

Summary
The development of C1 synthons to afford the products that add one extra carbon has become an important research theme in the past decade, and significant progress has been achieved with CO2, CO, HCOOH, and others as C1 units. Despite the great advance, the search for new C1 synthons that display unique reactivity, complement to the current C1 sources, and add more value to C1 chemistry is still desirable. Herein, we report a quadruple cleavage of chlorodifluoromethane to yield a C1 source, which was successfully employed in the construction of various N-containing compounds especially with pharmaceutical molecules under mild conditions. This strategy provides a useful method for late-stage modification of pharmaceutical compounds. Four bonds in ClCF2H were orderly cleaved under basic conditions in the absence of transition metals. Preliminary mechanistic studies revealed that (E)-N-phenylformimidoyl fluoride intermediate is involved in this process by in situ1H NMR studies and control experiments.
Subject Areas: Catalysis, Organic Chemistry, Organic Synthesis
Graphical Abstract

Highlights
-
•
Quadruple cleavage of ClCF2H to afford a C1 synthon
-
•
The cleavage of two stable C(sp3)-F bonds in aliphatic gem-difluoroalkanes
-
•
Enrich C1 chemistry, green chemistry, and fluorine chemistry
-
•
Various N-containing compounds were afforded via different role of ClCF2H
Catalysis; Organic Chemistry; Organic Synthesis
Introduction
The C1 chemistry has emerged as an elegant strategy for efficient preparation of homologous compounds, which added one extra carbon in modern chemical transformations (Aresta et al., 2014, Sakakura et al., 2007, Huang et al., 2011, Yan et al., 2012, Natte et al., 2017, Wakade et al., 2017, Senadi et al., 2019). There are ample significance and a plethora of characteristics for C1 chemistry, for instance, carbon chain increasing (Aresta et al., 2014), construction of importance functional groups (carboxylic or carbonyl groups) (Huang et al., 2011, Cokoja et al., 2011), incorporation of two or more organic small molecules to yield important products (Oh and Hu, 2013), and modification of the pharmaceutical or natural products for value-added bulk (Liu et al., 2015, Ma et al., 2018a). Among all known C1 synthons, CO2, CO, and formic acid are the most famous ones, which have been widely used in various reaction processes, and many beautiful transformations have been developed with them, which further attracted more and more chemists devoted to this field (Aresta et al., 2014, Sakakura et al., 2007, Aresta et al., 2014, Huang et al., 2011, Cokoja et al., 2011, Oh and Hu, 2013, Sordakis et al., 2018, Gibson, 1969, Enthaler et al., 2010). Despite the significance and great advance of C1 chemistry, the search for C1 synthons that display unique reactivity, complement to the current C1 sources, and add more value to C1 chemistry is still highly desirable. Thus, direct introduction of one extra carbon from cheap and available materials under mild conditions to provide a cost-efficient, pragmatic, and valuable alternative avenue would be popular in the field of synthetic and pharmaceutical communities, which might have deep impact on industry as well.
Chlorodifluoromethane (ClCF2H) is well known as an inexpensive and abundant industrial raw material (Hudlicky and Pavlath, 1995) for the construction of various fluorinated compounds (Wang et al., 2014, Fier and Hartwig, 2012, Gu et al., 2014, Yu et al., 2017, Wu et al., 2019, Miao et al., 2018, Zhang et al., 2019), featuring thermodynamic stability and kinetic inertness as well as atomic economy as fluorine source. Therefore, efficient transformations of this easily accessible raw material to create valuable chemicals have deservedly gained great attention. The most common transformation of ClCF2H involves the formation of difluorocarbene (:CF2) by the cleavage of both C-Cl and C-H bonds (Figure 1Aa) (Feng et al., 2017), usually under basic conditions with heteroatom nucleophiles, rendering the corresponding difluoromethylated heteroatom compounds (Hine and Porter, 1957, Nawrot and Jonczyk, 2007). Pyrolysis of ClCF2H at high temperature or pressure leads to the important raw industrial material tetrafluoroethylene (Hudlicky and Pavlath, 1995, Sung et al., 2004). Very recently a reaction by palladium-catalyzed cross-coupling between arylboronic acids and ClCF2H via a metal-difluorocarbene intermediate has been reported (Feng et al., 2017, Yu et al., 2019), representing the catalytic transformation of ClCF2H. Other conversion processes that do not involve difluorocarbene species were still difluoromethylation-related ones in which only one C-Cl bond was broken through a difluoromethyl radical pathway (Figure 1Ab) (Xu et al., 2018). Trifluoromethyl anion (CF3−) is readily derived from the difluorocarbene species and external fluorine source via double cleavage of ClCF2H (Figure 1Ac) (Zheng et al., 2015). Intriguingly and surprisingly, quadruple cleavage of ClCF2H to provide versatile C1 synthons, by breaking one C-Cl bond, two stable C-F bonds, and one C-H bond orderly in a single-vessel reaction (Figure 1B), has never been reported to date, probably mainly because of the high BDE of C(sp3)-F bonds (the bond dissociation energy of a single C-F bond: 485 KJ/mol) (O'Hagan, 2008).
