in the solid state.
Figure 1.

The molecular structure of bis(4-acetoxy-N,N-dimethyltryptammonium) fumarate, showing the atomic labeling. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen bonds are shown as dashed lines. Symmetry code: (i) 2 − x, 1 − y, 2 − z.
Supramolecular features
The 4-acetoxy-N,N-dimethyltryptammonium cations and fumarate dianions are held together in an infinite one-dimensional chain through N—H⋯O hydrogen bonds (Table 1 ▸) along the [111] direction. The anionic oxygen of the carboxylic acid possesses a hydrogen bond with the ammonium proton of the psilacetin molecule. Each of these oxygens also forms a hydrogen bond with the hydrogen of an indole nitrogen of a different psilacetin cation. Both anionic oxygens of the fumarate dianions form the same hydrogen-bonding interactions, generated through symmetry. The hydrogen-bonding interactions of a single fumarate dianion are shown in Fig. 2 ▸. The packing of the compound is shown in Fig. 3 ▸.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O4ii | 0.90 (2) | 1.91 (2) | 2.786 (2) | 165 (2) |
| N2—H2⋯O4 | 0.99 (2) | 1.61 (2) | 2.607 (2) | 179 (2) |
Symmetry code: (ii) 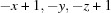 .
.
Figure 2.

The hydrogen bonding of the fumarate ion in the structure of the title compound. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms not involved in hydrogen bonds are omitted for clarity. Symmetry codes: (i) 2 − x, 1 − y, 2 − z, (iii) 1 − x, 1 + y, 1 + z, (iv) 1 − x, −y, 1 − z.
Figure 3.

The crystal packing of the title compound, viewed along the b axis. The N—H⋯O bonds (Table 1 ▸) are shown as dashed lines. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms not involved in hydrogen bonding are omitted for clarity
Database survey
We recently reported a closely related structure in which one 4-acetoxy-N,N-dimethyltryptammonium cation is charge balanced by one 3-carboxyacrylate anion (Chadeayne et al., 2019 ▸). The structure reported here has the same 4-acetoxy-N,N-dimethyltryptammonium cation, two of which are charge-balanced by a single fumarate dianion. The bond distances and angles observed in the compound reported here are consistent with our prior report. The two other reported 4-substituted tryptamine structures are those of the naturally occurring products of ‘magic’ mushrooms – psilocybin, C12H16N2PO4 (Weber & Petcher, 1974 ▸) and psilocin, C12H16N2O (Petcher & Weber, 1974 ▸). Psilocybin is the 4-phosphate-substituted variation of N,N-dimethyltryptamine, and exists as an ammonium/phosphate zwitterion in the solid state. Psilocin, 4-hydroxy-N,N-dimethyltryptamine, is believed to be a statistical mixture of a neutral molecule and an ammonium/phenoxide zwitterion. In both cases, the tryptamine components are structurally very similar to the title compound, but their arrangements in the solid state are substantially different as there are no counter-ions present.