Figure 1.

Various Transformations of ClCF2H
(A) Known transformations of ClCF2H.
(a) Double cleavage of ClCF2H to lead to difluorocarbene species.
(b) Single cleavage of ClCF2H to lead to difluoromethyl radical.
(c) Double cleavage of ClCF2H with external F− to lead to trifluoromethyl anion.
(B) Our work.
(d) Quadruple cleavage of ClCF2H as a C1 source.
Herein, we report a quadruple cleavage of chlorodifluoromethane as a type of C1 source to access valuable formimidamide derivatives that are widely employed as ligands or forming metal complexes as quasi-N-heterocyclic carbenes (NHCs) (Figure 1 (i) and (ii)) (Schröder et al., 2009, Schröder et al., 2010, Bitterlich et al., 2007, Boogaerts and Nolan, 2010, Ohishi et al., 2008, Hopkinson et al., 2014). Despite the importance of these compounds, their elegant syntheses are very rare. Therefore, expanding the toolbox of methods for their synthesis will enrich diversity of this kind of compounds. Amines are very common raw materials as well as crucial building blocks with rich chirality (France et al., 2014); we envision that formimidamides could be readily generated in a single-vessel synthesis from two amines with ClCF2H as C1 source due to the special reactivity of ClCF2H. These processes represent a significant reaction modality for ClCF2H, which might promote and enrich C1 chemistry, organic fluorine chemistry (Gouverneur and Seppelt, 2015), as well as green chemistry (Horváth, 2007). Meanwhile, ClCF2H provides a unique and alternative approach for the current known C1 sources: the control and comparison experiments with CO, CO2, and formic acid as C1 synthons were performed under our standard conditions as well as the reported procedures; notably, no desired formimidamides were ever formed or obtained under those reaction conditions. These results further underscore the uniqueness and peculiarity of ClCF2H as a C1 source (for further details, see also Schemes S1 and S2).
Results and Discussion
Optimization of Reaction Conditions
Our design is based on our recent discovery in which ethyl bromodifluoroacetate (BrCF2COOEt) (Ma et al., 2018b, Deng et al., 2019) could act as a C1 source and formylating reagent with amines via quadruple cleavage under basic conditions (Ma et al., 2018a, Ma et al., 2018c, Ma et al., 2018d); we postulated that the quadruple cleavage of ClCF2H could also be occurred under the similar sets since it is known that difluorocarbene could be readily accessible from ClCF2H under basic conditions. Moreover, compared with BrCF2COOEt, ClCF2H is obviously much cheaper and more atomic economical. We commenced our hypothesis by using low-cost and widely available aniline (1a) and N-methyl aniline (2a) as model substrates. To our delight, the yield of 76% of the anticipated product 3a (Zhao et al., 2005) was obtained from the reaction of 1a with 2a under the ClCF2H atmosphere without water (entry 1). More delightfully, the yield was significantly increased to 83%–92% with the increase of dosage of water (entries 2-3); notably, excess water caused deteriorated effect on the reaction, since some unknown by-products were observed when the amount of water was increased to 20–30 equivalents, and the yield of the desired product was dropped to 83% (entry 2 versus entry 3, and for further details, see also Table S2). Replacing KOH with either Cs2CO3 or K2CO3 as the base resulted in lower yields (entries 4-5). To our surprise, this transformation was completely suppressed with Na2CO3 and NaHCO3 as the base (entries 6-7). The solvent was so crucial that no reaction occurred in other solvents, such as in THF and dioxane (entries 8-9) (for further details, see also Table S3). In addition, the yield of the desired product was slightly higher at the ambient temperature than at 50°C. In terms of reaction time, the longer time (36 h) led to the best result (entry 11).
Substrate Scope for Intermolecular Transformation
With the optimal reaction conditions in hand (entry 2 in Table 1), we explored the generality and limitation of this transformation (Figure 2). First, a variety of para-substituted anilines with electron-donating groups (alkoxy, phenoxyl, alkyl, and N, N-dimethyl) (1b-1j), as well as electron-withdrawing groups, such as halogen (1k-1m) and nitro group (1n), delivered the desired formimidamide derivatives (3a-3n) in good to excellent yields under the standard conditions. We next examined [1,1′-biphenyl]-4-amine (1o) under this reaction condition to provide the desired product 3o in 77% yield. Besides, a large-scale (10 mmol) reaction of the N-methyl aniline (2a) has been carried out to afford 3a in 62% yield (for further details, see also Scheme S3). Similar result could be obtained for meta-substituted (m-Br) aniline (1p). Using the disubstituted 3,4-dimethylaniline (1q) and trisubstituted 2,4,6-trimethylaniline (1r), the corresponding products (3q and 3r) could be obtained in good yields (83%–85%). 5,6,7,8-Tetrahydronaphthalen-1-amine (1s) and 9H-fluoren-2-amine (1t) were carried out under the standard conditions to provide the target molecules (3s and 3t) in 80% and 76% yields, respectively. The absolute molecular structure of product 3t was unambiguously confirmed by X-ray crystallography analysis (Figure 2, and for further details, see also Table S1 and Data S1) (CCDC: 1874971). The fused polycyclic amines 9,9-diphenyl-9H-fluoren-2-amine (1u) and naphthalen-1-amine (1v) were subjected under the optimized reaction conditions, rendering the expected products (3u-3v) in moderate yields (68%–77%). Heterocyclic compounds, such as benzo[d]thiazol-2-amine (1w), were also amenable to this transformation, and the corresponding product was obtained in 62% yield. We then further investigated the scope of the N-substituted aniline derivatives with aniline (1a) under the viable reaction conditions. Delightedly, the corresponding products (3x-3z, 3aa-3ac) were obtained in good to excellent yields with good functional group tolerance. In addition, given the prevailing existence of amines in pharmaceutical molecules and natural products (Ma et al., 2018a, Brunet and Neibecker, 2001), we selected Benzocaine (1ad, local anesthetic), Amoxapine (2ae, antidepressant), 2-(piperazin-1-yl)-4-(trifluoromethyl)pyrimidine (2af, medical/material intermediates), and multi-functional Vildagliptin (2ag, inhibit glucagon/chiral reagent/medicinalintermediate) and exposed them under the standard conditions; the corresponding products were obtained in 59%, 76%, 61%, and 71% yields, respectively. Gratifyingly, the chiral molecule (S)-N-benzyl-1-phenylethan-1-amine (2ah) experienced the optimal reaction conditions to deliver (S,E)-N-benzyl-N′-phenyl-N-(1-phenylethyl)formimidamide (3ah) in 60% yield, which might be a potential chiral ligand to realize enantioselective-control reactions.
Table 1.
Representative Results for Optimization of the Formation of (E)-N-methyl-N,N′-diphenylformimidamide (3a)
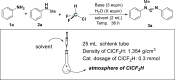 | |||||
|---|---|---|---|---|---|
| Entry | Base (3 Equiv) | H2O (X Equiv) | Solvent (2 mL) | T (°C) | Yield (%)a |
| 1 | KOH | 0 | CH3CN | r.t. | 76 |
| 2 | KOH | 5 | CH3CN | r.t. | 92 (88)b |
| 3 | KOH | 30 | CH3CN | r.t. | 83 |
| 4 | Cs2CO3 | 5 | CH3CN | r.t. | 76 |
| 5 | K2CO3 | 5 | CH3CN | r.t. | 79 |
| 6 | Na2CO3 | 5 | CH3CN | r.t. | N.D. |
| 7 | NaHCO3 | 5 | CH3CN | r.t. | N.D. |
| 8 | KOH | 5 | THF | r.t. | N.D. |
| 9 | KOH | 5 | Dioxane | r.t. | N.D. |
| 10 | KOH | 5 | CH3CN | 50 | 70 |
| 11c,d | KOH | 5 | CH3CN | r.t. | 62c (79)d |
| 12e | KOH | 5 | CH3CN | r.t. | N.D. |
Reaction condition: aniline (1a, 1.2 equiv. 0.12 mmol), N-methylaniline (2a, 0.1 mmol), the atmosphere of chlorodifluoromethane (ClCF2H) (cat. 0.3 mmol), base (3 equiv.), solvent (2 mL), for 36 h.
r.t., room temperature; N.D., not detected.
GC yields.
Isolated yields.
For 12 h.
For 24 h.
No ClCF2H.
Figure 2.

Synthesis of Formimidamide Derivatives
(A) Scope of the primary amines.
(B) Scope of the secondary amines.
(C) Scope of R groups.
(D) Scope of the pharmaceutical molecules.