Synthesis and crystallization
A commercial sample (The Indole Shop) of 4-acetoxy-N,N-dimethyltryptamine fumarate (100 mg, 0.16 mmol) was dissolved in 10 mL of water and treated with one equivalent of lead(II) acetate(53 mg, 0.16 mmol). Lead(II) fumarate precipitated and was filtered [the presence of lead(II) fumarate was confirmed by the unit cell of the precipitate]. Water was removed in vacuo and the resulting residue was picked up in acetone and filtered. The filtrate was allowed to evaporate slowly, resulting in single crystals suitable for X-ray analysis.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The methyl hydrogens on C2 were disordered over two positions and were refined at 50% occupancy with the C–C–H planes set at 60o to each other. The H atoms on N1 and N2 were found in the difference-Fourier map and refined freely. H atoms were placed in calculated positions (C—H = 0.95–0.99 Å) and refined as riding with U iso(H) = 1.5U eq(C-methyl) and 1.2U eq(C) for all other H atoms.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | 2C14H19N2O2 +·C4H2O4 2− |
| M r | 608.68 |
| Crystal system, space group | Triclinic, P
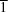
|
| Temperature (K) | 200 |
| a, b, c (Å) | 8.3965 (13), 8.9879 (14), 12.0126 (16) |
| α, β, γ (°) | 101.730 (5), 100.818 (5), 112.463 (5) |
| V (Å3) | 784.2 (2) |
| Z | 1 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.09 |
| Crystal size (mm) | 0.19 × 0.16 × 0.13 |
| Data collection | |
| Diffractometer | Bruker D8 Venture CMOS |
| Absorption correction | Multi-scan (SADABS; Bruker, 2016 ▸) |
| T min, T max | 0.714, 0.745 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 21581, 2877, 2087 |
| R int | 0.056 |
| (sin θ/λ)max (Å−1) | 0.604 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.045, 0.110, 1.03 |
| No. of reflections | 2877 |
| No. of parameters | 210 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.20 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989019007370/ff2159sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019007370/ff2159Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989019007370/ff2159Isup3.cml
CCDC reference: 1917404
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| 2C14H19N2O2+·C4H2O42− | Z = 1 |
| Mr = 608.68 | F(000) = 324 |
| Triclinic, P1 | Dx = 1.289 Mg m−3 |
| a = 8.3965 (13) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 8.9879 (14) Å | Cell parameters from 6407 reflections |
| c = 12.0126 (16) Å | θ = 3.3–25.1° |
| α = 101.730 (5)° | µ = 0.09 mm−1 |
| β = 100.818 (5)° | T = 200 K |
| γ = 112.463 (5)° | BLOCK, colourless |
| V = 784.2 (2) Å3 | 0.19 × 0.16 × 0.13 mm |
Data collection