In addition, we found that aliphatic secondary amine is compatible under the standard conditions as well; in terms of the substrate dicyclohexylamine (4), the corresponding product 5 was acquired in 76% yield (Equation 1 in Figure 3A). It is worth mentioning that our strategy is highly regio-selective, for example, when N1-isopropyl-N3-phenylbenzene-1,3-diamine (6) was investigated under viable reaction condition, only compound 7 was afforded with diphenylamine part intact (Equation 2). In addition, when the target 3a was hydrolyzing under 1 M HCl, the two original substrates (1a and 2a) as well as two formylated compounds 1a-1 and 2a-1 were obtained, respectively (Ma et al., 2018a) (for further details, see also Scheme S4), which infers that our strategy might be a potential method for drug delayed or sustainable release when two different pharmaceutical molecules are combined by one extra carbon with our strategy (Equation 3). We carried out, therefore, correlative experiment using Benzocaine (1ad) and Vildagliptin (2ag) as the substrates under standard reaction condition 1; to our delight, the corresponding product 3ai was obtained in 70% yield (Equation 4). More interestingly, a highly chemoselective process was disclosed with two primary amines, in which N,N′-diphenylformimidamide and N-(difluoromethyl)-N,N′-diphenylformimidamide were obtained, respectively, by careful control of reaction conditions. Bases and additives played key roles on these two successful transformations: with K2CO3 as base, phenol and water as additives (see condition 2 in Figure 3 and for further details, see also Table S4), N,N′-diphenylformimidamides (8 and 9) were obtained in moderate yields; with Cs2CO3 as the base and S8 as additive (see condition 3 in Figure 3, and for further details, see also Table S5) (Zheng et al., 2017), N-(difluoromethyl)-N,N′-diphenylformimidamides (10–15) were acquired in good yields. In the latter transformation, ClCF2H played a dual role as both C1 source and difluorocarbene source (Figure 3C).
Figure 3.

High Regio- and Chemoselectivity of Our Strategy and Intriguing Products
(A) High regio-selectivity.
(B) The dispose of target product and the combination of two pharmaceutical molecules.
(C) Selective synthesis of formimidamides with two primary amines.
(D) Selective synthesis of N-difluoromethyl-formimidamides from two primary amines with ClCF2H as both C1 source and difluoromethylating reagent.
Substrate Scope for Intramolecular Transformation
The success of the above-mentioned intermolecular transformation prompted us to exploit the intramolecular transformations, since the latter one always leads to cyclic compounds that are the essential skeletons in pharmaceutical and natural products (Sasaki et al., 2006, Kubo et al., 1993). Gratifyingly, when N1-methylbenzene-1,2-diamine (16) was subjected to the standard conditions for intermolecular transformation, benzimidazole 17 was obtained in 90% yield, which could be readily converted into 2-bromo-benzimidazole 18 in the presence of NBS. Then, after a series of transformation, Telmisartan, a potent angiotensin II receptor antagonist used in the treatment of essential hypertension, will be afforded (Figure 4A) (Martin et al., 2015). In addition, the transformation could be easily scaled up to 70 times from 16 to 17 without loss of the efficiency (for details, see also Scheme S6). Encouraged by this promising result, we next focused on the exploration of the formation of benzo[d]oxazoles and 1H-benzo[d]imidazoles compounds via intramolecular pattern, since it is well known that benzo[d]oxazoles and 1H-benzo[d]imidazoles are prevalent molecular scaffolds in various bioactive natural products, agrochemicals, and pharmaceuticals. After many attempts, an optimized condition was obtained (for further details, see also Tables S6 and S7). These transformations demonstrated a good functional group tolerance (Figure 4B). Different substituent groups on the benzene ring, including alkyl (19a, 19b), halo groups (19c, 19d), were all compatible, rendering the corresponding products (20a-20d) in moderate to good yield (68%–81%). Surprisingly, no desired products were detected when 2-amino-4-nitrophenol (19e) and 2-amino-5-nitrophenol (19f) underwent the same conditions, instead, the selective difluoromethylation of hydroxyl group occurred (20e′ and 20f′). Good yields were achieved on various benzene-1,2-diamine compounds under the standard Reaction Condition 5 with K2CO3 as base in CH3CN (2 mL) and H2O (0.5 mL) at 100°C for 16 h (Figure 4C). Remarkably, the products of difluoromethylation of benzimidazoles (21-29) were acquired in moderate yields via the slight adjustment of reaction condition (see reaction condition 6 for details); once again, ClCF2H played a dual role as both C1 source and difluorocarbene source in this transformation (Figure 4D).
Figure 4.

The Synthetic Route of the Telmisartan and the Intramolecular Reaction Scope
(A) Telmisartan synthesis with our strategy.
(B) Scope of benzoxazoles.
(C) Scope of benzimidazoles.
(D) Scope of N-difluoromethyl benzimidazoles.