| Bruker D8 Venture CMOS diffractometer | 2087 reflections with I > 2σ(I) |
| φ and ω scans | Rint = 0.056 |
| Absorption correction: multi-scan (SADABS; Bruker, 2016) | θmax = 25.4°, θmin = 3.3° |
| Tmin = 0.714, Tmax = 0.745 | h = −10→10 |
| 21581 measured reflections | k = −10→10 |
| 2877 independent reflections | l = −14→14 |
Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.045 | w = 1/[σ2(Fo2) + (0.0387P)2 + 0.3852P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.110 | (Δ/σ)max < 0.001 |
| S = 1.03 | Δρmax = 0.26 e Å−3 |
| 2877 reflections | Δρmin = −0.20 e Å−3 |
| 210 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.050 (4) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 0.0322 (2) | 0.33780 (19) | 0.50722 (14) | 0.0539 (4) | |
| O2 | 0.30616 (18) | 0.46535 (16) | 0.48485 (12) | 0.0413 (4) | |
| O3 | 0.6497 (2) | 0.3361 (2) | 1.04226 (12) | 0.0597 (5) | |
| O4 | 0.69101 (17) | 0.29711 (17) | 0.86294 (11) | 0.0386 (4) | |
| N1 | 0.2368 (2) | −0.0732 (2) | 0.29315 (15) | 0.0412 (4) | |
| N2 | 0.3416 (2) | 0.1243 (2) | 0.79890 (13) | 0.0375 (4) | |
| C1 | 0.2567 (4) | 0.5920 (3) | 0.6569 (2) | 0.0578 (6) | |
| H1A | 0.3818 | 0.6670 | 0.6650 | 0.087* | 0.5 |
| H1B | 0.1847 | 0.6558 | 0.6561 | 0.087* | 0.5 |
| H1C | 0.2531 | 0.5457 | 0.7240 | 0.087* | 0.5 |
| H1D | 0.1646 | 0.5787 | 0.6984 | 0.087* | 0.5 |
| H1E | 0.3617 | 0.5899 | 0.7073 | 0.087* | 0.5 |
| H1F | 0.2933 | 0.7000 | 0.6394 | 0.087* | 0.5 |
| C2 | 0.1817 (3) | 0.4512 (3) | 0.54365 (18) | 0.0406 (5) | |
| C3 | 0.2616 (3) | 0.3380 (2) | 0.37872 (17) | 0.0367 (5) | |
| C4 | 0.2330 (3) | 0.3748 (3) | 0.27365 (19) | 0.0482 (6) | |
| H4 | 0.2329 | 0.4804 | 0.2735 | 0.058* | |
| C5 | 0.2040 (3) | 0.2580 (3) | 0.1667 (2) | 0.0572 (7) | |
| H5 | 0.1839 | 0.2852 | 0.0945 | 0.069* | |
| C6 | 0.2041 (3) | 0.1050 (3) | 0.16395 (18) | 0.0491 (6) | |
| H6 | 0.1855 | 0.0261 | 0.0912 | 0.059* | |
| C7 | 0.2325 (2) | 0.0689 (3) | 0.27162 (16) | 0.0361 (5) | |
| C8 | 0.2611 (2) | 0.1833 (2) | 0.38128 (15) | 0.0324 (4) | |
| C9 | 0.2860 (3) | 0.1039 (2) | 0.47124 (16) | 0.0378 (5) | |
| C10 | 0.2697 (3) | −0.0505 (3) | 0.41319 (17) | 0.0428 (5) | |
| H10 | 0.2796 | −0.1313 | 0.4506 | 0.051* | |
| C11 | 0.3232 (4) | 0.1765 (3) | 0.60302 (17) | 0.0552 (7) | |
| H11A | 0.4523 | 0.2569 | 0.6377 | 0.066* | |
| H11B | 0.2518 | 0.2408 | 0.6160 | 0.066* | |
| C12 | 0.2802 (3) | 0.0483 (3) | 0.66691 (16) | 0.0446 (6) | |
| H12A | 0.3380 | −0.0264 | 0.6453 | 0.054* | |
| H12B | 0.1482 | −0.0222 | 0.6407 | 0.054* | |
| C13 | 0.3087 (4) | −0.0104 (3) | 0.8580 (2) | 0.0623 (7) | |
| H13A | 0.3654 | −0.0819 | 0.8298 | 0.094* | |
| H13B | 0.1786 | −0.0794 | 0.8389 | 0.094* | |