Mechanism Investigation
To gain more insights into the mechanism of the aforementioned transformations, some control experiments were performed. Initially, isotope labeling experiments were conducted, 84% (3a′) and 78% (20g′) of D atoms were incorporated into the final products correspondingly for intermolecular and intramolecular versions, and N-H of benzimidazole was replaced by N-D completely (Figures 5A and 5B). These results suggested that the hydrogen atom attached on the extra introduced carbon (from ClCF2H) was originated from H2O in this process possibly (for further details, see also Scheme S8). The trace amount of the desired product 3a was observed when benzimidazole was added into the reaction system as a difluorocarbene scavenger; instead, 1-(difluoromethyl)-1H-benzo[d]imidazole was detected by GC-MS (Figure 5C). When N-methyl-N-phenylformamide (30) or isocyanobenzene (31) was subjected to the earlier standard reaction conditions with amines, the corresponding target products 3a and 8 were not obtained (Figure 5D), indicating that the compounds 30 and 31 are not intermediates for this transformation, which is in sharp contrast to our previous reports in which isocyanides are the key intermediates for those transformations with BrCF2COOEt (Ma et al., 2018c, Ma et al., 2018d). To thoroughly understand the reaction sequence, two more control experiments were carried out, in which the primary amine and the secondary amine were added to the reaction mixture stepwise instead of one-pot to check which one is the first amine species interacting with ClCF2H. It turned out that primary amine might react with ClCF2H before the secondary one since 47% of the desired product was obtained in the primary-secondary amine sequence, whereas no desired product was detected with the secondary-primary amine sequence (Figures 5E and 5F). Finally, we carried out comparison experiments for CO2, CO and HCOOH as C1 source with amines 1a and 2a under various reaction conditions; no desired product 3a was detected (Figure 5G), which further highlighted the uniqueness of ClCF2H as the C1 source in these transformations.
Figure 5.

Mechanistic Studies
Proposed Mechanism
To thoroughly figure out the possible reactive intermediate, we carried out in situ 1H NMR studies between 4-ethoxyaniline (1c) and ClCF2H (Figure 6A). Since isocyanides have been ruled out to be the possible intermediates for this transformation, we envision that a type of intermediate 3c′ might be formed in this transformation. To our delight, in situ 1H NMR studies indeed indicated the formation of (E)-N-(4-ethoxyphenyl)formimidoyl fluoride (3c′), which was increased continually at the first 6 h, whereafter it started to decline probably owing to its volatile property and the existence of various nucleophiles in the reaction system, such as H2O and amines. The intermediate 3c′ was totally consumed after 18 h or during the process of striping the solvent (for further details, see also Schemes S9–S11). To validate its presence, various nucleophiles (phenols, alcohols, amines, and carboxylic acids) were added into the system after 2 h, and the corresponding desired products were detected in GC-MS (Equation 1 in Figure 6B, and for further details, Scheme S12). In addition, one more control experiment was carried out with a ClCF2H balloon for 5 h; the product 32, isocyanide 33, and N-(4-ethoxyphenyl)formamide (34) were obtained in 15%, 27%, and 38% yields, respectively (Equation 2 in Figure 6B), suggesting that the intermediate is a chemically active compound, which will further decompose into isocyanide by one more C-F bond cleavage easily. We have carried out the control experiments in the presence of radical scavengers; the reactions proceeded smoothly at room temperature to afford desired products in moderate yields. Those results suggest that the single electron transfer (SET) pathway could not be involved in this transformation (for further details, see also Scheme S13). On the basis of the above-mentioned results, a proposed mechanism for the reaction of ClCF2H as a C1 source is depicted in Figure 6C. The base coordinates with ClCF2H to generate difluorocarbene first. Then the primary amine traps the in situ generated difluorocarbene affording intermediate I, which is very sensitive under basic conditions to lead to monofluoroimine species via the cleavage of one C-F bond; subsequent inter- or intramolecular nucleophilic attack on the imine species (II and III) eventually delivers products 3, 5, 7, and 20 by SNAr substitution (path a) or nucleophilic addition (path b) (Ma et al., 2018a). As either R2 or R3 is H, the product could embark on capturing one more in situ generated difluorocarbene unceasingly to render the products (10–15). The products 21–29 were obtained in one-pot synthesis when compound 20 (X = NH) meets with excess ClCF2H in basic conditions as a difluorocarbene scavenger.
Figure 6.

In situ1H NMR, the Capture of Reaction Intermediates and the Proposed Mechanism
(A) In situ1H NMR studies.
(B) The capture of reaction intermediates.
(C) The plausible reaction mechanism.
Conclusion
In summary, we have disclosed a C1 source generated from chlorodifluoromethane (ClCF2H). This method allows the synthesis of a broad range of the formimidamides and benzo[d]oxazoles, benzo[d]imidazole derivatives via intermolecular and intramolecular reactions with good efficiency as well as high regio- and chemoselectivity under mild reaction condition. To our knowledge, this is the first example that ClCF2H proceeds quadruple cleavage to act as a C1 synthon and the valuable products were fabricated from readily available starting materials under transition-metal-free conditions. This process might enrich C1 chemistry, green chemistry, and fluorine chemistry as well as might partially solve the problem of the disposition of ODS. Preliminary mechanistic studies revealed that (E)-N-phenylformimidoyl fluoride intermediate is involved in this process, which is a distinct intermediate from BrCF2COOEt case. Further studies toward the detailed mechanism and transformations and applications as well as exploration on more intriguing methodologies with this unusual C1 source are under way in our laboratory.