| H13C | 0.3603 | 0.0415 | 0.9443 | 0.094* | |
| C14 | 0.2660 (3) | 0.2400 (4) | 0.84318 (19) | 0.0610 (7) | |
| H14A | 0.3073 | 0.2793 | 0.9303 | 0.092* | |
| H14B | 0.1341 | 0.1804 | 0.8159 | 0.092* | |
| H14C | 0.3063 | 0.3371 | 0.8128 | 0.092* | |
| C15 | 0.9426 (3) | 0.4767 (2) | 1.03029 (16) | 0.0346 (5) | |
| H15 | 0.9873 | 0.5193 | 1.1146 | 0.042* | |
| C16 | 0.7463 (3) | 0.3612 (2) | 0.97637 (16) | 0.0340 (5) | |
| H1 | 0.240 (3) | −0.159 (3) | 0.242 (2) | 0.055 (7)* | |
| H2 | 0.475 (3) | 0.191 (3) | 0.8236 (18) | 0.048 (6)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0506 (10) | 0.0427 (9) | 0.0626 (10) | 0.0148 (8) | 0.0223 (8) | 0.0098 (8) |
| O2 | 0.0381 (8) | 0.0330 (8) | 0.0418 (8) | 0.0099 (6) | 0.0069 (6) | 0.0048 (6) |
| O3 | 0.0445 (9) | 0.0788 (12) | 0.0310 (8) | 0.0068 (8) | 0.0124 (7) | 0.0053 (8) |
| O4 | 0.0332 (8) | 0.0447 (8) | 0.0248 (7) | 0.0120 (6) | 0.0032 (6) | −0.0008 (6) |
| N1 | 0.0449 (11) | 0.0422 (11) | 0.0307 (9) | 0.0198 (9) | 0.0081 (8) | 0.0003 (8) |
| N2 | 0.0323 (9) | 0.0398 (10) | 0.0267 (8) | 0.0065 (8) | 0.0070 (7) | 0.0022 (7) |
| C1 | 0.0716 (17) | 0.0518 (14) | 0.0449 (13) | 0.0323 (13) | 0.0071 (12) | 0.0028 (11) |
| C2 | 0.0472 (13) | 0.0356 (12) | 0.0406 (12) | 0.0208 (11) | 0.0097 (10) | 0.0125 (9) |
| C3 | 0.0318 (11) | 0.0356 (11) | 0.0347 (11) | 0.0086 (9) | 0.0082 (8) | 0.0083 (9) |
| C4 | 0.0514 (14) | 0.0464 (13) | 0.0463 (13) | 0.0165 (11) | 0.0143 (10) | 0.0230 (11) |
| C5 | 0.0671 (16) | 0.0699 (17) | 0.0386 (13) | 0.0257 (13) | 0.0204 (11) | 0.0283 (12) |
| C6 | 0.0501 (13) | 0.0634 (16) | 0.0291 (11) | 0.0189 (12) | 0.0169 (10) | 0.0114 (10) |
| C7 | 0.0287 (10) | 0.0423 (12) | 0.0308 (10) | 0.0106 (9) | 0.0097 (8) | 0.0070 (9) |
| C8 | 0.0254 (10) | 0.0364 (11) | 0.0270 (9) | 0.0082 (8) | 0.0050 (7) | 0.0055 (8) |
| C9 | 0.0436 (12) | 0.0359 (11) | 0.0269 (10) | 0.0170 (9) | 0.0019 (8) | 0.0036 (8) |
| C10 | 0.0511 (13) | 0.0416 (12) | 0.0306 (11) | 0.0217 (10) | 0.0034 (9) | 0.0055 (9) |
| C11 | 0.0909 (19) | 0.0408 (13) | 0.0269 (11) | 0.0334 (13) | −0.0004 (11) | 0.0031 (9) |
| C12 | 0.0386 (12) | 0.0459 (13) | 0.0265 (10) | 0.0036 (10) | 0.0053 (9) | −0.0018 (9) |
| C13 | 0.0717 (17) | 0.0533 (15) | 0.0392 (12) | 0.0026 (13) | 0.0183 (12) | 0.0149 (11) |
| C14 | 0.0548 (15) | 0.091 (2) | 0.0379 (12) | 0.0421 (14) | 0.0129 (11) | 0.0014 (12) |
| C15 | 0.0373 (11) | 0.0371 (11) | 0.0228 (9) | 0.0151 (9) | 0.0025 (7) | 0.0037 (8) |
| C16 | 0.0372 (11) | 0.0350 (11) | 0.0267 (10) | 0.0159 (9) | 0.0065 (8) | 0.0056 (8) |
Geometric parameters (Å, º)
| O1—C2 | 1.200 (2) | C5—H5 | 0.9500 |
| O2—C2 | 1.349 (2) | C5—C6 | 1.369 (3) |
| O2—C3 | 1.405 (2) | C6—H6 | 0.9500 |
| O3—C16 | 1.228 (2) | C6—C7 | 1.396 (3) |
| O4—C16 | 1.282 (2) | C7—C8 | 1.412 (3) |
| N1—C7 | 1.365 (3) | C8—C9 | 1.437 (3) |
| N1—C10 | 1.373 (3) | C9—C10 | 1.362 (3) |
| N1—H1 | 0.90 (2) | C9—C11 | 1.506 (3) |
| N2—C12 | 1.493 (2) | C10—H10 | 0.9500 |
| N2—C13 | 1.488 (3) | C11—H11A | 0.9900 |
| N2—C14 | 1.475 (3) | C11—H11B | 0.9900 |
| N2—H2 | 0.99 (2) | C11—C12 | 1.480 (3) |
| C1—H1A | 0.9800 | C12—H12A | 0.9900 |
| C1—H1B | 0.9800 | C12—H12B | 0.9900 |
| C1—H1C | 0.9800 | C13—H13A | 0.9800 |
| C1—H1D | 0.9800 | C13—H13B | 0.9800 |
| C1—H1E | 0.9800 | C13—H13C | 0.9800 |
| C1—H1F | 0.9800 | C14—H14A | 0.9800 |
| C1—C2 | 1.489 (3) | C14—H14B | 0.9800 |
| C3—C4 | 1.370 (3) | C14—H14C | 0.9800 |
| C3—C8 | 1.396 (3) | C15—C15i | 1.309 (4) |
| C4—H4 | 0.9500 | C15—H15 | 0.9500 |
| C4—C5 | 1.397 (3) | C15—C16 | 1.494 (3) |
| C2—O2—C3 | 118.62 (15) | C5—C6—C7 | 117.8 (2) |
| C7—N1—C10 | 108.50 (17) | C7—C6—H6 | 121.1 |
| C7—N1—H1 | 126.7 (15) | N1—C7—C6 | 129.45 (19) |
| C10—N1—H1 | 123.8 (15) | N1—C7—C8 | 107.93 (17) |
| C12—N2—H2 | 107.7 (12) | C6—C7—C8 | 122.6 (2) |
| C13—N2—C12 | 110.37 (16) | C3—C8—C7 | 117.04 (17) |
| C13—N2—H2 | 105.4 (12) | C3—C8—C9 | 136.10 (17) |
| C14—N2—C12 | 114.55 (17) | C7—C8—C9 | 106.85 (17) |
| C14—N2—C13 | 111.26 (18) | C8—C9—C11 | 127.06 (18) |
| C14—N2—H2 | 107.1 (12) | C10—C9—C8 | 106.02 (16) |
| H1A—C1—H1B | 109.5 | C10—C9—C11 | 126.91 (18) |
| H1A—C1—H1C | 109.5 | N1—C10—H10 | 124.7 |
| H1A—C1—H1D | 141.1 | C9—C10—N1 | 110.70 (19) |
| H1A—C1—H1E | 56.3 | C9—C10—H10 | 124.7 |
| H1A—C1—H1F | 56.3 | C9—C11—H11A | 108.7 |
| H1B—C1—H1C | 109.5 | C9—C11—H11B | 108.7 |
| H1B—C1—H1D | 56.3 | H11A—C11—H11B | 107.6 |
| H1B—C1—H1E | 141.1 | C12—C11—C9 | 114.06 (17) |
| H1B—C1—H1F | 56.3 | C12—C11—H11A | 108.7 |
| H1C—C1—H1D | 56.3 | C12—C11—H11B | 108.7 |
| H1C—C1—H1E | 56.3 | N2—C12—H12A | 109.0 |
| H1C—C1—H1F | 141.1 | N2—C12—H12B | 109.0 |
| H1D—C1—H1E | 109.5 | C11—C12—N2 | 112.99 (16) |
| H1D—C1—H1F | 109.5 | C11—C12—H12A | 109.0 |
| H1E—C1—H1F | 109.5 | C11—C12—H12B | 109.0 |
| C2—C1—H1A | 109.5 | H12A—C12—H12B | 107.8 |
| C2—C1—H1B | 109.5 | N2—C13—H13A | 109.5 |
| C2—C1—H1C | 109.5 | N2—C13—H13B | 109.5 |
| C2—C1—H1D | 109.5 | N2—C13—H13C | 109.5 |
| C2—C1—H1E | 109.5 | H13A—C13—H13B | 109.5 |
| C2—C1—H1F | 109.5 | H13A—C13—H13C | 109.5 |
| O1—C2—O2 | 122.94 (19) | H13B—C13—H13C | 109.5 |
| O1—C2—C1 | 126.3 (2) | N2—C14—H14A | 109.5 |
| O2—C2—C1 | 110.81 (19) | N2—C14—H14B | 109.5 |
| C4—C3—O2 | 118.17 (19) | N2—C14—H14C | 109.5 |
| C4—C3—C8 | 120.95 (19) | H14A—C14—H14B | 109.5 |
| C8—C3—O2 | 120.68 (17) | H14A—C14—H14C | 109.5 |
| C3—C4—H4 | 119.8 | H14B—C14—H14C | 109.5 |