Limitation of the Study
Primary aliphatic amines showed poor or no reactivity toward this reaction system. In addition, reactive intermediate (e.g., 3c’) was not isolated owing to its high reactivity.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Financial support from the National Natural Science Foundation of China (21772046) and the Natural Science Foundation of Fujian Province (2016J01064) are gratefully acknowledged. We also thank Instrumental Analysis Center of Huaqiao University for analysis support. X.M. thanks the Subsidized Project for Cultivating Postgraduates' Innovative Ability in Scientific Research of Huaqiao University. Finally, we also thank Zhuoni Xie for reproducing the results of 3a, 3c, 4a, 4b, and 11.
Author Contributions
X.M. and J.S. performed the experiments and developed the reactions. X.Z. checked the manuscript and came up with suggestions for this transformation. Q.S. designed and directed the project and wrote the manuscript with the feedback of X.M.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.005.
Contributor Information
Xingang Zhang, Email: xgzhang@mail.sioc.ac.cn.
Qiuling Song, Email: qsong@hqu.edu.cn.
Data and Code Availability
The structures of 3t reported in this article have been deposited in the Cambridge Crystallographic Data Centre under accession numbers CCDC: 1874971.
Supplemental Information
Document S1. Transparent Methods, Figures S1–S155, and Schemes S1–S13
References
- Aresta M., Dibenedetto A., Angelini A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014;114:1709–1742. doi: 10.1021/cr4002758. [DOI] [PubMed] [Google Scholar]
- Bitterlich B., Anilkumar G., Gelalcha G.G., Spilker B., Grotevendt A., Jackstell A., Tse M.K., Beller M. Development of a general and efficient iron-catalyzed epoxidation with hydrogen peroxide as oxidant. Chem. Asian J. 2007;2:521–529. doi: 10.1002/asia.200600407. [DOI] [PubMed] [Google Scholar]
- Boogaerts I.I.F., Nolan S.P. Carboxylation of C-H bonds using N-heterocyclic carbene gold(I) complexes. J. Am. Chem. Soc. 2010;132:8858–8859. doi: 10.1021/ja103429q. [DOI] [PubMed] [Google Scholar]
- Brunet J.J., Neibecker D. Catalytic hydroamination of unsaturated carbon-carbon bonds. In: Togni A., Grützmacher H., editors. Catalytic Heterofunctionalization from Hydroamination to Hydrozirconation. Wiley-VCH Verlag GmbH; 2001. pp. 91–141. [Google Scholar]
- Cokoja M., Bruckmeier C., Rieger B., Herrmann W.A., Kîhn F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011;50:8510–8537. doi: 10.1002/anie.201102010. [DOI] [PubMed] [Google Scholar]
- Deng S., Chen H., Ma X., Zhou Y., Lan Y., Song Q. S8-Catalyzed triple cleavage of bromodifluoro compounds for the assembly of N-containing heterocycles. Chem. Sci. 2019 doi: 10.1039/c9sc01333d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enthaler S., von Langermann J., Schmidt T. Carbon dioxide and formic acid the couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010;3:1207–1217. [Google Scholar]
- Feng Z., Min Q.Q., Fu X.P., An L., Zhang X. Chlorodifluoromethane triggered formation of difluoromethylated arenes catalysed by palladium. Nat. Chem. 2017;9:918–923. doi: 10.1038/nchem.2746. [DOI] [PubMed] [Google Scholar]
- Fier P.S., Hartwig J.F. Copper-mediated difluoromethylation of aryl and vinyl iodides. J. Am. Chem. Soc. 2012;134:5524–5527. doi: 10.1021/ja301013h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- France S., Guerin D.J., Miller S.J., Lectka T. Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem. Rev. 2014;103:2985–3012. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]
- Gibson H.W. Chemistry of formic acid and its simple derivatives. Chem. Rev. 1969;69:673–692. [Google Scholar]
- Gouverneur V., Seppelt K. Introduction: fluorine chemistry. Chem. Rev. 2015;115:563–565. doi: 10.1021/cr500686k. [DOI] [PubMed] [Google Scholar]
- Gu Y., Leng X., Shen Q. Cooperative dual palladium/silver catalyst for direct difluoromethylation of aryl bromides and iodides. Nat. Commun. 2014;5:5405–5412. doi: 10.1038/ncomms6405. [DOI] [PubMed] [Google Scholar]
- Hine J., Porter J.J. Methylene derivatives as intermediates in polar reactions. VIII. Difluoromethylene in the reaction of chlorodifluoromethane with sodium methoxide. J. Am. Chem. Soc. 1957;79:5493–5496. [Google Scholar]
- Hopkinson M.N., Richter C., Schedler M., Glorius F. An overview of N-heterocyclic carbenes. Nature. 2014;510:485–496. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Horváth I.T. Innovations and green chemistry. Chem. Rev. 2007;107:2169–2173. doi: 10.1021/cr078380v. [DOI] [PubMed] [Google Scholar]
- Huang K., Sun C.-L., Shi Z.-J. Transition-metal-catalyzed C-C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011;40:2435–2452. doi: 10.1039/c0cs00129e. [DOI] [PubMed] [Google Scholar]
- Hudlicky M., Pavlath A.E., editors. Chemistry of Organic Fluorine Compounds II: A Critical Review. DC American Chemical Society; 1995. pp. 1–1200. [Google Scholar]
- Kubo K., Imamiya E., Sugiura Y., Inada Y., Furukawa Y., Nishikawa K., Naka T. Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids. J. Med. Chem. 1993;36:2182–2195. doi: 10.1021/jm00067a016. [DOI] [PubMed] [Google Scholar]
- Liu Q., Wu L., Jackstell R., Beller M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015;6:5933. doi: 10.1038/ncomms6933. [DOI] [PubMed] [Google Scholar]
- Ma X., Deng S., Song Q. Halodifluoroacetates as formylation reagents for various amines via unprecedented quadruple cleavage. Org. Chem. Front. 2018;5:3505–3509. [Google Scholar]
- Ma X., Xuan Q., Song Q. N-H and O-H difluoromethylation of N-Heterocycles. Acta Chim. Sinica. 2018;76:972–976. [Google Scholar]
- Ma X., Zhou Y., Song Q. Synthesis of β-Aminoenones via cross-coupling of in-situ generated isocyanides with 1,3-Dicarbonyl compounds. Org. Lett. 2018;20:4777–4781. doi: 10.1021/acs.orglett.8b01888. [DOI] [PubMed] [Google Scholar]
- Ma X., Mai S., Zhou Y., Cheng G.-J., Song Q. Dual role of ethyl bromodifluoroacetate in the formation of fluorine-containing heteroaromatic compounds. Chem. Commun. (Camb.) 2018;54:8960–8963. doi: 10.1039/c8cc04298e. [DOI] [PubMed] [Google Scholar]
- Martin A.D., Siamaki A.R., Belecki K., Gupton B.F. A convergent approach to the total synthesis of telmisartan via a suzuki cross-coupling reaction between two functionalized benzimidazoles. J. Org. Chem. 2015;80:1915–1919. doi: 10.1021/jo5025333. [DOI] [PubMed] [Google Scholar]
- Miao W., Zhao Y., Ni C., Gao B., Zhang W., Hu J. Iron-catalyzed difluoromethylation of arylzincs with difluoromethyl 2-pyridyl sulfone. J. Am. Chem. Soc. 2018;140:880–883. doi: 10.1021/jacs.7b11976. [DOI] [PubMed] [Google Scholar]
- Natte K., Neumann H., Beller M., Jagadeesh R.V. Transition-metal-catalyzed utilization of methanol as a C1 source in organic synthesis. Angew. Chem. Int. Ed. 2017;56:6384–6394. doi: 10.1002/anie.201612520. [DOI] [PubMed] [Google Scholar]
- Nawrot E., Jonczyk A. Difluoromethyltrialkylammonium salts-their expeditious synthesis from chlorodifluoromethane and tertiary amines in the presence of concentrated aqueous sodium hydroxide. The catalytic process. J. Org. Chem. 2007;72:10258–10260. doi: 10.1021/jo701735n. [DOI] [PubMed] [Google Scholar]
- Oh Y., Hu X. Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction. Chem. Soc. Rev. 2013;42:2253–2261. doi: 10.1039/c2cs35276a. [DOI] [PubMed] [Google Scholar]
- O'Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- Ohishi T., Nishiura M., Hou Z. Carboxylation of organoboronic esters catalyzed by N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Ed. 2008;47:5792–5795. doi: 10.1002/anie.200801857. [DOI] [PubMed] [Google Scholar]
- Sakakura T., Choi J.-C., Yasuda H. Transformation of carbon dioxide. Chem. Rev. 2007;107:2365–2387. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- Sasaki H., Haraguchi Y., Itotani M., Kuroda H., Hashizume H., Tomishige T., Kawasaki M., Matsumoto M., Komatsu M., Tsubouchi H. Synthesis and antituberculosis activity of a novel series of optically active 6-Nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J. Med. Chem. 2006;49:7854–7860. doi: 10.1021/jm060957y. [DOI] [PubMed] [Google Scholar]
- Schröder K., Enthaler S., Join B., Junge K., Bellera M. Iron-catalyzed epoxidation of aromatic olefins and 1,3-dienes. Adv. Synth. Catal. 2010;352:1771–1778. [Google Scholar]