| C3—C4—C5 | 120.4 (2) | C15i—C15—H15 | 117.7 |
| C5—C4—H4 | 119.8 | C15i—C15—C16 | 124.7 (2) |
| C4—C5—H5 | 119.4 | C16—C15—H15 | 117.7 |
| C6—C5—C4 | 121.2 (2) | O3—C16—O4 | 124.80 (18) |
| C6—C5—H5 | 119.4 | O3—C16—C15 | 118.60 (16) |
| C5—C6—H6 | 121.1 | O4—C16—C15 | 116.59 (16) |
| O2—C3—C4—C5 | 174.36 (19) | C6—C7—C8—C3 | −0.6 (3) |
| O2—C3—C8—C7 | −173.84 (16) | C6—C7—C8—C9 | −179.64 (19) |
| O2—C3—C8—C9 | 4.9 (3) | C7—N1—C10—C9 | 0.4 (2) |
| N1—C7—C8—C3 | −179.97 (16) | C7—C8—C9—C10 | −0.7 (2) |
| N1—C7—C8—C9 | 1.0 (2) | C7—C8—C9—C11 | 179.2 (2) |
| C2—O2—C3—C4 | 107.9 (2) | C8—C3—C4—C5 | −0.5 (3) |
| C2—O2—C3—C8 | −77.2 (2) | C8—C9—C10—N1 | 0.2 (2) |
| C3—O2—C2—O1 | −2.8 (3) | C8—C9—C11—C12 | 159.8 (2) |
| C3—O2—C2—C1 | 176.73 (17) | C9—C11—C12—N2 | 172.04 (19) |
| C3—C4—C5—C6 | −0.3 (4) | C10—N1—C7—C6 | 179.8 (2) |
| C3—C8—C9—C10 | −179.5 (2) | C10—N1—C7—C8 | −0.8 (2) |
| C3—C8—C9—C11 | 0.3 (4) | C10—C9—C11—C12 | −20.4 (4) |
| C4—C3—C8—C7 | 0.9 (3) | C11—C9—C10—N1 | −179.7 (2) |
| C4—C3—C8—C9 | 179.6 (2) | C13—N2—C12—C11 | −175.2 (2) |
| C4—C5—C6—C7 | 0.6 (3) | C14—N2—C12—C11 | 58.3 (3) |
| C5—C6—C7—N1 | 179.1 (2) | C15i—C15—C16—O3 | −174.9 (3) |
| C5—C6—C7—C8 | −0.2 (3) | C15i—C15—C16—O4 | 4.1 (4) |
Symmetry code: (i) −x+2, −y+1, −z+2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O4ii | 0.90 (2) | 1.91 (2) | 2.786 (2) | 165 (2) |
| N2—H2···O4 | 0.99 (2) | 1.61 (2) | 2.607 (2) | 179 (2) |
Symmetry code: (ii) −x+1, −y, −z+1.
Funding Statement
This work was funded by National Science Foundation grant CHE-1429086.
References
- Bruker (2016). APEX3, SAINT, and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Carhart-Harris, R. L. & Goodwin, G. M. (2017). Neuropsychopharmacology, 42, 2105–2113. [DOI] [PMC free article] [PubMed]
- Chadeayne, A. R., Golen, J. A. & Manke, D. R. (2019). Psychedelic Science Review, https://psychedelicreview.com/the-crystal-structure-of-4-aco-dmt-fumarate/.
- Dinis-Oliveira, R. J. (2017). Drug Metab. Rev. 49, 84–91. [DOI] [PubMed]
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Nichols, D. E. & Frescas, S. (1999). Synthesis, pp. 935–938.
- Petcher, T. J. & Weber, H. P. (1974). J. Chem. Soc. Perkin Trans. 2, pp. 946–948.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Weber, H. P. & Petcher, T. J. (1974). J. Chem. Soc. Perkin Trans. 2, pp. 942–946.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989019007370/ff2159sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019007370/ff2159Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989019007370/ff2159Isup3.cml
CCDC reference: 1917404
Additional supporting information: crystallographic information; 3D view; checkCIF report