- Schröder K., Enthaler K., Bitterlich B., Schulz T., Spannenberg A., Tse M.K., Junge K., Beller M. Design of and mechanistic studies on a biomimetic iron–imidazole catalyst system for epoxidation of olefins with hydrogen peroxide. Chem. Eur. J. 2009;15:5471–5481. doi: 10.1002/chem.200802731. [DOI] [PubMed] [Google Scholar]
- Senadi G.C., Kudale V.S., Wang J.-J. Sustainable methine sources for the synthesis of heterocycles under metal- and peroxide-free conditions. Green Chem. 2019;21:979–985. [Google Scholar]
- Sordakis K., Tang C., Vogt L.K., Junge H., Dyson P.J., Beller M., Laurenczy G. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 2018;118:372–433. doi: 10.1021/acs.chemrev.7b00182. [DOI] [PubMed] [Google Scholar]
- Sung D., Moon D., Lee Y., Hong S.-I. Catalytic pyrolysis of difluorochloromethane to produce tetrafluoroethylene. Int. J. Chem. React. Eng. 2004;2:1542–6580. [Google Scholar]
- Wakade S.B., Tiwari D.K., Prabhakar Ganesh P.S.K., Phanindrudu M., Likhar P.R., Tiwari D.K. Transition-metal-free quinoline synthesis from acetophenones and anthranils via sequential one-carbon homologation/conjugate addition/annulation cascade. Org. Lett. 2017;19:4948–4951. doi: 10.1021/acs.orglett.7b02429. [DOI] [PubMed] [Google Scholar]
- Wang J., Sánchez-Roselló K., Aceña J.L., del Pozo J., Sorochinsky A.E., Fustero S., Soloshonok V.A., Liu H. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001-2011) Chem. Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Wu L., Wang F., Chen P., Liu G. Enantioselective construction of quaternary all-carbon centers via copper-catalyzed arylation of tertiary carbon-centered radicals. J. Am. Chem. Soc. 2019;141:1887–1892. doi: 10.1021/jacs.8b13052. [DOI] [PubMed] [Google Scholar]
- Xu C., Guo W.-H., He X., Gao Y.-L., Zhang X.-Y., Zhang X. Difluoromethylation of (hetero)aryl chlorides with chlorodifluoromethane catalyzed by nickel. Nat. Commun. 2018;9:1170–1179. doi: 10.1038/s41467-018-03532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y., Zhang Y., Feng C., Zha Z., Wang Z. Selective iodine-catalyzed intermolecular oxidative amination of C(sp3)-H bonds with ortho-carbonyl-substituted anilines to give quinazolines. Angew. Chem. Int. Ed. 2012;51:8077–8081. doi: 10.1002/anie.201203880. [DOI] [PubMed] [Google Scholar]
- Yu J., Lin J.-H., Xiao J.-C. Reaction of thiocarbonyl fluoride generated from difluorocarbene with amines. Angew. Chem. Int. Ed. 2017;129:16896–16900. doi: 10.1002/anie.201710186. [DOI] [PubMed] [Google Scholar]
- Yu C., Su J., Ma X., Zhou Y., Song Q. Difluoromethylation of tosylhydrazone compounds with chlorodifluoromethane under mild conditions. Asian J. Org. Chem. 2019;8:694–697. [Google Scholar]
- Zhang K.-F., Bian K.-J., Li C., Sheng J., Li Y., Wang X.-S. Nickel-catalyzed carbofluoroalkylation of 1,3-enynes to access structurally diverse fluoroalkylated allenes. Angew. Chem. Int. Ed. 2019;58:5069–5074. doi: 10.1002/anie.201813184. [DOI] [PubMed] [Google Scholar]
- Zhao P., Krug C., Hartwig J.F. Transfer of amido groups from isolated rhodium(i) amides to alkenes and vinylarenes. J. Am. Chem. Soc. 2005;127:12066–12073. doi: 10.1021/ja052473h. [DOI] [PubMed] [Google Scholar]
- Zheng J., Lin J.-H., Deng X.-Y., Xiao J.-C. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU)-Promoted decomposition of difluorocarbene and the subsequent trifluoromethylation. Org. Lett. 2015;17:532–535. doi: 10.1021/ol503548s. [DOI] [PubMed] [Google Scholar]
- Zheng J., Cheng R., Lin J.-H., Yu D.-H., Ma L., Jia L., Zhang L., Wang L., Xiao J.-C., Liang S.H. An unconventional mechanistic insight into SCF3 formation from difluorocarbene: preparation of 18F-Labeled α-SCF3 carbonyl compounds. Angew. Chem. Int. Ed. 2017;56:3196–3200. doi: 10.1002/anie.201611761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Transparent Methods, Figures S1–S155, and Schemes S1–S13
Data Availability Statement
The structures of 3t reported in this article have been deposited in the Cambridge Crystallographic Data Centre under accession numbers CCDC: 1874971.